

Abstract
The organization of eukaryotic DNA into nucleosomes and chromatin undergoes dynamic structure changes to regulate genome processing, including transcription and DNA repair. Critical chromatin rearrangements occur over a wide range of distances including the mesoscopic length scale of tens of nanometers. However, there is a lack of methodologies that probe changes over this mesoscopic length scale within chromatin. We have designed, constructed, and implemented a DNA-based nanocaliper that probes this mesoscopic length scale. We developed an approach of integrating nucleosomes into our nanocaliper at two attachment points with over 50% efficiency. Here, we focused on attaching the two DNA ends of the nucleosome to the ends of the two nanocaliper arms, so the hinge angle is a readout of the nucleosome end-to-end distance. We demonstrate that nucleosomes integrated with 6 bp, 26 bp and 51 bp linker DNA are partially unwrapped by the nanocaliper by an amount consistent with previously observed structural transitions. In contrast, the nucleosomes integrated with the longer 75 bp linker DNA remains fully wrapped. We found that the nanocaliper angle is a sensitive measure of nucleosome disassembly and can read out transcription factor (TF) binding to its target site within the nucleosome. Interestingly, the nanocaliper not only detects TF binding, but it significantly increases the probability of TF occupancy at its site by partially unwrapping the nucleosome. These studies demonstrate the feasibility of using DNA nanotechnology to both detect and manipulate nucleosome structure, which provides a foundation of future mesoscale studies of nucleosome and chromatin structural dynamics.
Keywords: DNA origami, nucleosome, mesoscale dynamics, transcription factors
Graphical abstract
Eukaryotic chromosomes are compacted into long chromatin fibers by repeatedly wrapping DNA into nucleosomes, the fundamental organizing unit of eukaryotic genomes.1, 2 The nucleosome contains 147 base pairs of DNA wrapped 1.65 times around a histone octamer core, which consists of an H3-H4 tetramer and two H2A-H2B heterodimers.3, 4 The structure and dynamics of these complexes regulate genomic DNA interactions with essential transcription,5 replication,6 and repair7 regulatory complexes. For example, nucleosomes regulate occupancy of transcription factors (TFs) within both promoters and enhancers, which are key gene regulatory regions that contain multiple nucleosomes and are at least tens of nanometers in size.8 TF target sequences are often located within nucleosomes,9 so TF binding to these sites within promoters and enhancers involves large scale DNA unwrapping and rearrangements of neighboring nucleosomes over mesoscopic lengths of 10 nm and greater. Nucleosome and chromatin structural changes occur spontaneously via thermal fluctuations10–12 and enzymatically by chromatin remodeling complexes,13, 14 both separately and in combination. Currently, there is a lack of methodologies that probe nucleosome and chromatin structural changes on length scales of tens of nanometers, yet this is a length scale over which essential regulatory DNA-protein interactions and chromatin conformational changes occur within gene regulatory regions.
Nucleosome dynamics has been extensively studied with Forster Resonance Energy Transfer (FRET), which has proven to be a highly effective method for investigating a wide range of nucleosome structural dynamics over the range of 3–8nm.15 These measurements have investigated thermally excited unwrapping fluctuations,16 nucleosome assembly/disassembly,17, 18 chromatin remodeling,19 protein binding within nucleosomes,20, 21 and H1 interactions with nucleosomes and chromatin.22, 23 However, larger mesoscopic structural changes that can often occur within gene regulatory regions are out of reach for FRET measurements. In addition, information on the structural transitions that cause changes in FRET efficiency is limited since the measurement reports on changes in the magnitude of the distance between the two fluorophores. FRET measurements usually do not provide information on the directionality or orientation of the structural change. Multiple label positions can help to determine the 3D direction of the structural change,24 but these are highly challenging measurements that are often difficult to interpret especially at the single molecule level.
There has also been great success in probing nucleosome and chromatin structural dynamics with single molecule force spectroscopy, including optical tweezers (OT),25–28 magnetic tweezers (MT),29–31 and atomic force microscopy (AFM).32 Here, a force is applied to the ends of a long (~1 μm) DNA molecule that contains one or more nucleosomes. These studies have determined the force required to increase length across the force attachment points, the interactions that stabilize single nucleosomes, chromatin compaction, and factors that regulate nucleosome and chromatin stability. However, in these measurements structural dynamics may be averaged over many nucleosomes within the DNA molecule and it is difficult to localize where structural changes occur within the sample. Furthermore, structural dynamics of the nucleosomes are typically convolved with the dynamics of a long flexible DNA tether necessary to anchor to a relatively large bead or a surface. Methods that directly probe structure and conformational changes over the mesoscopic length scale of tens of nanometers, which is larger than what FRET is sensitive to but smaller than what is probed by force spectroscopy, would provide synergistic information about nucleosome and chromatin structural dynamics and function.
Here, we report the development and implementation of a DNA nanodevice that is designed to probe nucleosome structure and stability on a length scale ranging over tens of nanometers. We constructed nanocalipers with scaffolded DNA origami,33 where each arm of the caliper can be attached to opposite ends of the DNA molecule that is wrapped into a nucleosome. Our attachment scheme couples the caliper angle to the end-to-end distance of the DNA wrapped into the nucleosome, such that the angle of the nanocaliper can be used as a readout of structural changes within the attached nucleosome. We optimized an assembly scheme to achieve over 50% integration efficiency of nucleosomes into a nanocaliper. We then used the nanocaliper angle as a reporter of DNA wrapped within the nucleosome. First, we found that increasing the DNA linker length results in increased caliper angle, demonstrating our ability to measure structures in the size range of 20–50nm. We then investigated the caliper angle sensitivity to monovalent salt induced nucleosome disassembly and found that as the nucleosome is disassembled the caliper angle significantly increases. Finally, we investigated the caliper angle sensitivity to a TF binding to its target site within the nucleosome that requires the DNA to partially unwrap from the histone octamer core. We found that the caliper angle detects TF binding to its target site. Interestingly, the concentration required for TF binding is much lower than that of free nucleosomes, suggesting that the nanocaliper is biasing the nucleosome to a more unwrapped and accessible conformation. These studies demonstrate the feasibility and promise of using DNA-based nanostructures for investigating nucleosome and chromatin structural dynamics to gain insight into DNA packaging and dynamic regulation occurring at mesoscopic length scales.
RESULTS
A single nucleosome can be efficiently integrated into a DNA origami nanocaliper
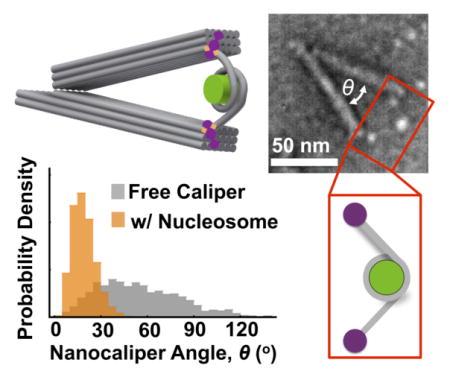
Structural DNA nanotechnology,34–36 and in particular scaffolded DNA origami,37–39 has enabled the construction of a wide range of highly complex nanostructures. More recently, a range of dynamic nanostructures have been developed33, 40–43 including hinges, pistons, and even containers that open in response to specific trigger molecules.44–47 Efforts to integrate dynamic DNA nanostructures with natural biomolecules have led to devices that can control bi-molecular interactions,42, 48 readout DNA or protein binding,49, 50 or probe DNA conformational changes.51, 52 Here, we developed a DNA device that reports on the structural dynamics of protein-DNA complexes on the scale of ~10–100nm. We use a recently developed DNA hinge (Figure 1)33 as a nanocaliper to detect changes in nucleosome structure through changes in the caliper angle. This nanocaliper exhibits a broad angular distribution, which ranges from 0 to 120 degrees (Figure 1c). The length of the caliper from the pivot point to the end of each arm is ~70 nm. So, the distance between the two ends of the caliper can vary from zero when the hinge is closed up to ~120nm when the hinge angle reaches 120°. This distance range is much larger than the range of FRET measurements. However, to use the nanocaliper to measure changes in the nucleosome structure, a nucleosome must be integrated into the origami device via two attachment points, one on each arm, so that the nucleosome structure changes are coupled to the nanocaliper angle.
Figure 1. Free nanocaliper characterization.

(a) TEM images revealed a range of angular conformations of the free DNA origami nanocalipers. (b) Zoomed in examples of nanocalipers were used to measure hinge angle, as indicated in the top left image. Scale bars = 50 nm. (c) The free nanocalipers exhibits a broad angular distribution ranging from 0° to ~120° with a relatively flat maximum ranging from ~25–60°.
