

Abstract
Both historical clinical and recent preclinical data suggest that the M1 muscarinic acetylcholine receptor is an exciting target for the treatment of Alzheimer’s disease and the cognitive and negative symptom clusters in schizophrenia; however, early drug discovery efforts targeting the orthosteric binding site have failed to afford selective M1 activation. Efforts then shifted to focus on selective activation of M1 via either allosteric agonists or positive allosteric modulators (PAMs). While M1 PAMs have robust efficacy in rodent models, some chemotypes can induce cholinergic adverse effects (AEs) that could limit their clinical utility. Here, we report studies aimed at understanding the subtle structural and pharmacological nuances that differentiate efficacy from adverse effect liability within an indole-based series of M1 ago-PAMs. Our data demonstrate that closely related M1 PAMs can display striking differences in their in vivo activities, especially their propensities to induce adverse effects. We report the discovery of a novel PAM in this series that is devoid of observable adverse effect liability. Interestingly, the molecular pharmacology profile of this novel PAM is similar to that of a representative M1 PAM that induces severe AEs. For instance, both compounds are potent ago-PAMs that demonstrate significant interaction with the orthosteric site (either bitopic or negative cooperativity). However, there are subtle differences in efficacies of the compounds at potentiating M1 responses, agonist potencies, and abilities to induce receptor internalization. While these differences may contribute to the differential in vivo profiles of these compounds, the in vitro differences are relatively subtle and highlight the complexities of allosteric modulators and the need to focus on in vivo phenotypic screening to identify safe and effective M1 PAMs.
Keywords: M1, muscarinic acetylcholine receptor, agonist, positive allosteric modulator (PAM), ago-PAM, seizure
Graphical abstract
INTRODUCTION
Both Alzheimer’s disease (AD) and schizophrenia represent significant unmet medical needs for which treatment options are limited and offer, at best, only palliative treatment of symptoms and often with intolerable side-effects.1,2 Dysregulation and reduction in cholinergic projections to cortical and forebrain regions is a hallmark of AD,3–9 and specific reductions in densities of M1 subtype of muscarinic acetylcholine receptor (mAChR) are present in postmortem schizophrenic brain.10,11 In addition, M1 functionally interacts with the N-methyl-D-aspartate subtype of the glutamate receptor (NMDAR)12 and has been shown to reverse behavioral pharmacological (phencyclidine and MK-801) and genetic (NR1 KD mice)NMDAreceptor deficits (i.e., the NMDA receptor hypofunction hypothesis of schizophrenia).13–23 Over the last 30 years, the M1 muscarinic acetylcholine receptor has emerged as an attractive molecular target for both of these devastating CNS disorders; however, while early efforts with orthosteric muscarinic agonists (activating M1–M5 at varying degrees) showed a trend toward cognitive enhancing efficacy in patients, they failed to meet significant end points for cognition enhancement, a result attributed to dose-limiting nonselective cholinergic agonist adverse effects mediated by the concomitant activation of peripheral M2 and M3 receptors.13–25 Similarly, functionally M1 selective allosteric agonists garnered interest early on. However, many of these compounds proved to interact with both the orthosteric and allosteric binding sites of the receptor and bound bitopically to the receptor.26–35 This resulted in a lack of trueM1 selectivity as potency was increased,33–35 as well as receptor reserve-dependent efficacy (i.e., brain region dependent pharmacology).36 Therefore, with the exception of a recent advancement of an M1 partial agonist, HTL9936, into clinical trials by Heptares Therapeutics, strategies of targeting the orthosteric binding site were largely abandoned.37
Based on these previous efforts, it has become increasingly clear that targeting a less-conserved, allosteric site on the M1 receptor, may represent the best path forward for this promising target.38–40 Fortunately, this approach proved fruitful with the discovery of BQCA (1), the first and prototypical M1 positive allosteric modulator (PAM).41,42 Within the BQCA series, structure–activity relationships (SARs) were steep, with little modifications tolerated in the context of the β-keto acid moiety, as in 1 and 2 (Figure 1).43 Subsequent optimization identified the (S,S)-hydroxycyclohexyl amide moiety in 3 as a replacement for the β-ketoacid,44 which was then parlayed into the key nonhuman primate tool compound PQCA (4)45,46 and the advanced M1 PAM (5).47 Recently, Scammels and co-workers48 further optimized 5 into a novel fused arylpyrimidone core with improved physiochemical properties.
Figure 1.

Conserved structural motifs present inM1 PAMs from Merck (1–5) and the Vanderbilt Center for Neuroscience Drug Discovery (VCNDD) (6–14).
In parallel, we conducted a high-throughput screening (HTS) campaign and identified a novel isatin hit 6 (VU0119498) as an M1,3,5 PAM (Figure 1)49 from which we were able to develop highly selective M1 PAMs 7 and 8,50,51 as well as the first selective M5 PAMs.52 Further optimization afforded 9 and, finally, 10 (VU0453595),22,53 an important in vivo proof-of-concept tool within this series that is structurally divergent from 5. A second HTS hit, VU0108370 (11), represented a third unique chemotype based on an indole core. Chemical optimization around 11 afforded ML169 (12) and by surveying azaindoles, diverse amides and sulfones, as well as significant heterobiaryl development, VU0456940 (14) was discovered.53–56 Overall, this series had significant liabilities that precluded it from further development as a clinical candidate, including modest to low brain penetration (Kp/Kp,uu), interaction with the orthosteric site, and varying degrees of allosteric agonist activity. Allosteric agonists are referred to as ago-PAMs or PAM-agonists and defined as an allosteric modulator with PAM pharmacology, but that also activates the receptor in the absence of the orthosteric endogenous ligand similar to an agonist. Typically, the allosteric agonist activity demonstrates weaker potency than the PAM activity and is a feature to be avoided with pro-convulsant CNS targets.57,58
Shortly thereafter, multiple M1 PAM programs were launched by generating chimeras 15 between the (S,S)-hydroxycylcohexyl amide and the indole/azaindole cores (Figure 2).59 Davoren et al. then published their series, highlighted by PF-06764427 (16), a potent M1 ago-PAM with reported efficacy in reversing amphetamine-induced hyperlocomotion (AHL) in mice.60 This report in particular caught our attention, as we, and others, have not reported efficacy in reversing hyperlocomotion induced by psychostimulants with M1 PAMs, and instead use cognition models to drive the optimization effort.33–45 Moreover, a 2009 study found that the attenuation of amphetamine-induced activity by xanomeline, an M1/M4 preferring agonist, was absent in M4 knockout mice, indicating that the efficacy of xanomeline in amphetamine-induced hyperlocomotion is driven by M4 activation.24,61 These results were further validated by reports of in vivo efficacy in reversing AHL withM4-selective PAMs.62,63
Figure 2.

Recent scaffold hopping exercises, employing 3 and 13, resulted in the patent applications listed above surrounding 15, and an intriguing publication focused on 16. Here, we disclose analogues 17, derived from 13, and insight into how to differentiate efficacy from adverse effect liability.
Previous studies provide strong evidence that overactivation of M1 can induce seizures and fully generalized convulsions in rodent models.58,64,65 These findings, coupled with the robust ago-PAM activity of some M1-selective PAMs, raises the possibility that direct agonist activity of some M1 PAMs could contribute to the ability of these compounds to induce seizure activity and other adverse effects.57 Furthermore, a propensity of some M1 PAMs to induce seizure activity, particularly at doses lower than those required to produce generalized behavioral convulsions, could potentially contribute to the reduction in AHL rather than the reflection of potential antipsychotic-like effects. Since the initial report of BQCA, all subsequent reports of M1 PAMs have been generated in cognition assays (e.g., reversing scopolamine-induced deficits in contextual fear conditioning). Moreover, there is building literature with structurally conserved M1 ago-PAMs, possessing M1 agonism-imparting (S,S)-hydroxycylcohexyl amide, regarding cholinergic toxicity and other adverse events.44,65 It is important to recognize that this class of M1 ago-PAMs, as well as the BQCA-derived ago-PAMs, possesses low Kp’s (<0.3) and Kp,uu’s (<0.1), requiring high peripheral exposure to achieve meaningful CNS levels.40–46,59 Thus, we performed a series of studies evaluating effects of these previously reported compounds, along with novel difluoro indole congeners 17, to gain a further understanding of the subtle structural and pharmacological nuances that differentiate efficacy from adverse effect liability within these indole-based M1 ago-PAMs.
RESULTS AND DISCUSSION
Chemistry and Structure–Activity Relationships (SARs)
The chemistry to access difluoro indole analogues 17 has deviated from that previously published for the sulfone congeners (Scheme 1).54,55 Here, commercial 4,6-difluoroindole 18 is a linchpin to access diversely functionalized derivatives 21, 24, and 27. Analogues 21 are readily prepared in four steps by first deprotonation of 18 and alkylation with the desired benzyl bromide to provide 19. Installation of the trifluoromethyl ketone was achieved in moderate to excellent yields by treatment with TFAA to deliver 20. Hydrolysis of the trifluoromethyl ketone to the corresponding carboxylic acid and BOP-mediated coupling affords analogues 21 in variable yields. Heterocyclic congeners 24 and 27 required four or five steps. In this case, indole 18 is alkylated with 2-bromo-5-(bromomethyl)pyridine to deliver 22 in 84% yield. A Suzuki coupling (exact conditions depend on the nature of the boronic acid or ester) then gives 23. Installation of the trifluoromethyl ketone, hydrolysis to the acid and BOP-mediated coupling generates analogues 24. Alternatively, the trifluoromethyl ketone can first be installed onto indole 18 to yield 25, followed by repetition of the aforementioned sequences delivering analogues 27.
Scheme 1. Synthesis of VU0456940 Hydrid Amide M1 PAMsa.

aReagents and conditions: (a) NaH, ArCH2Br, DMF, rt, 96–99%; (b) TFAA, DMF, rt, 38–82%; (c) NaOH, H2O/MeOH, 90–150 °C, then HCl, 14–83%; (d) NEt3, RNH2, BOP, DMF, rt, 7–77%; (e) Cs2CO3, 2-bromo-5-(bromomethyl)pyridine, DMF, rt, 84%; (f) Pd2(dba)3, Cy3P, Het-B(OH)2 or Het-B(OR)2, K3PO4, dioxane/H2O, 120 °C, 61–99%.
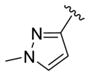
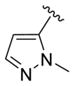
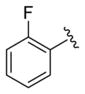
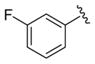
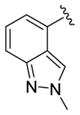
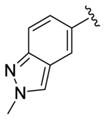
We elected to drive the SAR utilizing human M1 coupled with CNS penetration in a plasma/brain level (PBL) cassette approach.55,62 Table 1 highlights select SARs from analogues of 24 and 27, defined here as 28 in which the (S,S)-hydroxycylcohexyl amide is held constant. A diverse array of heterobiaryl motifs were surveyed and found to be well tolerated in terms of human M1 PAM potency (hM1 EC50s 210 nM to 3 μM), but Kp (0.11 to 0.18) and Kp,uu (0.01 to 0.027) were uniformly low (as expected). Deletion of the 2-hydroxy moiety in 28a led to a 50-fold loss in potency, but the Kp increased to 0.75, strongly suggesting that the critical (S,S)-hydroxycyclohexyl amide is responsible for the low CNS penetration of the class. In terms of SAR, both regioisomeric N-methyl pyrazoles, 28a and 28c, were equipotent and equi-CNS penetrant. All other fluorinated aryl congeners, 28e–g, were uniformly 4- to 8-fold less potent, and 6-membered heterocycles such as pyridine (28h), pyrimidine (28i), and pyridazine (28k) either lost M1 PAM potency or offered no advantage in terms of CNS penetration. From this first generation of analogs, 28a emerged as the most attractive compound.
Table 1.
