

Abstract
Fuel substrate supply and oxidative phosphorylation are key determinants of muscle performance. Numerous studies of mammalian mitochondria are carried out (i) with substrate supply that limits electron flow, and (ii) far below physiological temperature. To analyze potentially implicated biases, we studied mitochondrial respiratory control in permeabilized mouse myocardial fibers using high-resolution respirometry. The capacity of oxidative phosphorylation at 37 °C was nearly two-fold higher when fueled by physiological substrate combinations reconstituting tricarboxylic acid cycle function, compared with electron flow measured separately through NADH to Complex I or succinate to Complex II. The relative contribution of the NADH pathway to physiological respiratory capacity increased with a decrease in temperature from 37 to 25 °C. The apparent excess capacity of cytochrome c oxidase above physiological pathway capacity increased sharply under hypothermia due to limitation by NADH-linked dehydrogenases. This mechanism of mitochondrial respiratory control in the hypothermic mammalian heart is comparable to the pattern in ectotherm species, pointing towards NADH-linked mt-matrix dehydrogenases and the phosphorylation system rather than electron transfer complexes as the primary drivers of thermal sensitivity at low temperature. Delineating the link between stress and remodeling of oxidative phosphorylation is important for understanding metabolic perturbations in disease evolution and cardiac protection.
Introduction
Contractile activity in cardiac muscle mainly depends on mitochondrial (mt) energy transformed by oxidative phosphorylation (OXPHOS). The heart is highly sensitive to defects in OXPHOS1, stress-induced mitochondrial cytopathies and degenerative mitochondrial defects, including heart failure2, 3, acute ischemia and myocardial infarct4, ischemia-reperfusion5, type 2 diabetes6–9, aging10, 11, and inherited genetic diseases12–15. Patients present with functional impairment when the capacity of an enzyme is reduced below its threshold activity. This threshold effect is a function of the apparent excess enzyme activity above pathway capacity. To evaluate the threshold and excess capacity of a single step in OXPHOS, it is necessary not only to quantify the changes in enzyme activity, but determine the impact of these changes on respiratory pathway capacity16.
Respiration in the mammalian heart is supported by carbohydrates (10 to 40%)17 and fatty acids (60 to 90%)18. Electron transfer in the NADH- and succinate-linked pathway (NS-pathway) converges through Complex I and Complex II at the Q-junction19 (Fig. 1a). Downstream electron flow is catalyzed by Complex III and Complex IV (cytochrome c oxidase) to oxygen as the terminal electron acceptor. Conventional protocols in bioenergetics use either NADH-linked substrates (N-pathway) or succinate&rotenone (S-pathway), thereby separating the system into linear thermodynamic cascades, forming distinct electron transfer chains19–21. This experimental design aims at the measurement of biochemical coupling efficiency and proton stoichiometry, and is applied in the functional diagnosis of specific OXPHOS defects. As recognized in mitochondrial physiology, however, it does not allow estimation of maximal respiratory capacity under physiological conditions. Fuel substrates supporting convergent electron transfer at the Q-junction enhance respiratory capacity, as shown when succinate is added to NADH-linked substrates, reconstituting physiological tricarboxylic acid cycle function with combined NS-pathway flux. This effect of succinate varies depending on species, strains, organ and experimental conditions; stimulation is 1.6 to 2.0-fold in rat heart22, 23, 1.2 to 1.8-fold in rat skeletal muscle24–26, 1.4-fold in mouse skeletal muscle27, and 1.3 to 2.1-fold in human skeletal muscle (reviewed by Gnaiger28). Similarly, glycerol-3-phosphate (Fig. 1a) exerts an additive effect on respiration when combined with pyruvate&malate in rabbit skeletal muscle mitochondria29, and stimulates respiration beyond NS-pathway capacity in human lymphocytes30. Such substrate combinations do not exert completely additive effects on flux due to (i) intersubstrate competition for transport across the inner mt-membrane31, (ii) regulatory mechanisms in the tricarboxylate acid (TCA) cycle, and (iii) flux control by limiting enzyme capacities downstream of the Q-junction.
Figure 1.
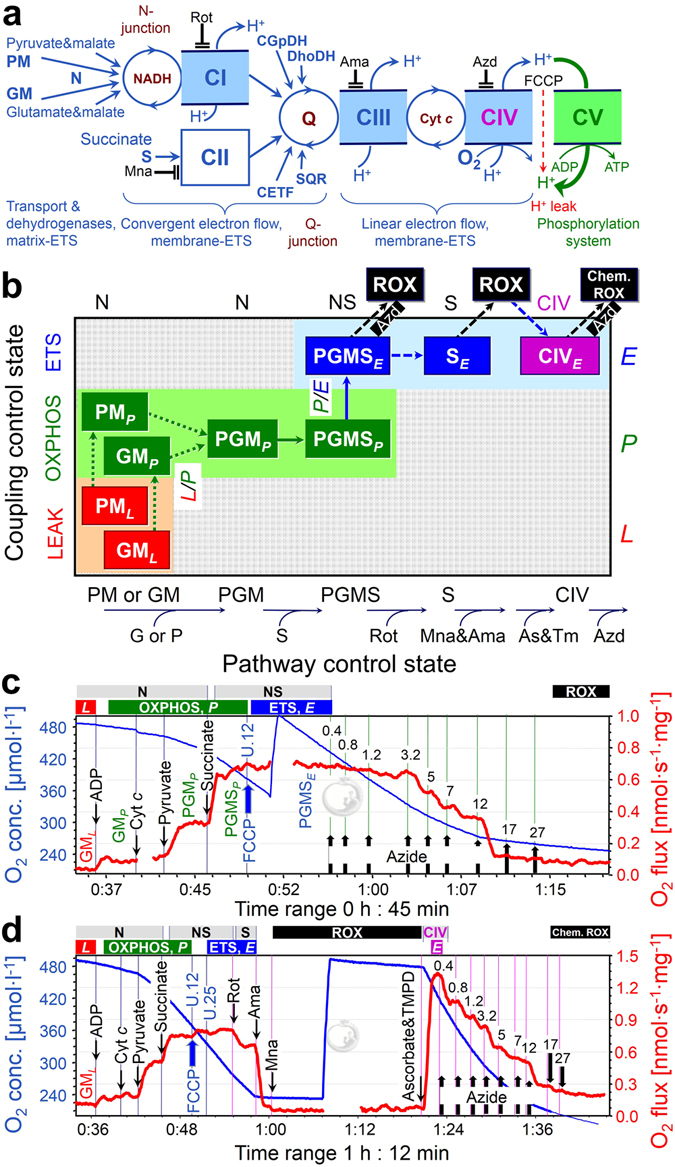
Mitochondrial pathways, substrate-uncoupler-inhibitor-titration (SUIT) protocols and respiration of permeabilized cardiac fibers. (a) Schematic representation of the electron transfer system (ETS) coupled to the phosphorylation system (ATP synthase, adenylate translocator and inorganic phosphate transporter). Electron flow from pyruvate&malate (PM) or glutamate&malate (GM) converges at the N-junction (NADH-cycle). Electrons converge at the Q-junction from Complex I (CI, NADH-ubiquinone oxidoreductase), Complex II (CII, succinate-ubiquinone oxidoreductase), glycerophosphate dehydrogenase Complex (CGpDH), electron-transferring flavoprotein Complex (CETF), dihydro-orotate dehydrogenase (DhoDH)92, sulfide-ubiquinone oxidoreductase (SQR)93, and choline dehydrogenase (not shown), followed by a linear downstream segment through Complexes III (CIII, ubiquinol-cytochrome c oxidoreductase) and CIV (cytochrome c oxidase), to the final electron acceptor oxygen. CI, CIII, and CIV are proton pumps generating an electrochemical potential difference across the inner mt-membrane. Coupling of the phosphorylation system with the ETS allows the proton potential to drive phosphorylation of ADP to ATP (coupled flow). Protonophores such as FCCP uncouple the ETS from ATP production. Rotenone, malonate and antimycin A are specific inhibitors of CI, CII and CIII, respectively, and were sequentially added at saturating concentrations. (b) Coupling/pathway control diagram illustrating the two protocols starting with either PM or GM (SUIT 1 and 2), convergent electron flow at the Q-junction in the NADH&succinate (NS) pathway, and azide titrations in the NS-pathway control state or single enzyme step of CIV. As&TM, Ascorbate&TMPD. (c) SUIT 2a with azide titration in the NS-pathway control state. (d) SUIT 2b with azide titration in the CIV single enzyme step as a basis of threshold plots.
With NADH-linked substrates, the major flux control of the N-pathway resides upstream in the dehydrogenases of the TCA cycle, with a high apparent excess capacity of respiratory complexes downstream16. Similarly, upstream electron supply limits respiratory capacity in the succinate-pathway. In the physiological state of combined NS-electron supply, flux control is shifted downstream. We therefore examined the apparent excess capacity of cytochrome c oxidase (CIV) over convergent NS-pathway flux, and determined the limitation of NS-OXPHOS capacity by the phosphorylation system (Fig. 1a). Mammalian mitochondria are frequently studied at 25 or 30 °C32. Temperature coefficients are used to extrapolate the results to 37 °C, but are available for only a few metabolic states33. We therefore determined respiration of mouse heart mitochondria in a variety of coupling/pathway control states at 37 °C and different levels of hypothermia34, i.e. at 30 °C (mild hypothermia), 25 °C (moderate hypothermia) and profound hypothermia (cold storage temperature, 4 °C), and extended our study to hyperthermia (40 °C). Moderate to profound hypothermia reduce myocardial metabolism temporarily and reversibly, and limit ischemic damage during cardiac surgery35–39, organ preservation40, 41, and preservation of mitochondrial function during preparation of isolated mitochondria and permeabilized fibers42.
