

Abstract
Squamous cell carcinoma (SCC) comprises 90% of all head and neck cancers and has a poor survival rate due to late-stage disease that is refractive to traditional therapies. Epidermal growth factor receptor (EGFR) is over-expressed in greater than 80% of head and neck SCC (HNSCC). However, EGFR targeted therapies yielded little to no efficacy in clinical trials. This study investigated the efficacy of co-targeting EGFR and the anaplastic lymphoma kinase (ALK) whose promoter is hypomethylated in late-stage oral SCC (OSCC). We observed increased ALK activity in late-stage human OSCC tumors and invasive OSCC cell lines. We also found that while ALK inhibition alone had little effect on proliferation, co-targeting ALK and EGFR significantly reduced OSCC cell proliferation in vitro. Further analysis showed significant efficacy of combined treatment in HSC3-derived xenografts resulting in a 30% decrease in tumor volumes by 14 days (p < 0.001). Western blot analysis showed that co-targeting ALK and EGFR significantly reduced EGFR phosphorylation (Y1148) in HSC3 cells but not Cal27 cells. ALK and EGFR downstream signaling interactions are also demonstrated by Western blot analysis in which lone EGFR and ALK inhibitors attenuated AKT activity whereas co-targeting ALK and EGFR completely abolished AKT activation. No effects were observed on ERK1/2 activation. STAT3 activity was significantly induced by lone ALK inhibition in HSC3 cells and to a lower extent in Cal27 cells. Together, these data illustrate that ALK inhibitors enhance anti-tumor activity of EGFR inhibitors in susceptible tumors that display increased ALK expression, most likely through abolition of AKT activation.
Keywords: Anaplastic lymphoma kinase, Epidermal growth factor receptor, Oral squamous cell carcinoma, Head and neck cancer
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the major malignancy of the oropharynx and the sixth most common cancer in the world [1]. The incidence of HNSCC is increasing worldwide. Currently, HNSCC patients are treated with surgery, chemotherapy, radiotherapy or combination. However, these treatment modalities are less effective with advanced disease involving lymph node invasion, which is associated with a five-year survival as low as 34% [2]. A major factor in decreased survival is resistance to chemotherapy. New lines of therapy are therefore needed to improve the survival rate of HNSCC patients.
In the effort to discover novel molecular factors that contribute to advanced HNSCC and may serve as therapeutic targets, our recent studies demonstrated that anaplastic lymphoma kinase (ALK) is epigenetically deregulated in late-stage, oral SCC (OSCC) and may play a role in OSCC invasiveness [3]. We found that invasive OSCC tumors with lymph node metastasis exhibited significantly lower ALK promoter methylation compared to noninvasive OSCC, suggesting that differential ALK promoter methylation affecting expression may predict the development of metastatic OSCC [3]. Correspondingly, our analysis of head and neck cancer cohort in The Cancer Genome Atlas revealed a significant increase (p < 0.01) of ALK expression in tumors compared to normal controls [3]. These studies suggested that ALK may be a potential therapeutic target for advanced OSCC.
ALK was identified as a therapeutic target for non-small cell lung cancer (NSCLC) in 2007 when Soda and colleagues discovered an inversion on chromosome arm 2p resulting in fusion of the 5′ end of echinoderm microtubule-associated protein-like 4 (EML4) to 3′ ALK yielding the EML4-ALK fusion gene [4,5]. However full-length ALK expression due to point mutations and constitutive activation were reported in numerous cancers including neuroblastoma, neuroectodermal tumors, melanoma and glioblastoma [5–12]. NSCLCs expressing EML4-ALK fusion protein respond very well to the ALK small molecule inhibitor, Crizotinib. However, a subgroup eventually develops resistance to ALK inhibition via induction of epidermal growth factor receptor (EGFR) bypass signaling which, like ALK, signals through AKT, RAS, and STAT3 [12–15].
Approximately 90% of HNSCC express EGFR, yet EGFR inhibitors have yielded little to no efficacy in clinical trials [16,17]. Studies evaluating Erlotinib, a small molecule inhibitor of EGFR, found that a mere 10–15% of oral cancer patients with stage I disease responded to Erlotinib [18]. Moreover, patients with stages II, III, and IV disease failed to respond entirely [18]. The reason for these unexpected failures of EGFR targeted therapies remains unclear. We hypothesize that parallel signaling pathways may compensate for EGFR inhibition rendering these treatments ineffective. In this study we tested the hypothesis that EGFR inhibition is thwarted by ALK parallel signaling in tumors that progress clinically. We assessed the effects of ALK and EGFR inhibition in ALK expressing OSCC cell lines and in a mouse OSCC xenograft model. We further demonstrated anti-proliferative, anti-mobility, and signaling effects in vitro and significant tumor growth inhibition in vivo.