The attachment approach (Figure 2) we used is based on a biotin-neutravidin-biotin linkage. We integrated into the nanocaliper single-stranded DNA extensions that positioned a biotin molecule at each end of the nanocaliper arms. We then separately prepared nucleosomes with DNA molecules that contained the 147 bp Widom-601 nucleosome positioning sequence (NPS)53 and additional linker DNA extending symmetrically out from the nucleosome (i.e. linker DNA). We made several nucleosome constructs with varying lengths of linker DNA: 6L (6 bp), 26L (26 bp), 51L (51 bp), and 75L (75 bp). The DNA was prepared with biotin attached to both 5 prime ends and then reconstituted into nucleosomes with purified histone octamer. Following the reconstitution, we incubated the nucleosomes with 5-fold molar excess neutravidin to saturate binding between neutravidin and biotin-labeled nucleosomes, and minimize neutravidin-mediated cross-linking between nucleosomes. Electrophoretic Mobility Shift Assays (EMSA) showed that all of the nucleosomes are bound by neutravidin and that they convert to a range of species that likely include multimers and loops of nucleosomes (Figure S1). We then imaged the neutravidin-bound nucleosome after sucrose gradient purification with Transmission Electron Microscopy (TEM) and found that a significant fraction of molecules were single nucleosomes bound with 2 neutravidin (Figure S2), and the excess neutravidin was efficiently purified away.
Figure 2. Assembly and integration of nucleosomes and nanocalipers.

(a) The top row schematically illustrates the assembly of nucleosome constructs starting with double biotin-labeled DNA that is reconstituted with purified histone octamer. The biotin molecules on either end of the reconstituted nucleosomes are saturated with neutravidin and then excess neutravidin is removed via sucrose gradient purification. The bottom row schematically illustrates the DNA origami self-assembly with biotin-labeled staples that ultimately form the two anchoring sites for the neutravidin-labeled nucleosomes. Excess staples are removed by PEG centrifugal purification. Finally, purified nucleosomes are integrated into nanocalipers in an equimolar assembly reaction. (b) Several examples of assembled complexes illustrate well-folded nanocalipers with two clearly incorporated neutravidin molecules with a visible nucleosome in between. Scale bar = 50 nm. (c) We achieve 55–62% good incorporation (purple) for varying constructs with different lengths of linker DNA. Our integration scheme also results in ~15–20% of nanocalipers that had a single nucleosome construct bound to only one arm (gray), ~10–15% of nanocalipers that had two separate constructs bound to each arm (pink), and ~5–13% of free nanocalipers (black) across the varying constructs.
To attach the nanocaliper to a neutravidin-labeled nucleosome, we incubated them at an equimolar ratio at room temperature in a physiological ionic strength buffer containing 50 mM Tris (pH 8.0) with 1 mM MgCl2 and 200 mM NaCl for 30 minutes. We then quantified the integration efficiency by analyzing TEM images of the samples (Figure 2b and Figure S3). The nanocalipers, nucleosomes, and neutravidin were all visible by TEM. Because the nucleosomes were pre-bound with neutravidin and the free neutravidin was purified away, the nanocalipers containing a nucleosome with a neutravidin located at the end of each nanocaliper arm is very likely a nanocaliper with a properly integrated nucleosome. TEM images revealed that over 50 percent of the calipers were attached to a single nucleosome through a biotin-neutravidin interaction at the end of each arm (Figure 2b, c). We also observed nanocalipers tethered to nucleosomes through single attachments (i.e. one nucleosome partially bound), nanocalipers tethered to two nucleosomes through single attachments (i.e. two nucleosomes partially bound), and free nanocalipers without a nucleosome at significantly lower frequencies (Figure 2c and S3). While some aggregation of nanocalipers was observed, many isolated single nanocalipers were easily identified. Furthermore, we did not observe nanocalipers with large nucleosome aggregates, which suggests that the large nucleosome aggregates did not integrate into the nanocaliper (Figure S3). These results demonstrate that the nanocaliper can be efficiently attached to a single nucleosome where opposite DNA ends of the nucleosome are separately attached to the ends of the caliper arms. Furthermore, the neutravidin attachments enable easy identification of calipers that contain a nucleosome attachment at the end of both caliper arms. This allows us to select by TEM the calipers that contain a single nucleosome with two attachment points.
The nanocaliper angle is an accurate measure of nucleosomal DNA end-to-end distance
The approach of using a nanocaliper to reliably measure distances depends on an accurate conversion from angle to distance. To determine if the nanocaliper angle is sensitive to a tethered nucleosome, we measured in TEM images the angle and neutravidin-neutravidin distance of nanocalipers that were doubly attached to nucleosomes that contained either a 6L, 26L, 51L, or 75L nucleosome. We analyzed at least 200 nanocalipers that were tethered to a single nucleosome with each linker DNA length. The angular distributions are significantly narrower than the angular distribution without a nucleosome, demonstrating that the nucleosome tethered across the arms constrains the nanocaliper angle (Figure 3a, S4, S5). The nanocaliper angle, θ, can be converted to an end-to-end distance, d, using the equation d = 2×Larm×sin(θ/2) where Larm is the length of the nanocaliper arms (68 nm). We determined if the distance inferred from the angle measurement is an accurate measure of end-to-end distance by comparing the average calculated and measured end-to-end distances for the 6L, 26L, 51L, and 75L nucleosomes (Figure 3b inset). We found that the calculated vs. measured average end-to-end distance fits to a line with a slope of 1.06 ± 0.09. In addition, scatter plots of calculated vs. measured end-to-end distances of individual nucleosome containing nanocalipers are highly correlated to a line with a slope of 1.08 ± 0.02, 1.01 ± 0.02, 0.99 ± 0.02 and 1.01 ± 0.02 for 6L, 26L, 56L and 75L nucleosome samples, respectively (Figure S6). These results demonstrate that the nucleosome end-to-end distance is accurately measured by the angle of individual nanocalipers.
Figure 3. The nanocaliper angle is a good measure of nucleosome structure.

(a) Nanocaliper angular distributions (orange) reveal a significant shift and strong narrowing upon integration of the 6L nucleosome relative to the free nanocalipers (gray). The 6L mean angle of 19° ± 2 is consistent with the conformation shown in the inset and also with the location of the nucleosome observed in TEM images (right). The 75L exhibits a wider distribution and larger mean angle of 41° ± 1. The mean angle and the location of the nucleosomes on TEM images (right) is consistent with the conformation shown in the inset. Scale bars = 50 nm. (b) The average angle of several nucleosomes with varying length of DNA linker exhibits an expected increase. The right side axis denotes the mean end-to-end length of the nucleosome calculated from the mean nanocaliper angle. The inset compares the end-do-end distance of the nucleosome calculated from the nanocaliper angle versus the end-to-end distance directly measured from TEM images illustrating they are in excellent agreement. (c) The schematics illustrate nucleosome configurations that satisfy the mean end-to-end distances of the various constructs revealing the 6L, 26L, and 51L nucleosomes are unwrapped by ~15–20bp, and the 75L is fully wrapped and likely wraps more than the normal 1.65 turns by ~10bp. Schematics are drawn to scale.
The nanocaliper angle is a reporter of nucleosome conformation
The nucleosome end-to-end distance within the nanocaliper is a function of the size and conformation of the nucleosome, and the length of DNA extending from the nucleosome including both DNA that has unwrapped from the histone core and the additional linker DNA. The nanocalipers with 6L nucleosomes have an average end-to-end distance of 23 ± 1 nm. A fully wrapped nucleosome with the Widom 601 positioning sequence (601-NPS) is ~10nm in size. The 6L DNA contains 6 bp of linker DNA on each side of the nucleosome. Given a length of 0.34 nm per basepair,54, 55 this is not sufficient to account for the additional 13 nm of end-to-end distance no matter how it extends out from the nucleosome. Therefore, there must be additional DNA length that is provided by partial nucleosome unwrapping. Based on the nucleosome crystal structure3, if the nucleosome is symmetrically unwrapped to just a single turn around the histone octamer, there would be an extra ~30 bp of DNA extending out of the nucleosome in antiparallel directions. With the additional 6 bp of linker DNA on both ends, this nucleosome conformation will have an end-to-end distance within the nanocaliper of 72 bp, or 24 nm. The size of the neutravidin molecules must also be considered since our end-to-end distance is measured from center-to-center of the neutravidin attachments on either side. Adding the full size of a neutravidin molecule, half on each end of the DNA, would give an end-to-end distance of ~29 nm. This length is larger than our measured end-to-end distance of 23.2 ± 0.4 nm, suggesting the DNA is unwrapped less than 30 bp on each end. Accounting for the angle at which the DNA exits the nucleosome, unwrapping by ~20 bp would give an end-to-end distance of 23 nm (Figure 3c and S7). As illustrated in the inset of Figure 3a, this conformation is consistent with the nucleosome being located just beyond the ends of the arms, which we observe in our TEM images (Figure 3a and S5). The location of the nucleosome varies from nanocaliper to nanocaliper suggesting there is fluctuation in the amount of wrapping. These results also agree reasonably well with single molecule force spectroscopy measurements26, 56 that have revealed that unwrapping occurs through discrete transitions with the first transition corresponding to unwrapping by ~30 bp on each end of the DNA.