Understanding the control of cell respiration in health and disease requires (i) measurement of mitochondrial pathway control at physiologically and clinically relevant temperatures and (ii) application of substrate combinations which are representative of cellular conditions. These criteria dramatically change our estimation of maximal respiratory capacity and control of mitochondrial respiration, providing a reference for physiological and pharmacological intervention strategies. Our results also provide new perspectives on evolutionary temperature adaptation, suggesting that key control of energy transformation and survival at low temperatures might not primarily be exerted by electron transfer complexes but by (i) upstream elements of the electron transfer system (ETS) including transport of substrates across the inner mitochondrial membrane and matrix dehydrogenases, and (ii) downstream elements of the phosphorylation system.
Results
Pathway control of mitochondrial respiratory capacity
We quantified mitochondrial respiratory capacity in substrate-uncoupler-inhibitor-titration (SUIT) protocols (Figs 1 and 2). Fuel substrates of the N-pathway reduce NAD+ to NADH at five key steps: pyruvate dehydrogenase, isocitrate dehydrogenase, glutamate dehydrogenase, oxoglutarate dehydrogenase and malate dehydrogenase. Addition of pyruvate (P) to glutamate&malate (GM) increased OXPHOS capacity (PGMP) by a factor of 2.5 (2.0–3.7) at 37 °C (Fig. 2a,b). In contrast, addition of glutamate (G) to pyruvate&malate (PM) exerted merely a slight stimulatory effect on respiration by a factor of 1.1 (1.0–1.2; Fig. 2b), also observed in rat heart (1.2)43 and skeletal muscle of the horse (1.2)44, rabbit (1.2)29 and humans (1.3)45. OXPHOS capacity (ADP-stimulated oxygen flux) was significantly higher with pyruvate&malate compared to glutamate&malate at physiological temperature (PMP versus GMP; Fig. 2b). This pattern was reversed at 4 °C, under which conditions N-OXPHOS capacity was higher with GM than PM (Fig. 2c).
Figure 2.
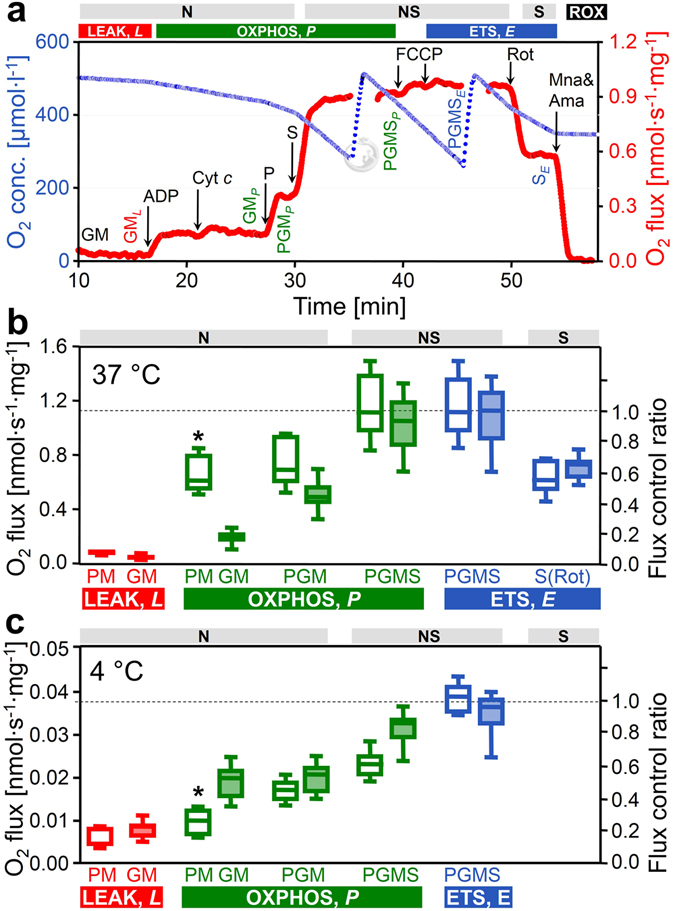
Respiration of permeabilized cardiac fibers (J O2, per wet weight), and flux control ratios normalized for NS-ETS capacity. (a) Oxygen consumption measured at 37 °C in SUIT 2 protocol: GM (N-LEAK respiration; GML), ADP (N-OXPHOS capacity; GMP), cytochrome c (integrity of outer mt-membrane), pyruvate (N-OXPHOS capacity; PGMP), succinate (NS-OXPHOS capacity; PGMSP), FCCP (NS-ETS capacity; PGMSE), rotenone (Rot; inhibition of CI, S-ETS capacity; SE), malonic acid and antimycin A (Mna and Ama; less than 2% residual oxygen consumption, ROX). (b,c) ROX-corrected respiration at 37 °C and 4 °C: LEAK respiration and OXPHOS capacity with PM (SUIT 1, open boxes, n = 7 and 6 at 37 and 4 °C) or GM (SUIT 2, filled boxes, n = 16 and 8 at 37 and 4 °C). Note the switch of the flux control pattern for N-OXPHOS capacity (PMP vs. GMP). The two SUIT protocols merge at state PGMP, but results are shown separately (not significantly different). Flux control ratios are normalized relative to median PGMSE of the combined protocols. Box plots indicate the minimum, 25th percentile, median, 75th percentile, and maximum. *Significant differences between the two SUIT protocols for the same state. For abbreviations see Fig. 1.
In isolated mitochondria or permeabilized fibers incubated with PM, GM or PGM, citrate and 2-oxoglutarate are formed and rapidly exchanged for malate by the tricarboxylate and 2-oxoglutarate carriers, thus limiting the formation of succinate. In addition, succinate is lost into the incubation medium through the active dicarboxylate carrier exchanging succinate for inorganic phosphate. The high malate concentration equilibrates with fumarate, inducing product inhibition of succinate dehydrogenase (CII). Taken together, this limits succinate-linked electron transfer46. To simultaneously activate CII and NADH-related dehydrogenases of the TCA cycle, a high exogenous succinate concentration is required in addition to the NADH-linked substrates47, 48, thus simulating the physiological condition of the NS-pathway with convergent electron flow into the Q-junction (Fig. 1a). In the NS-pathway control state (pyruvate&glutamate&malate&succinate; PGMSP, Fig. 1b), respiration almost doubled compared to OXPHOS capacity measured separately through the N- or S-pathway. The respiratory capacity through the S-pathway was similar to that observed with the NADH-linked substrate combinations. This provides evidence for an additive effect of convergent electron flow, expressed in terms of flux control ratios of N/NS = 0.53 (0.40–0.69), and S/NS = 0.61 (0.52–0.82) at 37 °C (Fig. 2b). This additive effect was less pronounced at 4 °C (Fig. 2c), but was similar between 25 °C and 40 °C (Fig. 3).
Figure 3.
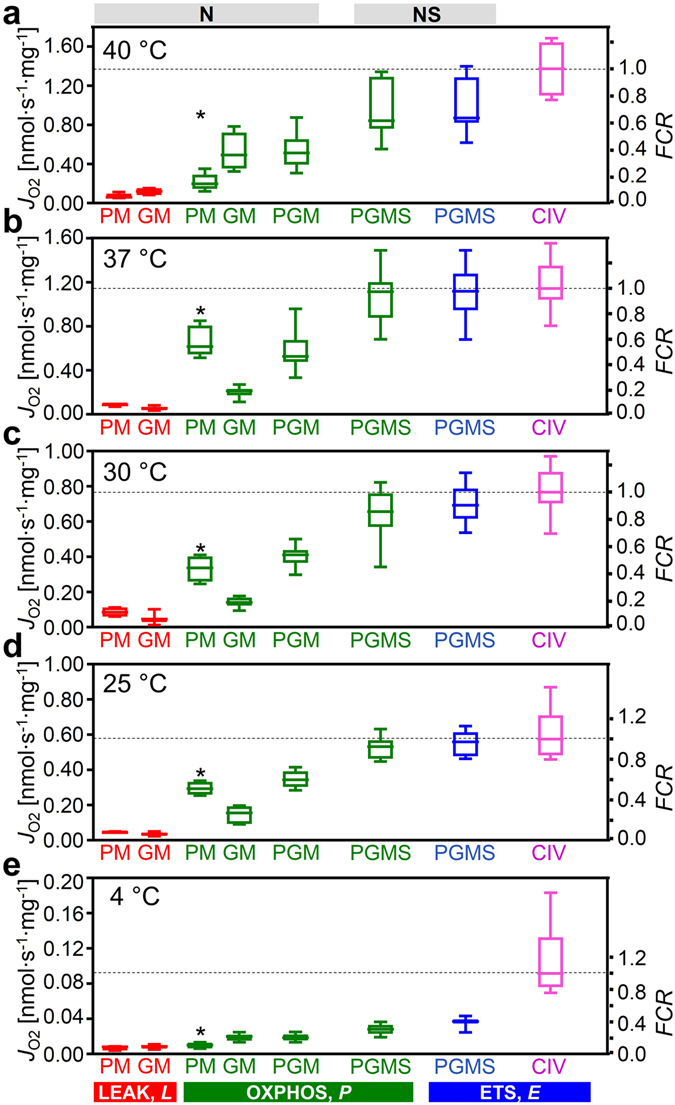
Respiration of permeabilized cardiac fibers (J O2, per wet weight), and flux control ratios, FCR, normalized for CIV activity at different temperatures. NL and NP with PM (SUIT 1; n = 4–7) or GM (SUIT 2; n = 5–16); NP with PGM, NSP and NSE with PGMS (both protocols combined; n = 9–23); normalized relative to median CIVE (n = 4–16). *Significant differences between the two SUIT protocols for the same state. For box plots and abbreviations see Figs 1 and 2.