Materials and methods
Human OSCC cell lines
OSCC cell lines, HSC3 and Cal27 were derived from human primary tongue OSCC. Cal27 cells were obtained from ATCC (Rockville, MD). HSC3 cells were kindly provided by Dr. Brian Schmidt (NYU) [19] and authenticated by Genetica DNA Laboratories (Cincinnati, OH). Cells were maintained in DMEM (Gibco, Carlsbad, CA) containing 10% FBS at 37 °C in 5% CO2.
Reagents
Gefitinib (EGFR inhibitor) and TAE684 (ALK inhibitor) were obtained from Selleck Chemicals (Houston, TX). TAE684 (50 mg/ml) and Gefitinib (5 mg/ml) were used for OSCC cell treatments in vitro. For animal studies we used TAE684 (10 mg/kg) and Gefitinib (100 mg/kg) dissolved in 0.05% propylene glycol.
Immunohistochemical (IHC) staining
Formalin fixed paraffin-embedded (FFPE) specimens (n = 3 per group) of HSC3-derived xenografts and human OSCC tissue arrays, containing stage I (T1N0M0) and stage IV (T4N0M0) OSCC biopsied from the tongue and normal tongue epithelium, were deparaffinized, rehydrated, and blocked with 3% normal goat serum. Immunohistochemistry was performed using the Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, CA) according to manufacturer’s protocol. Primary antibodies to p-ALK (1:100; MBS855239; MyBioSource; San Diego, CA) were used. Secondary anti-rabbit antibody Substrate interactions were visualized with DAB (Vector Laboratories, Burlingame, CA). Negative controls were incubated in pre-immune serum alone. IHC staining quantification was made using three non-overlapping fields per specimen, selected indiscriminately for analysis. Measurements were made with the use of NIS-Elements imaging software (Nikon, Brighton, MI) associated with an Nikon TE200U microscope and quantified using NIH ImageJ (v1.49) (http://rsb.info.nih.gov/ij) as previously described [20]. Briefly, the optical density (OD) of p-ALK immunostaining was evaluated with OD estimated using the following formula: OD = log (max intensity/mean intensity), where max intensity = 255 for 8-bit images [20].
Immunofluorescent (IF) staining
OSCC cells were cultured on coverslips (1 × 105), fixed with 4% paraformaldehyde and stained as previously described [21,22] with antibodies to total ALK antibody (1:100; #ABN263 EMD Millipore, Billerica, MA) and p-ALK (1:100); #MBS855239; MyBio-Source; San Diego, CA) and anti-rabbit Alexa Flour 488 secondary antibody (1:100; #A-11008, ThermoFisher, Waltham, MA) and DAPI 1 μg/ml (ThermoFisher) as a nuclear stain. Images were acquired with a Nikon TE2000U microscope using a 40×/1.3NA objective lens and processed for illustration purposes with NIS-Elements imaging software (Nikon, Brighton, MI).
Cell viability assays
Cytotoxicity was assessed using the Cell Titer 96® Aqueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI) according to manufacturer’s protocol. Absorbance values of test groups were normalized against controls (n = 4).
Cell migration assay
Cellular migration was performed using the IncuCyte ZOOM™ live cell image device (Essen Bioscience). After achieving cell confluence in 96 well format, the IncuCyte scratch system was used to generate simultaneous rectangular ‘wounds’ with a defined area in HSC3 and Cal27 cell layers. Cells were then subjected to treatment with Gefitinib and/or TAE684 (concentrations specified above) for the time course indicated in the figures. Images of cells moving within the wound area were taken by the automated Incu-Cyte system over intervals of 2 h (HSC3) or 3 h (Cal27). Migration was calculated as percent of cell confluence within the wound, starting at 0% confluence (i.e., no cells in the wound area). When the wound was completely filled with migrating cells, this was calculated at 100% confluence.
Flow cytometry
OSCC cell lines were cultured to 50% confluency and treated with Gefitinib (500 nM), TAE684 (500 nM) and combination treatments for 30 h. Cells were harvested, fixed in 70% EtoH, treated with RNase A, and stained with propidium iodide (PI). FACS analysis of DNA profiles were performed to quantitate cells in the sub-G1, G1, S + G2/M phases of the cell cycle.
