

Abstract
Background & objectives:
Human cryptosporidiosis is endemic worldwide, and at least eight species have been reported in humans; the most common being Cryptosporidium hominis and C. parvum. Detailed understanding of the epidemiology of Cryptosporidium is increasingly facilitated using standardized universal technique for species differentiation and subtyping. In this study micro- and minisatellite targets in chromosome 6 were used to assess genetic diversity of C. hominis by sequence length polymorphisms along with single nucleotide polymorphisms (SNPs).
Methods:
A total of 84 Cryptosporidium positive stool specimens were subjected to speciation and genotyping using small subunit (SSU) ribosomal RNA (rRNA) as the target gene. Genetic heterogeneity amongst C. hominis isolates was assessed by sequencing minisatellites, microsatellites and polymorphic markers including genes encoding the 60 kDa glycoprotein (GP60), a 47 kDa protein (CP47), a mucin-like protein (Mucin-1), a serine repeat antigen (MSC6-7) and a 56 kDa transmembrane protein (CP56).
Results:
Of the 84 Cryptosporidium positive stool specimens, 77 (92%) were positive by SSU rRNA gene polymerase chain reaction (PCR) assay. Of these 77 isolates, 54 were identified as C. hominis and 23 as C. parvum. Of all the loci studied by multilocus sequence typing (MLST), GP60 gene could reveal the highest genetic diversity. Population substructure analysis of C. hominis performed by combined sequence length and nucleotide polymorphism showed nine multilocus subtypes, all of which were distinct groups in the study population.
Interpretation & conclusions:
MLST, a powerful discriminatory test, demonstrated both variations and distribution pattern of Cryptosporidium species and its subtypes.
Keywords: Cryptosporidium, genetic diversity, multilocus sequence typing, population structure
Cryptosporidium is an apicomplexan protozoan, responsible for causing enteric diseases worldwide1. Two species, Cryptosporidium hominis and C. parvum, are of major significance to public health and are responsible for several sporadic diarrhoea and outbreaks2,3. C. parvum is zoonotic and infects a wide range of animal hosts including humans, whereas C. hominis infects only humans4. The genomes of both C. parvum and C. hominis (IOWA and TU502 reference strains, respectively) have been sequenced5,6. The genome sizes for C. parvum and C. hominis are 9.11 and 9.16 Mb, respectively. Genome comparison has shown that C. hominis and C. parvum are very similar and exhibit only 3-5 per cent sequence divergence, with no large insertions, deletions or rearrangements6. The genomic data for the genome representatives are available online (http://CryptoDB org., http://cryptodb.org/cryptodb/).
Various molecular tools are available to distinguish different Cryptosporidium spp. that are pathogenic to humans2. Molecular typing studies based on a single locus are limited in their abilities to capture the structure of a population, which is better explored using a multilocus approach. Hence, for better understanding of the transmission dynamics, genetic diversity and population structure of the Cryptosporidium spp., high-resolution markers are required to be studied. Multilocus sequence typing (MLST) that has been extensively used for population biology study of many microorganisms7,8 has been used for subtyping primarily for C. hominis and to a lesser extent for C. parvum9,10. Studies from Scotland and the United States have indicated that C. hominis shows a clonal population structure as it displays lower number of subtypes and much stronger linkage disequilibrium (LD) compared to C. parvum10. In this study, microsatellite and minisatellite targets in chromosome 6 were used to study genetic diversity with special reference to C. hominis by studying sequence length polymorphisms along with single nucleotide polymorphisms (SNPs).
Material & Methods
This study was conducted in the department of Microbiology, All India Institute of Medical Sciences, a tertiary care referral and teaching hospital in New Delhi, India. The study protocol was reviewed and approved by the Institutional Ethics Committee. All participants provided written informed consent before participating in the study. Stool specimens from 84 patients comprising 82 diagnosed as positive for Cryptosporidium species by microscopy11 and an additional two by polymerase chain reaction (PCR) assay using 18S ribosomal RNA (rRNA) gene12 were collected for the study. The clinical samples that were routinely submitted for relevant examination were obtained from both inpatient and outpatient departments of the hospital from June 2010 to November 2012. Clinical data such as age, sex and major clinical symptoms were recorded in a structured questionnaire for each patient and/or guardian in case of children.
DNA extraction and species identification: DNA was extracted from all positive clinical specimens using QIAamp DNA stool kit (QIAGEN Inc., USA) following the manufacturer's protocol except for one modification that the initial lysis duration was increased from 10 to 45 min at 95°C. Speciation and genotyping was performed for each specimen by nested PCR assay and restriction fragment length polymorphism (RFLP) analysis that amplifies an approximately (~) 830 bp fragment of the small subunit ribosomal RNA (SSU rRNA) gene13. Restriction analysis of PCR products was done with the enzymes SspI and AseI (New England Bio Labs, Beverly, MA, USA)13.