Structures and Activities of Representative VU0456940 Hybrid Amide Analogues 28 Surveying Diverse Heterobiaryl Motifsa
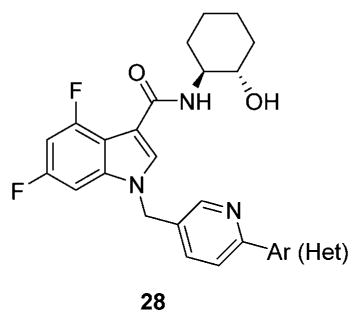
| |||||
|---|---|---|---|---|---|
| Compound | Ar (Het) | hM1 EC50 (μM) | hM1 pEC50 | hACh Max (%) | Rat Kp (Kp,uu)b |
| 28a |
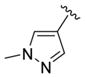
|
0.21 | 6.67±0.05 | 86±1 | 0.113 (0.025) |
| 28b |
|
3.0 | 5.53±0.07 | 77±8 | ND |
| 28c |
|
0.22 | 6.65±0.05 | 86±1 | 0.115 (0.027) |
| 28d |
|
0.85 | 6.07±0.05 | 78±5 | ND |
| 28e |
|
2.2 | 5.65±0.01 | 55±5 | ND |
| 28f |
|
1.3 | 5.90±0.02 | 36±4 | ND |
| 28g |
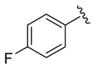
|
0.77 | 6.11±0.15 | 44±3 | ND |
| 28h |
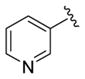
|
0.52 | 6.28±0.13 | 83±6 | 0.18 (0.04) |
| 28i |
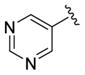
|
1.5 | 5.82±0.10 | 77±6 | ND |
| 28k |
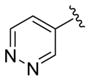
|
0.87 | 6.06±0.18 | 80±9 | ND |
| 28l |
|
0.48 | 6.32±0.07 | 78±7 | 0.05 (0.01) |
| 28m |
|
0.65 | 6.19±0.12 | 76±8 | ND |
Calcium mobilization assays with hM1-CHO cells performed in the presence of an EC20 fixed concentration of acetylcholine. Values are the average of at least three independent determinations performed in triplicate ± SEM.
Total and calculated unbound brain/plasma partition coefficients determined at 0.25 h post-administration of an IV cassette dose (0.20–0.25 mg/kg) to male, SD rat (n = 1), in conjunction with in vitro rat plasma protein and brain homogenate binding assay data.
ND = not determined.
Next, we held the southern heterobiaryl tail of 28a constant and surveyed other amide moieties (Table 2) evaluating both hM1 PAM potency and CNS penetration for analogues 29. Replacing the cyclohexyl moiety with regioisomeric pyrans, such as 29a and 29b, retained or improved hM1 PAM potency, but both Kp (0.04) and Kp,uu (0.01 to 0.02) diminished. Constriction to the cyclobutyl congener 29c resulted in the loss of ~10-fold PAM activity, and the cyclopentyl derivative 29d lost ~4-fold; moreover, both displayed reduced CNS penetration relative to 28a. Interestingly, expansion to the cycloheptyl ring (29e) retained potency and doubled the Kp (0.24) and Kp,uu (0.08); however, the predicted in vitro hepatic clearance (CLhep) was at rat hepatic blood flow. All other amides surveyed, such as 29f-h, were inactive.
Table 2.
Structures and Activities of Representative VU0456940 Hybrid Amide Analogues 29 Surveying Diverse Amide Moieties
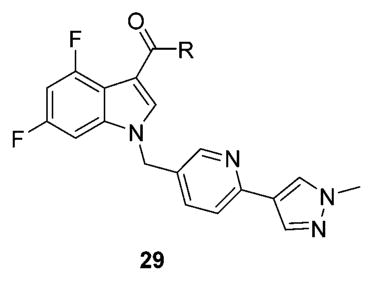
| |||||
|---|---|---|---|---|---|
| Compound | R | hM1 EC50 (μM)a | hM1 pEC50 | hACh Max (%)a | RatKp (Kp,uu)b |
| 28a |
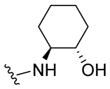
|
0.21 | 6.67±0.05 | 86±1 | 0.113 (0.025) |
| 29a |
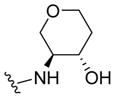
|
0.30 | 6.53±0.12 | 81±10 | 0.04 (0.02) |
| 29b |
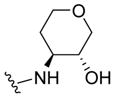
|
0.13 | 6.88±0.08 | 87±2 | 0.04 (0.01) |
| 29c |
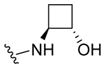
|
2.8 | 5.56±0.12 | 84±4 | 0.051 (0.051) |
| 29d |
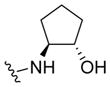
|
0.98 | 6.01±0.04 | 87±2 | 0.08 (0.05) |
| 29e |
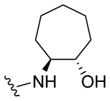
|
0.34 | 6.47±0.16 | 81±7 | 0.24 (0.08) |
| 29f |
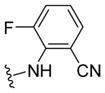
|
>10 | >5 | 28 | ND |
| 29g |
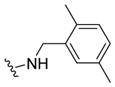
|
>10 | >5 | 34 | ND |
| 29h |
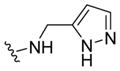
|
>10 | >5 | 29 | ND |
Calcium mobilization assays with hM1-CHO cells performed in the presence of a fixed EC20 concentration of acetylcholine. Values are the average of at least three independent determinations performed in triplicate ± SEM.
Total and calculated unbound brain/plasma partition coefficients determined at 0.25 h post-administration of an IV cassette dose (0.20–0.25 mg/kg) to male, SD rat (n = 1), in conjunction with in vitro rat plasma protein and brain homogenate binding assay data.
ND = not determined.
These data led us to one final library of analogues based on 28a, where we surveyed additional diversity in the southern tail with analogues 30 (Table 3; Figure 3). Replacing the central pyridine core of the heterobiaryl with a 2-fluorophenyl moiety (30a) resulted in a 4-fold loss in PAM potency, and replacement with a 2,6-difluorophenyl ring (30b) reduced potency ~10-fold. Moving the pyridine nitrogen, as in 30c, resulted in a ~ 5-fold reduction in potency and replacement of the N-methyl pyrazole with a methyl group (30d) reduced potency by ~8-fold. Interestingly, replacement of the heterobiaryl moiety with a simple 4-N-methylacetamide phenyl group (30e) retained, and slightly improved, hM1 PAM potency, but at the expense of CNS penetration. Replacement of the acetamide with a methyl sulfone (30f) resulted in a considerable loss of PAM activity. Therefore, 28a emerged as the key 2,6-difluoroindole analogue for further profiling and to delve into the pharmacodynamic (PD) data reported by Davoren et al.59 with 16. However, to ensure against structural subtleties underlying the PD phenomenon, we also prepared the regioisomeric N-methyl pyrazole congener of 28a (VU6004256) that matches 16 (PF-06764427), 28c (VU6006251). In addition, we also prepared 31, the trans-racemic congener of 16, and the regioisomeric N-methyl pyrazole congener of 16 that matches 28a, as both a trans-racemate (32) as well as the single trans-(1S,2S) analogue 33. Could a 3-versus 4-N-methyl pyrazole result in divergent pharmacology?
Table 3.
Structures and Activities of Representative VU0456940 Hybrid Amide Analogues 30 Surveying Alternate Southern Tailsa
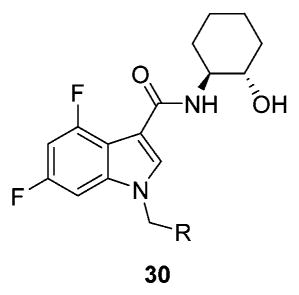
| ||||
|---|---|---|---|---|
| Compound | R | hM1 EC50 (μM) | hM1 pEC50 | hACh Max (%) |
| 30a |
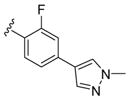
|
0.95 | 6.02±0.03 | 82±6 |
| 30b |
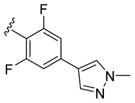
|
3.0 | 5.51±0.04 | 83±4 |
| 30c |
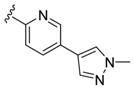
|
1.0 | 5.98±0.06 | 82±7 |
| 30d |
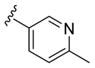
|
1.66 | 5.78±0.06 | 83±6 |
| 30e |
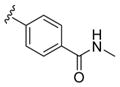
|
0.13 | 6.90±0.13 | 73±8 |
| 30f |
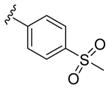
|
3.47 | 5.46±0.06 | 77±5 |
Calcium mobilization assays with hM1-CHO cells performed in the presence of a fixed EC20 concentration of acetylcholine. Values are the average of at least three independent determinations performed in triplicate ± SEM.
Figure 3.

Structures of the six M1 PAMs, both aza-indoles (16, 31–33) and 4,6-difluoro indoles (28a and 28c) that will be the subject of in-depth in vitro and in vivo characterization to understand subtle nuances that differentiate efficacy from adverse effects.
Assessment of Adverse Effects
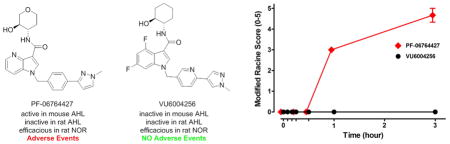
From pioneering pharmacology in the 1990s, studies with the muscarinic agonist pilocarpine and M1–M5 knockout (KO) mice have clearly demonstrated the pro-convulsive/seizure liabilities of excessive M1 activation.58,64,65 It has also been shown that potent allosteric agonist activity must be avoided to minimize adverse effect liability with another pro-convulsant GPRC, mGlu5, suggesting that a similar strategy might also provide a higher safety window for ligands targeting M1.57 One strategy to allow for early detection of adverse events (AEs) is to administer M1 PAMs/ago-PAMs at 100 mg/kg intraperitoneally (i.p.) in mice (a species with high susceptibility to cholinergic seizure activity)58 within compound workflow to eliminate compounds or chemotypes that may engender AEs in development. Previous unpublished work identified key AEs (pro-convulsive activity) with BQCA and related M1 ago-PAM congeners in mice. Therefore, we administered 100 mg/kg i.p. of 1, 16, 28a, 28c, 31, 32, and 33 and behavioral convulsions were assessed for 3 h (Figure 4) using the modified Racine scale (0–5).57 As anticipated, BQCA (1) induced robust seizure activity, reaching stage 4–5 behavioral convulsions on the Racine scale. Likewise, multiple other M1 PAMs in this class induced fully generalized behavioral convulsions (Figure 4).
Figure 4.

Induction of behavioral convulsions by structurally distinct M1 ago-PAMs. (A) Mice were administered 100 mg/kg compound, and seizure activity was measured for 3 h using the modified Racine scale (0–5). Compounds were formulated in 10% Tween 80 (10 mg/mL, pH 7.0) and injected intraperitoneally. BQCA (1), PF-06764427 (16), VU6004877 (32), and VU6006270 (33) induced robust behavioral convulsions, while VU6004256 (28a), VU6006251 (28c), and VU6005263 (31) did not display observable behavioral seizure activity. Data represent mean ± SEM (n = 3). B) Dose-dependent induction of behavioral convulsions by M1 PAM PF-06764427 (16). Mice were administered 10, 30, or 60 mg/kg compound, and seizure activity was measured for 2 h using the modified Racine scale (0–5). PF-06764427 (16) induced dose-dependent behavioral convulsions. Data represent mean ± SEM (n = 4).
Interestingly, neither difluoro indole 28a nor 28c (both regioisomeric N-Me pyrazoles) induced behavioral convulsions during the 3 h assessment period, nor did the trans-racemic congener 31 (VU6005263) of 16 (PF-06764427) (Figure 4). The trans-racemic analogue of 31 with the 4-versus 3-N-Me pyrazole, 32, induced robust seizure activity in contrast to both 31 and 28a. The single trans-(1S,2S)-isomer 33 of racemic 32 also induced seizure activity in mice. Importantly, 16 (PF-06764427) also induced pronounced behavioral convulsions between 30 min and 1 h post-administration, and resulted in Racine scale 5 seizures by 3 h. As shown in Figure 4B, the seizure activity was dose-dependent, with no measurable behavioral convulsions at 10 mg/kg, but becoming significant at 30 mg/kg and 60 mg/kg. However, mice given the 10 mg/kg dose were already displaying significant decreases in spontaneous locomotor activity (Figure 7), raising the possibility that this dose may induce behavioral alterations impacting the interpretation of the AHL data previously published.59,65 Additionally, we observed seizure activity with PF-06764427 at a dose as low as 30 mg/kg, i.p., which correlates to total and unbound brain concentrations of 1.95 μM and 195 nM, respectively, 2 h following administration. This correlates to unbound brain levels of 4-fold and 1.9-fold higher than the M1 PAM and M1 allosteric agonist potency (mEC50 = 100 nM, 91% ACh max), respectively. However, a 100 mg/kg, i.p. dose of VU6004256 results in total and unbound maximum brain concentrations of 49.9 μM and 649 nM, respectively. The free brain levels also correlate to a 4-fold and 1.4-fold increase in PAM and allosteric agonist potency (mEC50 = 450 nM, 86% ACh max), respectively. These data suggest that the brain levels achieved relative to the potency at M1 does not appear to contribute to the adverse effect liability observed with some M1 PAMs.