Temperature sensitivity of respiration depends on metabolic state
We determined temperature coefficients for evaluation of metabolic depression by hypothermia and specific remodeling of pathway control of OXPHOS (Table 1). Mitochondrial content, evaluated by the mitochondrial matrix marker citrate synthase activity, did not differ among tissue preparations. Citrate synthase activity [IU per mg fibers] measured at 30 °C was 0.25 (0.17–0.30), 0.23 (0.16–0.30), 0.24 (0.19–0.26), and 0.22 (0.18–0.28) in the 40, 37, 30 and 25 °C cohort, respectively. Therefore, divergences in mitochondrial content can be ruled out as a confounding factor. NS-OXPHOS capacity declined at 30 and 25 °C by 1.7- and 2.1-fold of the normothermic level (37 °C), and decreased slightly from 37 to 40 °C (Fig. 3; Table 1). The thermal sensitivity of respiration was strongly dependent on coupling/pathway control states (Fig. 4). The Q 10 is the factor by which the reaction velocity increases for a rise in temperature of 10 °C. It varied between 1.2 and 2.4 across metabolic states in the temperature range of 25 to 37 °C (Fig. 4b; Table 1). The Q 10 for S-ETS capacity was 1.9 between 30 and 37 °C (Table 1), similar to results reported for heart mitochondria from guinea pigs (2.3) and rabbits (1.9)33. In this temperature range, Q 10 values were close to 2.0 for NS-OXPHOS and ETS capacity, but an unexpectedly low Q 10 of 1.4 was observed for PGMP. The N/NS flux control ratio (PGM/PGMS) increased with a decrease of temperature, from 0.53 (0.40–0.69) at 37 °C to 0.66 (0.58–0.72) at 25 °C, and remained high at 0.68 (0.55–0.79) at 4 °C (Fig. 2c).
Table 1.
Effect of temperature on mitochondrial respiration as a function of metabolic state in permeabilized mouse heart fibers.
| State | Q 10 (x MF) from T 1 to T 2 | ||||
|---|---|---|---|---|---|
| 4 °C to 25 °C | 25 °C to 37 °C | 30 °C to 37 °C | 37 °C to 40 °C | ||
| CIVE0 | extrapolated | 1.84 (x 3.61) | 1.66 (x 1.83) | 2.17 (x 1.72) | 0.39 (x 0.76) |
| CIVE | measured | 2.40 (x 6.27) | 1.77 (x 1.99) | 1.77 (x 1.49) | 1.84 (x 1.20) |
| NSE | PGMSE | 3.64 (x 15.1) | 1.78 (x 2.00) | 1.98 (x 1.61) | 0.43 (x 0.78) |
| NSP | PGMSP | 4.06 (x 19.0) | 1.85 (x 2.10) | 2.13 (x 1.70) | 0.39 (x 0.76) |
| SE | PGMS (Rot)E | — | 1.93 (x 2.20) | 2.19 (x 1.73) | 0.25 (x 0.66) |
| NP | PGMP | 4.03 (x 18.7) | 1.42 (x 1.53) | 1.43 (x 1.28) | 0.91 (x 0.97) |
| NP | PMc P | 5.01 (x 29.5) | 1.86 (x 2.10) | 2.36 (x 1.82) | 0.48 (x 0.80) |
| NP | PMP | 4.24 (x 20.7) | 1.95 (x 2.22) | 2.39 (x 1.84) | 0.55 (x 0.84) |
| NP | GMc P | 2.67 (x 7.85) | 1.29 (x 1.36) | 1.79 (x 1.51) | 0.78 (x 0.93) |
| NP | GMP | 2.49 (x 6.81) | 1.39 (x 1.48) | 2.07 (x 1.67) | 0.65 (x 0.88) |
| NL | PML | 2.23 (x 5.41) | 1.76 (x 1.97) | 1.01 (x 1.01) | 3.13 (x 1.41) |
| NL | GML | 1.93 (x 3.99) | 1.45 (x 1.54) | 1.23 (x 1.16) | 1.38 (x 1.10) |
Q 10 is the multiplication factor for an increase by 10 °C, calculated for respiratory flux, J, in the experimental temperature intervals, T 1 to T 2. Numbers in parentheses are multiplication factors (MF = J T2/J T1) to convert J from T 1 to T 2. Q 10 and MF are medians (n = 4–16).
Q 10 = MF exp (10/(T 2 − T 1)). Cytochrome c oxidase activity was measured with 2 mM ascorbate and 500 µM TMPD (CIVE) or extrapolated from threshold plots (CIVE0; Fig. 5). For abbreviations see Figs 1 and 2.
Figure 4.
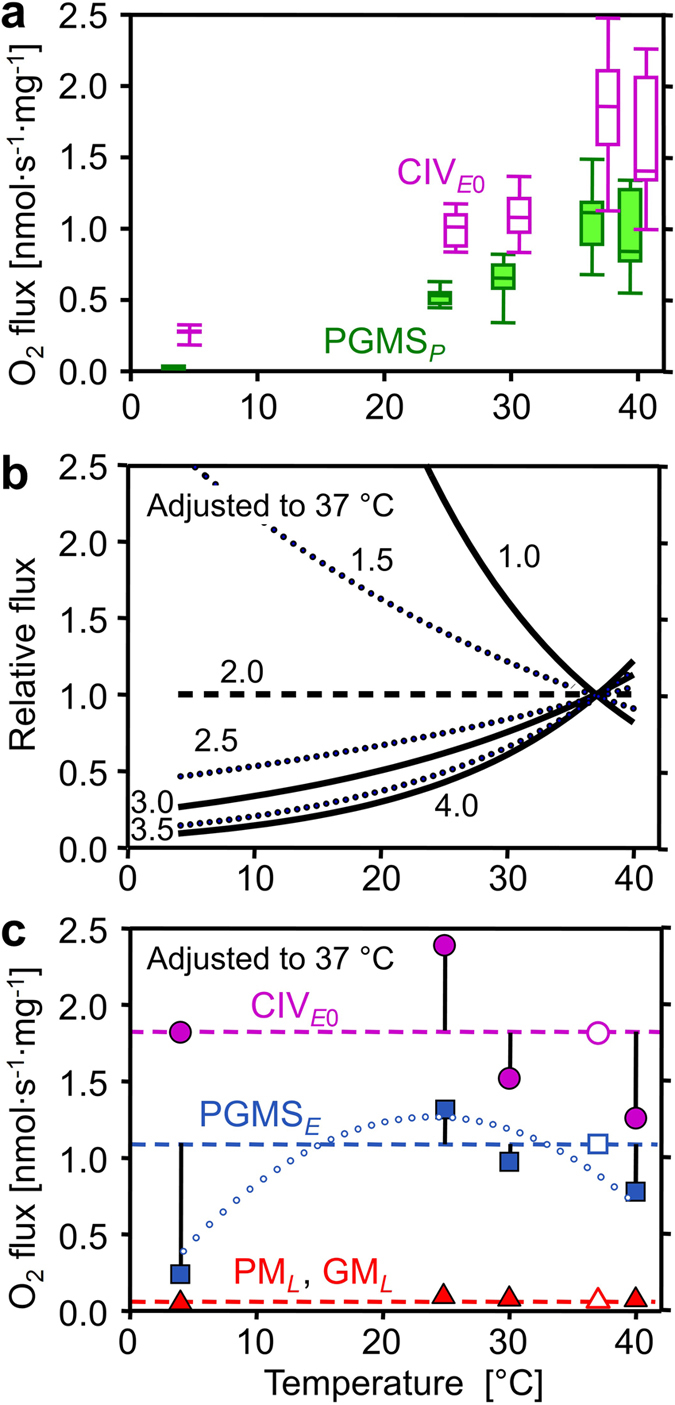
Effect of temperature on mitochondrial respiration. (a) Mass-specific oxygen flux (J O2) in respiratory states PGMSP (filled boxes) and CIVE0 extrapolated from the threshold plots in Fig. 5 (empty boxes). (b) Respiratory flux (j O2) relative to a simple temperature reference model: the reference flux is defined at 37 °C as 1.0 and at other temperatures as 1.0 if Q 10 is constant at 2.0 (horizontal dashed line). Non-linear deviations from standard conditions (full and dotted lines) are obtained when Q 10 differs from 2.0 (Q 10 shown by numbers) and is constant throughout the entire temperature range. (c) j O2 relative to standard temperature correction of flux at 37 °C (Q 10 = 2.0; horizontal lines): CIVE0 (● extrapolated from the threshold plots, Fig. 5), ETS capacity (■ PGMSE), and LEAK respiration (▲ pooled GML and PML). Vertical bars show deviations of experimental results (means, n = 5 to 14) from the theoretical line for a Q 10 of 2.0. The dotted trend line illustrates the change of temperature sensitivity for ETS, particularly at 4 °C. For box plots and abbreviations see Figs 1 and 2.
OXPHOS at 4 °C was only 3% of normothermic flux. This pronounced metabolic arrest resulted from the high thermal sensitivity of respiration with pyruvate (Table 1). Q 10 for OXPHOS capacity with pyruvate&malate and PGMS increased up to 4 and 5 under hypothermia from 25 to 4 °C. OXPHOS capacity with glutamate&malate was much less temperature dependent, with a Q 10 of 1.4 from 37 to 25 °C, and 2.7 from 25 to 4 °C. The OXPHOS capacity with PM was more than 2-fold higher than for GM at physiological temperature, but was depressed at 4 °C to a level even below GM-supported respiration (Fig. 2c).