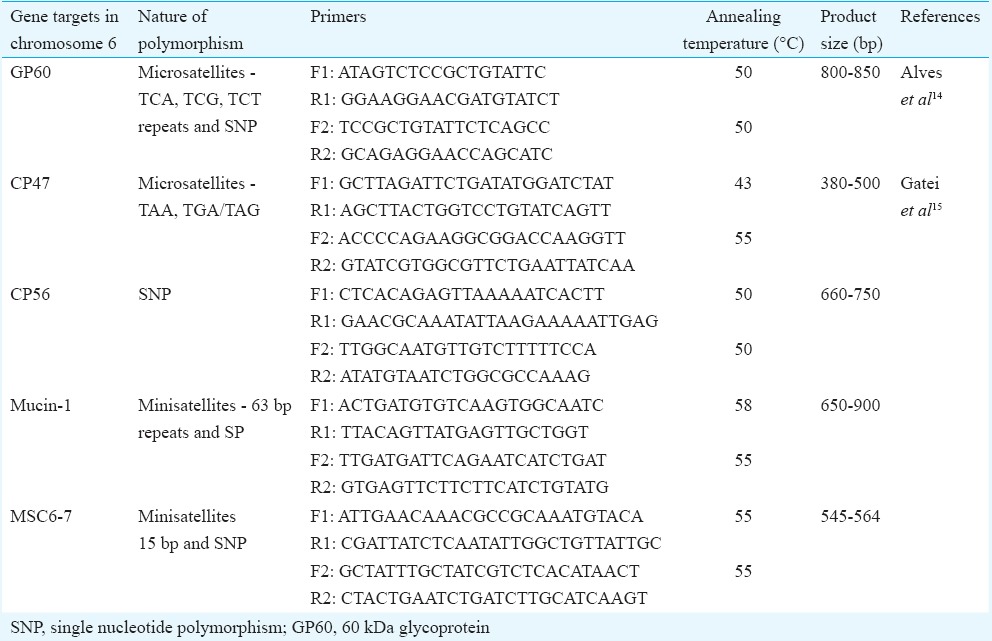
Targets for multilocus sequence typing (MLST): Five gene loci in chromosome 6 were targeted for the subtype analysis of C. hominis. The genetic loci included 60 kDa glycoprotein (GP60) (accession no. JF495139), a 47 kDa protein (CP47) (accession no. AAM46174), a serine repeat antigen (MSC6-7) (XM_660698), a mucin-like protein (Mucin-1) (accession no. XM_661200) and a 56 kDa transmembrane protein (CP56) (accession no. XM_661161). GP60 and CP47 are the microsatellites, Mucin-1 and MSC6-7 are the minisatellites and CP56 is a SNP marker. The GP60 gene encodes glycoproteins gp15 and gp45, which are implicated in attachment to and invasion of host cells14. Microsatellites and minisatellites are DNA sequences that consist of tandemly repeated sequence motifs of 1-4 base pair (microsatellites) or more (minisatellites). These markers evolve at higher rates than nuclear genes because these are variations in the number of tandem repeats and also in the form of SNPs. This makes them ideal targets to be used in the study of population genetics15. Nested PCR assay was performed for all gene targets. The sequences of primary and secondary primers, annealing temperatures used and size of the expected PCR products are given in Table I.
Table I.
Primers used in the multilocus sequence typing, annealing temperatures used in the polymerase chain reaction assay and expected product sizes
Multilocus sequence typing (MLST) polymerase chain reaction (PCR) assays: For PCR assay, the total volume of PCR mixture was 100 μl, which contained 2 μl of DNA (for primary PCR) and 1 μl of the primary PCR product (for secondary PCR), primer pair at a concentration of 0.4 μM (for both the primary and secondary PCR) (Integrated DNA Technologies, USA), 0.2 mM deoxyribonucleotide triphosphate mix (Fermentas, USA), 3 mM MgCl2, 1x PCR buffer and 2.5 U of Taq DNA polymerase (Bangalore Genei, India). The primary PCR reaction also contained 400 ng/ml of nonacetylated bovine serum albumin (Sigma-Aldrich, USA). PCR amplification was carried out with an initial denaturation at 94°C for five minutes, 35 cycles of 94°C for 45 sec, specific annealing temperature for each target gene (Table I) for 45 sec, and 72°C for one minute and a final extension of the PCR product at 72°C for 10 min15. PCR products were visualized under ultraviolet light by ethidium bromide staining of two per cent agarose gel (Sigma-Aldrich, USA).
DNA sequence analysis: Gel-based extraction of the PCR products was performed as per the manufacturer's instructions using MinElute Gel Extraction Kit (Qiagen, USA). Sequencing for the study isolates was performed in both forward and reverse direction on ABI 3500xL Genetic Analyzer from Chromous Biotech, India. Nucleotide sequences were read using the software ChromasPro (www.technelysium.com.au/ChromasPro.html). Alignment of consensus sequences obtained and those from the GenBank database was done using ClustalX (http://bips.ustrasbg.fr/fr/Documentation/ClustalX/) after manual editing of the alignments using the BioEdit program version 7.0.4 (http://www.mbio.ncsu.edu/BioEdit/bioedit.html).