Figure 7.

Effects of M1 PAMs on spontaneous locomotor activity and amphetamine-induced hyperlocomotion. (A) PF-06764427 caused a dose-dependent reduction in basal locomotor activity with significant decreases observed beginning at the lowest dose of 1 mg/kg. (B) VU6004256 decreased locomotor activity at the highest dose of 10 mg/kg. Data represent mean distance traveled (cm) per 5 min intervals ± SEM (n = 12). *p < 0.05, **p < 0.01, ***p < 0.001 (C, D) PF-06764427 dose-dependently reversed amphetamine-induced hyperlocomotion (AHL) in mice. Data are expressed as the mean total distance traveled (cm) per 5 min intervals ± SEM (n = 12). (E) Reversal of AHL by PF-06764427 is absent in M1 KO mice.
Overall, these data highlight how subtle structural variations in this series of M1 PAMs can lead to dramatic differences in adverse effect liability.66 When comparing 31 to 32, the regiosiomeric N-Me pyrazole is critical, as is the aza-versus the difluoro indole core. However, these structural elements alone do not complete the picture, and detailed molecular and behavioral pharmacology studies, as well as assessments in exposure, are required to further elucidate the origins of the AE versus safe profiles.
Molecular and Drug Metabolism Studies
Based on the SARs for the proconvulsant activity, we elected to evaluate the molecular pharmacological profiles of 16, 28a, 28c, 31, 32, and 33 across human, rat, and mouse M1 in both PAM and agonist modes (Tables 4–6) to determine if these M1 PAMs can be differentiated based on their effects on M1 signaling.60 As shown in Table 4, the difluoro indoles 28a and 28c were potent M1 PAMs on both human (EC50 = 210 nM and EC50 = 224 nM, respectively) and rat (EC50 = 141 nM and EC50 = 191 nM, respectively) receptors with comparable efficacies (86–92% ACh Max) and rat CNS penetration (Kp’s = 0.11, Kp,uu’s = 0.02); however, they were both 5- to 14-fold less potent than 16 (hEC50 = 31 nM, rEC50 = 30 nM), 31 (hEC50 = 59 nM, rEC50 = 39 nM), 32 (hEC50 = 48 nM, rEC50 = 24 nM), and 33 (hEC50 = 17 nM, rEC50 = 18 nM) but with similar efficacies and Kp’s/Kp,uu’s. Thus, absolute M1 PAM potency was a differentiating point; however, 31 was similarly potent to the M1 ago-PAMs (16, 32, and 33) that induced seizures. Therefore, M1 PAM potency alone or in combination with CNS exposure could not account for the AE liability. In addition, all six ago-PAMs were completely selective for rat and human M1 over rat and human M2–M5 (see Supporting Information Figures 1 and 2); thus, it appears unlikely that AEs are derived from off-target mAChR activity.
Table 4.
Human and Rat M1 PAM Potency Coupled with Rat CNS Penetration
| compd | hM1 EC50 (nM)a | hM1 pEC50 | hACh Max (%)a | rM1 EC50 (nM)b | rM1 pEC50 | rACh Max (%)b | rat Kp (Kp,uu)c |
|---|---|---|---|---|---|---|---|
| 28a | 210 | 6.67 ± 0.05 | 86 ± 1 | 141 | 6.86 ± 0.05 | 92 ± 4 | 0.113 (0.025) |
| 28c | 224 | 6.65 ± 0.05 | 86 ± 1 | 191 | 6.72 ± 0.01 | 92 ± 4 | 0.115 (0.027) |
| 16 | 30 | 7.52 ± 0.05 | 82 ± 3 | 30 | 7.53 ± 0.01 | 90 ± 5 | 0.21 (0.05) |
| 31 | 59 | 7.23 ± 0.03 | 90 ± 2 | 39 | 7.41 ± 0.03 | 91 ± 4 | 0.26 (0.21) |
| 32 | 48 | 7.33 ± 0.05 | 88 ± 3 | 24 | 7.61 ± 0.02 | 94 ± 4 | 0.20 (0.06) |
| 33 | 16 | 7.79 ± 0.07 | 83 ± 1 | 18 | 7.76 ± 0.07 | 89 ± 4 | 0.13 (0.05) |
Calcium mobilization assays with hM1-CHO cells performed in the presence of a fixed EC20 concentration of acetylcholine. Values are the average of at least three independent determinations performed in triplicate ± SEM.
Calcium mobilization assays with rM1-CHO cells performed in the presence of a fixed EC20 concentration of acetylcholine. Values are the average of at least three independent determinations performed in triplicate ± SEM.
Total and calculated unbound brain/plasma partition coefficients determined at 0.25 h post-administration of an IV cassette dose (0.20–0.25 mg/kg) to male, SD rat (n = 1), in conjunction with in vitro rat plasma protein and brain homogenate binding assay data.
Table 6.
Mouse M1 PAM Potency Coupled with Mouse CNS Penetration
| compd | mM1 EC50 (nM)a | mM1 pEC50 | mACh Max (%)a | Mouse Kp (Kp,uu)b |
|---|---|---|---|---|
| 28a | 155 | 6.81 ± 0.03 | 79 ± 1 | 4.84 (2.61) |
| 28c | 263 | 6.58 ± 0.02 | 77 ± 1 | ND |
| 16 | 46 | 7.34 ± 0.02 | 79 ± 1 | 0.21 (0.16) |
| 31 | 83 | 7.08 ± 0.02 | 73 ± 1 | ND |
| 32 | 46 | 7.34 ± 0.02 | 75 ± 2 | 0.21 (0.16) |
| 33 | 23 | 7.63 ± 0.02 | 78 ± 1 | ND |
Calcium mobilization assays with mM1-CHO cells performed in the presence of a fixed EC20 fixed concentration of acetylcholine. Values are the average of at least three independent determinations performed in triplicate ± SEM.
Total and calculated unbound brain:plasma partition coefficients determined at 0.25 h post-administration of a discrete dose of compound (10 mg/kg, i.p.) to male mice (n = 3 or 4), in conjunction with in vitro mouse plasma protein and brain homogenate binding assay data.
Interestingly, differences between the allosteric agonist activity of these compounds at M1 may provide insight regarding adverse effect liability as opposed to differences in PAM potencies (Table 5), and the raw calcium traces highlight the robust agonist activity of these ago-PAMs (see Supporting Information Figures 3 and 4). Both 28a and 28c display modest M1 agonist activity at both human (EC50 = 4.8 μM and EC50 = 5.9 μM, respectively) and rat (EC50 = 4.0 μM and EC50 = 4.6 μM, respectively) with comparable efficacies (48–68% ACh Max, depending on the species). The other four ago-PAMs all displayed sub-micromolar agonist activity in our human and rat M1 cell lines with high agonist efficacy (47–81% ACh Max, depending on the species). Importantly, 16 was an ~600 nM agonist (65% and 77% ACh Max, respectively) at human and rat M1; in contrast, 28a was 7-fold weaker M1 agonist.
Table 5.
Human and Rat M1 Agonist Potency
| compd | hM1 EC50 (μM)a | hM1 pEC50 | hACh Max (%)a | rM1 EC50 (μM)b | rM1 pEC50 | rACh Max (%)b |
|---|---|---|---|---|---|---|
| 28a | 4.8 | 5.33 ± 0.15 | 56 ± 14 | 4.0 | 5.40 ± 0.05 | 68 ± 9 |
| 28c | 5.9 | 5.23 ± 0.09 | 48 ± 12 | 4.6 | 5.34 ± 0.08 | 65 ± 9 |
| 16 | 0.59 | 6.23 ± 0.19 | 65 ± 10 | 0.58 | 6.24 ± 0.12 | 77 ± 2 |
| 31 | 0.65 | 6.19 ± 0.10 | 50 ± 13 | 0.40 | 6.40 ± 0.14 | 71 ± 8 |
| 32 | 0.44 | 6.36 ± 0.25 | 47 ± 11 | 0.44 | 6.36 ± 0.14 | 71 ± 11 |
| 33 | 0.63 | 6.20 ± 0.10 | 66 ± 7 | 0.33 | 6.48 ± 0.24 | 81 ± 4 |
Calcium mobilization assays with hM1-CHO cells performed in agonist mode. Values are the average of at least three independent determinations performed in triplicate ± SEM.
Calcium mobilization assays with rM1-CHO cells performed in agonist mode. Values are the average of at least three independent determinations performed in triplicate ± SEM.
Since the AEs were noted in mouse, we also profiled the six ago-PAMs on a murine M1 cell line (Table 6). Once again, both 28a and 28c were potent mouse M1 PAMs (EC50s of 155 nM and 263 nM, respectively) with robust PAM efficacy (77–79% ACh Max), but 3- to 5-fold less potent than 16 (EC50 = 46 nM, 79% ACh Max) and 31–33. Mouse CNS penetration was consistent for 16 (Kp = 0.21, Kp,uu = 0.16), but was significantly, and unexpectedly, enhanced for 28a (Kp = 4.84, Kp,uu = 2.61). Quickly, our efforts centered on a direct head-to-head comparison of 16 to 28a as these two ago-PAMs began to clearly diverge in vitro and in vivo. Next, we assessed mouse agonist activity and found both 16 (EC50 = 100 nM, 91% ACh Max) and 28a (EC50 = 450 nM, 86% ACh Max) had agonist activity at mouse M1 at sub-micromolar concentrations, yet 16 remained 4.5-fold more potent. To confirm, we also assessed mouse M1 agonist in a low expressing mouse M1 cell line, and found similar data, but with diminished ACh Max (15–18%). Therefore, these compounds are both potent M1 mouse agonists and PAMs.
Previous studies suggest that structurally related M1 PAMs can also display differences with regards to stimulus bias, and that this can contribute to different in vivo profiles.49 Interestingly, in a Discover X bias panel,67 both 16 and 28a displayed comparable M1 PAM potency and efficacy in a β-arrestin recruitment assay (see Supporting Information Table 1), suggesting no signaling bias in this pathway. However, 28a had no effect on receptor internalization (EC50 > 10 μM), whereas 16 induced M1 internalization with an EC50 of 2.1 μM. Another question concerned the mechanism of the M1 agonism and whether this was due to orthosteric or allosteric agonism. To assess the interactions of these M1 PAMs with antagonist binding to the orthosteric site, we preformed the radioligand binding assay with [3H]-NMS in human M1 membranes (Figure 5). Interestingly, both ago-PAMs induced slight inhibition of binding of the orthosteric radioligand at high concentrations.
Figure 5.

Inhibition of orthosteric radioligand binding with [3H]-NMS by 16 and 28c. Both 16 and 28a weakly displaced the orthosteric radioligand, [3H]-NMS (0.075 nM) in competition binding studies. As a positive control, the orthosteric antagonist, atropine, potently inhibited [3H]-NMS binding with a Ki of 0.9 nM (in agreement with the literature). These data suggest that both PAMs have some interaction with the orthosteric site at higher concentrations, suggesting, perhaps, a bitopic mode of activity. Data represent the mean ± SEM of three independent experiments.
Mechanistically, we found that both 16 and 28a act as PAMs by significantly increasing the affinity of ACh for the M1 receptor (Figure 6). Here as well, 16 was a more efficacious M1 PAM, shifting the ACh Ki a maximum of 430-fold to the left (at 10 μM) versus only 178-fold for 28a. Even at micromolar concentrations of the ago-PAMs, 16 increased ACh affinity to a 3-fold greater extent than 28a; however, both showed a significant impact on ACh affinity.
Figure 6.

Evaluating the ability of M1 PAMs 16 and 28a to increase ACh affinity at the M1 receptor. (A) In the presence of increasing fixed concentrations (0.3–10.0 μM) of 16 (PF-06764427), the potency of ACh to displace [3H]-NMS binding is shifted leftward, yielding Ki values of 4847 ± 1230 (DMSO), 149 ± 49 nM (0.3 μM16), 35 ± 4 nM(1.0 μM16), and 11 ± 1 nM(10 μM16), which represent 32-fold, 139-fold, and 430-fold shifts in ACh potency, respectively, when compared to the DMSO control. (B) In the presence of increasing fixed concentrations (0.3–10.0 μM) of 28a (VU6004256), the potency of ACh to displace [3H]-NMS binding is shifted leftward, yielding Ki values of 4599 ± 720 (DMSO), 426 ± 45 nM (0.3 μM 28a), 80 ± 10 nM (1.0 μM28a), and 26 ± 1 nM(10 μM28a), which represent 5-fold, 11-fold, 58-fold, and 178-fold shifts in ACh potency, respectively, when compared to the DMSO control. Data represent the mean ± SEM of three independent experiments, performed in duplicate.