Immunoblotting
OSCC cell lines treated with vehicle, Gefitinib (500 nM) and/or TAE684 (500 nM) for 6 h were harvested and lysed in 1% Triton-PBS. Cell lysates (100units/A280) were used for protein electrophoresis in 10% SDS–PAGE. SDS–PAGE separated proteins were transferred to PVDF membrane and the membrane blocked in 5% milk. Rabbit monoclonal antibodies against STAT3 (1:2000; #12640), p-STAT3(Tyr705) (1:4000; #9145) and ERK1/2 (1:2000; #4695) were used (Cell Signaling; Danvers, MA). Rabbit polyclonal antibodies against AKT (1:2000; #9272), p-AKT(Ser473) (1:4000; #9271), EGFR (1:1000, #4267), p-EGFR(Y1148) (1:4000; #4404) and p-ERK1/2 (1:5000; #9101) were also used (Cell Signaling; Danvers, MA). GAPDH rabbit polyclonal antibodies (1:1000; Rockland, #600-401-A33, Limerick, PA) were used to confirm equal loading of protein. Primary antibodies were diluted in a total of 5 ml diluent (1% milk in PBS-0.1% Tw-20) and the membrane incubated overnight at 4 °C. The membrane was washed 3× with PBS-Tw-20, incubated with ECL Plus detection solution (GE Healthcare, South San Francisco, CA) for 1 min and signal detected by exposure to radiograph film for 30 sec. Band intensities were quantified using ImageJ software.
OSCC mouse xenograft models
All studies were approved by the UTHSCSA Institutional Animal Care and Use Committee. Six week-old female athymic nude mice (Harlan, Indianapolis, IN) were used in a laminar air-flow cabinet under pathogen-free conditions. Mice were provided with a 12 h light/dark schedule at controlled temperature and humidity with food and water ad libitum. Mice were acclimated for one week prior to study initiation.
Mice were injected subcutaneously in the right flank with 3 × 106 HSC3 cells in 0.1 ml of sterile PBS. Two weeks post-inoculation, tumors grew to an average volume of 125 mm3. Mice were stratified into four experimental groups (n = 5 per group), which received the following treatments via oral gavage: group A, vehicle control (170 μl); group B, TAE684 (10 mg/kg; 170 μl); group C, Gefitinib (100 mg/kg; 170 μl), and group D, TAE684 (10 mg/kg) + Gefitinib (100 mg/kg); 170 μl). Treatments were repeated every day for a total of 14 days. Mice were monitored daily for tumor growth (using digital calipers), cachexia, and weight loss. Tumor volumes were calculated by the elliptical formula: 1/2(Length × Width2) [23]. At experimental conclusion, tumors were fixed and processed for histological analysis. Hematoxylin and eosin (H&E) staining and IHC studies were performed by the UTHSCSA Cancer Therapy and Research Center (CTRC) core pathology laboratory.
Statistical analysis
Statistical analysis was performed using GraphPad Prism4 (San Diego, California). Cell viability assays (n = 4 per group), cell migration assays (n = 4 per group), Western blot band intensities (n = 3 per group), and IHC staining (n = 3 per group) were analyzed by one-way ANOVA and Bonferroni’s post hoc test Statistical analyses of tumor growth were made using analysis of variance with repeated measures with Bonferroni’s post hoc test (n = 5 per group). A p value less than 0.05 was considered statistically significant.
Results
Activated ALK is expressed in late-stage human OSCC and in OSCC cell lines/xenografts
Our previous studies showed differential expression in ALK was associated with late-stage OSCC [3]. We also showed that OSCC cell lines exhibit different levels of ALK mRNA expression, which corresponded with cell invasiveness [3]. To further examine the role of ALK in OSCC, we examined expression of activated ALK (the phosphorylated form, phospho-ALK) in a human OSCC tissues containing early-stage (stage I) and late-stage (stage 4) OSCCs and in OSCC cell lines, HSC3 and Cal27. IHC analysis of human OSCC showed that activated phospho-ALK was expressed at relatively high levels in stage 4 OSCCs (Fig. 1A, panels a, b, & f; p < 0.01), whereas staining for phospho-ALK was negative in early-stage OSCC (Fig. 1A, panels c, d & f) and normal control (Fig. 1A, panel e). Nonspecific staining in normal oral epithelium is due to tissue folding at the periphery. Immunofluorescent staining revealed expression of total and activated ALK in both OSCC cell lines (Fig. 1B). Total ALK exhibited membrane and cytoplasmic staining as well as more prominent nuclear staining (Fig. 1B, panels a & c). Activated phospho-ALK demonstrated mainly membrane/cytoplasmic localization with some nuclear staining (Fig. 1B, panels b & d). Our immunostaining analysis revealed that HSC3-derived tumors also express high levels of activated phospho-ALK in vivo (Fig. 1C, panel b).
Fig. 1.

Panel A Immunohistochemical analysis of activated, phosphorylated ALK (phospho-ALK) in human OSCC tumors (4×). High phospho-ALK expression is illustrated in Stage IV OSCCs (panels a & b). Low phospho-ALK expression is illustrated in Stage I OSCCs (panels c & d). Normal tongue tissue (panel e) shows some artefactual staining due to tissue folding (panel e). Relative quantification is shown in panel f. Panel B: Immunofluorescent detection of total and activated (phospho-ALK) in OSCC cell lines (40×). Cal27 cells stained for total and phospho-ALK, panels a & b respectively; HSC3 cells stained for total and phospho-ALK, panels c & d respectively. Panel C: Immunohistochemical analysis of HSC3-derived tumors (20×). Representative H & E staining of HSC3-derived tumors is shown in panel a and corresponding phospho-ALK staining is shown in panel b.