The 26L nucleosome has an average end-to-end distance of 32 ± 2 nm. A nucleosome with ~30 bp of DNA unwrapped on each end to a single turn and the additional 26 bp linkers and neutravidin would give an end-to-end distance of ~42nm. This is larger than our measured end-to-end distance for the 26L nucleosome. Similarly, for the 51L nucleosome, unwrapping by ~30 bp on each end to a single turn would give a total end-to-end distance of 59 nm including the neutravidin, which is larger than our measured end-to-end distance of 38 ± 2 nm. One possible conformation is the nucleosome has increased wrapping from 1.65 to two full turns. This requires an additional ~15 bp of DNA wrapping on each end. That would leave ~11bp and ~36 bp of DNA extending to each side of a double-wrapped 26L or 51L nucleosome, respectively, resulting in end-to-end distances of ~12 nm and ~29 nm, which is significantly less than the measured end-to-end distances and rules out this possible conformation. Instead, a partial unwrapping of ~19 bp and ~17 bp gives the correct end-to-end distances for the 26L and 51L nucleosomes after accounting for the angle at which the DNA exits the nucleosome (Figure 3c and S7). While the end-to-end distance may also be a function of other factors such as nucleosome orientation and DNA deformations, these results suggest that both the 26L and 51L nucleosomes are partially unwrapped by the nanocaliper similarly to the 6L nucleosome.
In contrast, a 75L nucleosome that is wrapped by an additional ~15 bp of DNA on each end such that the DNA is wrapped two full turns around the histone octamer leaves 60 bp of DNA extending out from the nucleosome. This results in a total end-to-end distance of 120 bp, or 45 nm, including neutravidin. This is close to the measured end-to-end distance of 41 ± 1 nm suggesting the 75L is fully wrapped and likely wraps around the histone octamer by more than 1.65 turns (Figure 3c and S7). The inset of Figure 3a depicts the 75L nucleosome within the calipers in a configuration close to 2 full turns.
While DNA bending, twisting, stretching, and nucleosome orientation likely impact the measured angles and end-to-end distances, the end-to-end distributions indicate that when integrated into the nanocalipers, the 6L, 26L, and 51L nucleosomes fluctuate in the amount of unwrapped DNA with an average of about 20 bp unwrapped. This result is consistent with the nucleosome unwrapping free energy landscape, which predicts that the first 20 bp of the nucleosome requires about a kBT to unwrap.57 In contrast, the 75L nucleosome is primarily fully wrapped and on average adopts a configuration close to two full turns, which suggests that a total linker length of about 150 bp, the persistence length of dsDNA, is needed to allow for the nucleosome to remain fully wrapped in the nanocalipers.
The nanocaliper angle is sensitive to nucleosome stability
Nucleosome stability and disassembly is required for essential processes including RNA transcription and DNA repair.58 Nucleosome stability is often investigated with monovalent salt titrations since the increase of NaCl from 0.2 to 2M results in complete nucleosome disassembly.59 Salt induced disassembly occurs stepwise, where NaCl concentrations between 0.5 M and 1.0 M induce dissociation of H2A-H2B heterodimers, while higher NaCl concentrations of 1.0 M to 2.0 M cause H3-H4 tetramers to dissociate. To test if the nanocaliper can detect nucleosome disassembly, we investigated the influence of NaCl concentration on the angle of nanocalipers containing 6L nucleosomes (Figure 4 and S8). We found that as the NaCl concentration is increased from 0.2 M to 2.0 M the caliper angle increases significantly, which converts to an increase in the average end-to-end distance from 22 ± 3 nm to 45 ± 1 nm. At 2.0 M NaCl, the average caliper angle converges to the average angle of nanocalipers attached to the 6L DNA alone (Figure 4b, S9 and S10), which is consistent with progressive disassembly with increasing NaCl concentration and the known result that 2.0 M NaCl causes nucleosomes to fully disassemble.
Figure 4. The nanocaliper can quantify nucleosome disassembly.

(a) The angular distribution of the nanocalipers with the 6L nucleosome is distinctly different than the nanocaliper control sample with the same DNA and no histone core at physiological salt conditions (0.2 M NaCl and 1 mM MgCl2). TEM images (right) illustrate a clearly visible nucleosome that is slightly larger than the neutravidin molecules. Increasing the concentration of NaCl drives a shift towards the control distribution, suggesting the nucleosome is unwrapping, which is consistent with nucleosomes in the TEM images being less apparent at 1.0 M NaCl. The nucleosomes are largely not visible at 2.0 M NaCl, suggesting the histone core is disassembling. Scale bars = 50 nm. (b) The top graph shows the mean nanocaliper angle with the 6L construct increases as a function of NaCl concentration illustrating the salt-mediated nucleosome disassembly. The bottom graph illustrates the fraction of nucleosomes with no visible histone core increases as a function of NaCl concentration up to 68% at 2.0 M revealing salt-mediated dissociation of the histone core. At physiological concentrations of NaCl (0.2 M), all nanocalipers with an integrated nucleosome have a clearly visible histone core.
In addition to measuring the nanocaliper angle, we separately investigated nucleosome-nanocaliper complexes through visual inspection of TEM images to determine the presence of a fully or partially assembled nucleosome between the neutravidin attachments. Partially assembled nucleosomes consist of hexasomes, which contain the H3-H4 tetramer and one H2A-H2B heterodimer, and tetrasomes, which contain only the H3-H4 tetramer. We found that 100 percent of the nanocalipers in 0.2 M NaCl contain histone cores that can clearly be identified by TEM (Figure 4 and S9) and appear to be fully assembled histone octamers. Since it was difficult to quantitatively differentiate tetrasomes, hexasomes, and fully assembled octamers by visual inspection we only quantified whether any visible histone core, fully or partially assembled, was present (Figure 4b). As the salt concentration was increased, we determined the fraction of calipers that retained a visible histone core between the two neutravidin attachments. At 0.5 M and 1.0 M NaCl, 10% and 35% of the calipers, respectively, have fully lost the histone core between the neutravidin attachments indicating that disassembly starts to occur (Figure 4, S8, and S9), but the nucleosomes do not fully disassemble at these NaCl concentrations. Furthermore, while the mean caliper angle at these conditions increases, it remains less than the control nanocaliper containing the same DNA as the 6L construct without any histones (Figure 4 and S8–S10). These combined results are consistent with previous observations that at 0.5 M and 1.0 M NaCl the H2A-H2B heterodimers dissociate, but the H3-H4 tetramer remains bound.59 In contrast, only 32% of the nanocalipers have a visible histone core between neutravidin attachments in 2.0 M NaCl (Figure 4 and S9). This significant reduction in visible histones and the observation that the 6L nucleosome nanocaliper angles converge to the DNA only control at 2.0 M NaCl (Figure 4 and S10) implies that the H3-H4 tetramers have largely dissociated from the DNA. This result is also consistent with the known disassembly of nucleosomes at 2.0 M NaCl. Overall, these results demonstrate that our nanocaliper can quantify nucleosome disassembly.
The nanocaliper angle detects transcription factor occupancy within the nucleosome
Because the nanocalipers detected nucleosomal structural changes due to disassembly, we hypothesized that TF binding within the nucleosome could be detected if TF binding influences the structure in a manner that results in a change in the end-to-end distance of the nucleosome. To investigate if our nanocalipers can detect TF occupancy within the nucleosome, we prepared nucleosomes with the 6L DNA that contains a Gal4 recognition sequence within the Widom 601 NPS (Figure 1 and 5). The 6L construct was chosen because it exhibited the narrowest distribution. The Gal4 site is located between the 8th and 26th base pairs of the NPS. In the average configuration, the 6L construct is unwrapped by ~20 bp, so the end of the binding site would still be wrapped. Hence, we anticipated the steric bulk of the TF would require a structural change that could be detected. We carried out binding titrations with Gal4-VP16, which has been shown to function as a transcriptional activator in mammalian cells.60 We found that the average angle of nucleosome-labeled nanocalipers increases from 19 ± 3 degrees to 28 ± 2 degrees (Figure 5, S11–S12). The change in angle for increasing Gal4-VP16 concentrations fits to a binding isotherm with an S1/2 of 5 ± 1 nM (Figure 5b). The S1/2 is the concentration of Gal4-VP16 required to change the angle by 50 percent. To control for nonspecific Gal4-VP16 binding, we separately carried out Gal4-VP16 titrations with nucleosome containing nanocalipers, where the nucleosome did not contain the Gal4 recognition sequence (Figure 5b, S11 and S13). We found that without the Gal4 binding site, there is a negligible change in the caliper angle. This control shows that non-specific binding of Gal4-VP16 to either the nucleosome or the nanocaliper does not induce a significant change in the nanocaliper angle. Instead, the mean nanocaliper angle and nucleosome end-to-end distance is sensitive to Gal4-VP16 binding specifically to its target sequence within a nucleosome. Furthermore, Gal4-VP16 binding alters the nucleosome structure so that the mean end-to-end distance increases from 23 ± 3 nm to 33 ± 2 nm. This could be due to an increase in unwrapping, a change in nucleosome orientation, and/or a change in the angle the DNA exits the nucleosome.
Figure 5. The nanocaliper is sensitive to transcription factor binding within the nucleosome.

(a) The DNA constructs used in the nanocaliper measurements that detect binding of the transcription factor, Gal4-VP16, to its target site within the nucleosome. The 6L DNA contains the Gal4 binding site that is located from the 8th to the 26th base pairs of the Widom 601 NPS. The 6LN does not contain the Gal4 binding site. (b) The nanocaliper angle distributions with 1nM, 10nM, and 100nM Gal4-VP16. The teal distributions are for measurements done with 6L DNA, while the blue distributions are done with the nucleosome containing 6LN DNA. The grey distributions are measurements with the 6L (left columns) and 6LN (right column) without Gal4-VP16. (c) A plot of the average nanocaliper angle (left axis) and the nucleosome end-to-end distance (right axis) for the 6L (teal) and the 6LN (blue) nucleosomes with increasing concentrations of Gal4-VP16. The data were fit to a binding isotherm with an S1/2 of 5 ± 1 nM for the 6L nucleosome that contains the Gal4 binding site.