To evaluate other potential differences between 16 and 28a, we performed a battery of in vitro and in vivo DMPK assays (see Supporting Information Table 2) and found no major differences in terms of plasma protein binding across species, CYP450 profiles or predicted hepatic clearance. Soft-spot analysis in mouse, rat, and human S9 liver microsomes also showed very similar stability and conserved metabolism to the amide headgroup and dealkylation of the pyrazole moiety (see Supporting Information Figures 5 and 6). These metabolites are considerably more polar and thus unlikely to cross the blood-brain barrier. In addition, both ago-PAMs were profiled in a Eurofin Lead Profiling radioligand binding panel (see Supporting Information Tables 3 and 4), against 68 GPCRs, ion channels, and transporters, and found to have no significant ancillary pharmacology (no percent inhibition > 50% at 10 μM).68,69 Thus, we evaluated 16 and 28a in rodent AHL and other pharmacodynamic assays for which M1 PAMs have been reported to be efficacious.
Behavioral Pharmacology Assessment
In their recent paper,59 Davoren et al. reported pharmacodynamic efficacy of 16 in mouse AHL at dose of 3 and 10 mg/kg, but discussed that there was no dose–response despite increased drug exposure. It is possible that the effect noted may not represent a pharmacological reversal of a hyperdopaminergic state, but may be mediated by an overall decreased basal locomotor activity, thereby not affording a dose–response. Therefore, (Figure 7), we repeated the design of the published study59 and found that 16 (PF-06764427) has pronounced effects on spontaneous locomotor activity (Figure 7A) that correlate with reversal of AHL in mice (Figure 7C). In contrast, 28a, a weaker ago-PAM shows significantly less effect on spontaneous locomotor activity (Figure 7B), and did not reverse AHL in mice (Figure 7D). The effect of 16 is mediated by M1, as the effect is lost in M1 knockout (KO) mice (Figure 7E). While these studies do not provide definitive insights into the mechanism by which 16 reduces AHL, it is possible that doses of this compound that are below doses required for inducing fully generalized convulsions result in seizure activity below the threshold required for behavioral convulsions but that could result in disruption of locomotor activity. The finding that 28a is devoid of measurable pro-convulsant activity yet has no activity in the AHL assay is consistent with that possibility, and suggests that the ability to decrease AHL is not a common feature ofM1 PAMs.
As mice are more sensitive to cholinergic activation of seizure activity, we next evaluated the two ago-PAMs in rat AHL. As shown in Figure 8, neither M1 ago-PAM significantly reversed AHL in rats at a dose of 30 mg/kg, which is consistent with the possibility that the proconvulsant activity of 16 in mice may play a role in the reduced locomotor activity in AHL.
Figure 8.

Reversal of amphetamine-induced hyperlocomotion in rats. PF-06764427 (16) slightly reversed AHL in SD rats, whereas VU6004256 (28a) has no effect. Data are represented as the mean ambulations ± SEM.
As we and others have demonstrated, selective activation ofM1 is well validated for cognitive enhancement in wild-type animals, upon pharmacological challenge, or in genetic models of either AD or NMDA hypofunction.22,23,40–44,54 To ensure sufficient potentiation of M1 was present in the aforementioned rat AHL studies (dosed at 30 mg/kg), we evaluated 16 and 28a in the novel object recognition paradigm in rats (Figure 9). Here, at doses of 1, 3, and 10 mg/kg, 16 did not enhance cognitive performance in this task (p = 0.0529), but did trend toward significance. In contrast, 28a dose-dependently enhanced recognition memory in rats. Thus, at doses of 28a that are fully efficacious in rat models of cognitive performance, there is no effect on AHL.
Figure 9.

Novel object recognition. VU6004256 (28a) dose-dependently enhanced recognition memory in rats. Pretreatment with 3 and 10 mg/kg VU6004256 (i.p., 10% Tween 80 in water, 30 min) prior to exposure to identical objects significantly enhanced recognition memory assessed 24 h later. PF-06764427 (16) did not enhance cognitive performance in this task. Data are expressed as mean ± SEM (n = 12). Statistical analysis was completed using a one-way analysis of variance. If significant (p < 0.05), comparison of group effects relative to the vehicle group was completed using a Dunnett’s test, *p < 0.05.
Finally, we performed a Modified Irwin Toxicology Battery test (Figure 10) in mice to assess any potential adverse effects of the ago-PAMs 16 and 28a on the CNS and GI effects to compare with the seizure data in mice (Figure 4) shown previously. Here, 28a displayed no pronounced cholinergic effects. In contrast, 16 immediately produced effects on the corneal and pinnae reflex loss, salivation and motor activity. Additionally, the best battery was terminated due to significant behavioral convulsions. These data, coupled with the proconvulsant effects of 16, suggest that these two M1 PAMs can be clearly differentiated in terms of their adverse effect liabilities, despite highly conserved chemotypes and only subtle variations in pharmacological profiles.
Figure 10.

Modified Irwin Toxicology Battery test in rodents was used to assess any potential adverse effects of novel compounds on the CNS. To assess CNS and GI effects, animals were assessed prior to treatment to obtain baseline measurements. After administration of vehicle or compound, animals were evaluated at 15, 30, 50, 180, and 360 min for a number of behavioral and physiological changes from pretreatment baselines. An observer blinded to the compound treatment evaluated a series of autonomic nervous system, somatomotor system assessments and scored them on a scale from 0 (normal) to 3 (severe). The mean Modified Irwin Toxicology Battery test scores are classified as follows: − no effect, + 0.01–0.25, +0.251–0.50, (++) 0.51–1.0, + + 1.01–1.50, (+++) 1.51–2.0, +++ 2.01–2.5, (++++) 2.51–3.0. Pretreatment of VU6004256 (100 mg/kg, i.p.) did not cause significant observable adverse effects in normal, healthy mice as assessed by the modified Irwin Toxicology Battery test. However, the previously published M1 PAM, PF-06764427, caused significant prolonged behavioral convulsions in mice beginning at 60 min, preventing continuation of the CNS assessment.
CONCLUSIONS
We have reported a novel series ofM1 ago-PAMs, represented by 28a, that have robust activity in potentiating M1 responses to acetylcholine and in enhancing recognition memory. However, this compound is devoid of the severe adverse effect liability, including behavioral convulsions and peripheral cholinergic AEs that are observed with previously reported M1 ago-PAMs, such as 16. Furthermore, our studies suggest that the reported efficacy of M1 ago-PAMs, such as 16, in reversing AHL in mice is not observed with other M1 PAMs and could be related to actions that are not shared by other PAMs, such as proconvulsant activity. Unfortunately, the current data do not provide a clear mechanistic basis for the fundamental differences between these closely related M1 ago-PAMs. These compounds do display subtle differences in their effects on M1 signaling that could be relevant for these differences in in vivo activities. For instance, 16 has slightly higher in vitro efficacy as a PAM than does 28a. Furthermore, 16 has more potent agonist activity when compared with 28a. Thus, it is possible that 16 has a tendency to induce more excessive activation of M1 than does 28a and that this contributes to the striking differences in the AE liabilities of these two compounds. However, the differences between these compounds in the assays included here are relatively subtle and are not likely to fully explain the differences of these compounds in terms of adverse effect liability. Recent advances make it increasingly clear that structurally related allosteric modulators can have diverse effects on different aspects of GPCR signaling that can influence their in vivo activity. In addition to differences in agonist activity reported here, this can include differential effects on coupling of GPCRs to different signaling cascades, or stimulus bias, different effects on homodimeric versus heterodimeric forms of receptors, different effects on receptor desensitization, and/or different effects on receptor signaling that are impacted by differences in cell type specific receptor reserve. For instance, we have found that we can avoid adverse effect liability of mGlu5 PAMs by optimizing compounds that are fully devoid of allosteric agonist activity and display stimulus bias so that they do not potentiate coupling of mGlu5 to potentiation of NMDA receptor currents. Based on these complexities and data such as those reported in this paper, it is prudent to incorporate in vivo assays of AE liability as an early screening tier when optimizing M1 PAMs. Furthermore, the potential liabilities of excessive activation of M1 encourage optimization of compounds that are completely devoid of allosteric agonist activity, as we have done for optimization of mGlu5 PAMs. Based on the strong data suggesting that M1 PAMs may have robust therapeutic effects in improving cognitive function in AD and schizophrenia and reducing negative symptoms in schizophrenia, it will be important to optimize drug candidates that are devoid of major adverse effect liabilities and are suitable for advancing through clinical development for treatment of these devastating brain disorders. Further studies to elucidate more definitive underpinnings that give rise to AEs with M1 ago-PAMs are underway and will be reported in due course.
METHODS
Chemical Synthesis and Purification
All 1H and 13C NMR spectra were recorded on Bruker AV-400 (400 MHz) or Bruker AV-NMR (600 MHz) instrument. Chemical shifts are reported in ppm relative to residual solvent peaks as an internal standard set to δH7.26 or δC 77.0 (CDCl3) and δH 3.31 or δC 49.0 (CD3OD). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), integration, coupling constant (Hz). IR spectra were recorded as thin films and are reported in wavenumbers (cm−1). Low resolution mass spectra were obtained on an Agilent 1200 LCMS with electrospray ionization. High resolution mass spectra were recorded on a Waters Qtof-API-US plus Acquity system. The value Δ is the error in the measurement (in ppm) given by the equation Δ = [(ME − MT)/MT] × 106, where ME is the experimental mass and MT is the theoretical mass. The HRMS results were obtained with ES as the ion source and leucine enkephalin as the reference. Optical rotations were measured on a PerkinElmer-341 polarimeter. Analytical thin layer chromatography was performed on 250 μM silica gel 60 F254 plates. Visualization was accomplished with UV light, and/or the use of ninhydrin, anisaldehyde and ceric ammonium molybdate solutions followed by charring on a hot-plate. Chromatography on silica gel was performed using Silica Gel 60 (230–400 mesh) from Sorbent Technologies. Analytical HPLC was performed on an Agilent 1200 analytical LCMS with UV detection at 214 and 254 nm along with ELSD detection. Solvents for extraction, washing, and chromatography were HPLC grade. All reagents were purchased from Aldrich Chemical Co. and were used without purification. All polymer-supported reagents were purchased from Biotage, Inc. Flame-dried (under vacuum) glassware was used for all reactions. All reagents and solvents were commercial grade and purified prior to use when necessary. High-resolution mass spectrometry (HRMS) data were obtained using a Micromass Q-T of API-US mass spectrometer. For full experimental procedures, please see the Supporting Information.
4,6-Difluoro-N-(1S,2S)-2-hydroxycyclohexyl-1-((6-(1-methyl-1H-pyrazol-4-yl)pyridine-3-yl)methyl)-1H-indole-3-carboxamide (VU6004256, 28a). 1-((6-Bromopyridin)-3-yl)methyl)-4,6-difluoro-1H-indole (22)
To a round-bottom flask was added at room temperature 4,6-difluoroindole 18 (750 mg, 4.90 mmol) dissolved in DMF (9.8 mL). To the solution was added sodium hydride (392 mg, 9.80 mmol, 60% oil dispersion) at 0 °C. Then 2-bromo-5-(bromomethyl)pyridine (1.48 g, 5.88 mmol) was added to the reaction mixture, followed by stirring at room temperature for 18 h. The reaction was monitored by TLC analysis and upon completion, an aqueous NH4Cl solution (10 mL) and EtOAc (20 mL) were added to the reaction mixture. The reaction mixture was extracted with EtOAc (20 mL × 2). The organic layers were combined, dried over MgSO4, filtered, and concentrated in vacuo. The crude material was purified on SiO2 gel by flash column chromatography eluting with 0 to 100% hexane/EtOAc to afford 1.52 g (96% yield) of 1-[(6-bromo-3-pyridyl)methyl]-4,6-difluoro-1H-indole, 22. LCMS: Rt = 1.128 min; >93% pure at 254 nm, m/z = 323 (M + H). 1H NMR (400 MHz, CDCl3) δ ppm): 8.25 (d, J = 2.1 Hz, 1H), 7.42 (d, J = 8.2 Hz, 1H), 7.16 (dd, J = 8.2 Hz, 2.1 Hz, 1H), 7.05 (d, J = 3.2 Hz, 1H), 6.70 (d, J = 9.1 Hz, 1H), 6.65–6.58 (m, 2H), 5.23 (s, 2H).