Electron transfer system capacity and coupling
Uncoupler titrations were performed in the ADP-activated state, to evaluate ETS capacity (E, noncoupled) in relation to OXPHOS capacity (P, coupled). P and E were numerically almost identical, indicating that the capacity of the phosphorylation system did not exert a limiting effect on respiration, with control located mainly at the level of the dehydrogenases (Fig. 1a). LEAK respiration is an acronym for the resting oxygen flux compensating for proton leak, proton slip and cation cycling. LEAK respiration is an inverse function of the proton/electron stoichiometry49 and is therefore higher for the S- versus N-pathway with two and three coupling sites, respectively. LEAK respiration, L, was measured in the presence of PM or GM before addition of ADP (NL) and OXPHOS capacity was obtained after stimulation by ADP (NP). OXPHOS coupling efficiencies19, 1-L/P, were independent of temperature in the range of 25 to 40 °C, and higher for PM than GM: 0.85 (0.78–0.89), 0.73 (0.57–0.80) and 0.82 (0.79–0.82) for PM, compared to 0.72 (0.54–0.81), 0.64 (0.39–0.89) and 0.69 (0.67–0.85) for GM at 37 °C, 30 °C, and 25 °C, respectively. At 4 °C, OXPHOS coupling efficiencies declined with GM to 0.58 (0.47–0.73), but even more with PM to 0.48 (0.28–0.60). The Q 10 of LEAK respiration with GM and PM remained close to 2.0 down to 4 °C (Fig. 4b; triangles).
Apparent cytochrome c oxidase excess capacity
Since electron transfer capacity is limited when electrons are supplied only through the N-pathway, the major control resides under these conditions upstream in the dehydrogenases of the TCA cycle, with a correspondingly high apparent excess capacity of respiratory complexes downstream (for review see ref. 16). In physiological states with simultaneous NS-electron flow, the apparent excess capacity of downstream electron transfer is lower and flux control is shifted towards CIII and CIV. We therefore examined the apparent excess capacity of CIV at maximum convergent pathway flux through the ETS. Azide titrations resulted in a hyperbolic inhibition of CIV (Fig. 5a–e). The threshold plots display NS-pathway flux as a function of CIV activity (Fig. 5f–j). The two distinct phases are related to (1) the elimination of excess capacity above the threshold (initial slope; dotted lines) and (2) the flux control coefficient below the threshold, where further inhibition of CIV causes a linear inhibition of pathway flux (full lines). Inhibition of CIV activity to 41% of controls exerted only a minor effect on respiratory capacity (37 °C; Fig. 5g). The apparent excess capacity of CIV, j ExCIV (for definition see Fig. 5), at 37 °C was significant (median 0.72, range 0.28–1.11) with reference to convergent NS-electron flow, which provides the basis for a low flux control coefficient of CIV and a high functional threshold (Fig. 5g). CIV excess capacities based on threshold plots were 0.6 to 0.8 at 40 to 25 °C (Fig. 5f–i). The steep increase of j ExCIV to 6.2 at 4 °C (Fig. 5j) was associated with the high temperature sensitivity of ETS capacity upstream of CIV (Fig. 3e).
Figure 5.
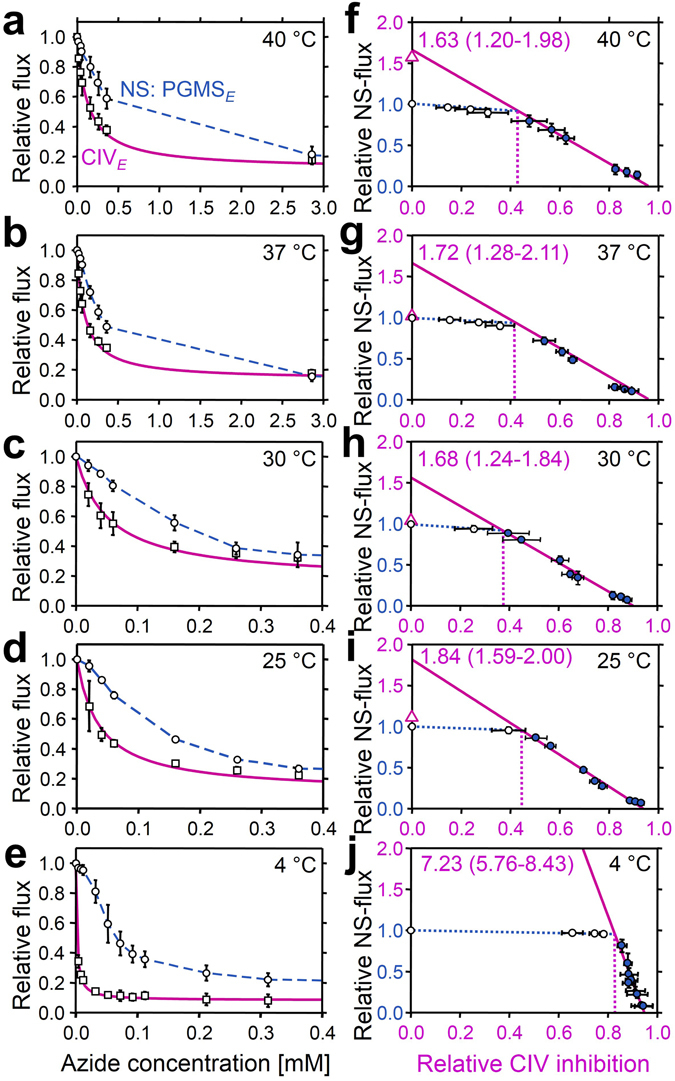
Azide titration and Complex IV threshold in permeabilized cardiac fibers at 40 to 4 °C. (a to e) Effect of azide titration on relative NS-pathway ETS capacity (PGMSE; circles, dashed line: linear interpolations) and velocity of the single enzyme cytochrome c oxidase (CIVE; squares, solid line: hyperbolic fit). (f to j) Threshold plots of relative NS-pathway flux as a function of relative inhibition of CIV at identical azide concentrations. Data up to the threshold of inhibition are shown by open symbols. The CIVE0/NSE flux ratio is calculated as the intercept at zero CIV inhibition of a linear regression through the data above the inflection point (closed symbols; r 2 ≥ 0.99). CIVE0/NSE values are listed in the graphs as medians (min to max). The apparent excess capacity of CIV is j ExCIV = CIVE0/NSE − 1. Triangles on Y axes show medians of relative CIV activities measured directly with ascorbate and TMPD (CIVE/NSE = 3.0 at 4 °C; not shown). The threshold of inhibition is at the intercept between the linear regression and the extrapolated line drawn from the control to the first inhibited flux (dotted vertical lines). Circles are means ± SD (n = 4–5).
Discussion
Mitochondrial respiration in the living cell is supported by fuel substrates supplying electrons to multiple dehydrogenases followed by convergent electron entry into the Q-junction. Physiological respiratory capacity is underestimated in isolated mitochondria and permeabilized fibers when using simple substrate combinations such as pyruvate&malate supporting the NADH-pathway (N), or a single substrate for the succinate-pathway (S). N-OXPHOS capacity (PGMP = 0.52 nmol O2·s−1 mg−1 wet weight) from the present study of permeabilized cardiac fibers of the mouse (Fig. 2b), is in the same order of magnitude as ex vivo maximal myocardial oxygen consumption measured with cardiac perfusion after stimulation to maximal workload (0.2–0.7 nmol O2·s−1 mg−1 wet weight)50, 51. However, PGMP reached only 54% of NS-OXPHOS capacity (PGMSP = 1.02 nmol O2·s−1 · mg−1 wet weight), which actually exceeds maximal oxygen consumption of the perfused working heart. Similarly, oxygen consumption of the perfused dog heart has been compared with N-OXPHOS capacity of isolated mitochondria expressed per tissue mass (0.42 and 0.50 nmol O2∙s−1 mg−1 wet weight, respectively)52. Under conditions of separate N- or S-pathway control, flux is limited artificially by selective substrate supply and electron gating, thus effectively under-utilizing the apparent excess capacity of respiratory complexes downstream19 and shifting flux control to dehydrogenases upstream53. As expected on the basis of this respiratory flux control pattern, the artificial excess capacity of CIV with respect to N-OXPHOS capacity is high (2.2; unpublished observation), similar to artificial CIV excess capacities in permeabilized fibers from human skeletal muscle54 and isolated mitochondria from rat heart and skeletal muscle (1.0 to 2.0)55, 56. Our data provide a rationale suggesting that these high apparent CIV excess capacities represent in vitro experimental artifacts which can be easily avoided. The additive effect of convergent electron flow has profound consequences for optimization of mitochondrial respiratory control. The apparent CIV excess capacity was lower when related to maximum ETS capacity of the NS-pathway (0.7 at 37 °C; Fig. 5), consistent with apparent CIV excess capacities in intact, uncoupled cells (0.0 to 0.4)57–59 and permeabilized human skeletal muscle fibers with NS-pathway control (0.4)54. Therefore, previously suggested discrepancies of CIV excess capacities in isolated mitochondria versus permeabilized fibers or intact cells54, 57, 58 can be explained by NS- versus N- or S-pathway control states in mt-preparations and the concept of the Q-junction. Rocher et al.60 indicated that the quantity of mtDNA in human cell lines is tightly correlated to CIV activity. Importantly, apparent excess of catalytic capacity does not signify that it is not functionally required, considering the importance for high affinity of mitochondria to oxygen55, 61 and the role of CIV in the control of cytochrome reduction levels62.