Phylogenetic analysis: Phylogeny was inferred by constructing an unweighed pair group method with arithmetic mean (UPGMA) tree for length polymorphism using ClustalW (www.ebi.ac.uk/Tools/msa/clustalw2/) and MEGA5 software (www.megasoftware.net). The evolutionary distances were also computed by MEGA5 software using the Kimura 2-parameter method. The rate variation amongst sites was modelled with a gamma distribution. The phylogram for the MLST loci for C. hominis was inferred by constructing neighbour-joining tree using MEGA5 software. The evolutionary distances were computed using the Kimura 2-parameter method.
Results
Eighty four patients with cryptosporidiosis presenting with chronic diarrhoea comprised 40 children (26 males) and 44 adults (28 males). The mean age of children was 1.5 ± 1.1 yr and of adults was 36.5 ± 15.5 yr. Of the total 84 isolates, 72 (92%) were positive by SSU rRNA gene PCR assay. The seven isolates could not be amplified probably because of the presence of inhibitors in the stool specimens, insufficient DNA content in the clinical sample and/or degradation or shearing of DNA due to repeated freeze and thaw. Of the total 77 isolates, 54 were identified as C. hominis and 23 as C. parvum.
Multilocus analysis: Multilocus analysis could be performed for nearly 50 per cent (n=24) of representative samples of C. hominis isolates. Sequence length polymorphisms were observed only at GP60, CP47 and MSC6-7 loci, whereas the Mucin-1 and CP56 locus were monomorphic in size. For all markers, locus fragment sizes were determined from the overall sequence length rather than the number of repeats as deletions and insertions seen in non-repeat regions15.
Classification of subtypes: Based on the length polymorphism of 24 isolates of C. hominis, five subtypes were identified at the CP47 locus at a frequency of 8.3 per cent (n=2) for subtype 1 (406 bp), 8.3 per cent (n=2) for subtype 2 (484 bp), 8.3 per cent (n=2) for subtype 3 (490 bp), 12.5 per cent (n=3) for subtype 4 (496 bp) and 62.5 per cent (n=15) for subtype 5 (526 bp). To identify the subtypes, trinucleotide TAA was coded as ‘A’ while the TAG/TGA was coded as ‘G’ with the following digit showing the number of each trinucleotide repeat. The five subtypes identified in this study were IA30G16 (8.3%), IA44G28 (8.3%), IA45G29 (8.3%), IA46G30 (12.5%) and IA51G35 (62.5%), respectively (Table II).
Table II.
Length polymorphism (in base pairs) and frequencies (%) of multilocus sequence typing-based subtypes at each locus (n=24)
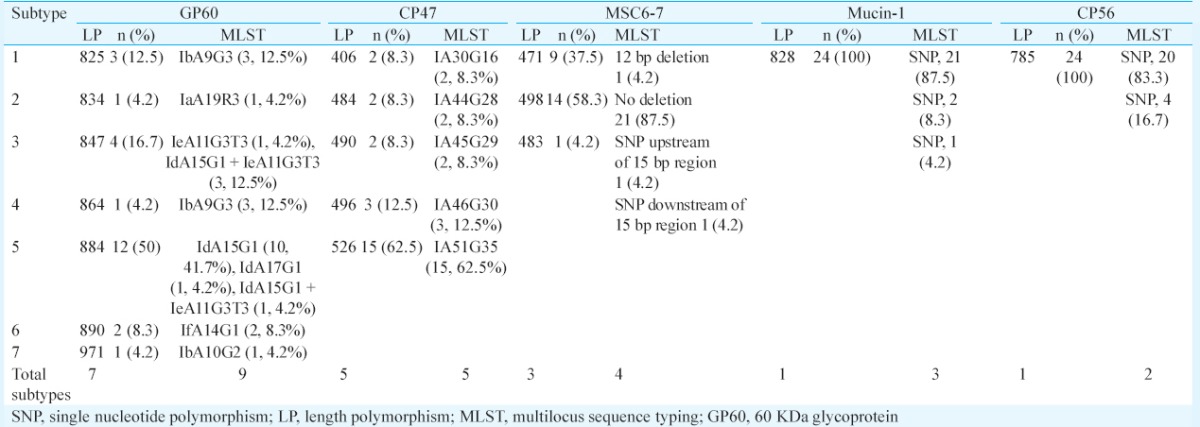
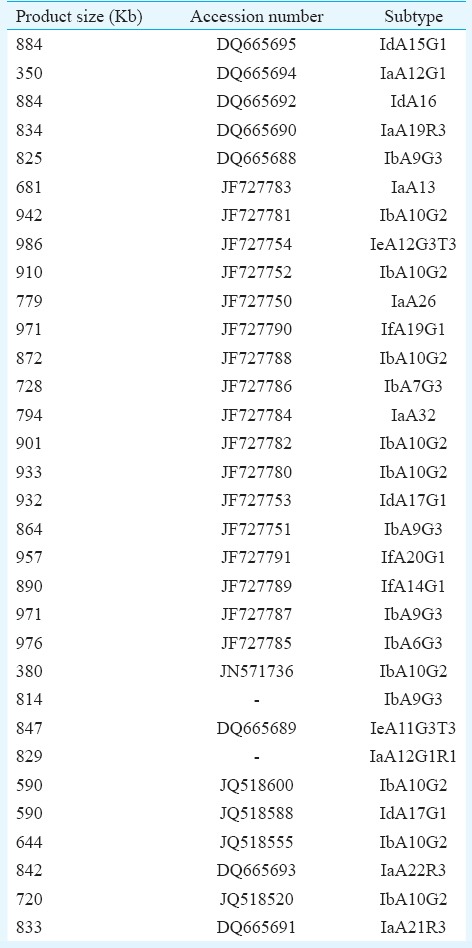