4,6-Difluoro-1-((6-Methyl-1H-pyrazol-4-yl)pyridine-3-yl)methyl)-1H-indole (23)
In a microwave vial, 1-[(6-bromo-3-pyridyl)methyl]-4,6-difluoro-1H-indole 22 (1.50 g, 4.64 mmol), 1-methylpyrazole-4-boronic acid pinacol ester (1.16 g, 5.52 mmol), potassium phosphate tribasic monohydrate (1.07 g, 4.46 mmol), tricyclohexylphosphine (32 mg, 0.116 mmol), and Pd2(dba)3 (42 mg, 0.0464 mmol) were added. The vessel was charged with a stir bar and dioxane/H2O (2:1, 15.5 mL), and then sealed with a crimped-cap. The reaction was heated to 120 °C for 30 min in the microwave. After the reaction had cooled to ambient temperature, the reaction mixture was diluted with 1:1 CH2Cl2/water (20 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude material was purified on SiO2 gel by flash column chromatography eluting with 0 to 100% hexane/EtOAc to afford 1.39 g (93% yield) of 4,6-difluoro-1-((6-methyl-1H-pyrazol-4-yl)pyridine-3-yl)methyl)-1H-indole, 23. LCMS: Rt = 0.826 min; >98% pure at 254 nm, m/z = 325 (M + H). 1H NMR (400 MHz, CDCl3) δ ppm): 8.45 (s, 1H), 8.28 (br, 1H), 7.95 (s, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.47 (d, J = 8.0 Hz, 1H), 7.08 (d, J = 3.2 Hz, 1H), 6.74 (d, J = 8.9 Hz, 1H), 6.68–6.60 (m, 2H), 5.30 (s, 2H), 3.97 (s, 3H).
1-(4,6-Difluoro-1-((6-(1-methyl-1H-pyrazol-4-yl)pyridin-3-yl)-methyl)-1H-indol-3-yl-2,2,2-trifluoroethan-1-one
To a round-bottom flask was added at room temperature 4,6-difluoro-1-((6-methyl-1H-pyrazol-4-yl)pyridine-3-yl)methyl)-1H-indole, 23 (1.39 g, 4.29 mmol) dissolved in DMF (4.3 mL) and trifluoroacetic anhydride (894 μL, 6.43 mmol). The resultant mixture was stirring at room temperature for 48 h. Upon completion of the reaction by LC/MS, a NaHCO3 (saturated aqueous solution, 20 mL) and CH2Cl2 (20 mL) were added to the reaction mixture. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo to afford 1.48 g (82% yield) of 1-(4,6-difluoro-1-((6-(1-methyl-1H-pyrazol-4-yl)pyridine-3-yl)methyl)-1H-indol-3-yl-2,2,2-trifluoroethan-1-one. The residue was used in subsequent reactions without further purification. LCMS: Rt = 0.888 min; >98% pure at 254 nm, m/z = 421 (M + H). 1H NMR (400 MHz, CDCl3) δ ppm): 8.53 (s, 1H), 8.09 (br, 1H), 8.00 (s, 1H), 7.95 (s, 1H), 7.50 (d, J = 8.2 Hz, 1H), 7.45 (d, J = 8.2 Hz, 1H), 6.89–6.82 (m, 2H), 5.38 (s, 2H), 3.96 (s, 3H).
4,6-Difluoro-1-((6-(1-methyl-1H-pyrazol-4-yl)pyridin-3-yl)-methyl)-1H-indol-3-carboxylic acid
To a microwave vial was added 1-(4,6-difluoro-1-((6-(1-methyl-1H-pyrazol-4-yl)pyridine-3-yl)methyl)-1H-indol-3-yl-2,2,2-trifluoroethan-1-one (1.28 g, 3.06 mmol). The vessel was charged with a stir bar and water (7.6 mL) was added, followed by sodium hydroxide (134 mg, 3.36 mmol) and then sealed with a crimped-cap. The reaction was heated to 150 °C for 30 min in the microwave. The reaction was cooled to ambient temperature. Upon completion of the reaction by LC/MS, the reaction mixture was concentrated and the residue was taken up in water (10 mL). Then 12N HCl was added dropwise to adjust the pH to 2. The resultant solid was filtered, washed with water, and dried in vacuo to provide 833 mg (74% yield) of 4,6-difluoro-1-((6-(1-methyl-1H-pyrazol-4-yl)pyridin-3-yl)-methyl)-1H-indol-3-carboxylic acid. LCMS: Rt = 0.636 min; >95% pure at 254 nm, m/z = 369 (M + H). 1HNMR (400 MHz, (CD3)2SO) δ ppm): 8.60 (s, 1H), 8.36 (s, 1H), 8.27 (s, 1H), 7.98 (s, 1H), 7.70 (d, J = 8.3 Hz, 1H), 7.63 (d, J = 8.3 Hz, 1H), 7.52 (d, J = 9.1 Hz, 1H), 7.01 (t, J = 9.1 Hz, 1H), 5.51 (s, 2H), 3.90 (s, 3H).
4,6-Difluoro-N-(1S,2S)-2-hydroxycyclohexyl-1-((6-(1-methyl-1H-pyrazol-4-yl)pyridine-3-yl)methyl)-1H-indole-3-carboxamide (VU6004256, 28a)
To a vial at room temperature were added 4,6-difluoro-1-((6-(1-methyl-1H-pyrazol-4-yl)pyridin-3-yl)methyl)-1H-indol-3-carboxylic acid (250 mg, 0.679 mmol), (1S, 2S)-2-aminocyclohexanol (94 mg, 0.814 mmol), BOP reagent (benzotriazole-1-yl-oxy-tris(dimethylamino)-phosphonium hexafluorophosphate) (390 mg, 0.882 mmol), and triethylamine (378 uL, 2.72 mmol), which were then dissolved in DMF (2.5 mL). This mixture was stirred for 20 min. The reaction was monitored by LC/MS and upon completion, the reaction mixture was diluted with 1:1 CH2Cl2/water (10 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (5 mL × 2). The organic layers were combined, passed through a phase separator, and concentrated in vacuo. The crude material was purified on SiO2 gel by flash column chromatography eluting with 0 to 10% CH2Cl2/MeOH and then via reverse-phase preparative HPLC eluting with 24 to 79% H2O/acetonitrile with 0.1% TFA. The obtained residue was stirred for 1 h in hexane/Et2O (1:1). Then, the deposited solid was collected by filtration and dried under reduced pressure to afford 219 mg (69% yield) of 4,6-difluoro-N-(1S,2S)-2-hydroxycyclohexyl-1-((6-(1-methyl-1H-pyrazol-4-yl)pyridine-3-yl)methyl)-1H-indole-3-carboxamide (VU6004256, 28a). Specific rotation [α]23D = −18.8° (c = 1.0, CHCl3); LCMS: Rt = 0.814 min; 1H NMR (400 MHz, CD3OD) δ ppm): 8.46 (s, 1H), 8.15 (s, 1H), 8.01 (s, 2H), 7.64 (s, 2H), 7.21 (dd, J = 10.8 Hz, 1.6 Hz, 1H), 6.86 (dt, J = 1.6 Hz, 10.8 Hz, 1H), 5.49 (s, 2H), 3.96 (s, 3H), 3.86–3.77 (m, 1H), 3.54–3.46 (m, 1H), 2.20–2.02 (m, 2H), 1.85–1.71 (m, 2H), 1.50–1.23 (m, 4H); 13C NMR (100.6 MHz, (CD3)2SO) δ ppm): 163.5, 159.4 (dd, JC–F = 237 Hz, 12 Hz), 155.9 (dd, JC–F = 249 Hz, 15 Hz), 152.3, 149.6, 138.9 (t, JC–F = 15 Hz), 138.0, 137.0, 134.1, 130.5, 130.4, 123.3, 120.1, 112.4, 111.4 (d, JC–F = 19 Hz), 97.6 (t, JC–F = 28 Hz), 95.0 (d, JC–F = 31 Hz), 72.3, 55.7, 47.9, 39.7, 35.0, 31.9, 25.1, 24.8 HRMS: calcd for C25H25N5O2F2, 465.1976; found, 465.1979.
Cell Lines
Chinese hamster ovary (CHO) cells stably expressing human and rat M1 were maintained in Ham’s F-12 growth medium containing 10% FBS, 20 mM HEPES, antibiotic/antimycotic, and 500 μg/mL G418 in the presence of 5% CO2 at 37 °C as previously described.49 To determine the functional activity at mouse M1, the mouse M1 full-length open reading frame (ORF) was amplified from a mouse brain cDNA library (Clontech Laboratories, Mountain View, CA) and subcloned into the BamH1 and EcoR1 sites of pcDNA3.1 (+) vector (Life Technologies, Carlsbad, CA). Sequencing of the mouseM1-pcDNA3.1 (+) plasmid confirmed the presence of mouse M1 ORF (NM_007698). CHO cells were transfected with mouse M1 expression plasmid using Fugene 6 (Promega, Madison, WI), and the transfected cells were used for calcium mobilization to assess the functional activity at mouse M1. Also, the transfected cells were incubated with the selection medium containing 1 mg/mL G418 for 2 weeks, and the resulting polyclones were then plated in a 96-well plate for monoclonal generation. The positive monoclones of mouse M1 expression were identified in the calcium mobilization assay described below.
Calcium Mobilization Assay
To determine the potency and efficacy of M1 ago-PAMs, calcium flux was measured using the Functional Drug Screening System (FDSS7000, Hamamatsu, Japan) as previously described.62 Briefly, the M1-CHO cells were plated in black-walled, clear-bottomed 384 well plates (Greiner Bio-One, Monroe, NC) at 20 000 cells/well in 20 μL of growth medium without G418 the day before assay. The following day, cells were washed with assay buffer (Hank’s balanced salt solution, 20mMHEPES, and 2.5mM probenecid) and immediately incubated with 20 μL of 1.15 μM Fluo-4-acetomethoxyester (Fluo-4 AM) dye solution prepared in assay buffer for 45 min at 37 °C. During the incubation time, all compounds were serial diluted (1:3) in DMSO for 10 point concentration–response curves (CRC), and further diluted in assay buffer at starting final concentration 30 μM using Echo liquid handler (Labcyte, Sunnyvale CA). Dye was removed and replaced with assay buffer. Immediately, calcium flux was measured using the FDSS7000 as previously described.49 The CRC of compounds or vehicle was added to cells for 2.5 min and then an EC20 concentration of acetylcholine (ACh) was added and incubated for 1 min. ECmax concentration was also added to cells that were incubated with DMSO vehicle to ensure the EC20 calcium response. Using a four point logistical equation in GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA), the concentration response curves were generated for determination of the potency and efficacy of the agonist and PAM.
Radioligand Binding Assay
Competition binding assay was performed using [3H]-N-methylscopolamine ([3H-]NMS, PerkinElmer. Boston, MA) as previously described.48 Briefly, compounds were serial diluted 1:3 in DMSO for 11 point CRC, then further diluted at starting final concentration at 10 μMin binding buffer (20 mM HEPES, 10 mM MgCl2, and 100 mM NaCl, pH 7.4). During the dilution of the concentrated compound stocks in the binding buffer, stock solutions more than ≥20 μM were sonicated due to insolubility issue before adding to the binding assay plate. Membranes from human M1-CHO cells (10 μg) were incubated with the serial diluted compounds in the presence of Kd concentration of [3H-]NMS, 0.075 nM, at room temperature for 1 h with constant shaking. Nonspecific binding was determined in the presence of 10 μM atropine. Binding was terminated by rapid filtration through GF/B Unifilter plates (PerkinElmer) using a Brandel 96-well plate Harvester (Brandel Inc., Gaithersburg, MD), followed by three washes with ice-cold harvesting buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl). Plates were air-dried overnight, 50 μL of Microscint20 was added to the plate, and radioactivity was counted using a TopCount Scintillation Counter (PerkinElmer Life and Analytical Sciences).
For the measurement of the acetylcholine affinity at hM1, all binding conditions were the same as those for competition binding with the exception of addition of varying concentration of acetylcholine (serial diluted at 11 point CRC) in the presence of vehicle or theM1 ago-PAMs (10, 1, and 0.3 μM).