Co-targeting ALK and EGFR has additive effects on cell growth in OSCC cell lines
Because ALK was previously shown to interact with the EGFR pathway, which may explain the poor response of OSCCs to drugs targeting EGFR, we examined the response of HSC3 and Cal27, two OSCC cell lines with high ALK expression and activity, to inhibitors of ALK and EGFR singly and in combination. Based upon preliminary data from dose response curves (data not shown), OSCC cell lines were treated with 500 nM of the ALK inhibitor TAE684, 500 nM of the EGFR inhibitor Gefitinib, and a combination of both for 48, 72, and 96 h. A progressive decrease of OSCC cell growth was observed over a 96 h time course with these treatments (Fig. 2A and B). HSC3 and Cal27 cells demonstrated significant anti-proliferative effects in response to single anti-ALK or anti-EGFR treatments in vitro. Co-treatment with Gefitinib and TAE684 resulted in a significant additive reduction in cell growth at all time-points (p < 0.001). To determine whether decrease in cellular proliferation was due to cell cycle effects, we performed flow cytometry cell staining with PI. Our data suggest that cell cycle arrest at the G1 phase associated with a decrease in S phase and an increase in the SubG1 phase may, at least in part, account for decreased proliferation of HSC3 and Cal27 in single treatments, but no additive effects were observed combination treatment (Tables 1 and 2).
Fig. 2.

Panel A Cell viability assay of HSC3 cells treated with Gefitinib (500 nM), TAE684 (500 nM), or both. Panel B: Cell viability assay of Cal27 cells treated with Gefitinib (500 nM), TAE684 (500 nM), or both. Panel C: Wound-healing assay of HSC3 cells treated with Gefitinib (500 nM), TAE684 (500 nM), or both. Panel D: Wound-healing assay of Cal27 cells treated with Gefitinib (500 nM), TAE684 (500 nM), or both. *p < 0.05, **p < 0.01, ***p < 0.001 treatment compared to control. #p < 0.05, ##p < 0.01, ###p < 0.001 Gefitinib compared to combination treatment. This data represents four replicates and error bars indicate SD.
Table 1.
Cal27 cell cycle analysis.
| Treatment | SubG1 | G1 | S | G2 |
|---|---|---|---|---|
| Control | 0.95 | 86.78 | 5.12 | 7.15 |
| Gefitinib | 2.53 | 90.79 | 2.59 | 4.44 |
| TAE684 | 1.68 | 88.11 | 2.25 | 8.40 |
| Combination | 1.94 | 88.7 | 2.32 | 7.65 |
Values represent % of cell distribution.
Table 2.
HSC3 cell cycle analysis.
| Treatment | SubG1 | G1 | S | G2 |
|---|---|---|---|---|
| Control | 0.84 | 85.67 | 8.21 | 4.91 |
| Gefitinib | 2.03 | 95.60 | 0.0 | 1.87 |
| TAE684 | 0.34 | 95.23 | 0.84 | 3.73 |
| Combination | 1.90 | 96.25 | 0.59 | 2.26 |
Values represent % of cell distribution.
We then examined the effects of ALK and/or EGFR inhibition on HSC3 and Cal27 cell migration using the IncuCyte in vitro ‘wound’ healing assay. HSC3 and Cal27 filled the ‘wound’ (i.e., reaching 100% migration density) within 6 and 24 h, respectively, after the scratch [3]. ALK inhibition had little effect on migration of HSC3 cells yet significantly reduced Cal27 migration from 9 to 24 h. Mobility was significantly reduced with EGFR inhibition in both cell lines. The highly invasive HSC3 cells displayed significant reduction from 2 to 8 h and completely filling the wound by 10 h (p < 0.001, Fig. 2C). In contrast, migration of the less invasive Cal27 cells was significantly reduced at 9 h and remained significantly low for 48 h (Fig. 2D; p < 0.001). Co-treatment of both HSC3 and Cal27 cells resulted in greater reduction in cell migration than the effects seen with Gefitininb or TAE684 alone (Fig. 2C and D; p < 0.001); however the difference between the combination treatments and single treatments with Gefitinib was not statistically significant.