At the GP60 locus, seven subtypes were observed based on length polymorphism at a subtype frequency of 12.5 per cent (n=3), 4.2 per cent (n=1), 16.7 per cent (n=4), 4.2 per cent (n=1), 50 per cent (n=12), 8.3 per cent (n=2) and 4.2 per cent (n=1) for subtypes 1 (825 bp); 2 (834 bp), 3 (847 bp), 4 (864 bp), 5 (884 bp), 6 (890 bp) and 7 (971 bp), respectively, for all 24 isolates. At the GP60 locus, subtype classification was based on a combination of the number of trinucleotide TCA, TCG or TCT repeats in the microsatellite region and the SNP in the rest of the sequence. Using this method, seven subtypes were identified of some of the reference subtypes that have been listed in Table III. The subtypes identified were IdA15G1 (41.7%, 10/24), IbA9G3 (16.7%, 4/24), IeA11G3T3 (4.2%, 1/24), IfA14G1 (8.3%, 2/24), IbA10G2 (4.2%, 1/24), IdA17G1 (4.2%, 1/24) and IaA19R3 (4.2%, 1/24) (Table II). Four (16.7%) patients, however, had mixed infections with IdA15G1 and IeA11G3T3 subtypes.
Table III.
60 kDa glycoprotein gene subtypes with their product size
In addition, initially, three subtypes were identified at the MSC6-7 locus at a frequency of 37.5 per cent (n=9), 58.3 per cent (n=14) and 4.2 per cent (n=1) for subtypes 1 (471 bp), 2 (498 bp) and 3 (483 bp), respectively. The MSC6-7 locus had a 15 bp TGATGATGAT(G)GAACC(T) minisatellite repeat region from position 91-105 bp and an additional insertion or deletion of a 12 bp fragment of TTCATCTTCATT between positions 202 and 213 amongst all the sequences (Fig. 1). One extra subtype was identified, resulting in a total of four subtypes due to the presence of SNP outside the repeat region of the two isolates. The isolate CH1 had an insertion of one base pair at 24th position upstream of the repeat region, whereas the isolate CH4 had a deletion of one base pair at 422nd position downstream of the repeat region (Fig. 2). Subtype frequencies for subtypes 1-4 were, therefore, 33.3 per cent (n=8), 8.3 per cent (n=2), 54.2 per cent (n=13) and 4.2 per cent (n=1), respectively.
Fig. 1.

The boxed area indicates the deletion of 12 bp region between position 201-213 bp of MSC-6 gene amongst Cryptosporidium hominis isolates.
Fig. 2.

The boxed area indicates the 15bp repeat region and the circled area refers to the single nucleotide polymorphisms amongst MSC6-7 gene isolates.
The mucin-1 locus has a 63 bp minisatellite repeat region (CCAAAACCTGAAAAAGATTCTA AGTCATCATG TGCTTGCTCTAAATCAGATAAA) in addition to SNP within the minisatellite region of the gene fragment. There are 9-11 copies of the repeat in C. hominis Mucin-1 gene sequence. All the Mucin-1 gene sequences in our study had 11 copies of the minisatellite repeats. Diversity was observed due to SNP, which resulted in the formation of three subtypes at a frequency of 87.5 per cent (n=21), 8.3 per cent (n=2) and 4.2 per cent (n=1). CP56 locus is a highly polymorphic subtelomere target in chromosome 6. Polymorphisms in this locus occur due to SNPs across approximately 785 bp fragment. Of the known three subtypes, subtype 3 was present at a frequency of 83.3 per cent (n=20) followed by 16.7 per cent subtype 2 (n=4) (Table II). Subtype 1 was not observed in the present study.