The nanocaliper facilitates transcription factor binding within the nucleosome
The integration of the 6L nucleosome into the nanocaliper significantly constrains the nanocaliper as indicated by the reduction of the angular distribution average and width (Figure 3). Furthermore, as discussed above, the average end-to-end distance of 23 nm implies that the 6L nucleosomes are partially unwrapped by the nanocaliper (Figure 3c). These results suggest that the nanocaliper could facilitate Gal4-VP16 binding. To investigate if the nanocaliper influences Gal4-VP16 binding, we determined the Gal4-VP16 binding isotherms with 6L nucleosomes that were not integrated into nanocalipers with FRET efficiency measurements as we have previously done with LexA61 and the Gal4 DNA binding and dimerization domains.21 To do this, we attached a Cy3 fluorophore to the 7th bp of the 6L DNA molecule. This places the Cy3 molecule at the DNA entry-exit region of the nucleosome on the same side as the Gal4 binding site (Figure 6). We then labeled the histone octamer with Cy5 fluorophore at H2AK119C, which positions one of the Cy5 molecules close to the Cy3 fluorophore. At these positions within the fully wrapped nucleosomes, Cy3 and Cy5 exhibit significant FRET efficiency (Figure 6).61
Figure 6. Gal4-VP16 binding to unconstrained nucleosomes.

(a) The DNA construct for detecting the binding of Gal4-VP16 to its site in a nucleosome using FRET. The DNA molecule is the 6L construct with a Cy3 fluorophore attached to the second base pair of the Widom 601 sequence. (b) The crystal structure of the nucleosome highlighting the FRET labeling sites (PDB: 1KX5). The histone octamer is in green and the DNA molecule is in gray with the Gal4 binding site in red. H2A(K119C) is shown in magenta, which is where Cy5 is attached. The DNA base in green is the site where Cy3 is attached. (c) The three state model of Gal4 binding to its target site within the nucleosome. (d) The fluorescence emission spectra of the Cy3-Cy5 labeled nucleosome with 0nM (blue), 3000 nM (orange) and 10000 nM (gray) of Gal4-VP16. As the concentration of Gal4-VP16 is increased, the stimulated emission of Cy5 decreases and the emission of Cy3 increases. The fluorescence spectra were analyzed by the (ratio)A method. (e) The plot of the FRET efficiency for increasing concentrations of Gal4-VP16 without (diamonds) and with (circles) neutravidin. The FRET efficiencies were fit to a binding isotherm with an S1/2 of 3.5 ± 0.2 μM and 7.2 ± 0.2 μM without and with neutravidin, respectively.
We measured the FRET efficiency as a function of Gal4-VP16 concentration. As Gal4-VP16 binds to partially unwrapped nucleosomes, it traps the nucleosome in a partially unwrapped, low FRET efficiency state. We fitted this to a binding isotherm and found that Gal4-VP16 binds with an S1/2 = 3.5 ± 0.2 μM (Figure 6). This is about 700 fold higher than the S1/2 measured by the nanocaliper. We also determined if this reduction in FRET efficiency is due to Gal4-VP16 binding to its target site within the nucleosome. We found that there is no Gal4-VP16 induced reduction in FRET efficiency with nucleosomes that do not contain the Gal4 binding site (Figure S14), which demonstrates that the reduction in FRET efficiency by Gal4-VP16 is due to specific binding to the Gal4 target site.
The presence of the neutravidin at each end of the nucleosome could itself impact Gal4-VP16 occupancy due to steric interactions, so we also carried out Gal4-VP16 binding experiments with nucleosomes pre-bound by neutravidin. We found that with neutravidin the Gal4-VP16 S1/2 is increased to 7.2 ± 0.2 μM (Figure 6), but the binding remains site specific (Figure S14). This result demonstrates that neutravidin does not increase Gal4-VP16 occupancy and thus does not account for the 700-fold difference in the Gal4-VP16 S1/2 between binding to nucleosomes with and without the nanocaliper. Instead, our results indicate that the nanocaliper itself significantly increases the Gal4-VP16 binding to its target sequence within the nucleosome and that the nanocaliper can function to perturb DNA accessibility within nucleosomes.
DISSCUSION
Scaffolded DNA origami allows for the fabrication of nanodevices with nanometer resolution that are inherently biocompatible. We utilized this emerging technology to engineer a nanocaliper device to monitor structural changes within single nucleosomes at distance scales that range from 20 to 50 nm, distances an order of magnitude larger than those accessible by FRET efficiency measurements. We developed a robust approach to efficiently integrate nucleosomes into the nanocaliper device and then demonstrated their utility by using the device to both detect and induce nucleosome structural changes. We found that the nanocalipers can quantify the amount of DNA unwrapped from the histone octamer, nucleosome disassembly and TF binding within the nucleosome. Furthermore, by using short DNA linkers (6 bp, 26 bp, and 51 bp), nucleosomes integrated into the nanocaliper are shifted into a partially unwrapped state, which can facilitate interactions between the exposed DNA and DNA-binding proteins. Many regulatory factors, including chromatin architectural proteins and chromatin remodeling complexes, regulate nucleosome wrapping and could preferentially interact with partially unwrapped nucleosomes. Hence, the nanocalipers can be used to investigate protein interactions with partially unwrapped nucleosomes by altering the nucleosome into a pre-designed partially unwrapped state.
The partial unwrapping indicates the nanocaliper applies a force to the nucleosome, which could cause deformation of the DNA. Force spectroscopy studies have shown ~3 pN partially unwraps a nucleosome to a single turn,26, 56 suggesting the force applied by the nanocalipers is less than 3 pN. The extensional stiffness of double-stranded DNA is 1100 pN,62–64 so at 3pN there is only a 0.3% change in length, which is less than the resolution of our measurement. In addition, assuming 0.34nm per base pair,54, 55 the 6L, 26L, 51L, and 75L linkers have lengths of ~2 nm, 9 nm, 17, and 26 nm. These lengths are all shorter than the 50 nm bending54 and ~40–100 nm twisting65 persistence lengths of double-stranded DNA, implying that bending and twisting of the linker DNA likely do not significant impact our measurements. However, deformation of the DNA may be a significant component to consider for longer linkers or nanocaliper designs that apply more force
Our measurements of Gal4-VP16 binding to 6L nucleosomes are an example of how the nanocalipers can be used to study protein interactions within a partially unwrapped nucleosome. We found that the 6L nucleosome tethered into the nanocaliper is on average unwrapped by ~20 bp (Figure 3), which exposes approximately two-thirds of the Gal4 target site and in turn dramatically increases the probability of Gal4-VP16 binding to its target sequence (Figure 5 and 6). In addition, the increasing concentrations of Gal4-VP16 alter the distribution of end-to-end distances where a second population with an average end-to-end distance of 45 ± 6 nm appears at high Gal4-VP16 concentrations (Figure S15). This much larger end-to-end distance is consistent with a nucleosome that is largely unwrapped since the full DNA length is 53 nm (Figure 7). This suggests that Gal4-VP16 binding can induce a transition from a partially unwrapped to a nearly fully unwrapped nucleosome. The end-to-end distance distribution at saturating concentrations of Gal4-VP16 indicates that when Gal4-VP16 is bound to its target sequence, the majority of nucleosomes are in the partially unwrapped state, but ~30% of the nucleosomes transition into the fully unwrapped state (Figure S15). Interestingly, force spectroscopy measurements have revealed an unwrapping transition between a partially and fully unwrapped nucleosome at about 15 pN of applied force resulting in 22 nm length changes.25, 26, 56 Here, we observe a transition of similar length change resulting from TF binding. It appears that by tethering the nucleosome within the nanocaliper, the binding of Gal4-VP16 to its partially exposed recognition site can facilitate transitions into an almost fully unwrapped state. This suggests that when nucleosomes must satisfy tethered boundary constraints, protein binding can facilitate relatively large conformational changes, which could be important for DNA accessibility and nucleosome disassembly; however, future studies will be required to better understand these nucleosome structural transitions driven by TF binding.
Figure 7. Model of Gal4-VP16 binding to a nucleosome integrated into a nanocaliper.

Our nanocaliper studies of the 6L nucleosome indicate that in the presence of Gal4-VP16 there are three possible states. (State I) The nucleosome without Gal4-VP16 bound is unwrapped ~20 bp by the nanocaliper with an end-to-end distance of 23 nm. (State II) The nucleosome is unwrapped by a little more than 20 bp so Gal4-VP16 can bind and the end-to-end distance increases to 26 nm. In this state, the DNA still wraps about 1 full turn around the histone octamer. (State III) While Gal4-VP16 is bound to the nucleosome, the nucleosome reorients so that it is almost completely unwrapped. This significantly increases the end-to-end distance of the nucleosome to 45 nm.