Co-targeting ALK and EGFR has additive effects on tumor growth in vivo
To further investigate the effects of inhibiting ALK and EGFR in vivo, we generated mouse OSCC xenografts using HSC3 cells (Fig. 3). The findings in this model recapitulated the in vitro studies in that HSC3-derived tumor growth was significantly reduced by EGFR inhibition alone as early as day 6 and co-treatments at day 4 (Fig. 3). Notably, HSC3-derived tumor growth was unaffected by single anti-ALK treatments. However, co-treating with Gefitinib and TAE684 further reduced tumor growth (p < 0.001) and, in fact, caused a significant and dramatic shrinkage in tumor volumes by day 14 with mean volumes of 60 mm3 (co-treatment), vs 431 mm3 (vehicle control), 381 mm3 (TAE684), and 138 mm3 (Gefitinib). The effects of co-treating with Gefitinib and TAE684 were statistically significantly compared to single Gefitinib treatment, suggesting that while ALK inhibition alone is not efficacious, it does enhance the anti-tumorigenic effects of the EGFR inhibitor (p < 0.001; Fig. 3).
Fig. 3.
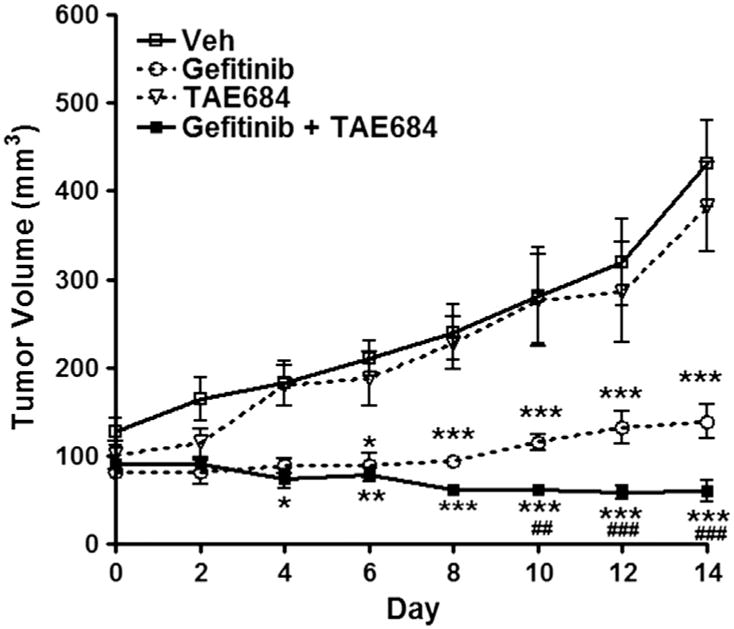
HSC3 mouse xenografts treated daily with Gefitinib (100 mg/kg); TAE684 (10 mg/kg), or both. No change in tumor growth is seen with TAE684 alone. Significant reduction in tumor volume compared to control is seen as early as day 6 with Gefitinib alone and day 4 with co-treatments. By day 10 a significant reduction in tumor volume between Gefitinib and co-treatments is seen. *p < 0.05, **p < 0.01, ***p < 0.001 treatments compared to control. ##p < 0.01, ###p < 0.001 Gefitinib compared to co-treatments. This data represents five replicates and error bars indicate SD.
Targeting ALK and EGFR parallel signaling pathways in OSCC cells abolishes AKT signaling in vitro
To investigate the molecular mechanisms behind parallel signaling between ALK and EGFR in OSCC, we examined the effects of ALK and/or EGFR inhibition on the activity of common cellular signaling pathways, using Western blot analysis of HSC3 and Cal27 cell lines. Both Gefitinib and TAE684 significantly reduced AKT activation in HSC3 cells; however co-treatments completely abolished phospho-AKT (Fig. 4A). Cal27 cells treated with Gefitinib alone and in combination with TAE684 also demonstrated undetectable levels of phospho-AKT (Fig. 4B). TAE684 treatment significantly decreased phospho-AKT in both OSCC cell lines but seemed to be less effective than Gefitinib. Levels of total and phospho-ERK1/2, another downstream effector of EGFR and ALK, were unaffected by any of the treatments. Notably, activation of STAT-3 was induced by TAE684 treatment in HSC3 cells (p < 0.01) and slightly induced in Cal27 cells. Taken together, EGFR and ALK inhibitors effects on these parallel pathways appear to converge upon AKT signaling in OSCC cell lines.
Fig. 4.