Drug Metabolism Methods
In Vitro. Protein binding ofM1 PAMs were determined in rat plasma via equilibrium dialysis employing Single-Use RED Plates with inserts (ThermoFisher Scientific, Rochester, NY). Briefly, plasma (220 μL) was added to the 96-well plate containing test article (5 μL) and mixed thoroughly. Subsequently, 200 μL of the plasma-test article mixture was transferred to the cis chamber (red) of the RED plate, with an accompanying 350 μL of phosphate buffer (25 mM, pH 7.4) in the trans chamber. The RED plate was sealed and incubated 4 h at 37 °C with shaking. At completion, 50 μL aliquots from each chamber were diluted 1:1 (50 μL) with either plasma (cis) or buffer (trans) and transferred to a new 96-well plate, at which time ice-cold acetonitrile (2 volumes) was added to extract the matrices. The plate was centrifuged (3000 rpm, 10 min), and supernatants transferred to a new 96-well plate. The sealed plate was stored at −20 °C until LC/MS/MS analysis. The metabolism of M1 PAMs was investigated in multispecies hepatic microsomes (BD Biosciences, Billerica, MA) using substrate depletion methodology (percent of test article remaining). A potassium phosphate-buffered reaction mixture (0.1 M, pH 7.4) of test article (1 μM) and microsomes (0.5 mg/mL) was preincubated (5 min) at 37 °C prior to the addition of NADPH (1 mM). The incubations, performed in 96-well plates, were continued at 37 °C under ambient oxygenation and aliquots (80 μL) were removed at selected time intervals (0, 3, 7, 15, 25, and 45 min). Protein was precipitated by the addition of chilled acetonitrile (160 μL), containing glyburide as an internal standard (50 ng/mL), and centrifuged at 3000 rpm (4 °C) for 10 min. Resulting supernatants were transferred to new 96-well plates in preparation for LC/MS/MS analysis. The in vitro half-life (t1/2, min, eq 1), intrinsic clearance (CLint, mL/min/kg, eq 2), and subsequent predicted hepatic clearance (CLhep, mL/min/kg, eq 3) were determined employing the following equations:
| (1) |
where k represents the slope from linear regression analysis (percent of test article remaining).
| (2) |
where the a notes scale-up factors of 20 (human) and 45 (rat) and Q is heptatic perfusion.
| (3) |
In Vivo
Male Sprague–Dawley rats (n = 2) weighing around 300 g were purchased from Harlon laboratories (Indianapolis, IN) and implanted with catheters in the carotid artery and jugular vein. The cannulated animals were acclimated to their surroundings for approximately 1 week before dosing and were provided food and water ad libitum. M1 PAMs were administered intravenously (i.v.) to rats in cassette format via the jugular vein catheter in EtOH/PEG400/Saline at a total dose of 1 mg/kg (0.20 or 0.25 mg/kg per compound) and a dose volume of 1 mL/kg. Brain dissection and blood collections via the carotid artery were performed at 0.25 h post dose. Samples were collected into chilled, EDTA-fortified tubes, centrifuged for 10 min at 3000 rpm (4 °C), and resulting plasma aliquoted into 96-well plates for LC/MS/MS analysis. The brain samples were rinsed in PBS, snap frozen and stored at −80 °C. Prior to LC/MS/MS analysis, brain samples were thawed to room temperature and subjected to mechanical homogenation employing a Mini-Beadbeater and 1.0 mm Zirconia/Silica Beads (BioSpec Products). All animal studies were approved by the Vanderbilt University Medical Center Institutional Animal Care and Use Committee. The animal care and use program is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International.
Liquid Chromatography/Mass Spectrometry Analysis. In Vivo Experiments
M1 PAMs were analyzed via electrospray ionization (ESI) on an AB Sciex API-4000 (Foster City, CA) triple-quadrupole instrument that was coupled with Shimadzu LC-10AD pumps (Columbia, MD) and a Leap Technologies CTC PAL autosampler (Carrboro, NC). Analytes were separated by gradient elution using a Fortis C18 2.1 × 50 mm, 3.5 μm column (Fortis Technologies Ltd., Cheshire, U.K.) thermostated at 40 °C. HPLC mobile phase A was 0.1% NH4OH (pH unadjusted), and mobile phase B was acetonitrile. The gradient started at 30% B after a 0.2 min hold and was linearly increased to 90% B over 0.8 min; held at 90% B for 0.5 min and returned to 30% B in 0.1 min followed by a re-equilibration (0.9 min). The total run time was 2.5 min, and the HPLC flow rate was 0.5 mL/min. The source temperature was set at 500 °C and mass spectral analyses were performed using multiple reaction monitoring (MRM) utilizing a Turbo-Ionspray source in positive ionization mode (5.0 kV spray voltage). All data were analyzed using AB Sciex Analyst 1.4.2 software.
In Vitro Experiments
M1 PAMs were analyzed similarly to that described above (in vivo) with the following exceptions: LC/MS/MS analysis was performed employing a TSQ QuantumULTRA instrument that was coupled to a ThermoSurveyor LC system (Thermoelectron Corp., San Jose, CA) and a Leap Technologies CTC PAL autosampler (Carrboro, NC). Chromatographic separation of analytes was achieved with an Acquity BEH C18 2.1 × 50 mm, 1.7 μm column (Waters, Taunton, MA).
Mouse Plasma-Brain Exposure
M1 PAMs were dissolved in 10% Tween 80 at the concentration of 1–10 mg/mL (base form) and administered intraperitoneally to male C57BL/6J (Jackson Laboratory, Sacramento, CA) mice aged 7–9 weeks at a volume of 10 mL/kg. The blood and brain were collected at 0.25 h. Animals were euthanized and decapitated, and the brains were removed, thoroughly washed with cold saline and immediately frozen on dry ice. Trunk blood was collected in EDTA coated Eppendorf tubes, and plasma was separated by centrifugation and stored at −80 °C until processed for LC/MS/MS analysis as previously described. Three animals were used for each time point.
In Vivo Studies
Animals
All present studies used 7–8 week old male C57Bl/6 mice (Jackson Laboratory, Sacramento, CA) or male Sprague–Dawley rats weighing 275–300 g (Envigo, Indianapolis, IN), which were cared for in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experimental procedures were approved by the Vanderbilt University Animal Care and Use Committee.
Spontaneous Locomotor Activity
The effects of M1 PAMs on basal locomotor activity were assessed. Mice were administered vehicle orM1 PAM (1–10 mg/kg, intraperitoneal (i.p.), 10% Tween 80 in water, 10 mL/kg, n = 12) and placed into an open field arena (MedAssociates, St. Albans, VT) following a 30 min pretreatment. Locomotor activity was then measured for 90 min. Changes in locomotor activity were measured as the total distance traveled (cm). Data are expressed as mean ± SEM and analyzed using one-way analysis of variance (ANOVA) with a Dunnett’s post hoc test comparing all dosing groups to vehicle + amphetamine-treated controls. Statistical significance was determined as p < 0.05.
Amphetamine-Induced Hyperlocomotion
The effects ofM1 PAMs on amphetamine-induced hyperlocomotion were determined as previously described.62 Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol. Pharmacol. 78, 1105–1123). For studies in mice, animals were placed in an open field arena (MedAssociates, St. Albans, VT) for 90 min. At t = 90, mice were administered vehicle or M1 PAM (1–10 mg/kg, i.p., 10% Tween 80 in water, 10 mL/kg, n = 12) or vehicle followed by amphetamine (3 mg/kg, subcutaneous (s.c.), saline, 10 mL/kg). Locomotor activity was then measured for an additional 60 min. Changes in locomotor activity were measured as the total distance traveled (cm) per 5 min bins. For locomotor activity assessment in rats, rats were placed in the activity chambers (KinderScientific, San Diego, CA) to habituate. At t = 30, rats were administered M1 PAM (30 mg/kg i.p., 10% Tween 80 in water, 5 mL/kg) or vehicle and placed back in the activity chambers. Five minutes later the animals were injected with amphetamine (1 mg/kg s.c.; 1 mL/kg) or saline and placed back in the activity chambers. Locomotor activity was then measured for an additional 60 min. Changes in locomotor activity were measured as the total number of beam breaks per 5 min bins. Data are expressed as mean ± SEM and analyzed using one-way ANOVA (last 60 min) with a Dunnett’s post hoc test comparing all dosing groups to vehicle + amphetamine-treated controls. Statistical significance was determined as p < 0.05.
Behavioral Manifestations of Seizure Activity
To evaluate induction of behavioral manifestation of seizure activity, C57Bl/6 mice received administration of vehicle or 100 mg/kg M1 PAM. Compounds were formulated in 10% Tween 80 (pH 7.0) at a concentration of 10 mg/mL and injected i.p. (n = 3). Animals were monitored continuously and scored for behavioral manifestations of seizure activity at 5, 10, 15, and 30 min, and 1 and 3 h. Behavioral manifestations of seizures were scored using a modified Racine scoring system.70 Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281). Briefly, a score of 0 represents no behavior alterations; score 1, immobility, mouth and facial movements, or facial clonus; score 2, head nodding, tail extension; score 3, forelimb clonus, repetitive movements; score 4, rearing and tonic clonic seizure; and score 5, continuous rearing and falling, severe generalized tonic clonic seizure.
Modified Irwin Toxicology Battery
The potential CNS adverse effects ofM1 PAMs were evaluated using the Modified Irwin Toxicology Battery. Baseline assessments were conducted prior to administration of compounds. Mice were then administered vehicle orM1 PAM (100 mg/kg, 10% Tween 80, 10 mL/kg, i.p., n = 3), placed back into their home cages and evaluated by an observed blinded to dosing conditions at 15, 30, 60, 180, and 360 min for autonomic nervous and somatomotor system parameters outlined in Figure 10. Mice were assigned a score for each parameter of 0 (normal), 1 (slight/light), 2 (moderate), or 3 (marked) relative to vehicle-treated controls.
Novel Object Recognition Task
Rats were habituated for 10 min for 2 consecutive days in an empty novel object recognition (NOR) arena consisting of dark-colored plexiglass box (40 × 64 × 33 cm3). On day 3, rats were administered vehicle (10% tween 80) or M1 PAM (1–10 mg/kg, i.p., 3 mL/kg, n = 12) and returned to their home cage for 30 min. Rats were then placed in the NOR arena containing two identical objects for 10 min. Following the exposure period, rats were placed back into their home cages for 24 h. The rats were then returned to the arena in which one of the previously exposed (familiar) objects was replaced by a novel object and were video recorded for 5 min while they explored the two objects. Time spent exploring each object was scored by an observer blinded to the experimental conditions and the recognition index was calculated as [(time spent exploring novel object) – (time spent exploring familiar object)]/total time exploring objects.
Supplementary Material
Acknowledgments
Funding
This work was funded by the NIH and NIMH (MH082867 and MH106839).
We thank William K. Warren, Jr. and the William K. Warren Foundation who funded the William K. Warren, Jr. Chair in Medicine (to C.W.L.).
ABBREVIATIONS
- i.p
intraperioneal
- p.o
oral dosing
- LTS, GPCR
G protein-coupled receptor
- M1
muscarinic acetylcholine receptor subtype 1
- NOR
novel object recognition
- AHL
amphetamine-induced hyperlocomotion
Footnotes
ORCID
Craig W. Lindsley: 0000-0003-0168-1445
Author Contributions
J.M.R., M.A., and H.P.C. contributed equally to this work. C.W.L., P.J.C., and C.M.N., and J.M.R. and C.K.J., and A.L.B. oversaw and designed the chemistry, molecular pharmacology, behavioral pharmacology and DMPK, respectively. M.A. and D.W.E., and J.L.E. performed synthetic/medicinal chemistry and scaled-up key compounds for in vivo studies. H.P.C. designed and executed advanced molecular pharmacology assays. K.D.N. and P.M.G.-B. performed molecular pharmacology assays. S.C. and J.J.F. performed DMPK and bioanalysis. C.W.L., P.J.C., and J.M.R. wrote the manuscript.
Notes
The authors are developing M1 PAMs for the treatment of schizophrenia and AD, and have an open-IND for the same as well as a patent portfolio of M1 PAMs.
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications Web site at DOI: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.6b00429.