Respiratory complexes form supercomplexes modulated by assembly factors, contributing to respiratory control and optimization of cellular metabolism63. Electrons are channeled through supercomplexes (CI + III and CI + III + IV in the mouse heart)64, 65 which restrict exchange with the free Q- and cytochrome c-pools, thus limiting random collisions64, 66, 67. Under conditions of tight electron channeling, activation of an additional convergent pathway would exert a completely additive effect on ETS capacity. Our results provide evidence against maximally tight supercomplex channeling in the mouse heart, since the combined NS-pathway capacity was significantly less than predicted from a completely additive effect of the N- and S-pathway fluxes measured separately. Two other mechanisms could reduce the additive effect on ETS capacity: (i) substrate competition for transport across the inner mt-membrane31, and (ii) limitation of electron transfer by deficient enzyme capacities downstream of the Q-cycle in the absence of tight channeling. The additive effect on flux of convergent electron transfer at the Q-junction varies between species, strains, tissues, age, and pathophysiological conditions, unraveling an unexpected diversity of mitochondrial respiratory control patterns28.
The phosphorylation system represents another functional unit potentially contributing to the limitation of OXPHOS capacity, P, relative to ETS capacity, E. The extent of this limitation, E − P, is expressed by the excess E − P capacity factor, j ExP = (E − P)/E = 1 − P/E, ranging from zero (no limitation) to the upper limit of 1.019. j ExP was very low but significantly different from zero in mouse heart (0.03 at 25, 37, and 40 °C, and 0.05 at 30 °C; P < 0.05, pooled protocols; Fig. 3), while it is zero in mouse skeletal muscle27 and rat heart23, and 0.04 and 0.02 in rat soleus and extensor digitorum longus muscle, respectively26. In human skeletal muscle, however, the limitation by the phosphorylation system is highly significant, j ExP(NS) = 0.05 to 0.27, 68 and j ExP(N) = 0.2 with glutamate&malate as substrates69, and is even more pronounced in the human heart, with j ExP(N) increasing from 0.52 in healthy controls to 0.59 in heart failure2.
Temperature exerts complex effects on coupling control of mitochondrial respiration70. Proton leak is a property of the inner mt-membrane and depends on mt-membrane potential, whereas proton slip is a property of the proton pumps and depends on enzyme turnover. Fierce controversies on proton leak versus slip, which collectively control LEAK respiration, could be resolved by considering physiological (37 °C) versus conventional ‘bioenergetic’ temperature (25 °C)70. Temperature coefficients vary between different enzyme-catalyzed reactions involved in mitochondrial respiration (Table 1). Relatively small differences of Q 10 among the different enzymes of a pathway may have dramatic impact on metabolic organization and OXPHOS remodeling over a wide range of temperatures. The excess E − P capacity factor in the mouse heart increased at 4 °C to 0.36 (0.33–0.51) and 0.09 (0.03–0.16) in SUIT 1 and 2, respectively (Fig. 2c), consistent with an increase of the flux control coefficient of the phosphorylation system at low temperature in rat liver mitochondria70, 71. Furthermore, the apparent CIV excess capacity in mouse heart increased strikingly at 4 °C compared to normothermic values (Fig. 5). The excess of CIV relative to NS-pathway capacity increases in peritoneal but not in alveolar murine macrophage-derived cell lines at 25 °C compared to 37 °C72. Although a decline of CI activity with temperature is possibly involved in the shift of CIV excess capacity72, our results suggest a different mechanism. The highest OXPHOS Q 10 of 5.0 was obtained with pyruvate&malate between 4 and 25 °C, in contrast to the lower Q 10 with glutamate&malate (Table 1). Both substrate combinations fuel the N-pathway with electron entry into the Q-junction through CI. Hence the activities of CI and downstream respiratory complexes cannot explain the observed hypothermic response pattern. The most likely candidates responsible for the high temperature sensitivity of the pyruvate&malate pathway, therefore, are pyruvate dehydrogenase, the pyruvate transporter and possibly citrate synthase. This is supported by a previous study on rat heart mitochondria showing a high thermal sensitivity of pyruvate-supported respiration and activity of pyruvate dehydrogenase at low temperature73.
The thermal sensitivity of pyruvate supported respiration was not as pronounced in a cold adapted fish species (Anarhicas lupus)74, suggesting that pyruvate dehydrogenase plays not only an important role in the response of OXPHOS to temperature in the murine heart73, but is also a potential site of key adaptation to upgrade mitochondrial capacity at low temperature in ectotherm species75. This is in line with evidence of control of convergent OXPHOS at upstream steps of electron supply in Fundulus heteroclitus heart mitochondria76, 77. A higher control of the phosphorylation system is observed at lower temperature (17 °C compared to 25 °C) in permeabilized heart fibers from the triplefin fish (Forsterygion lapillum)78. N- or S-pathway capacity appeared to be more affected by temperature changes at lower temperature compared to CIV in the freshwater turtle Trachemys scripa 79. Similarly, permeabilized muscle fibers from Drosophila simulans show an increase of CIV excess capacity at low temperature80. Taken together, NADH-linked mt-matrix dehydrogenases and the phosphorylation system rather than electron transfer complexes appear to be the primary modulators of respiratory control patterns at low temperature in mitochondria from endotherm and ectotherm species.
The conventional respiratory acceptor control ratio, RCR (State 3/State 4 or P/L ratio)20 was 6.7 with PM (37 °C), and much lower (3.6) with GM. For statistical and conceptual reasons, the RCR is replaced by the OXPHOS coupling efficiency19, 1 − L/P = 1-RCR−1. The low OXPHOS coupling efficiency for GM of 0.72 (compared to 0.85 for PM) reflects the low OXPHOS capacity supported by specific substrates, rather than low coupling efficiency. Evaluation of coupling must be based on pathway control states supporting a high ETS capacity under conditions not limited by substrates or by the phosphorylation system. Hence, pyruvate&malate supporting a ‘fast’ N-pathway at 37 °C yields a more appropriate index of coupling. Under deep hypothermia (4 °C), however, estimation of coupling is complicated by the strong thermal sensitivity of ETS capacity compared to LEAK respiration (Fig. 3b) and by the limitation imposed on OXPHOS capacity by the phosphorylation system.
Our results imply that relevant experimental models of pathophysiological metabolic states can be established by reconstitution of bioenergetic pathways in mitochondrial preparations19. In addition to the core bioenergetic N- and S-pathway, fatty acid oxidation contributes to convergent electron flow to the Q-junction through the electron transferring flavoprotein complex2, 19 (Fig. 1). In human hearts, ATP demand is fueled up to 50–70% by fatty acid oxidation, predominating over glucose oxidation (see review81). The temperature dependence of substrate competition versus additivity of mitochondrial fatty acid and glucose-linked oxidative capacity, therefore, will represent an important extension of the present study. In addition, the convergent glycerophosphate pathway (Fig. 1) and the malate-aspartate shuttle play key roles in linking the redox biochemistry of the cytosolic and mitochondrial compartments. Mapping the additive effects of convergent bioenergetic pathways on respiratory capacities sets the stage to integrate further analyses of respiratory states (mitochondrial membrane potential, cytochrome redox states82) and generation of reactive oxygen species into a systems analysis of mitochondrial respiratory control.
Estimation of maximal respiratory capacity should be performed at physiological temperature and with substrate combinations appropriate to ensure suitable operation of the TCA cycle and multiple entries of electrons into the Q-cycle. Those conditions define the physiological reference state for mitochondrial respiratory control. This is of major significance to better explain the pathological effects of genetic mutations and acquired CIV deficiencies. Furthermore, our results on the key modulators of thermal sensitivity of mitochondrial metabolism will allow to determine how cells and tissues are impaired by temperature changes, including mild to deep hypothermia in cardiac surgery. This novel aspect of mitochondrial medicine may provide a basis for intervention strategies to limit damages72. Consideration of a standardized nomenclature for categories of hypothermia will improve conceptually the experimental design and reporting in the mitochondrial field, and facilitate translational research from mitochondrial physiology to the clinic34. Application of comprehensive OXPHOS analysis for diagnosis of mitochondrial preservation will contribute to optimize the degree of therapeutic hypothermia in the clinical setting, including cardiac arrest during surgery and resuscitation83, 84. From an evolutionary perspective, our study allows to pinpoint candidate loci that are potentially under selective pressure and therefore represent targets for seeking mechanisms of adaptation to different temperature regimes. In summary, we propose the hypothesis that the likely modulators of adaptation and acclimatization to low temperature are compensatory mechanisms to counteract upstream limitations of ETS capacity at the entry to and within the TCA cycle (particularly pyruvate dehydrogenase) and downstream limitation of flux by the phosphorylation system.
Methods
Preparation of permeabilized fibers
Adult male mice C57 BL/6 N were housed under standard conditions according to the Austrian Animal Care Law. At 8 to 10 weeks of age (23 ± 3 g), animals were anaesthetized with ketamine and xylazine (80 and 10 mg∙kg−1, respectively) given intramuscularly and checked for absence of the toe-pinch reflex before performing a cervical dislocation. The heart was excised and placed in 5 ml of ice-cold relaxing solution BIOPS2. After rapid mechanical permeabilization of the left ventricle (~40 mg wet weight), bundles of fibers were agitated gently (30 min, 4 °C) in BIOPS supplemented with 50 μg∙ml−1 saponin85. Fibers were washed by agitation (10 min, 4 °C) in mitochondrial respiration medium MiR0542, immediately blotted, weighed, and used for respirometric measurements.