Western blot analysis of EGFR signaling components in OSCC cell lines treated for six hours with Gefitinib (500 nM), TAE684(500 nM), or both. Panel A: HSC3 cells treated with Gefitinib demonstrate a reduced ratio of phosphorylated EGFR (p-EGFR) to total EGFR. An additive reduction of p-EGFR/EGFR is seen with a combination treatment (Combo) of TAE684 and Gefitinib. The ratio of phosphorylated AKT (p-AKT) to total AKT is significantly reduces with individual treatments and abolished with combination treatments. No change in the ratio of phosphorylated ERK1/2 (p-ERK1/2) to total ERK1/2 is seen. Lone treatment with the ALK inhibitor, TAE684, significantly induces activation of STAT3 as reflected in the ratio of phosphorylated STAT3 (p-STAT3) to total STAT3. This induction is reversed with combination treatments of TAE684 and Gefitinib. Panel B: Cal27 cells treated with Gefitinib demonstrate no significant changes in the ratio of p-EGFR to total EGFR. Gefitinib alone abolished p-AKT and TAE684 alone significantly reduced the ratio of p-AKT to total AKT. Similarly, combination treatment of Gefitinib and TAE684 (Combo) abolished p-AKT and no change in the ratio of p-ERK1/2 to total ERK1/2 is seen in Cal27 cells. Although a trend is seen with an increase in the p-STAT3 to total STAT3 ratio is seen with all treatments, no significant changes were detected. *p < 0.05, **p < 0.01, ***p < 0.001 denote significance between treatments.
Discussion
The invasion of OSCC into loco-regional structures is a deadly consequence for patients with this malignancy. As of yet, effective therapies that target invasive OSCC are lacking. Although EGFR is expressed in the majority of OSCCs, drugs targeting this tyrosine kinase receptor have surprisingly led to little effect. Although this might be due to increased copy number of EGFR, which is associated with poor prognosis in head and neck cancer, studies showed that EGFR gene amplification, polysomy and truncation (EGFRvIII) do not predict response to EGFR inhibitors [24–26]. Thus, the reasons behind the lack of efficacy of EGFR targeted therapy in OSCC remain unclear.
Our analysis of EGFR RNA expression detected the full receptor, and to a much lower extent the truncated variant EGFRvIII (data not shown); correspondingly, protein expression of full EGFR (Fig. 4) was detected but not EGFRvIII (data not shown; antibody used to internal EGFR domain can detect both full and truncated EGFR based on differences in size), suggesting that EGFRvIII likely does not play a role in decreased EGFR inhibitor efficacy. Another mechanism that may account for the lack of efficacy of EGFR inhibitors is the activity of compensating pathways that may diminish efficacy of these drugs. We have previously shown that ALK may be induced in OSCC, and ALK activity may lead to enhanced invasiveness in OSCC [3]. In this study we examined the interaction between ALK and EGFR in OSCC. This was based on the evidence that induction of EGFR signaling confers resistance to anti-ALK treatment in NSCLC and that anti-EGFR treatment is not effective in OSCC. We hypothesized that co-targeting ALK and EGFR would lead to enhanced anti-oncogenic response compared to single treatments. To pursue this hypothesis, we tested the effects of ALK and EGFR inhibitors, TAE684 and Gefitinib, respectively, on two OSCC cell lines HSC3 and Cal27 expressing both ALK and EGFR.
Our in vitro and in vivo assays using HSC3 and Cal27 cells reveal that while single treatments against EGFR or ALK have varying effects on growth, combination treatments, on the other hand, were very effective in significantly reducing growth in vitro and in vivo. While decrease in proliferation from single treatments may be in part due to arrest at the G phase, apoptotic effects cannot be ruled out. By examining downstream pathways, including AKT, ERK1/2, and STAT3, we found that AKT may be the mechanistic target in combination treatments. Interestingly, while AKT activity was diminished by EGFR inhibition in Cal27, the effect was less prominent in HSC3 cells. Nonetheless, co-treatment with ALK and EGFR inhibitors resulted in equivalent effect in abolishing activated levels of AKT in both HSC3 and Cal27 cells. Also, Cal27 was more responsive to decreased growth and motility by ALK inhibition than HSC3 cells, which was consistent with a stronger response in decreasing AKT activity, suggesting that AKT regulation by ALK may modulate aggressive growth of OSCC. Further studies are required to understand the regulation of AKT by ALK and their downstream role in OSCC growth and invasion.
Importantly, the data highlight that the AKT pathway may be a downstream therapeutic target that modulates both ALK and EGFR activity in OSCC. Previous studies suggest that phosphorylation of AKT predicts poor clinical outcomes in OSCC and significantly correlates with local recurrences and low five-year survival rates [27,28]. AKT activation is also shown to be a significant prognostic indicator for OSCC with lymph node metastasis [29]. Several PI3/AKT pathway inhibitors have been tested in preclinical and clinical trials, which may provide new avenues for the use of these drugs in the treatment of OSCC [30]. While our data suggest that anti-AKT drugs may be a therapeutic strategy in the treatment of OSCC, further examination of this pathway in invasive OSCC may lead to a more efficacious and targeted therapeutic intervention that may also include combinations of EGFR or ALK inhibitors.