Phylogenetic analysis: As the sequence length polymorphisms were detected only at GP60, CP47 and MSC6-7 loci, the relationship between multilocus subtypes based on length polymorphisms was inferred by constructing an UPGMA tree for the three loci using MEGA5 software. The evolutionary distances were computed using the Kimura 2-parameter method. Multilocus length polymorphism data from 12 isolates at the three loci inferred by UPGMA method resulted in the generation of nine multilocus subtypes (Fig. 3). Likewise, phylogenetic tree was constructed by concatenating all five MLST loci for 12 C. hominis isolates and was inferred by neighbour-joining method. The isolates showed nine major subtype groups (Fig. 4).
Fig. 3.
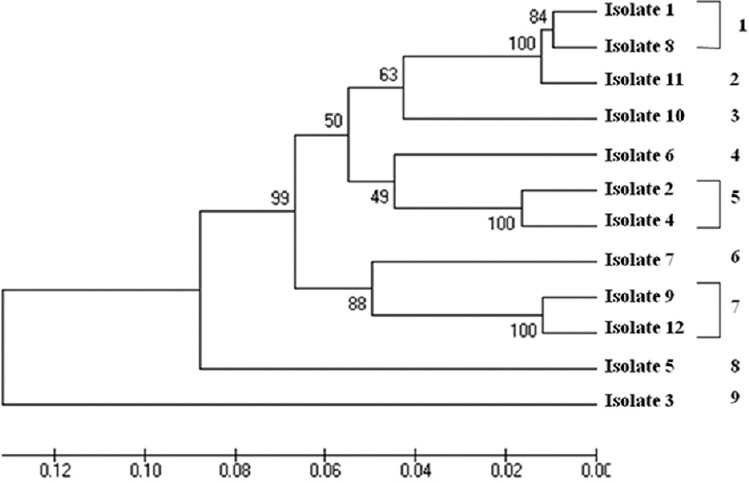
Unweighed pair group method with arithmetic mean analysis showing relationship between multilocus subtypes based on length polymorphism. The optimal tree with the sum of branch length = 0.65904592 is shown. The percentage of replicate trees, in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Kimura 2-parameter method and are in the units of the number of base substitutions per site. The rate variation amongst sites was modelled with a gamma distribution (shape parameter=1). The analysis involved 12 nucleotide sequences. Evolutionary analyses were conducted in MEGA5.
Fig. 4.
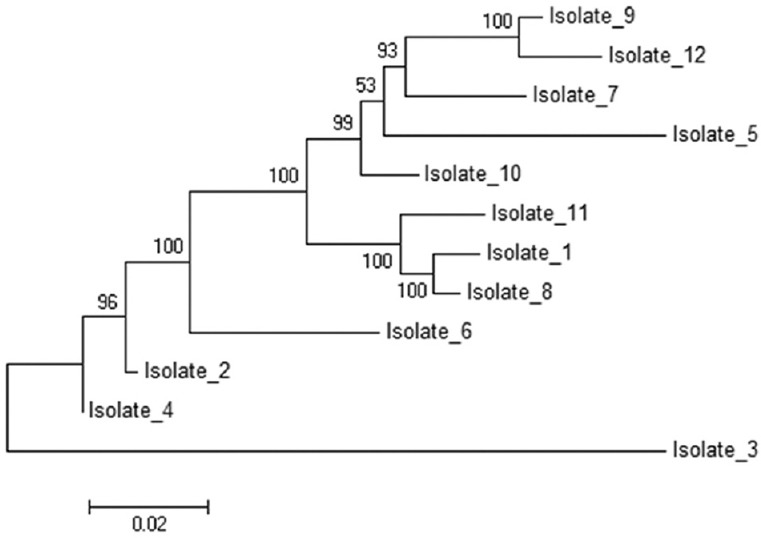
Phylogram for multilocus sequence typing genes for Cryptosporidium hominis isolates (n=12). The evolutionary history was inferred using the neighbour-Joining method. The percentage of replicate trees, in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Kimura 2-parameter method and are in the units of the number of base substitutions per site. The rate variation amongst sites was modelled with a gamma distribution (shape parameter = 1). The analysis involved 12 nucleotide sequences. Codon positions included were 1st+2nd+3rd+Noncoding. All ambiguous positions were removed for each sequence pair. Evolutionary analyses were conducted in MEGA5.
Discussion
The present study confirmed that cryptosporidiosis was endemic in India as also evident in other studies from India16,17. In Delhi, a prevalence of 12-20 per cent of Cryptosporidium spp. by microscopy has been documented in HIV seropositive patients18,19,20,21. In a multisite study conducted in India in children with cryptosporidiosis from Delhi, Trichy and Vellore, C. hominis was the commonly identified species (88%, 59/67), followed by C. parvum and C. meleagridis22. In another study from south India, 81 per cent (47/58) of the species were C. hominis23. The present study also showed C. hominis as the predominant species. This signifies that anthroponotic transmission plays an important role in the epidemiology of cryptosporidiosis in India15,24,25. Clinical data such as age, sex and major clinical symptoms were obtained from each patient and/or guardian in case of children. No significant association of the genotypes or subgenotypes with clinical history could be inferred from the present study.