CONCLUSIONS
These proof-of-principle studies demonstrate the feasibility and advantages to using the DNA origami nanocaliper to investigate nucleosome structural changes. However, additional improvements to this methodology should increase the impact of this approach. For example, the geometry of the arms could be varied so that nucleosome arrays could be integrated and studied, which would provide insight into higher levels of the chromatin hierarchical structure. In the context of these nanocalipers, fluorophores could be integrated on separate arms of the nanocaliper or within nucleosomes to probe end-to-end distance and/or local structural changes in real-time using single molecule FRET (smFRET) studies. Finally, there are numerous factors that regulate nucleosome and chromatin dynamics and function including: histone post-translational modifications and their binding proteins,66 chromatin architectural proteins,67 histone chaperones,68 and chromatin remodeling complexes.14 These complexes influence nucleosome unwrapping, nucleosome disassembly, nucleosome sliding, and chromatin higher order compaction. Since the nanocalipers function well in the physiological ionic conditions we used here, this device in conjunction with other approaches including single molecule and TEM methodologies can contribute significantly to understanding how these factors regulate nucleosome and chromatin structural dynamics on the 10 to 100 nanometer length scale.
More generally, we demonstrate the use of a dynamic DNA origami device to probe the structure and conformational changes of a large biomolecular complex. A key aspect to using the nanocalipers to quantify structural changes is that the macromolecular complex needs to be attached to both arms of the nanocaliper. This is essential to couple the end-to-end distance of the nucleosome to the nanocaliper angle. In fact, this is a general challenge for using structurally dynamic DNA origami structures to investigate biomolecular structural dynamics because a double attachment is crucial to couple the movement of the device to the structural changes of the biomolecule. Previous studies have integrated proteins and protein complexes into DNA origami nanostructures69, 70 and used dynamic devices to regulate bimolecular interactions.42, 48 However, these studies used a single attachment site. Previous work to study conformational changes has only focused on DNA structures that can be integrated at multiple locations via base-pairing.51, 52 Here, we used biotin-neutravidin-biotin for two attachment points on the same biomolecule. Adding the neutravidin to the biomolecule sample first and then removing unbound neutravidin by sucrose gradient purification prior to integration in the nanocalipers gave optimal incorporation efficiencies. This is a general approach that can work with any biomolecular complex that is significantly larger than the neutravidin tetramer.
METHODS
Preparation of the DNA origami nanocaliper
The basic design of the DNA origami nanocalipers used here was initially presented by Marras et al.33 The caliper arms consist of 18 dsDNA helices bundled into a 3 × 6 helix-bundle (hb) cross-section. The arms are joined together at one edge via 6 single-stranded DNA connections. Three short connections (2 nucleotides long) form the hinge axis of rotation, and 3 longer connections (30 nucleotides long) influence the hinge flexibility. The arms are 200 bp or ~70 nm from the hinge vertex to the opposite end. For this work, the design was modified to include 2 biotinylated overhangs at the ends of the arms. The caDNAno71 design diagram is presented in Figure S16. The design is based on an 8064nt single-stranded scaffold derived from the M13MP18 bacteriophage virus and prepared in our laboratory as described in37 and 184 oligonucleotide staples that were ordered from a commercial vendor (Table S1, Eurofins, Huntsville, AL). The self-assembly reaction was also carried out as previously described.37 The folding reaction contained 20 nM scaffold and 200 nM of each staple in a solution containing ddH2O, 5 mM Tris, 5 mM NaCl, 1 mM EDTA, and 18 mM MgCl2, pH 8.0. An initial screen of MgCl2 concentrations revealed 18 mM yielded the best folding results (Figure S17). This folding reaction was subjected to a thermal annealing in a thermal cycler (Bio-Rad, Hercules, CA) using folding protocols modified from a previously published method.72 Specifically, the thermal ramp consisted of rapidly heating the solution to 65°C for 15 minutes, cooling down to 52°C for 4 hours, and cooling it for 10 minutes at 4°C.
Folding results were characterized via agarose gel electrophoresis (Figure S18) as described in a previously published method.37 Bands of well-folded structures were also characterized on a transmission electron microscope (TEM). For integration with nucleosome constructs, the DNA origami structures folded in 18 mM MgCl2 were purified via centrifugation using protocols modified from73 after the folding reaction. Briefly, after thermal annealing, folded nanostructures were mixed with 15% PEG8000 (Sigma Life Science) diluted in ddH20 with 200 mM NaCl. The combined solution was centrifuged at 16,000g for 30 minutes to pellet nanostructures. The supernatant was removed and structures were resuspended in 59mM Tris, 1.7mM MgCl2, 240 mM NaCl, pH 8.0. This buffer was selected to achieve the desired final buffer concentrations upon integration with nucleosome constructs. After resuspension, the nanostructure concentration was tested on the NanoDrop (NanoDrop 2000C Spectrophotometer, Thermo Scientific), and samples were analyzed via TEM.
Preparation of DNA Molecules and Nucleosomes
Biotinylated DNA molecules (Table S2) were prepared by PCR using oligonucleotides containing a biotin label on the 5′-end (Table S3) and a plasmid containing the Widom 601 NPS with or without the Gal4 binding site (CCGGAGGGCTGCCCTCCGG) at the 8th – 26th base pairs. The oligonucleotides used to prepare the DNA molecules for the FRET measurements also contained an amine-modified thymine at the 2nd base of the 601 NPS, which was then labeled with Cy3-NHS-ester (GE Healthcare). The Cy3-labeled DNA oligo was then purified by reverse phase HPLC on a 218TP C 18 column (Grace/Vydac). PCR-synthesized DNA molecules were phenol-extracted and then purified by an anion-exchange HPLC using a Gen-Pak Fax column (Waters).
Recombinant X. laevis histones H2A(K15C), H2A(K119C), H2B, H3(C110A) and H4 were expressed, purified and refolded into histone octamers (HOs) as previously described.74 The H2A(K15C) or H2A(K119C) containing HOs were then labeled with Cy5-maleamide (GE Healthcare) and purified following the protocol as detailed in previous publication.61
Nucleosomes were reconstituted as previously described with the double salt dialysis method61 by mixing purified HO and DNA at a molar ratio of 0.85:1 (HO: DNA) in a 50 μl volume small dialysis chamber containing 0.5x TE pH 8.0, 1 mM Benzamidine hydrochloride (BZA) and 2 M NaCl. The small dialysis chamber was then placed into an 80mL dialysis tube containing 0.5x TE pH 8.0 and 1 mM BZA. The dialysis tube was dialyzed for 12 hours against 4L of buffer containing 0.5x TE pH 8.0 and 1 mM BZA. The buffer was changed and dialysis was done for another 16 hours. The recovered nucleosomes were then analyzed by Electrophoretic Mobility Shift Assay (EMSA).
Preparation of Neutravidin-Bound DNA/Nucleosome Complexes
Biotinylated DNA molecules and nucleosomes were mixed with neutravidin at a molar ratio of 5:1 (neutravidin: DNA/nucleosome) to form a 2-neutravidin pre-bound complex prior to incorporation into DNA origami calipers. For DNA molecules and nucleosomes used in the TEM imaging experiments, Rhodamine Red X-Conjugate neutravidin (Life Technologies #A6378) was used. For nucleosomes used in the FRET experiment, non-fluorescent neutravidin (Molecular Probes by Life Technologies #A2666) was used. The mixed complexes were loaded onto sucrose gradients and purified by ultracentrifugation (Beckman Coulter, Optima L-90K) at 4°C, 41000 rpm for 22 hrs in a SW41 rotor. All lengths of DNA linkers and 6L and 26L nucleosomes were purified on a sucrose gradient from 5% to 30% (w/v) and 51L and 75L nucleosomes were purified on a sucrose gradient of 5% to 35% (w/v). The gradient was analyzed by gel electrophoresis after ultracentrifugation and fractions containing DNA or nucleosome bound with 2 neutravidin molecules were collected and concentrated.
The DNA and nucleosome molecules with and without neutravidin bound were analyzed by EMSA. 200 fmole DNA and/or nucleosome samples were diluted into 20 μl in 0.5x TE, loaded onto a native 5% polyacrylamide gel in 0.3x TBE and resolved by running the gel at 300V for 1hr. The gel was imaged on a Typhoon scanner (GE Healthcare Life Sciences, Typhoon FLA 9500) for Rhodamine Red-labeled neutravidin and Cy5-labeled HO, and then stained by ethidium bromide and imaged by UV illumination for detecting unlabeled DNA.