High-resolution respirometry
Respiration was measured simultaneously in 10 respiration chambers (O2k; Oroboros Oxygraph-2k, Innsbruck, Austria), one O2k with two chambers for each of the following temperatures: 4, 25, 30, 37, and 40 °C. Permeabilized fibers (0.7–1.3 mg at 25 to 40 °C and 7–8 mg at 4 °C) were used in each chamber containing 2 ml of MiR05. Respiratory flux was expressed per mg wet weight of fibers. Instrumental and chemical oxygen background fluxes were calibrated as a function of oxygen concentration and subtracted from the total volume-specific oxygen flux (Datlab software, Oroboros Instruments)55, 82, 86. An oxygen regime of 500 to >200 µM was maintained at 30 to 40 °C, but up to 700 and 900 µM at 25 and 4 °C, to avoid artificial oxygen diffusion limitation of flux86, 87. In the first substrate-uncoupler-inhibitor titration protocol (SUIT 1), the following final concentrations were added sequentially: P (5 mM), M (5 mM), G (10 mM), ADP (1 mM), cytochrome c (10 μM), S (10 mM), FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone; optimum concentration, 0.125 to 0.375 μM), rotenone (Rot; 0.5 μM), antimycin A (Ama; 2.5 μM), malonic acid (Mna; 5 mM), ascorbate (As; 0.5 mM) and TMPD (N,N,N’,N’-tetramethyl-p-phenylenediamine; 2 mM). In SUIT 2 addition of P and G were inversed (Fig. 1b). An increase of respiration due to cytochrome c addition after ADP was observed at 30 to 40 °C, with cytochrome c control factors (change of respiration divided by cytochrome c stimulated respiration)19 in the range of 0.05 to 0.15, with higher values of 0.11 to 0.20 at 25 °C. At 4 °C, N-OXPHOS capacity showed a trend to decline during the experiment particularly with PM, and no stimulation could be observed with cytochrome c. Thus the integrity of the outer mitochondrial membrane in mouse heart permeabilized fibers was comparable to rat heart fibers studied at 30 °C88. Residual oxygen consumption (ROX), evaluated after inhibition of CI, CII and CIII with Rot, Mna and Ama was a small fraction (0.01 to 0.02) of NS-ETS capacity at 25 to 40 °C, but increased to 0.04 to 0.10 at 4 °C. Nevertheless, correction of fluxes in all respiratory states for ROX was significant, particularly in the resting state of LEAK respiration, when ROX was as high as 0.12 to 0.32 of total oxygen consumption in the N-LEAK state at 25 to 40 °C.
Apparent CIV excess capacities were determined by azide titrations of CIV activity and of NS-ETS capacity at 4, 25, 30, 37, and 40 °C. Threshold plots of relative respiration rate against the fraction of inhibited CIV activity at the same azide concentration were made as previously described57, 89. Azide titrations were performed at optimum uncoupler concentration supporting maximum flux, preventing the effect of inhibition of ATP synthase90 and eliminating any contribution of the phosphorylation system to flux control. The following azide concentrations were used [mM]: 0.02, 0.04, 0.06, 0.16, 0.26, 0.36, 2.9, 5.4, 10.4 between 25 and 40 °C, and 0.004, 0.008, 0.012, 0.032, 0.052, 0.072, 0.092, 0.11, 0.21, 0.31, 2.8, 5.3, 10.3 at 4 °C (not all points visible in Fig. 5 due to overlap).
The contents of the chambers were removed at the end of each experimental run and the chamber was rinsed twice with 500 μl of respiration medium. The fibers were homogenized for 2 × 30 s with an Ultra-Turrax homogenizer at maximum speed and immediately frozen in liquid nitrogen and stored at −80 °C for subsequent measurement of citrate synthase at 30 °C91.
Statistical analysis
Statistica software© was used for statistical analyses. Data were log transformed to meet the requirement for heteroscedasticity according to Levene’s test. A three-factor ANOVA (protocol, state and temperature) followed by a posteriori Tukey multiple comparison tests were used to test for differences between protocols at a specific state and temperature. To determine the effects of addition of substrates, cytochrome c, inhibitors, or uncoupler, a t-test for dependent samples was used. Significance was considered at P < 0.05. Results are presented without transformation as medians (min-max) unless specified otherwise.
Acknowledgements
We kindly appreciate the support by J.-C. Tardif and R. Margreiter. M. Schneider performed CS analysis. We thank C.L. Hoppel and B. Tandler for helpful comments. H.L. received scholarships from the Canadian NSERC and the Quebec FQRNT. Funding for this work was provided by an NSERC discovery grant to P.U.B. and H.L. (RGPIN 155926 and 402636), K-Regio project MitoFit (EG) and Oroboros Instruments. Contribution to COST Action CA15203 MITOEAGLE. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions
H.L. performed the experiments, analyzed the data, wrote the manuscript and was involved in funding acquisition. P.U.B. gave suggestions on the discussion section and was responsible for funding acquisition. E.G. designed the study, analyzed the data, wrote and revised the manuscript and was responsible for the resources and project administration.
Competing Interests
E.G. is founder and CEO of OROBOROS INSTRUMENTS, Innsbruck, Austria.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Schwerzmann K, Hoppeler H, Kayar SR, Weibel ER. Oxidative capacity of muscle and mitochondria: Correlation of physiological, biochemical, and morphometric characteristics. Proc. Natl. Acad. Sci. USA. 1989;86:1583–1587. doi: 10.1073/pnas.86.5.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lemieux H, Semsroth S, Antretter H, Höfer D, Gnaiger E. Mitochondrial respiratory control and early defects of oxidative phosphorylation in the failing human heart. Int. J. Biochem. Cell Biol. 2011;43:1729–1738. doi: 10.1016/j.biocel.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 3.Li P, et al. Mitochondrial respiratory dysfunctions of blood mononuclear cells link with cardiac disturbance in patients with early-stage heart failure. Sci. Rep. 2015;5:10229. doi: 10.1038/srep10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marín-García, J. Mitochondria and the heart (Springer Science+Business Media, Inc., 2005).
- 5.Kuznetsov AV, et al. Mitochondrial defects and heterogeneous cytochrome c release after cardiac cold ischemia and reperfusion. Am. J. Physiol.-Heart Circul. Physiol. 2004;286:H1633–H1641. doi: 10.1152/ajpheart.00701.2003. [DOI] [PubMed] [Google Scholar]
- 6.Schrauwen P, Hesselink MKC. Oxidative capacity, lipotoxicity and mitochondrial damage in type 2 diabetes. Diabetes. 2004;53:1412–1417. doi: 10.2337/diabetes.53.6.1412. [DOI] [PubMed] [Google Scholar]
- 7.Boushel R, et al. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia. 2007;50:790–796. doi: 10.1007/s00125-007-0594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bombicino SS, et al. Diabetes impairs heart mitochondrial function without changes in resting cardiac performance. Int. J. Biochem. Cell Biol. 2016;S1357–2725:30280–30281. doi: 10.1016/j.biocel.2016.09.018. [DOI] [PubMed] [Google Scholar]
- 9.Warfel JD, et al. Mitochondrial fat oxidation is essential for lipid-induced inflammation in skeletal muscle in mice. Sci. Rep. 2016;28:37941. doi: 10.1038/srep37941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lesnefsky EJ, Chen Q, Hoppel CL. Mitochondrial metabolism in aging heart. Circ. Res. 2016;118:1593–1611. doi: 10.1161/CIRCRESAHA.116.307505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martín-Fernández B, Gredilla R. Mitochondria and oxidative stress in heart aging. Age (Dordr) 2016;8:225–238. doi: 10.1007/s11357-016-9933-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chinnery PF, Turnbull DM. Clinical features, investigation, and management of patients with defects of mitochondrial DNA. J. Neurol. Neurosurg. Psychiatry. 1997;63:559–563. doi: 10.1136/jnnp.63.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Kleist-Retzow JC, Schauseil-Zipf U, Michalk DV, Kunz WS. Mitochondrial diseases - an expanding spectrum of disorders and affected genes. Exp. Physiol. 2003;88:155–166. doi: 10.1113/eph8802509. [DOI] [PubMed] [Google Scholar]
- 14.Wallace DC. Mitochondrial defects in cardiomyopathy and neuromuscular disease. Am. Heart J. 2000;139:S70–85. doi: 10.1067/mhj.2000.103934. [DOI] [PubMed] [Google Scholar]
- 15.Sameni S, Syed A, Marsh JL, Digman MA. The phasor-FLIM fingerprints reveal shifts from OXPHOS to enhanced glycolysis in Huntington Disease. Sci. Rep. 2016;6:4755. doi: 10.1038/srep34755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rossignol R, et al. Mitochondrial threshold effects. Biochem. J. 2003;370:751–762. doi: 10.1042/bj20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gertz EW, Wisneski JA, Stanley WC, Neese RA. Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J. Clin. Invest. 1988;82:2017–2025. doi: 10.1172/JCI113822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Vusse GJ, Glatz JF, Stam HC, Reneman RS. Fatty acid homeostasis in the normoxic and ischemic heart. Physiol. Rev. 1992;72:881–940. doi: 10.1152/physrev.1992.72.4.881. [DOI] [PubMed] [Google Scholar]
- 19.Gnaiger, E. Mitochondrial pathways and respiratory control. An introduction to OXPHOS analysis. 4th ed. Mitochondr Physiol Network 19.12. OROBOROS MiPNet Publications, Innsbruck, 80 pp (2014).