Another factor that may affect efficacy of response to receptor tyrosine kinase receptor inhibitors is the translocation of these receptors into the nucleus [31]. Previous studies have shown that EGFR and receptor family member ErbB2 translocate into the nucleus where they act as transcription factors [31]. Similarly, ALK has been shown to be present in the nucleus, which has so far been associated with the NPM-ALK fusion but not the wild-type form [32]. In the case of EGFR and ERbB2, full receptors (no fusions) are transported to the nucleus via endocytosis [31]. It is yet to be seen if this is also true for ALK. Our IF data suggest that ALK is translocated in the nucleus in HSC3 and Cal27 OSCC cells, which raises the possibility that it could exert transcriptional effects in addition to the cell signaling effects described above. Interestingly, ErbB2 nuclear interaction with STAT3 and actions of this complex as a transcription factor was observed in human breast cancer cells [31]. Additional studies are needed to examine the mechanisms underlying the translocation of ALK and consequent interactions at the nuclear level and regulation of STAT3 at the signal transduction level.
In summary, our study suggests that co-targeting ALK and EGFR signaling may enhance the efficacy of EGFR targeted therapies. Treatment with both the ALK and EGFR inhibitors led to additive effects in OSCC cells exhibiting elevated ALK activity. AKT may be a converging point for ALK and EGFR signaling, and STAT3 may modulate ALK activity. Additional studies are required to examine the role of AKT as an effector of ALK and EGFR and the potential of its utility as a therapeutic target alone or in combination with ALK and/or EGFR inhibitors.
Acknowledgments
This work was supported by the University of Texas Health Science Center of San Antonio (UTHSCSA), Cancer Therapy and Research Center (CTRC), Experimental and Developmental Therapeutics Pilot Award and The Owen’s Foundation Cancer Research Pilot Award. Immunohistochemistry was performed by the CTRC Core Pathology Laboratory supported by the NCI Cancer Center Support Grant (P30 CA054174).
Abbreviations
- ALK
anaplastic lymphoma kinase
- EGFR
epidermal growth factor receptor
- OSCC
oral squamous cell carcinoma
Footnotes
Conflict of interest statement
The authors have no conflict of interest.
References
- 1.Cancer facts and figures. Atlanta: American Cancer Society; 2015. [Google Scholar]
- 2.Cancer facts and figures. Atlanta: American Cancer Society; 2012. [Google Scholar]
- 3.Huang TT, Gonzales CB, Gu F, Hsu YT, Jadhav RR, Wang CM, et al. Epigenetic deregulation of the anaplastic lymphoma kinase gene modulates mesenchymal characteristics of oral squamous cell carcinomas. Carcinogenesis. 2013 doi: 10.1093/carcin/bgt112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horn L, Pao W. EML4-ALK: honing in on a new target in non-small-cell lung cancer. J Clin Oncol. 2009;27:4232–5. doi: 10.1200/JCO.2009.23.6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Mol Diag. 2013 doi: 10.1016/j.jmoldx.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Webb TR, Slavish J, George RE, Look AT, Xue L, Jiang Q, et al. Anaplastic lymphoma kinase: role in cancer pathogenesis and small-molecule inhibitor development for therapy. Expert Rev Anticancer Ther. 2009;9:331–56. doi: 10.1586/14737140.9.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powers C, Aigner A, Stoica GE, McDonnell K, Wellstein A. Pleiotrophin signaling through anaplastic lymphoma kinase is rate-limiting for glioblastoma growth. J Biol Chem. 2002;277:14153–8. doi: 10.1074/jbc.M112354200. [DOI] [PubMed] [Google Scholar]
- 8.Miyake I, Hakomori Y, Shinohara A, Gamou T, Saito M, Iwamatsu A, et al. Activation of anaplastic lymphoma kinase is responsible for hyperphosphorylation of ShcC in neuroblastoma cell lines. Oncogene. 2002;21:5823–34. doi: 10.1038/sj.onc.1205735. [DOI] [PubMed] [Google Scholar]
- 9.Shao CK, Su ZL, Feng ZY, Rao HL, Tang LY. Significance of ALK gene expression in neoplasms and normal tissues. Ai Zheng. 2002;21:58–62. [PubMed] [Google Scholar]
- 10.Wellstein A. ALK receptor activation, ligands and therapeutic targeting in glioblastoma and in other cancers. Front Oncol. 2012;2:192. doi: 10.3389/fonc.2012.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grzelinski M, Steinberg F, Martens T, Czubayko F, Lamszus K, Aigner A. Enhanced antitumorigenic effects in glioblastoma on double targeting of pleiotrophin and its receptor ALK. Neoplasia. 2009;11:145–56. doi: 10.1593/neo.81040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang YW, Tu PH, Lin KT, Lin SC, Ko JY, Jou YS. Identification of oncogenic point mutations and hyperphosphorylation of anaplastic lymphoma kinase in lung cancer. Neoplasia. 2011;13:704–15. doi: 10.1593/neo.11222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012;4:120ra117. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci USA. 2011;108:7535–40. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14:4275–83. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moon C, Chae YK, Lee J. Targeting epidermal growth factor receptor in head and neck cancer: lessons learned from cetuximab. Exp Biol Med (Maywood) 2010;235:907–20. doi: 10.1258/ebm.2009.009181. [DOI] [PubMed] [Google Scholar]