Compound characterization, additional methods and supplemental pharmacology and drug metabolism data (PDF)
References
- 1.Perese EF, Wu YB. Shortfalls of Treatment for Patients with Schizophrenia: Unmet Needs, Obstacles to Recovery. Int J Psychosoc Rehab. 2010;14:43–56. [Google Scholar]
- 2.Citron M. Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discovery. 2010;9:387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- 3.Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci. 1991;11:3218–3226. doi: 10.1523/JNEUROSCI.11-10-03218.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levey AI, Edmunds SM, Koliatsos V, Wiley RG, Heilman CJ. Expression of M1–M4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervations. J Neurosci. 1995;15:4077–4092. doi: 10.1523/JNEUROSCI.15-05-04077.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levey AI. Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of Alzheimer disease. Proc Natl Acad Sci U S A. 1996;93:13541–13546. doi: 10.1073/pnas.93.24.13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Felder CC, Porter AC, Skillman TL, Zhang L, Bymaster FP, Nathanson NM, Hamilton SE, Gomeza J, Wess J, McKinzie DL. Elucidating the role of muscarinic receptors in psychosis. Life Sci. 2001;68:2605–2613. doi: 10.1016/s0024-3205(01)01059-1. [DOI] [PubMed] [Google Scholar]
- 7.Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, Silva AJ. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor in mutant mice. Nat Neurosci. 2002;6:51–58. doi: 10.1038/nn992. [DOI] [PubMed] [Google Scholar]
- 8.Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, LaFerla FM. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49:671–682. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 9.Caccamo A, Fisher A, LaFerla FM. M1 agonists as a potential disease-modifying therapy for Alzheimer’s disease. Curr Alzheimer Res. 2009;6:112–117. doi: 10.2174/156720509787602915. [DOI] [PubMed] [Google Scholar]
- 10.Dean B, Hopper S, Conn PJ, Scarr E. Changes in BQCA allosteric modulation of [3H]-NMS binding to human cortex within schizophrenia and by divalent cations. Neuropsychopharmacology. 2016;41:1620–1628. doi: 10.1038/npp.2015.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scarr E, Udawela M, Thomas EA, Dean B. Changed gene expression in subjects with schizophrenia and low cortical muscarinic M1 receptors predict disrupted upstream pathways interacting with that receptor. Mol Psychiatry. 2016 doi: 10.1038/mp.2016.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marino MJ, Rouse ST, Levey AI, Potter LT, Conn PJ. Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 1998;95:11465–11470. doi: 10.1073/pnas.95.19.11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balu DT. The NMDA Receptor and Schizophrenia: From Pathophysiology to Treatment. Adv Pharmacol. 2016;76:351–382. doi: 10.1016/bs.apha.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kantrowitz JT, Javitt DC. Thinking glutamatergically: changing concepts of schizophrenia based upon changing neurochemical models. Clin Schizophr Relat Psychoses. 2010;4:189–200. doi: 10.3371/CSRP.4.3.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kantrowitz JT, Javitt DC. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010;83:108–121. doi: 10.1016/j.brainresbull.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Catts VS, Lai YL, Weickert CS, Weickert TW, Catts SV. A quantitative review of the postmortem evidence for decreased cortical N-methyl-d-aspartate receptor expression levels in schizophrenia: How can we link molecular abnormalities to mismatch negativity deficits? Biol Psychol. 2016;116:57–67. doi: 10.1016/j.biopsycho.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 17.Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jadi MP, Behrens MM, Sejnowski TJ. Abnormal Gamma Oscillations in N-Methyl-D-Aspartate Receptor Hypofunction Models of Schizophrenia. Biol Psychiatry. 2016;79:716–726. doi: 10.1016/j.biopsych.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- 20.Milenkovic M, Mielnik CA, Ramsey AJ. NMDA receptor-deficient mice display sexual dimorphism in the onset and severity of behavioural abnormalities. Genes Brain Behav. 2014;13:850–862. doi: 10.1111/gbb.12183. [DOI] [PubMed] [Google Scholar]
- 21.Gregory KJ, Herman EJ, Ramsey AJ, Hammond AS, Byun NE, Stauffer SR, Manka JT, Jadhav S, Bridges TM, Weaver CD, Niswender CM, Steckler T, Drinkenburg WH, Ahnaou A, Lavreysen H, Macdonald GJ, Bartolome JM, Mackie C, Hrupka BJ, Caron MG, Daigle TL, Lindsley CW, Conn PJ, Jones CK. N-aryl piperazine metabotropic glutamate receptor 5 positive allosteric modulators possess efficacy in preclinical models of NMDA hypofunction and cognitive enhancement. J Pharmacol Exp Ther. 2013;347:438–457. doi: 10.1124/jpet.113.206623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghoshal A, Rook JM, Dickerson JW, Roop GN, Morrison RD, Jalan-Sakrikar N, Lamsal A, Noetzel MJ, Poslusney MS, Wood MR, Melancon BJ, Stauffer SR, Xiang Z, Daniels JS, Niswender CM, Jones CK, Lindsley CW, Conn PJ. Potentiation of M1 Muscarinic Receptor Reverses Plasticity Deficits and Negative and Cognitive Symptoms in a Schizophrenia Mouse Model. Neuropsychopharmacology. 2016;41:598–610. doi: 10.1038/npp.2015.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grannan MD, Mielnik CA, Moran SP, Gould RW, Ball J, Bubser M, Ramsey AJ, Abe M, Cho HP, Nance KD, Blobaum AL, Niswender CM, Conn PJ, Lindsley CW, Jones CK. Prefrontal cortex-mediated impairments in a genetic model of NMDA receptor hypofunction are reversed by the novel M1 PAM VU6004256. ACS Chem Neurosci. 2016;7:1706–1716. doi: 10.1021/acschemneuro.6b00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bridges TM, LeBois EP, Hopkins CR, Wood MR, Jones JK, Conn PJ, Lindsley CW. Antipsychotic potential of muscarinic allosteric modulation. Drug News Perspect. 2010;23:229–240. doi: 10.1358/dnp.2010.23.4.1416977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conn PJ, Jones C, Lindsley CW. Subtype selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders’. Trends Pharmacol Sci. 2009;30:148–156. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melancon BJ, Tarr JC, Panarese JD, Wood MR, Lindsley CW. Allosteric modulation of the M1 muscarinic acetylcholine receptor: improving cognition and a potential treatment for schizophrenia and Alzheimer’s disease. Drug Discovery Today. 2013;18:1185–1199. doi: 10.1016/j.drudis.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keov P, Valant C, Devine SM, Lane JR, Scammels PJ, Sexton PM, Christopoulos A. Reverse engineering of the selective agonist TBPB unveils both orthosteric and allosteric modes of action at the M1 muscarinic acetylcholine receptor. Mol Pharmacol. 2013;84:425–437. doi: 10.1124/mol.113.087320. [DOI] [PubMed] [Google Scholar]
- 28.Digby GJ, Utley TJ, Lamsal A, Sevel C, Sheffler DJ, Lebois EP, Bridges TM, Wood MR, Niswender CM, Lindsley CW, Conn PJ. Chemical modification of the M1 agonist VU0364572 reveals molecular switches in pharmacology as well as a bitopic binding mode. ACS Chem Neurosci. 2012;3:1025–1036. doi: 10.1021/cn300103e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheffler DJ, Sevel C, Le U, Lovell KM, Tarr JC, Cho HP, Digby GJ, Niswender CM, Conn PJ, Hopkins CR, Wood MR, Lindsley CW. Further exploration of M1 allosteric agonists. Subtle structural changes abolish M1 allosteric agonism and result in pan-mAChR orthosteric antagonism. Bioorg Med Chem Lett. 2013;23:223–227. doi: 10.1016/j.bmcl.2012.10.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daval SB, Valant C, Bonnet D, Kellenberger E, Hibert M, Galzi JL, Ilien B. Fluorescent derivatives of AC-42 to probe bitopic orthosteric/allosteric binding mechanisms on muscarinic M1 receptors. J Med Chem. 2012;55:2125–2143. doi: 10.1021/jm201348t. [DOI] [PubMed] [Google Scholar]
- 31.Avlani VA, Langmead CJ, Guida E, Wood MD, Tehan BG, Herdon HJ, Watson JM, Sexton PM, Christopoulos A. Orthosteric and allosteric modes of interaction of novel selective agonists of the M1 muscarinic acetylcholine receptor. Mol Pharmacol. 2010;78:94–104. doi: 10.1124/mol.110.064345. [DOI] [PubMed] [Google Scholar]
- 32.Thomas RL, Langmead CJ, Wood MD, Challiss RA. Contrasting effects of allosteric and orthosteric agonists on m1 muscarinic acetylcholine receptor internalization and down-regulation. J Pharmacol Exp Ther. 2009;331:1086–1095. doi: 10.1124/jpet.109.160242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lebois EP, Bridges TM, Dawson ES, Kennedy JP, Xiang Z, Jadhav SB, Yin H, Meiler J, Jones CK, Conn PJ, Weaver CD, Lindsley CW. Discovery and development of novel subtype-selective M1 allosteric agonists for the investigation of M1 receptor function. ACS Chem Neurosci. 2010;1:104–121. doi: 10.1021/cn900003h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.LeBois EP, Sheffler DJ, Digby GJ, Melancon BJ, Tarr JC, Cho HP, Morrison R, Bridges TM, Xiang Z, Daniels JS, Wood MR, Conn PJ, Lindsley CW. Development of a Highly Selective, Orally Bioavailable and CNS Penetrant M1 Agonist Derived from the MLPCN Probe ML071. Bioorg Med Chem Lett. 2011;21:6451–6455. doi: 10.1016/j.bmcl.2011.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melancon BJ, Gogliotti RD, Tarr JC, Saleh SA, Chauder BA, LeBois EP, Cho HP, Utley TJ, Sheffler DJ, Bridges TM, Daniels JS, Niswender CM, Conn PJ, Lindsley CW, Wood MR. Continued optimization of the MLPCN probe ML071 into highly potent agonists of the hM1 muscarinic acetylcholine receptor. Bioorg Med Chem Lett. 2012;22:3467–3472. doi: 10.1016/j.bmcl.2012.03.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Digby GJ, Noetzel MJ, Bubser M, Utley TJ, Walker AG, Byun NB, LeBois EP, Xiang Z, Sheffler DJ, Niswender CM, Plumley HC, Davis AA, Nemirovsky NE, Mennenga S, Camp BW, Bimonte-Nelson HA, Morrison R, Daniels S, Lindsley CW, Olive MF, Conn PJ. Novel allosteric agonists of the M1 muscarinic acetylcholine receptor induce brain region-specific responses and correspond with behavioral effects in animal models. J Neurosci. 2012;32:8532–8544. doi: 10.1523/JNEUROSCI.0337-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.For information on the heptares M1 agonist, see: http://www.heptares.com/news/261/74/Heptares-Announces-Positive-Results-from-Phase-1b-Clinical-Trial-with-HTL9936-a-First-In-Class-Selective-Muscarinic-M1-Receptor-Agonist-for-Improving-Cognition-in-Dementia-and-Schizophrenia.html.