Preparation of Gal4-VP16
N-terminal His6-tagged Gal4-VP16 was used in the transcription factor binding experiments. The expression vector was a generous gift from Prof. Aaron Johnson (University of Colorado, Denver). His6-tagged Gal4-VP16 was expressed in E.coli Rosetta(DE3)pLysS cells (Millipore) by inducing with 1mM IPTG and 100 μM Zinc Acetate for 3 hours. The cells were harvested by centrifugation, resuspended in Buffer A (50 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM DTT, 20 μM ZnCl2, 1 mM phenylmethanesulfonyl fluoride (PMSF), 20 μg/ml leupeptin, and 20 μg/ml pepstatin) and lysed by sonication. The cell debris was removed by centrifugation. The clarified supernatant was mixed with Nickel-NTA agarose (Qiagen) and loaded into a C10/10 FPLC column (GE Healthcare). The proteins bound to the column were washed using Buffer B (25 mM Tris-HCl pH 7.5, 200 mM NaCl, 0.2% Tween-20 (v/v), 20 μM Zinc Acetate, 1mM DTT, 1mM PMSF) with 10mM imidazole and then eluted using Buffer B with 200mM imidazole. The eluate was analyzed by SDS-PAGE and the fractions containing Gal4-VP16 were pooled and dialyzed against Buffer C (25 mM Tris-HCl pH 7.5, 20 μM Zinc Acetate, 1 mM DTT, 1mM PMSF) with 200 mM NaCl. The dialyzed proteins were FPLC purified using a Mono Q anion-exchange column (GE Healthcare Life Sciences, #9122097) with a linear gradient of Buffer C from 200 mM NaCl to 800 mM NaCl. Fractions containing high purity Gal4-VP16 were pooled, dialyzed against Buffer D (10 mM HEPES pH 7.5, 200 mM NaCl, 10% glycerol, 1 mM DTT, 10 μM ZnCl2, 1 mM PMSF) and stored at −80°C.
Integration and stability of nucleosome constructs in the DNA nanocaliper
After PEG-based centrifugal purification, nanocalipers were resuspended to 8 nM in 59 mM Tris, 1.7 mM MgCl2, 240 mM NaCl, pH 8.0. Nucleosomes samples were diluted to 40 nM in 0.5x TE pH 8.0 after sucrose gradient purification. Then, 5 μl of the 8 nM calipers were combined with the 1μl of 40nM nucleosomes and an additional 2 μl of a 50mM Tris and 200 mM NaCl solution for an equimolar final concentration 5nM:5nM (nanocalipers: nucleosomes) in a final buffer with 50 mM Tris, 1 mM MgCl2, and 200 mM NaCl. We confirmed the stability of the nanocalipers in the buffer over the timescale of our measurements (Figure S18). The mixed solution was incubated on ice for 30 minutes and then immediately deposited on the TEM grid.
To test the stability of integrated nucleosome constructs under varying salt conditions, the calipers were resuspended to 12 nM in the same buffer as above (59 mM Tris, 1.7 mM MgCl2, 240 mM NaCl, pH 8.0). Nucleosome samples were diluted down to 60 nM in 0.5x TE pH 8.0 after sucrose gradient purification. 5 μl of the 12nM calipers were combined with 1 μl of the 60 nM nucleosomes for an equimolar concentration of 10nM:10nM in a buffer of 50 mM Tris, 1.3 mM MgCl2, and 200 mM NaCl. The mixed solution was incubated for 30 minutes on ice. 6μl of a specific salt buffer was then added to the nucleosome-caliper assembly for a final equimolar concentration of 5nM:5nM in a solution. The varying salt buffers (SB#) were designed to achieve final buffer conditions of 50 mM Tris, 1 mM MgCl2, and the final NaCl concentration (0.5, 1.0, 1.5, or 2.0 M). SB500 contained 50 mM Tris, 0.67 mM MgCl2, and 0.8 M NaCl pH 8.0. SB1000 contained 50 nM Tris, 0.67 mM MgCl2, and 1.8 M NaCl pH 8.0. SB1500 contained 50 mM Tris, 0.67 mM MgCl2, and 2.8 M NaCl pH 8.0. SB2000 contained 50 mM Tris, 0.67 mM MgCl2, and 3.8 M NaCl pH 8.0. The mixed solution was incubated on ice for 2 minutes and then immediately deposited on a TEM grid as described below.
We further tested the ability to detect structural changes of the nucleosome mediated by binding of the transcription factor Gal4-VP16. For desired final concentrations of 1, 5, 10, 30, and 100 nM, the FPLC-purified Gal4-VP16 was initially diluted to 4x concentrations of 4, 20, 40, 120, and 400 nM in a dilution buffer containing 50 mM Tris and 200 mM NaCl at pH 8.0. For the Gal4-VP16 concentration data, the respective Gal4-VP16 concentrations were added to the nucleosome-caliper assemblies for the final concentrations of 5nM nucleosome-caliper assemblies and 1, 5, 10, 30, or 100 nM Gal4-VP16 with final buffer conditions of 50 mM Tris, 1 mM MgCl2, and 200 mM NaCl at pH 8.0. The assemblies were then incubated on ice for 2 minutes and then immediately deposited on a TEM grid as described subsequently.
Sample imaging and analysis by transmission electron microscopy
Purified structures and nanocaliper-nucleosome assemblies were imaged using transmission electron microscopy (TEM) for structural feedback and quantification. Samples were prepared using protocols modified from.37 3.4 μl of sample volume was deposited on Formvar-coated copper grids, stabilized with evaporated carbon film (Electron Microscopy Sciences; Hatfield, PA). The sample was incubated on the grid for 4 minutes and then wicked away. 10 μl of 2% uranyl formate (SPI, West Chester, PA) stain with 25 mM NaOH was immediately added and wicked away as a wash step, and then 20 μl the same stain was added and incubated for 15 seconds before being wicked off. Stained grids were allowed to dry for at least 30 minutes prior to imaging. TEM was carried out at the OSU Campus Microscopy and Imaging Facility on a FEI Tecnai G2 Spirit TEM at an acceleration voltage of 80 kV at a magnification of either 68000x or 110000x.
A combination of MATLAB, and the open source softwares ImageJ and eman2 were used to quantify the conformation of all samples. Raw TEM images were initially organized into image galleries of individual nanocalipers using the open source software eman2. Representative image galleries are shown in the supplemental methods for all cases of experiments (Figures S5, S9, S10, S12 and S13). These image galleries were imported into ImageJ for analysis. Angular measurements were performed manually in ImageJ by drawing lines along the inner edges of the hinge arms. Similar angle measurements were performed for the free calipers and nucleosome-caliper assemblies with varying nucleosome constructs and solution conditions. End-to-end distance measurements of samples were also manually performed in ImageJ by measuring the distance from center of neutravidin to center to neutravidin across the caliper arms. Angle and distance measurements were only carried out on well-folded calipers with nucleosomes clearly incorporated between the ends of the two arms. For the varying size linker constructs, we compared the end-to-end calculated from angle measurements based on the caliper geometry to end-to-end distance directly measured from the images using a custom MATLAB code. In this code, images of individual calipers were extracted from the image galleries. Four points were manually selected to obtain the coordinates of the hinge vertex, the two neutravidin molecules, and the nucleosome. The coordinates of these 4 points were then used to calculate the caliper angle, the end-to-end distance calculated from the caliper angle, and the directly measured end-to-end distance.
To quantify the error from the manual angle and distance measurements, we measured the angle of a representative structure multiple times. The standard deviation of the caliper angle measurement is 0.72 degrees, which corresponds to a standard deviation in calculated end-to-end distance of 0.85 nm. We also measured the end-to-end distance of a nucleosome construct integrated into a caliper device multiple times, which yielded a standard deviation of 0.86 nm (Figure S19).
FRET Measurements
FRET experiments were used to determine Gal4-VP16 binding to its target site within the nucleosome as previously described.21, 61 Gal4-VP16 titrations were done with 5 nM Cy3-Cy5-labeled nucleosomes in 50 μl volume of buffer containing 49 mM Tris-HCl pH 8.0, 200 mM NaCl, 1.08 mM MgCl2, and 0.1mM EDTA using a FluoroMax4 fluorescence spectrometer (Horiba). The FRET efficiency was calculated using the (ratio)A method15 using the fluorescence emission spectra of Cy3 and Cy5 fluorophores. The apparent binding affinity S1/2 was determined by fitting the FRET efficiency with a sigmoidal binding curve with a Hill coefficient of 1. Gal4-VP16 titrations were performed using nucleosomes with and without neutravidin pre-bound and the S1/2 is not significantly altered (Figure 6). Control experiments were done using FRET nucleosomes that do not contain a Gal4 binding site (Figure S14). These FRET efficiency data were also fit to a binding curve where the results show extremely similar initial and final FRET efficiency, implying that the observed reduction in FRET is due to Gal4-VP16 binding to the target sequence within the nucleosome.
Supplementary Material
Acknowledgments
We thank Ralf Bundschuh, Dengke Zhao, the Castro Lab and Poirier Lab for feedback on this work. This work was supported by grants from the NSF (1235060 to CEC; 1516976 to MGP and CEC), the NIH (R01-GM083055 to MGP; R21-CA174583 to CEC and MGP), and seed funding from the Center for Emergent Materials (NSF funded MRSEC, grant 0820414), the Ohio State University Institute for Materials Research and the Center for Exploration of Novel Complex Materials (ENCOMM). CRL is a recipient of a National Institutes of Health T32 Award in Oncology Training Fellowship at The Ohio State University Comprehensive Cancer Center, 5T32CA009338-37. We also thank the Campus Microscopy and Imaging Facility at The Ohio State University.
Footnotes
The authors declare no competing financial interests.
SUPPORTING INFORMATION AVAILABLE
Assays and TEM images of nucleosome constructs, additional nanocaliper measurements at each condition, additional TEM images of each version, geometric analysis of DNA wrapping, FRET measurements of Gal4-VP16, DNA origami layouts, staple sequences, and DNA origami stability. This material is available free of charge via the Internet at https://pubs.acs.org.