- 17.Petrelli F, Coinu A, Riboldi V, Borgonovo K, Ghilardi M, Cabiddu M, et al. Concomitant platinum-based chemotherapy or cetuximab with radiotherapy for locally advanced head and neck cancer: a systematic review and metaanalysis of published studies. Oral Oncol. 2014 doi: 10.1016/j.oraloncology.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 18.Tsien CI, Nyati MK, Ahsan A, Ramanand SG, Chepeha DB, Worden FP, et al. Effect of erlotinib on epidermal growth factor receptor and downstream signaling in oral cavity squamous cell carcinoma. Head Neck. 2012 doi: 10.1002/hed.23128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saghafi N, Lam DK, Schmidt BL. Cannabinoids attenuate cancer pain and proliferation in a mouse model. Neurosci Lett. 2011;488:247–51. doi: 10.1016/j.neulet.2010.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mustafa HN, El Awdan SA, Hegazy GA, Abdel Jaleel GA. Prophylactic role of coenzyme Q10 and Cynara scolymus L. on doxorubicin-induced toxicity in rats: biochemical and immunohistochemical study. Indian J Pharmacol. 2015;47:649–56. doi: 10.4103/0253-7613.169588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siddiqa A, Cavazos D, Chavez J, Long L, Marciniak RA. Modulation of telomeres in alternative lengthening of telomeres type I like human cells by the expression of Werner protein and telomerase. J Oncol. 2012:806382. doi: 10.1155/2012/806382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeske NA, Por ED, Belugin S, Chaudhury S, Berg KA, Akopian AN, et al. A-kinase anchoring protein 150 mediates transient receptor potential family V type 1 sensitivity to phosphatidylinositol-4,5-bisphosphate. J Neurosci. 2011;31:8681–8. doi: 10.1523/JNEUROSCI.0020-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jensen MM, Jorgensen JT, Binderup T, Kjaer A. Tumor volume in subcutaneous mouse xenografts measured by microCT is more accurate and reproducible than determined by 18F-FDG-microPET or external caliper. BMC Med Imaging. 2008;8:16. doi: 10.1186/1471-2342-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung CH, Ely K, McGavran L, Varella-Garcia M, Parker J, Parker N, et al. Increased epidermal growth factor receptor gene copy number is associated with poor prognosis in head and neck squamous cell carcinomas. J Clin Oncol. 2006;24:4170–6. doi: 10.1200/JCO.2006.07.2587. [DOI] [PubMed] [Google Scholar]
- 25.Fukuoka M, Wu YL, Thongprasert S, Sunpaweravong P, Leong SS, Sriuranpong V, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS) J Clin Oncol. 2011;29:2866–74. doi: 10.1200/JCO.2010.33.4235. [DOI] [PubMed] [Google Scholar]
- 26.Szabo B, Nelhubel GA, Karpati A, Kenessey I, Jori B, Szekely C, et al. Clinical significance of genetic alterations and expression of epidermal growth factor receptor (EGFR) in head and neck squamous cell carcinomas. Oral Oncol. 2011;47:487–96. doi: 10.1016/j.oraloncology.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 27.Yu Z, Weinberger PM, Sasaki C, Egleston BL, Speier WFt, Haffty B, et al. Cancer Epidemiol Biomark Prev. 2007;16:553–8. doi: 10.1158/1055-9965.EPI-06-0121. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Wang J, Wang F, Wang H, Zeng X, Liao G, et al. Tissue microarray analysis reveals the expression and prognostic significance of phosphorylated AktThr (3)(0)(8) in oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;116:591–7. doi: 10.1016/j.oooo.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 29.Knowles JA, Golden B, Yan L, Carroll WR, Helman EE, Rosenthal EL. Disruption of the AKT pathway inhibits metastasis in an orthotopic model of head and neck squamous cell carcinoma. Laryngoscope. 2011;121:2359–65. doi: 10.1002/lary.22180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pal I, Mandal M. PI3K and Akt as molecular targets for cancer therapy: current clinical outcomes. Acta Pharmacol Sin. 2012;33:1441–58. doi: 10.1038/aps.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang YN, Hung MC. Nuclear functions and subcellular trafficking mechanisms of the epidermal growth factor receptor family. Cell Biosci. 2012;2:13. doi: 10.1186/2045-3701-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duyster J, Bai RY, Morris SW. Translocations involving anaplastic lymphoma kinase (ALK) Oncogene. 2001;20:5623–37. doi: 10.1038/sj.onc.1204594. [DOI] [PubMed] [Google Scholar]