- 38.Conn PJ, Lindsley CW, Meiler J, Niswender CM. Opportunities and challenges in the discovery of allosteric modulators of GPCRs for the treatment of CNS disorders. Nat Rev Drug Discovery. 2014;13:692–708. doi: 10.1038/nrd4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conn PJ, Christopoulos A, Lindsley CW. Allosteric Modulators of GPCRs as a Novel Approach to Treatment of CNS Disorders’. Nat Rev Drug Discovery. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. Allosteric Modulation of 7 Transmembrane Spanning Receptors: Theory, Practice and Opportunities for CNS Drug Discovery’. J Med Chem. 2012;55:1445–1464. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma L, Seager MA, Wittmann M, Jacobson M, Bickel D, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danziger A, Regan C, Flick R, Pascarella D, Garson S, Doran S, Kreatsoulas C, Veng L, Lindsley CW, Shipe W, Kuduk S, Sur C, Kinney G, Seabrook GR, Ray WJ. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci U S A. 2009;106:15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Jadhav SB, Menon U, Christain EP, Doherty JJ, Quirk MC, Snyder DH, Levey AI, Watson ML, Nicholle MM, Lindsley CW, Conn PJ. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J Neurosci. 2009;29:14271–14286. doi: 10.1523/JNEUROSCI.3930-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang FV, Shipe WD, Bunda JL, Nolt MB, Wisnoski DD, Zhao Z, Barrow JC, Ray WJ, Ma L, Wittmann M, Seager M, Koeplinger K, Hartman GD, Lindsley CW. Parallel synthesis of N-Birayl Quinolone Carboxylic Acids as selective M1 positive allosteric modulators. Bioorg Med Chem Lett. 2010;20:531–536. doi: 10.1016/j.bmcl.2009.11.100. [DOI] [PubMed] [Google Scholar]
- 44.Kuduk SD, Chang RK, Greshock TJ, Ray WJ, Ma L, Wittmann M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MT. Identification of Amides as Carboxylic Acid Surrogates for Quinolizidinone-Based M1 Positive Allosteric Modulators. ACS Med Chem Lett. 2012;3:1070–1074. doi: 10.1021/ml300280g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.(a) Kuduk SD, Chang RK, Di Marco CN, Ray WJ, Ma L, Wittmann M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MT. Quinolizidinone carboxylic acids as CNS penetrant, selective m1 allosteric muscarinic receptor modulators. ACS Med Chem Lett. 2010;1:263–267. doi: 10.1021/ml100095k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kuduk SD, Di Marco CN, Cofre V, Pitts DR, Ray WJ, Ma L, Wittmann M, Veng L, Seager MA, Koeplinger K, Thompson CD, Hartman GD, Bilodeau MT. Nheterocyclic derived M1 positive allosteric modulators. Bioorg Med Chem Lett. 2010;20:1334–1337. doi: 10.1016/j.bmcl.2010.01.013. [DOI] [PubMed] [Google Scholar]; (c) Kuduk SD, Di Marco CN, Cofre V, Ray WJ, Ma L, Wittmann M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MT. Fused heterocyclic M1 positive allosteric modulators. Bioorg Med Chem Lett. 2011;21:2769–2772. doi: 10.1016/j.bmcl.2010.10.028. [DOI] [PubMed] [Google Scholar]
- 46.(a) Puri V, Wang X, Vardigan JD, Kuduk SD, Uslaner JM. The selective positive allosteric M1 muscarinic receptor modulator PQCA attenuates learning and memory deficits in the Tg2576 Alzheimer’s disease mouse model. Behav Brain Res. 2015;287:96–99. doi: 10.1016/j.bbr.2015.03.029. [DOI] [PubMed] [Google Scholar]; (b) Uslaner JM, Eddins D, Puri V, Cannon CE, Sutcliffe J, Chew CS, Pearson M, Vivian JA, Chang RK, Ray WJ, Kuduk SD, Wittmann M. The muscarinic M1 receptor positive allosteric modulator PQCA improves cognitive measures in rat, cynomolgus macaque, and rhesus macaque. Psychopharmacology. 2013;225:21–30. doi: 10.1007/s00213-012-2788-8. [DOI] [PubMed] [Google Scholar]
- 47.(a) Kuduk SD, Dimarco CN, Yang Z-Q. Patent WO 2012158474. N-linked quinolineamide M1 receptor positive allosteric modulators. 2012 May 11;; (b) Kuduk SD, Beshore DC, Yang Z, Shu Y. Fused isoindolone M1 receptor positive allosteric modulators and their preparation and use for the treatment of M1-mediated diseases. Patent WO2012003147. 2011 Jun 27;
- 48.Mistry SN, Jorg M, Lim H, Vinh NB, Sexton PM, Capuano B, Christopoulos A, Lane RJ, Scammells PJ. 4-Phenylpyridin-2-one Derivatives: A Novel Class of Positive Allosteric Modulator of the M1 Muscarinic Acetylcholine Receptor. J Med Chem. 2016;59:388–409. doi: 10.1021/acs.jmedchem.5b01562. [DOI] [PubMed] [Google Scholar]
- 49.Marlo JE, Niswender CM, Luo Q, Brady AE, Shirey JK, Rodriguez AL, Bridges TM, Williams R, Days E, Nalywajko NT, Austin C, Williams M, Xiang Y, Orton D, Brown HA, Kim K, Lindsley CW, Weaver CD, Conn PJ. Identification and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of activity. Mol Pharm. 2009;75:577–588. doi: 10.1124/mol.108.052886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bridges TM, Kennedy JP, Cho HP, Conn PJ, Lindsley CW. Chemical optimization of anM1, M3, M5 positive allosteric modulator (PAM) lead. Part II. Development of a highly selective M1 PAM. Bioorg Med Chem Lett. 2010;20:1972–1975. doi: 10.1016/j.bmcl.2010.01.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Melancon BJ, Poslusney MS, Gentry PR, Tarr JC, Mattmann ME, Bridges TM, Utley TJ, Sheffler DJ, Daniels JS, Niswender CM, Conn PJ, Lindsley CW, Wood MR. Isatin replacements applied to the highly selective, muscarinic M1 PAM ML137: the continued optimization of an MLPCN probe molecule. Bioorg Med Chem Lett. 2013;23:412–416. doi: 10.1016/j.bmcl.2012.11.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bridges TM, Kennedy JP, Cho HP, Conn PJ, Lindsley CW. Chemical optimization of anM1, M3, M5 positive allosteric modulator (PAM) lead. Part I. Development of a highly selective M5 PAM. Bioorg Med Chem Lett. 2010;20:558–562. doi: 10.1016/j.bmcl.2009.11.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Panarese JD, Cho HP, Adams JJ, Nance KD, Garcia-Barrantes PM, Chang S, Morrison RD, Blobaum AL, Niswender CM, Stauffer SR, Conn PJ, Lindsley CW. Further optimization of the M1 PAM VU0453595: Discovery of novel heterobicyclic core motifs with improved CNS penetration. Bioorg Med Chem Lett. 2016;26:3822–3825. doi: 10.1016/j.bmcl.2016.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reid PR, Bridges TM, Sheffler DA, Cho HP, Lewis LM, Days E, Daniels JS, Jones CK, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Wood MR. Discovery and optimization of a novel, selective and brain penetrant M1 positive allosteric modulator (PAM): the development of ML169, an MLPCN Probe. Bioorg Med Chem Lett. 2011;21:2697–2701. doi: 10.1016/j.bmcl.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tarr JC, Turlington ML, Reid PR, Utely TJ, Sheffler DJ, Cho HP, Klar R, Pancani T, Klein MT, Bridges TM, Morrison RD, Xiang Z, Daniels SJ, Niswender CM, Conn PJ, Wood MR, Lindsley CW. Targeting selective activation of M1 for the treatment of Alzheimer’s disease: further chemical optimization and pharmacological characterization of the M1 positive allosteric modulators ML169. ACS Chem Neurosci. 2012;3:884–895. doi: 10.1021/cn300068s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lindsley CW, Conn PJ, Wood MR, Tarr JC, Bridges TM. Indole compounds as positive allosteric modulators of the muscarinic receptor. US 8,772,509 US Patent. 2014
- 57.(a) Bridges TM, Rook JM, Noetzel MJ, Morrison RD, Zhou Y, Gogliotti RD, Vinson PN, Jones CK, Niswender CM, Lindsley CW, Stauffer SR, Conn PJ, Daniels JS. Biotransformation of a novel positive allosteric modulator of metabotropic glutamate receptor subtype 5 contributes to seizures in rats involving a receptor agonism-dependent mechanism. Drug Metab Dispos. 2013;41:1703–1714. doi: 10.1124/dmd.113.052084. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Noetzel MJ, Gregory KJ, Vinson JT, Manka SR, Stauffer SR, Daniels SJ, Lindsley CW, Niswender CM, Xiang Z, Conn PJ. A novel metabotropic glutamate receptor 5 positive allosteric modulator acts at a unique site and confers stimulus bias to mGlu5 signaling. Mol Pharmacol. 2013;83:835–847. doi: 10.1124/mol.112.082891. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rook JM, Xiang Z, Lv X, Ghosal A, Dickerson J, Bridges TM, Johnson KA, Bubser M, Gregory KJ, Vinson PN, Byun N, Stauffer SR, Daniels JS, Niswender CM, Lavreysen H, Mackie C, Conde-Ceide S, Alcazar J, Bartolome JM, Macdondald GJ, Steckler T, Jones CK, Lindsley CW, Conn PJ. Biased mGlu5 positive allosteric modulators provide in vivo efficacy without potentiating mGlu5 modulation of NMDAR currents. Neuron. 2015;86:1029–1040. doi: 10.1016/j.neuron.2015.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hamilton SE, Loose MD, Qi M, Levey AI, Hille B, McKnight GS, Idzerda RL, Nathanson NM. Disruption of the m1 receptor gene ablates muscarinic receptor-dependent M current regulation and seizure activity in mice. Proc Natl Acad Sci U S A. 1997;94:13311–13316. doi: 10.1073/pnas.94.24.13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.(a) Payne A, Castro-Pineiro JL, Birch LM, Khan A, Braunton AJ, Kitulagoda JE, Soejima M. Patent WO 15049574. Preparation of 4-azaindole derivatives as muscarinic M1 receptor modulators for treatment of cognitive deficits. 2015 Apr 2;; (b) Ballard TM, Flohr A, Groebke-Zbinden K, Pinard E, Rychmans T, Schaffhauser H. WO15028483. Preparation of pyrrolopyridine and pyrazolopyridine derivatives as muscarinic M1 receptor modulators. 2015 Mar 5;; (c) Ballard TM, Groebke-Zbinden K, Pinard E, Rychmans T, Schaffhauser H. Preparation of indole and indazole derivatives as muscarinic M1 receptor pos. allosteric modulators. PatentWO15044072. 2015 Apr 2;
- 60.Davoren JE, O’Neil SV, Anderson DP, Brodney MA, Chenard L, Dlugolenski K, Edgerton JR, Green M, Garnsey M, Grimwood S, Harris AR, Kauffman GW, LaChapelle E, Lazzaro JT, Lee CW, Lotarski SM, Nason DM, Obach RS, Reinhart V, Salomon-Ferrer R, Steyn SJ, Webb D, Yan J, Zhang L. Design and optimization of selective azaindole amide M1 positive allosteric modulators. Bioorg Med Chem Lett. 2016;26:650–655. doi: 10.1016/j.bmcl.2015.11.053. [DOI] [PubMed] [Google Scholar]
- 61.Woolley ML, Carter HJ, Gartlon JE, Watson JM, Dawson LA. Attenuation of amphetamine-induced activity by the non-selective muscarinic receptor agonist, xanomeline, is absent in muscarinic M4 receptor knockout mice and attenuated in muscarinic M1 receptor knockout mice. Eur J Pharmacol. 2009;603:147–149. doi: 10.1016/j.ejphar.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 62.Bubser M, Bridges TM, Dencker D, Gould RW, Grannan M, Noetzel MJ, Lamsal A, Niswender CM, Daniels JS, Poslusney MS, Melancon BJ, Tarr JC, Byers FW, Wess J, Duggan ME, Dunlop J, Wood MW, Brandon NJ, Wood MR, Lindsley CW, Conn PJ, Jones CK. Selective activation of M4 muscarinic acetylcholine receptors reverses MK-801-induced behavioral impairments and enhances associative learning in rodents. ACS Chem Neurosci. 2014;5:920–942. doi: 10.1021/cn500128b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Byun NE, Grannan M, Bubser M, Barry RL, Thompson A, Rosanelli J, Gowrishankar R, Kelm ND, Damon S, Bridges TM, Melancon BJ, Tarr JC, Brogan JT, Avison MJ, Deutch AY, Wess J, Wood MR, Lindsley CW, Gore JC, Conn PJ, Jones CK. Antipsychotic drug-like effects of the selective M4 muscarinic acetylcholine receptor positive allosteric modulator VU0152100. Neuropsychopharmacology. 2014;39:1578–1593. doi: 10.1038/npp.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cruickshank JW, Brudzynski SM, McLachlan RS. Involvement of M1 muscarinic receptors in the initiation of cholinergically induced epileptic seizures in the rat brain. Brain Res. 1994;643:125–129. doi: 10.1016/0006-8993(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 65.Mingo NS, Cottrell GA, Mendonca A, Gombos Z, Eubanks JH, Burnham WM. Amygdala-kindled and electroconvulsive seizures alter hippocampal expression of the M1 and M3 muscarinic cholinergic receptor genes. Brain Res. 1998;810:9–15. doi: 10.1016/s0006-8993(98)00748-3. [DOI] [PubMed] [Google Scholar]
- 66.Davoren JE, Lee C-W, Garnsey M, Brodney MA, Cordes J, Dlugolenski K, Edgerton JR, Harris AR, Helal CJ, Jenkinson S, Kauffman GW, Kenakin TP, Lazzaro JT, Lotarski SM, Mao Y, Nason DM, Northcott C, Nottebaum L, O’Neil SV, Pettersen B, Popiolek M, Reinhart V, Salomon-Ferrer R, Steyn SJ, Webb D, Zhang L, Grimwood S. Discovery of the Potent and Selective M1 PAM-Agonist N-[(3R,4S)-3-Hydroxytetrahydro-2Hpyran-4-yl]-5-methyl-4-[4-(1,3-thiazol-4-yl)benzyl]pyridine-2-carboxamide (PF-06767832): Evaluation of Efficacy and Cholinergic Side Effects. J Med Chem. 2016;59:6313–6328. doi: 10.1021/acs.jmedchem.6b00544. [DOI] [PubMed] [Google Scholar]
- 67.For information, see: https://www.discoverx.com/.
- 68.For information, see: https://eurofins.com/.
- 69.See the Supporting Information for full Eurofin data.
- 70.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32(3):281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.