References
- 1.Kornberg RD. Chromatin Structure: A Repeating Unit of Histones and DNA. Science. 1974;184:868–871. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 2.Kornberg RD. Structure of Chromatin. Annu Rev Biochem. 1977;46:931–954. doi: 10.1146/annurev.bi.46.070177.004435. [DOI] [PubMed] [Google Scholar]
- 3.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal Structure of the Nucleosome Core Particle at 2.8 a Resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 4.Tan S, Davey CA. Nucleosome Structural Studies. Curr Opin Struct Biol. 2011;21:128–136. doi: 10.1016/j.sbi.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li B, Carey M, Workman JL. The Role of Chromatin During Transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 6.Almouzni G, Cedar H. Maintenance of Epigenetic Information. Cold Spring Harb Perspect Biol. 2016;8:a019372. doi: 10.1101/cshperspect.a019372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinha M, Peterson CL. Chromatin Dynamics During Repair of Chromosomal DNA Double-Strand Breaks. Epigenomics. 2009;1:371–385. doi: 10.2217/epi.09.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maston GA, Landt SG, Snyder M, Green MR. Characterization of Enhancer Function from Genome-Wide Analyses. Annu Rev Genomics Hum Genet. 2012;13:29–57. doi: 10.1146/annurev-genom-090711-163723. [DOI] [PubMed] [Google Scholar]
- 9.North JA, Shimko JC, Javaid S, Mooney AM, Shoffner MA, Rose SD, Bundschuh R, Fishel R, Ottesen JJ, Poirier MG. Regulation of the Nucleosome Unwrapping Rate Controls DNA Accessibility. Nucleic Acids Res. 2012;40:10215–10227. doi: 10.1093/nar/gks747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polach KJ, Widom J. Mechanism of Protein Access to Specific DNA Sequences in Chromatin: A Dynamic Equilibrium Model for Gene Regulation. J Mol Biol. 1995;254:130–149. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- 11.Widom J. Role of DNA Sequence in Nucleosome Stability and Dynamics. Q Rev Biophys. 2001;34:269–324. doi: 10.1017/s0033583501003699. [DOI] [PubMed] [Google Scholar]
- 12.Chereji RV, Morozov AV. Functional Roles of Nucleosome Stability and Dynamics. Brief Funct Genomics. 2015;14:50–60. doi: 10.1093/bfgp/elu038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. Mechanisms and Functions of Atp-Dependent Chromatin-Remodeling Enzymes. Cell. 2013;154:490–503. doi: 10.1016/j.cell.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Becker PB, Workman JL. Nucleosome Remodeling and Epigenetics. Cold Spring Harb Perspect Biol. 2013;5:a017905. doi: 10.1101/cshperspect.a017905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clegg RM. Fluorescence Resonance Energy Transfer and Nucleic Acids. Methods Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 16.Wei S, Falk SJ, Black BE, Lee TH. A Novel Hybrid Single Molecule Approach Reveals Spontaneous DNA Motion in the Nucleosome. Nucleic Acids Res. 2015;43:e111. doi: 10.1093/nar/gkv549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park YJ, Dyer PN, Tremethick DJ, Luger K. A New Fluorescence Resonance Energy Transfer Approach Demonstrates That the Histone Variant H2az Stabilizes the Histone Octamer within the Nucleosome. J Biol Chem. 2004;279:24274–24282. doi: 10.1074/jbc.M313152200. [DOI] [PubMed] [Google Scholar]
- 18.Andrews AJ, Chen X, Zevin A, Stargell LA, Luger K. The Histone Chaperone Nap1 Promotes Nucleosome Assembly by Eliminating Nonnucleosomal Histone DNA Interactions. Mol Cell. 2010;37:834–842. doi: 10.1016/j.molcel.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang JG, Narlikar GJ. Fret-Based Methods to Study Atp-Dependent Changes in Chromatin Structure. Methods. 2007;41:291–295. doi: 10.1016/j.ymeth.2006.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li G, Widom J. Nucleosomes Facilitate Their Own Invasion. Nat Struct Mol Biol. 2004;11:763–769. doi: 10.1038/nsmb801. [DOI] [PubMed] [Google Scholar]
- 21.Luo Y, North JA, Rose SD, Poirier MG. Nucleosomes Accelerate Transcription Factor Dissociation. Nucleic Acids Res. 2014;42:3017–3027. doi: 10.1093/nar/gkt1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caterino TL, Fang H, Hayes JJ. Nucleosome Linker DNA Contacts and Induces Specific Folding of the Intrinsically Disordered H1 Carboxyl-Terminal Domain. Mol Cell Biol. 2011;31:2341–2348. doi: 10.1128/MCB.05145-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernier M, Luo Y, Nwokelo KC, Goodwin M, Dreher SJ, Zhang P, Parthun MR, Fondufe-Mittendorf Y, Ottesen JJ, Poirier MG. Linker Histone H1 and H3k56 Acetylation Are Antagonistic Regulators of Nucleosome Dynamics. Nat Commun. 2015;6:10152. doi: 10.1038/ncomms10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorenz M, Diekmann S. Distance Determination in Protein-DNA Complexes Using Fluorescence Resonance Energy Transfer. Methods Mol Biol. 2006;335:243–255. doi: 10.1385/1-59745-069-3:243. [DOI] [PubMed] [Google Scholar]
- 25.Brower-Toland BD, Smith CL, Yeh RC, Lis JT, Peterson CL, Wang MD. Mechanical Disruption of Individual Nucleosomes Reveals a Reversible Multistage Release of DNA. Proc Natl Acad Sci U S A. 2002;99:1960–1965. doi: 10.1073/pnas.022638399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mihardja S, Spakowitz AJ, Zhang Y, Bustamante C. Effect of Force on Mononucleosomal Dynamics. Proc Natl Acad Sci U S A. 2006;103:15871–15876. doi: 10.1073/pnas.0607526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gemmen GJ, Sim R, Haushalter KA, Ke PC, Kadonaga JT, Smith DE. Forced Unraveling of Nucleosomes Assembled on Heterogeneous DNA Using Core Histones, Nap-1, and Acf. J Mol Biol. 2005;351:89–99. doi: 10.1016/j.jmb.2005.05.058. [DOI] [PubMed] [Google Scholar]
- 28.Hall MA, Shundrovsky A, Bai L, Fulbright RM, Lis JT, Wang MD. High-Resolution Dynamic Mapping of Histone-DNA Interactions in a Nucleosome. Nat Struct Mol Biol. 2009;16:124–129. doi: 10.1038/nsmb.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kruithof M, Chien FT, Routh A, Logie C, Rhodes D, van Noort J. Single-Molecule Force Spectroscopy Reveals a Highly Compliant Helical Folding for the 30-Nm Chromatin Fiber. Nat Struct Mol Biol. 2009;16:534–540. doi: 10.1038/nsmb.1590. [DOI] [PubMed] [Google Scholar]
- 30.Yan J, Maresca TJ, Skoko D, Adams CD, Xiao B, Christensen MO, Heald R, Marko JF. Micromanipulation Studies of Chromatin Fibers in Xenopus Egg Extracts Reveal Atp-Dependent Chromatin Assembly Dynamics. Mol Biol Cell. 2007;18:464–474. doi: 10.1091/mbc.E06-09-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simon M, North JA, Shimko JC, Forties RA, Ferdinand MB, Manohar M, Zhang M, Fishel R, Ottesen JJ, Poirier MG. Histone Fold Modifications Control Nucleosome Unwrapping and Disassembly. Proc Natl Acad Sci U S A. 2011;108:12711–12716. doi: 10.1073/pnas.1106264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leuba SH, Zlatanova J. Single-Molecule Studies of Chromatin Fibers: A Personal Report. Arch Histol Cytol. 2002;65:391–403. doi: 10.1679/aohc.65.391. [DOI] [PubMed] [Google Scholar]
- 33.Marras AE, Zhou L, Su HJ, Castro CE. Programmable Motion of DNA Origami Mechanisms. Proc Natl Acad Sci U S A. 2015;112:713–718. doi: 10.1073/pnas.1408869112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linko V, Dietz H. The Enabled State of DNA Nanotechnology. Curr Opin Biotechnol. 2013;24:555–561. doi: 10.1016/j.copbio.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 35.Seeman NC. Nucleic Acid Junctions and Lattices. J Theor Biol. 1982;99:237–247. doi: 10.1016/0022-5193(82)90002-9. [DOI] [PubMed] [Google Scholar]
- 36.Seeman NC. DNA in a Material World. Nature. 2003;421:427–431. doi: 10.1038/nature01406. [DOI] [PubMed] [Google Scholar]
- 37.Castro CE, Kilchherr F, Kim DN, Shiao EL, Wauer T, Wortmann P, Bathe M, Dietz H. A Primer to Scaffolded DNA Origami. Nat Methods. 2011;8:221–229. doi: 10.1038/nmeth.1570. [DOI] [PubMed] [Google Scholar]