Significant variation in minisatellite loci is in the form of SNP, which may get missed by typing techniques based on length polymorphism alone. MLST is based on both length polymorphism and SNP and, therefore, increases the resolution of typing methods. In the present study, genetic diversity in C. hominis isolates (n=24) was studied exclusively based on sequence length polymorphism at each locus and subsequently based on the combined length polymorphism and SNP by concatenating all sequences, and the data were analyzed as a single multilocus gene. Sequence length polymorphism was analyzed at GP60, CP47 and MSC6-7 loci, CP56 and Mucin-1 loci being monomorphic in size. Amongst all the markers studied, GP60 showed highest gene diversity as shown earlier26,27. Many subtypes in GP60 family had the same length even though the sequences were different. The most common C. hominis subtype family in the present study was Id which had two subtypes of the same length (884 bp) while DNA sequencing revealed that these were two different subtypes, namely IdA15G1 (41.7%) and IdA17G1 (4.2%).
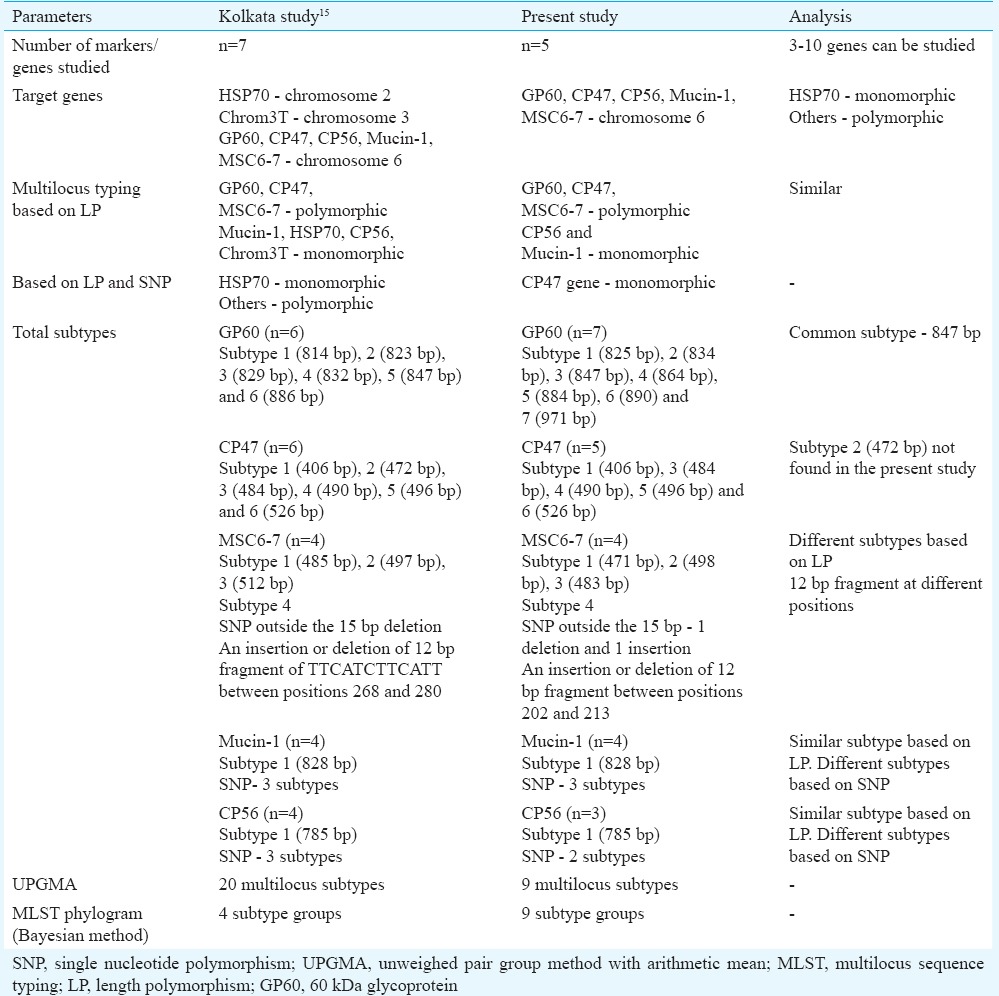
MLST analysis of isolates from our study differed from isolates analyzed from the eastern part of the country i.e., National Institute of Cholera and Enteric Diseases, Kolkata, in many aspects. MLST based on both length polymorphism and SNPs in our study showed that only CP47 gene was monomorphic, whereas the study conducted by Gatei et al15 reported all other loci to be polymorphic except HSP70. Six CP47 subtypes were identified by Gatei et al15 but the present study could identify only five CP47 subtypes. CP47 subtype 2 corresponding to 472 bp was not found in our study isolates. Six GP60 subtypes were reported in their study based on length polymorphism; however, we observed seven GP60 subtypes and all except subtype 847 bp were different. Based on SNP, two different subtypes were identified which were similar in ours as well as their study. Although three subtypes were observed based on length polymorphism at MSC6-7 locus in both the studies, these were different from each other. In addition, one extra subtype was identified due to the presence of SNP outside the 15 bp repeat region in two of our isolates (1 deletion and 1 insertion). An insertion or deletion of a 12 bp fragment of TTCATCTTCATT was reported by Gatei et al15 between positions 268 and 280 in the C. hominis sequence; we, however, observed the 12 bp deletion between positions 202 and 213 in one of our isolates. The Mucin-1 locus had 11 copies of 63 bp minisatellite repeat in both the studies. Three subtypes were observed based on SNP in both the studies. Based on this analysis, the UPGMA tree of Kolkata isolates generated twenty multilocus subtypes and the MLST phylogram inferred by Bayesian method showed four major subtype groups. The UPGMA tree in the present study generated nine multilocus subtypes and the MLST phylogram inferred by neighbour-joining method also showed nine subtype groups (Table IV).