- 20.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J. Biol. Chem. 1955;217:383–393. [PubMed] [Google Scholar]
- 21.Estabrook R. Mitochondrial respiratory control and the polarographic measurement of ADP:O ratios. Methods Enzymol. 1967;10:41–47. doi: 10.1016/0076-6879(67)10010-4. [DOI] [Google Scholar]
- 22.Costa LE, Boveris A, Koch OR, Taquini AC. Liver and heart mitochondria in rats submitted to chronic hypobaric hypoxia. Am. J. Physiol.-Cell Physiol. 1988;255:C123–C129. doi: 10.1152/ajpcell.1988.255.1.C123. [DOI] [PubMed] [Google Scholar]
- 23.Lemieux H, Vazquez EJ, Fujioka H, Hoppel CL. Decrease in mitochondrial function in rat cardiac permeabilized fibers correlates with the aging phenotype. J. Gerontol. A. Biol. Sci. Med. Sci. 2010;65:1157–1164. doi: 10.1093/gerona/glq141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garait B, et al. Fat intake reverses the beneficial effects of low caloric intake on skeletal muscle mitochondrial H2O2 production. Free Radic. Biol. Med. 2005;39:1249–1261. doi: 10.1016/j.freeradbiomed.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 25.Llesuy S, et al. Oxidative stress in muscle and liver of rats with septic syndrome. Free Rad. Biol. Med. 1994;16:445–451. doi: 10.1016/0891-5849(94)90121-X. [DOI] [PubMed] [Google Scholar]
- 26.Warren BE, et al. Early mitochondrial dysfunction in glycolytic muscle, but not oxidative muscle, of the fructose-fed insulin-resistant rat. Am. J. Physiol. Endocrinol. Metab. 2014;306:E658–E667. doi: 10.1152/ajpendo.00511.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aragonés J, et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat. Genet. 2008;40:170–180. doi: 10.1038/ng.2007.62. [DOI] [PubMed] [Google Scholar]
- 28.Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle. New perspectives of mitochondrial physiology. Int. J. Biochem. Cell. Biol. 2009;41:1837–1845. doi: 10.1016/j.biocel.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 29.Jackman MR, Willis WT. Characteristics of mitochondria isolated from type I and type IIb skeletal muscle. Am. J. Physiol.-Cell Physiol. 1996;39:C673–C678. doi: 10.1152/ajpcell.1996.270.2.C673. [DOI] [PubMed] [Google Scholar]
- 30.Pecina P, et al. Noninvasive diagnostics of mitochondrial disorders in isolated lymphocytes with high resolution respirometry. Biochim. Biophys. Acta Clinical. 2014;2:62–71. doi: 10.1016/j.bbacli.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haslam JM, Krebs HA. Substrate competition in the respiration of animal tissues. The metabolic interactions of pyruvate and alpha-oxoglutarate in rat liver homogenates. Biochem. J. 1963;86:432–446. doi: 10.1042/bj0860432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.N’Guessan B, et al. Evaluation of quantitative and qualitative aspects of mitochondrial function in human skeletal and cardiac muscles. Mol. Cell. Biochem. 2004;256:267–280. doi: 10.1023/B:MCBI.0000009874.14649.ca. [DOI] [PubMed] [Google Scholar]
- 33.Lindenmayer GE, Sordahl LA, Schwartz A. Reevaluation of oxidative phosphorylation in cardiac mitochondria from normal animals and animals in heart failure. Circ. Res. 1968;23:439–450. doi: 10.1161/01.RES.23.3.439. [DOI] [PubMed] [Google Scholar]
- 34.Yan TD, et al. Consensus on hypothermia in aortic arch surgery. Ann. Cardiothorac. Surg. 2013;2:163–168. doi: 10.3978/j.issn.2225-319X.2013.03.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hale SL, Kloner RA. Myocardial hypothermia: A potential therapeutic technique for acute regional myocardial ischemia. J. Cardiovasc. Electrophysiol. 1999;10:405–413. doi: 10.1111/j.1540-8167.1999.tb00689.x. [DOI] [PubMed] [Google Scholar]
- 36.Ning XH, Chen SH. Mild hypothermic cross adaptation resists hypoxic injury in hearts: A brief review. Chin. J. Physiol. 2006;49:213–222. [PubMed] [Google Scholar]
- 37.Schwartz DS, et al. Regional topical hypothermia of the beating heart: preservation of function and tissue. Ann. Thorac. Surg. 2001;72:804–809. doi: 10.1016/S0003-4975(01)02822-3. [DOI] [PubMed] [Google Scholar]
- 38.Tissier R, et al. Rapid cooling preserves the ischaemic myocardium against mitochondrial damage and left ventricular dysfunction. Cardiovasc. Res. 2009;83:345–353. doi: 10.1093/cvr/cvp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang CH, et al. Activation of mitochondrial STAT-3 and reduced mitochondria damage during hypothermia treatment for post-cardiac arrest myocardial dysfunction. Basic Res. Cardiol. 2015;110:59. doi: 10.1007/s00395-015-0516-3. [DOI] [PubMed] [Google Scholar]
- 40.Jahania MS, Sanchez JA, Narayan P, Lasley RD, Mentzer RMJ. Heart preservation for transplantation: principles and strategies. Ann. Thorac. Surg. 1999;68:1983–1987. doi: 10.1016/S0003-4975(99)01028-0. [DOI] [PubMed] [Google Scholar]
- 41.Schipper DA, et al. The Critical Role of Bioenergetics in Donor Cardiac Allograft Preservation. J. Cardiovasc. Transl. Res. 2016;9:176–183. doi: 10.1007/s12265-016-9692-2. [DOI] [PubMed] [Google Scholar]
- 42.Gnaiger, E. et al. In Life in the cold (eds G. Heldmaier & M. Klingenspor) 431–442 (Springer Berlin Heidelberg, 2000).
- 43.Digerness SB, Reddy WJ. The malate-aspartate shuttle in heart mitochondria. J. Mol. Cell. Cardiol. 1976;8:779–785. doi: 10.1016/0022-2828(76)90084-5. [DOI] [PubMed] [Google Scholar]
- 44.Lemieux H, et al. Mitochondrial function is altered in horse atypical myopathy. Mitochondrion. 2016;30:35–41. doi: 10.1016/j.mito.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 45.Winkler-Stuck K, et al. Re-evaluation of the dysfunction of mitochondrial respiratory chain in skeletal muscle of patients with Parkinsons disease. J. Neural Transm. 2005;112:499–518. doi: 10.1007/s00702-004-0195-y. [DOI] [PubMed] [Google Scholar]
- 46.Lemasters JJ. The ATP-to-oxygen stoichiometries of oxidative phosphorylation by rat liver mitochondria. J. Biol. Chem. 1984;259:13123–13130. [PubMed] [Google Scholar]
- 47.Rasmussen HN, Rasmussen UF. Small scale preparation of skeletal muscle mitochondria, criteria of integrity, and assays with reference to tissue function. Mol. Cell. Biochem. 1997;174:55–60. doi: 10.1023/A:1006851705996. [DOI] [PubMed] [Google Scholar]
- 48.Rasmussen UF, et al. Aerobic metabolism of human quadriceps muscle: in vivo data parallel measurements on isolated mitochondria. Am. J. Physiol.-Endocrinol. Metab. 2001;280:E301–E307. doi: 10.1152/ajpendo.2001.280.2.E301. [DOI] [PubMed] [Google Scholar]
- 49.Brand MD, Chien LF, Ainscow EK, Rolfe DF, Porter RK. The causes and functions of mitochondrial proton leak. Biochim. Biophys. Acta. 1994;1187:132–139. doi: 10.1016/0005-2728(94)90099-X. [DOI] [PubMed] [Google Scholar]
- 50.Yan J, et al. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation. 2009;119:2818–2828. doi: 10.1161/CIRCULATIONAHA.108.832915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boudina S, et al. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 52.Mootha VK, Arai AE, Balaban RS. Maximum oxidative phosphorylation capacity of the mammalian heart. Am. J. Physiol.-Heart Circul. Physiol. 1997;41:H769–H775. doi: 10.1152/ajpheart.1997.272.2.H769. [DOI] [PubMed] [Google Scholar]
- 53.Ainscow EK, Brand MD. Top-down control analysis of ATP turnover, glycolysis and oxidative phosphorylation in rat hepatocytes. Eur. J. Biochem. 1999;263:671–685. doi: 10.1046/j.1432-1327.1999.00534.x. [DOI] [PubMed] [Google Scholar]
- 54.Kunz WS, et al. Flux control of cytochrome c oxidase in human skeletal muscle. J. Biol. Chem. 2000;275:27741–27745. doi: 10.1074/jbc.M007296200. [DOI] [PubMed] [Google Scholar]
- 55.Gnaiger E, Lassnig B, Kuznetsov A, Reiger G, Margreiter R. Mitochondrial oxygen affinity, respiratory flux control and excess capacity of cytochrome c oxidase. J. Exp. Biol. 1998;201:1129–1139. doi: 10.1242/jeb.201.8.1129. [DOI] [PubMed] [Google Scholar]
- 56.Rossignol R, Malgat M, Mazat JP, Letellier T. Threshold effect and tissue specificity - Implication for mitochondrial cytopathies. J. Biol. Chem. 1999;274:33426–33432. doi: 10.1074/jbc.274.47.33426. [DOI] [PubMed] [Google Scholar]
- 57.Villani G, Attardi G. In vivo control of respiration by cytochrome c oxidase in wild-type and mitochondrial DNA mutation-carrying human cells. Proc. Natl. Acad. Sci. USA. 1997;94:1166–1171. doi: 10.1073/pnas.94.4.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Villani G, Greco M, Papa S, Attardi G. Low reserve of cytochrome c oxidase capacity in vivo in the respiratory chain of a variety of human cell types. J. Biol. Chem. 1998;273:31829–31836. doi: 10.1074/jbc.273.48.31829. [DOI] [PubMed] [Google Scholar]