- 38.Douglas SM, Dietz H, Liedl T, Hogberg B, Graf F, Shih WM. Self-Assembly of DNA into Nanoscale Three-Dimensional Shapes. Nature. 2009;459:414–418. doi: 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rothemund PW. Folding DNA to Create Nanoscale Shapes and Patterns. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 40.Gerling T, Wagenbauer KF, Neuner AM, Dietz H. Dynamic DNA Devices and Assemblies Formed by Shape-Complementary, Non-Base Pairing 3d Components. Science. 2015;347:1446–1452. doi: 10.1126/science.aaa5372. [DOI] [PubMed] [Google Scholar]
- 41.Kuzyk A, Schreiber R, Zhang H, Govorov AO, Liedl T, Liu N. Reconfigurable 3d Plasmonic Metamolecules. Nat Mater. 2014;13:862–866. doi: 10.1038/nmat4031. [DOI] [PubMed] [Google Scholar]
- 42.Liu M, Fu J, Hejesen C, Yang Y, Woodbury NW, Gothelf K, Liu Y, Yan H. A DNA Tweezer-Actuated Enzyme Nanoreactor. Nat Commun. 2013;4:2127. doi: 10.1038/ncomms3127. [DOI] [PubMed] [Google Scholar]
- 43.Wei B, Ong LL, Chen J, Jaffe AS, Yin P. Complex Reconfiguration of DNA Nanostructures. Angew Chem Int Ed Engl. 2014;53:7475–7479. doi: 10.1002/anie.201402437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andersen ES, Dong M, Nielsen MM, Jahn K, Subramani R, Mamdouh W, Golas MM, Sander B, Stark H, Oliveira CL, Pedersen JS, Birkedal V, Besenbacher F, Gothelf KV, Kjems J. Self-Assembly of a Nanoscale DNA Box with a Controllable Lid. Nature. 2009;459:73–76. doi: 10.1038/nature07971. [DOI] [PubMed] [Google Scholar]
- 45.Douglas SM, Bachelet I, Church GM. A Logic-Gated Nanorobot for Targeted Transport of Molecular Payloads. Science. 2012;335:831–834. doi: 10.1126/science.1214081. [DOI] [PubMed] [Google Scholar]
- 46.Zadegan RM, Jepsen MD, Thomsen KE, Okholm AH, Schaffert DH, Andersen ES, Birkedal V, Kjems J. Construction of a 4 Zeptoliters Switchable 3d DNA Box Origami. ACS Nano. 2012;6:10050–10053. doi: 10.1021/nn303767b. [DOI] [PubMed] [Google Scholar]
- 47.Banerjee A, Bhatia D, Saminathan A, Chakraborty S, Kar S, Krishnan Y. Controlled Release of Encapsulated Cargo from a DNA Icosahedron Using a Chemical Trigger. Angew Chem Int Ed Engl. 2013;52:6854–6857. doi: 10.1002/anie.201302759. [DOI] [PubMed] [Google Scholar]
- 48.Ke Y, Meyer T, Shih WM, Bellot G. Regulation at a Distance of Biomolecular Interactions Using a DNA Origami Nanoactuator. Nat Commun. 2016;7:10935. doi: 10.1038/ncomms10935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuzuya A, Sakai Y, Yamazaki T, Xu Y, Komiyama M. Nanomechanical DNA Origami ‘Single-Molecule Beacons’ Directly Imaged by Atomic Force Microscopy. Nat Commun. 2011;2:449. doi: 10.1038/ncomms1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamamoto S, De D, Hidaka K, Kim KK, Endo M, Sugiyama H. Single Molecule Visualization and Characterization of Sox2-Pax6 Complex Formation on a Regulatory DNA Element Using a DNA Origami Frame. Nano Lett. 2014;14:2286–2292. doi: 10.1021/nl4044949. [DOI] [PubMed] [Google Scholar]
- 51.Sannohe Y, Endo M, Katsuda Y, Hidaka K, Sugiyama H. Visualization of Dynamic Conformational Switching of the G-Quadruplex in a DNA Nanostructure. J Am Chem Soc. 2010;132:16311–16313. doi: 10.1021/ja1058907. [DOI] [PubMed] [Google Scholar]
- 52.Endo M, Takeuchi Y, Suzuki Y, Emura T, Hidaka K, Wang F, Willner I, Sugiyama H. Single-Molecule Visualization of the Activity of a Zn(2+)-Dependent Dnazyme. Angew Chem Int Ed Engl. 2015;54:10550–10554. doi: 10.1002/anie.201504656. [DOI] [PubMed] [Google Scholar]
- 53.Lowary PT, Widom J. New DNA Sequence Rules for High Affinity Binding to Histone Octamer and Sequence-Directed Nucleosome Positioning. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 54.Marko JF, Siggia ED. Stretching DNA. Macromolecules. 1995;28:8759–8770. [Google Scholar]
- 55.Mandelkern M, Elias JG, Eden D, Crothers DM. The Dimensions of DNA in Solution. J of Mol Biol. 1981;152:153–161. doi: 10.1016/0022-2836(81)90099-1. [DOI] [PubMed] [Google Scholar]
- 56.Meng H, Andresen K, van Noort J. Quantitative Analysis of Single-Molecule Force Spectroscopy on Folded Chromatin Fibers. Nucleic Acids Res. 2015;43:3578–3590. doi: 10.1093/nar/gkv215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Forties RA, North JA, Javaid S, Tabbaa OP, Fishel R, Poirier MG, Bundschuh R. A Quantitative Model of Nucleosome Dynamics. Nucleic Acids Res. 2011;39:8306–8313. doi: 10.1093/nar/gkr422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cutter AR, Hayes JJ. A Brief Review of Nucleosome Structure. FEBS Lett. 2015;589:2914–2922. doi: 10.1016/j.febslet.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wolffe A. Chromatin: Structure and Function. 2. Academic Press; London: 1995. p. xii.p. 299. [Google Scholar]
- 60.Sadowski I, Ma J, Triezenberg S, Ptashne M. Gal4-Vp16 Is an Unusually Potent Transcriptional Activator. Nature. 1988;335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- 61.Shimko JC, North JA, Bruns AN, Poirier MG, Ottesen JJ. Preparation of Fully Synthetic Histone H3 Reveals That Acetyl-Lysine 56 Facilitates Protein Binding within Nucleosomes. J Mol Biol. 2011;408:187–204. doi: 10.1016/j.jmb.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cluzel P, Lebrun A, Heller C, Lavery R, Viovy JL, Chatenay D, Caron F. DNA: An Extensible Molecule. Science. 1996;271:792–794. doi: 10.1126/science.271.5250.792. [DOI] [PubMed] [Google Scholar]
- 63.Smith SB, Cui Y, Bustamante C. Overstretching B-DNA: The Elastic Response of Individual Double-Stranded and Single-Stranded DNA Molecules. Science. 1996;271:795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 64.Wang MD, Yin H, Landick R, Gelles J, Block SM. Stretching DNA with Optical Tweezers. Biophys J. 1997;72:1335–1346. doi: 10.1016/S0006-3495(97)78780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lipfert J, Kerssemakers JW, Jager T, Dekker NH. Magnetic Torque Tweezers: Measuring Torsional Stiffness in DNA and Reca-DNA Filaments. Nat Methods. 2010;7:977–980. doi: 10.1038/nmeth.1520. [DOI] [PubMed] [Google Scholar]
- 66.Rothbart SB, Strahl BD. Interpreting the Language of Histone and DNA Modifications. Biochim Biophys Acta. 2014;1839:627–643. doi: 10.1016/j.bbagrm.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li G, Reinberg D. Chromatin Higher-Order Structures and Gene Regulation. Curr Opin Genet Dev. 2011;21:175–186. doi: 10.1016/j.gde.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Das C, Tyler JK, Churchill ME. The Histone Shuffle: Histone Chaperones in an Energetic Dance. Trends Biochem Sci. 2010;35:476–489. doi: 10.1016/j.tibs.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Busuttil K, Rotaru A, Dong M, Besenbacher F, Gothelf KV. Transfer of a Protein Pattern from Self-Assembled DNA Origami to a Functionalized Substrate. Chem Commun (Camb) 2013;49:1927–1929. doi: 10.1039/c3cc37408d. [DOI] [PubMed] [Google Scholar]
- 70.Sacca B, Meyer R, Erkelenz M, Kiko K, Arndt A, Schroeder H, Rabe KS, Niemeyer CM. Orthogonal Protein Decoration of DNA Origami. Angew Chem Int Ed Engl. 2010;49:9378–9383. doi: 10.1002/anie.201005931. [DOI] [PubMed] [Google Scholar]
- 71.Douglas SM, Marblestone AH, Teerapittayanon S, Vazquez A, Church GM, Shih WM. Rapid Prototyping of 3d DNA-Origami Shapes with Cadnano. Nucleic Acids Res. 2009;37:5001–5006. doi: 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sobczak JP, Martin TG, Gerling T, Dietz H. Rapid Folding of DNA into Nanoscale Shapes at Constant Temperature. Science. 2012;338:1458–1461. doi: 10.1126/science.1229919. [DOI] [PubMed] [Google Scholar]
- 73.Stahl E, Martin TG, Praetorius F, Dietz H. Facile and Scalable Preparation of Pure and Dense DNA Origami Solutions. Angew Chem Int Ed Engl. 2014;53:12735–12740. doi: 10.1002/anie.201405991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Luger K, Rechsteiner TJ, Richmond TJ. Preparation of Nucleosome Core Particle from Recombinant Histones. Methods Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.