Table IV.
Comparison of the multilocus sequence typing findings between the present study and Kolkata study
In conclusion, our study demonstrated the population structure of C. hominis by MLST typing. MLST is a better tool to study transmission dynamics of Cryptosporidium subtypes due to its high resolution. However, to further understand the significance of distinct subtypes with clinical manifestations of the disease and transmission risks, it will be necessary to analyze a large number of samples and if possible from different geographical areas. The limitation of this study was that the population structure for C. hominis based on MLST could not be assessed by measuring intragenic and intergenic LD and recombination rates due to inability in using some bioinformatics software.
Acknowledgment
Authors thank the Indian Council of Medical Research (ICMR), Department of Health Research, New Delhi, India, for granting financial support for the research work. The first author (PY) received Senior Research Fellowship of the ICMR, New Delhi to conduct this research.
Footnotes
Conflicts of Interest: None.
References
- 1.Fayer R, editor. Cryptosporidium and cryptosporidiosis. Boca Raton, FL: CRC Press Inc; 1997. The general biology of Cryptosporidium; pp. 1–41. [Google Scholar]
- 2.Xiao L, Ryan UM. Cryptosporidiosis: An update in molecular epidemiology. Curr Opin Infect Dis. 2004;17:483–90. doi: 10.1097/00001432-200410000-00014. [DOI] [PubMed] [Google Scholar]
- 3.Cacciò SM, Pozio E. Advances in the epidemiology, diagnosis and treatment of cryptosporidiosis. Expert Rev Anti Infect Ther. 2006;4:429–43. doi: 10.1586/14787210.4.3.429. [DOI] [PubMed] [Google Scholar]
- 4.Robertson LJ, Gjerde BK. Cryptosporidium oocysts: Challenging adversaries? Trends Parasitol. 2007;23:344–7. doi: 10.1016/j.pt.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, et al. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science. 2004;304:441–5. doi: 10.1126/science.1094786. [DOI] [PubMed] [Google Scholar]
- 6.Xu P, Widmer G, Wang Y, Ozaki LS, Alves JM, Serrano MG, et al. The genome of Cryptosporidium hominis. Nature. 2004;431:1107–12. doi: 10.1038/nature02977. [DOI] [PubMed] [Google Scholar]
- 7.Pérez-Losada M, Browne EB, Madsen A, Wirth T, Viscidi RP, Crandall KA. Population genetics of microbial pathogens estimated from multilocus sequence typing (MLST) data. Infect Genet Evol. 2006;6:97–112. doi: 10.1016/j.meegid.2005.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Urwin R, Maiden MC. Multi-locus sequence typing: A tool for global epidemiology. Trends Microbiol. 2003;11:479–87. doi: 10.1016/j.tim.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 9.Mallon M, MacLeod A, Wastling J, Smith H, Reilly B, Tait A. Population structures and the role of genetic exchange in the zoonotic pathogen Cryptosporidium parvum. J Mol Evol. 2003;56:407–17. doi: 10.1007/s00239-002-2412-3. [DOI] [PubMed] [Google Scholar]
- 10.Tanriverdi S, Widmer G. Differential evolution of repetitive sequences in Cryptosporidium parvum and Cryptosporidium hominis. Infect Genet Evol. 2006;6:113–22. doi: 10.1016/j.meegid.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 11.Garcia LS, Bruckner DA, Brewer TC, Shimizu RY. Techniques for the recovery and identification of Cryptosporidium oocysts from stool specimens. J Clin Microbiol. 1983;18:185–90. doi: 10.1128/jcm.18.1.185-190.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orlandi PA, Lampel KA. Extraction-free, filter-based template preparation for rapid and sensitive PCR detection of pathogenic parasitic protozoa. J Clin Microbiol. 2000;38:2271–7. doi: 10.1128/jcm.38.6.2271-2277.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiao L, Escalante L, Yang C, Sulaiman I, Escalante AA, Montali RJ, et al. Phylogenetic analysis of Cryptosporidium parasites based on the small-subunit rRNA gene locus. Appl Environ Microbiol. 