- 59.Dalmonte ME, et al. Control of respiration by cytochrome c oxidase in intact cells: role of the membrane potential. J. Biol. Chem. 2009;284:32331–32335. doi: 10.1074/jbc.M109.050146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rocher C, et al. Influence of mitochondrial DNA level on cellular energy metabolism: implications for mitochondrial diseases. J. Bioenerg. Biomembr. 2008;40:59–67. doi: 10.1007/s10863-008-9130-5. [DOI] [PubMed] [Google Scholar]
- 61.Larsen FJ, et al. Mitochondrial oxygen affinity predicts basal metabolic rate in humans. FASEB J. 2011;25:2843–2852. doi: 10.1096/fj.11-182139. [DOI] [PubMed] [Google Scholar]
- 62.Harrison DK, Fasching M, Fontana-Ayoub M, Gnaiger E. Cytochrome redox states and respiratory control in mouse and beef heart mitochondria at steady-state levels of hypoxia. J. Appl. Physiol. 2015;119:1210–1218. doi: 10.1152/japplphysiol.00146.2015. [DOI] [PubMed] [Google Scholar]
- 63.Cogliati S, et al. Mechanism of super-assembly of respiratory complexes III and IV. Nature. 2016;539:579–582. doi: 10.1038/nature20157. [DOI] [PubMed] [Google Scholar]
- 64.Bianchi C, Genova ML, Castelli GP, Lenaz G. The mitochondrial respiratory chain is partially organized in a supercomplex assembly. Kinetic evidence using flux control analysis. J. Biol. Chem. 2004;279:36562–36569. doi: 10.1074/jbc.M405135200. [DOI] [PubMed] [Google Scholar]
- 65.Schagger H, Pfeiffer K. The ratio of oxidative phosphorylation complexes I-V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J. Biol. Chem. 2001;276:37861–37867. doi: 10.1074/jbc.M106474200. [DOI] [PubMed] [Google Scholar]
- 66.Hackenbrock CR, Chazotte B, Gupte SS. The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. J. Bioenerg. Biomembr. 1986;18:331–368. doi: 10.1007/BF00743010. [DOI] [PubMed] [Google Scholar]
- 67.Schägger H. Respiratory chain supercomplexes of mitochondria and bacteria. Biochim. Biophys. Acta. 2002;1555:154–159. doi: 10.1016/S0005-2728(02)00271-2. [DOI] [PubMed] [Google Scholar]
- 68.Pesta D, et al. Similar qualitative and quantitative changes of mitochondrial respiration following strength and endurance training in normoxia and hypoxia in sedentary humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011;301:R1078–R1087. doi: 10.1152/ajpregu.00285.2011. [DOI] [PubMed] [Google Scholar]
- 69.Rasmussen UF, Rasmussen HN. Human quadriceps muscle mitochondria: A functional characterization. Mol. Cell. Biochem. 2000;208:37–44. doi: 10.1023/A:1007046028132. [DOI] [PubMed] [Google Scholar]
- 70.Dufour S, Rousse N, Canioni P, Diolez P. Top-down control analysis of temperature effect on oxidative phosphorylation. Biochem. J. 1996;314:743–751. doi: 10.1042/bj3140743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Quentin E, Averet N, Rigoulet M, Guerin B. Temperature dependence of the coupling efficiency of rat liver oxidative phosphorylation - role of adenine nucleotide translocator. Biochem. Biophys. Res. Commun. 1994;202:816–821. doi: 10.1006/bbrc.1994.2003. [DOI] [PubMed] [Google Scholar]
- 72.Groeger M, et al. Temperature and cell-type dependency of sulfide effects on mitochondrial respiration. Shock. 2012;38:367–374. doi: 10.1097/SHK.0b013e3182651fe6. [DOI] [PubMed] [Google Scholar]
- 73.Lemieux H, Tardif JC, Blier PU. Thermal sensitivity of oxidative phosphorylation in rat heart mitochondria: Does pyruvate dehydrogenase dictate the response to temperature? J. Therm. Biol. 2010;35:105–111. doi: 10.1016/j.jtherbio.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 74.Lemieux H, Tardif JC, Dutil JD, Blier PU. Thermal sensitivity of cardiac mitochondrial metabolism in an ectothermic species from a cold environment, Atlantic wolffish (Anarhichas lupus) J. Exp. Mar. Biol. Ecol. 2010;384:113–118. doi: 10.1016/j.jembe.2009.12.007. [DOI] [Google Scholar]
- 75.Blier PU, Lemieux H, Pichaud N. Holding our breath in our modern world: will mitochondria keep the pace with global changes? Can. J. Zool. 2014;92:591–601. doi: 10.1139/cjz-2013-0183. [DOI] [Google Scholar]
- 76.Baris TZ, Blier PU, Pichaud N, Crawford DL, Oleksiak MF. Gene by environmental interactions affecting oxidative phosphorylation and thermal sensitivity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016;311:R157–R165. doi: 10.1152/ajpregu.00008.2016. [DOI] [PubMed] [Google Scholar]
- 77.Baris TZ, Crawford DL, Oleksiak MF. Acclimation and acute temperature effects on population differences in oxidative phosphorylation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016;310:R185–R196. doi: 10.1152/ajpregu.00421.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Khan JR, Iftikar FI, Hebert NA, Gnaiger E, Hickey AJ. Thermal plasticity of skeletal muscle mitochondrial activity and whole animal respiration in a common intertidal triplefin fish, Forsterygion lapillum (Family: Tripterygiidae) J. Comp. Physiol. B. 2014;184:991–1001. doi: 10.1007/s00360-014-0861-9. [DOI] [PubMed] [Google Scholar]
- 79.Galli GLJ, Richards JG. The effect of temperature on mitochondrial respiration in permeabilized cardiac fibres from the freshwater turtle. Trachemys scripta. J. Therm. Biol. 2012;37:195–200. doi: 10.1016/j.jtherbio.2011.12.012. [DOI] [Google Scholar]
- 80.Pichaud N, et al. Thermal sensitivity of mitochondrial metabolism in two distinct mitotypes of Drosophila simulans: evaluation of mitochondrial plasticity. J. Exp. Biol. 2010;213:1665–1675. doi: 10.1242/jeb.040261. [DOI] [PubMed] [Google Scholar]
- 81.Liu Y, Neumann D, Glatz JF, Luiken JJ. Molecular mechanism of lipid-induced cardiac insulin resistance and contractile dysfunction. Prostaglandins Leukot Essent Fatty Acids. 2016;S0952–3278:30082–30085. doi: 10.1016/j.plefa.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 82.Harrison DK, Fasching M, Fontana-Ayoub M, Gnaiger E. Cytochrome redox states and respiratory control in mouse and beef heart mitochondria at steady-state levels of hypoxia. J. Appl. Physiol. 2015;119:1210–1218. doi: 10.1152/japplphysiol.00146.2015. [DOI] [PubMed] [Google Scholar]
- 83.Gong P, et al. Hypothermia-induced neuroprotection is associated with reduced mitochondrial membrane permeability in a swine model of cardiac arrest. J. Cereb. Blood Flow Metab. 2013;33:928–934. doi: 10.1038/jcbfm.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wood T, et al. Treatment temperature and insult severity influence the neuroprotective effects of therapeutic hypothermia. Sci. Rep. 2016;6:23430. doi: 10.1038/srep23430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Veksler VI, Kuznetsov AV, Sharov VG, Kapelko VI, Saks VA. Mitochondrial respiratory parameters in cardiac tissue: a novel method of assessment by using saponin-skinned fibers. Biochim. Biophys. Acta. 1987;892:191–196. doi: 10.1016/0005-2728(87)90174-5. [DOI] [PubMed] [Google Scholar]
- 86.Gnaiger E, Steinlechner-Maran R, Méndez G, Eberl T, Margreiter R. Control of mitochondrial and cellular respiration by oxygen. J. Bioenerg. Biomemb. 1995;27:583–596. doi: 10.1007/BF02111656. [DOI] [PubMed] [Google Scholar]
- 87.Pesta D, Gnaiger E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012;810:25–58. doi: 10.1007/978-1-61779-382-0_3. [DOI] [PubMed] [Google Scholar]
- 88.Kuznetsov AV, Brandacher G, Steurer W, Margreiter R, Gnaiger E. Isolated rat heart mitochondria and whole rat heart as models for mitochondrial cold ischemia-reperfusion injury. Transplant. Proc. 2000;32:45. doi: 10.1016/S0041-1345(99)00869-6. [DOI] [PubMed] [Google Scholar]
- 89.Letellier T, Heinrich R, Malgat M, Mazat JP. The kinetic basis of threshold effects observed in mitochondrial diseases - A systemic approach. Biochem. J. 1994;302:171–174. doi: 10.1042/bj3020171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bowler MW, Montgomery MG, Leslie AGW, Walker JE. How azide inhibits ATP hydrolysis by the F-ATPases. Proc. Natl. Acad. Sci. USA. 2006;103:8646–8649. doi: 10.1073/pnas.0602915103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Srere PA. Citrate synthase. Methods Enzymol. 1969;13:3–11. doi: 10.1016/0076-6879(69)13005-0. [DOI] [Google Scholar]
- 92.Fang J, et al. Dihydro-orotate dehydrogenase is physically associated with the respiratory complex and its loss leads to mitochondrial dysfunction. Biosci. Rep. 2013;33:e00021. doi: 10.1042/BSR20120097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bouillaud F, Blachier F. Mitochondria and sulfide: a very old story of poisoning, feeding, and signaling? Antioxid. Redox Signal. 2011;15:379–391. doi: 10.1089/ars.2010.3678. [DOI] [PubMed] [Google Scholar]