1999;65:1578–83. doi: 10.1128/aem.65.4.1578-1583.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alves M, Xiao L, Sulaiman I, Lal AA, Matos O, Antunes F. Subgenotype analysis of Cryptosporidium isolates from humans, cattle, and zoo ruminants in Portugal. J Clin Microbiol. 2003;41:2744–7. doi: 10.1128/JCM.41.6.2744-2747.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gatei W, Das P, Dutta P, Sen A, Cama V, Lal AA, et al. Multilocus sequence typing and genetic structure of Cryptosporidium hominis from children in Kolkata, India. Infect Genet Evol. 2007;7:197–205. doi: 10.1016/j.meegid.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 16.Sethi S, Sehgal R, Malla N, Mahajan RC. Cryptosporidiosis in a tertiary care hospital. Natl Med J India. 1999;12:207–9. [PubMed] [Google Scholar]
- 17.Kaur R, Rawat D, Kakkar M, Uppal B, Sharma VK. Intestinal parasites in children with diarrhea in Delhi, India. Southeast Asian J Trop Med Public Health. 2002;33:725–9. [PubMed] [Google Scholar]
- 18.Gupta K, Bala M, Deb M, Muralidhar S, Sharma DK. Prevalence of intestinal parasitic infections in HIV-infected individuals and their relationship with immune status. Indian J Med Microbiol. 2013;31:161–5. doi: 10.4103/0255-0857.115247. [DOI] [PubMed] [Google Scholar]
- 19.Uppal B, Kashyap B, Bhalla P. Enteric pathogens in HIV/AIDS from a tertiary care hospital. Indian J Community Med. 2009;34:237–42. doi: 10.4103/0970-0218.55291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gautam H, Bhalla P, Saini S, Uppal B, Kaur R, Baveja CP, et al. Epidemiology of opportunistic infections and its correlation with CD4 T-lymphocyte counts and plasma viral load among HIV-positive patients at a tertiary care hospital in India. J Int Assoc Physicians AIDS Care (Chic) 2009;8:333–7. doi: 10.1177/1545109709346881. [DOI] [PubMed] [Google Scholar]
- 21.Gupta S, Narang S, Nunavath V, Singh S. Chronic diarrhoea in HIV patients: Prevalence of coccidian parasites. Indian J Med Microbiol. 2008;26:172–5. doi: 10.4103/0255-0857.40536. [DOI] [PubMed] [Google Scholar]
- 22.Ajjampur SS, Liakath FB, Kannan A, Rajendran P, Sarkar R, Moses PD, et al. Multisite study of cryptosporidiosis in children with diarrhea in India. J Clin Microbiol. 2010;48:2075–81. doi: 10.1128/JCM.02509-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ajjampur SS, Gladstone BP, Selvapandian D, Muliyil JP, Ward H, Kang G. Molecular and spatial epidemiology of cryptosporidiosis in children in a semiurban community in South India. J Clin Microbiol. 2007;45:915–20. doi: 10.1128/JCM.01590-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das P, Roy SS, MitraDhar K, Dutta P, Bhattacharya MK, Sen A, et al. Molecular characterization of Cryptosporidium spp. from children in Kolkata, India. J Clin Microbiol. 2006;44:4246–9. doi: 10.1128/JCM.00091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tumwine JK, Kekitiinwa A, Bakeera-Kitaka S, Ndeezi G, Downing R, Feng X, et al. Cryptosporidiosis and microsporidiosis in Ugandan children with persistent diarrhea with and without concurrent infection with the human immunodeficiency virus. Am J Trop Med Hyg. 2005;73:921–5. [PubMed] [Google Scholar]
- 26.Robinson G, Chalmers RM. Assessment of polymorphic genetic markers for multi-locus typing of Cryptosporidium parvum and Cryptosporidium hominis. Exp Parasitol. 2012;132:200–15. doi: 10.1016/j.exppara.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 27.Quílez J, Vergara-Castiblanco C, Monteagudo L, Del Cacho E, Sánchez-Acedo C. Multilocus fragment typing and genetic structure of Cryptosporidium parvum isolates from diarrheic preweaned calves in Spain. Appl Environ Microbiol. 2011;77:7779–86. doi: 10.1128/AEM.00751-11. [DOI] [PMC free article] [PubMed] [Google Scholar]