

Abstract
Since arsenic trioxide was first approved as the front line therapy for acute promyelocytic leukemia 25 years ago, its anti-cancer properties for various malignancies have been under intense investigation. However, the clinical successes of arsenic trioxide in treating hematological cancers have not been translated to solid cancers. This is due to arsenic's rapid clearance by the body's immune system before reaching the tumor site. Several attempts have henceforth been made to increase its bioavailability toward solid cancers without increasing its dosage albeit without much success. This review summarizes the past and current utilization of arsenic trioxide in the medical field with primary focus on the implementation of nanotechnology for arsenic trioxide delivery to solid cancer cells. Different approaches that have been employed to increase arsenic's efficacy, specificity and bioavailability to solid cancer cells were evaluated and compared. The potential of combining different approaches or tailoring delivery vehicles to target specific types of solid cancers according to individual cancer characteristics and arsenic chemistry is proposed and discussed.
Keywords: arsenic trioxide, solid cancer, nanotechnology, drug delivery, liposome
Introduction
Arsenic is a naturally occurring metalloid found as a trace element in the earth's crust[1]. Instead of being present in its elemental forms; it exists as highly toxic arsenates of metals like calcium, sodium and potassium or as oxides and sulphides. Among its three inorganic forms, arsenic trioxide (ATO) manifests as white arsenic and is industrially produced as a by-product while roasting arsenic containing ores and purifying the smoke[2]. The transformation of ATO from being the "king of poisons" to a "broad spectrum anti-cancer drug" is one of the most exciting stories in clinical oncology[3].
Despite being the mainstay of materia medica since the past 2,400 years, ATO faced an escalating decline in its use as a therapeutic agent in the twentieth century due to an increasing awareness of its toxicity[4]. Its resurgence transpired in the late twentieth century when a group of Chinese physicians recognized it as the main anti-leukemic ingredient in traditional Chinese medicine and decided to evaluate its therapeutic potency in treating patients with acute promyelocytic leukemia (APL), a subtype of acute myeloid leukemia[5]. Clinical trials using ATO to treat APL patients as a single agent or in combination with conventional chemotherapy yielded highly encouraging results[5]. ATO was consequently approved by the Food and Drug Administration (FDA) as frontline therapy for APL in 2000[6]. Similar clinical successes were subsequently translated for other hematological malignancies, for instance, acute myeloid leukemia, chronic myelogenous leukemia, Hodgkins disease and multiple myeloma although not significantly for solid tumors[7]. Rapid clearance of ATO and its products from the blood by the reticuloendothelial system (RES) limits the therapeutic dose reaching the tumor site. Increasing ATO dosage could not be a solution as it leads to systemic toxicities such as long-term memory damage, liver failure, cardiac failure and peripheral neuropathy[8].
Since then, several attempts have henceforth been carried out to utilize arsenic's significant anti-cancer properties for treating solid tumors by increasing its bioavailability and specificity for carcinoma cells while reducing its administered dosage[9]. These include sensitizing the cells prior to ATO treatment, employing it in combination therapy with other conventional chemotherapeutic agents to explore their synergistic actions or employing nanotechnology to increase bioavailability of ATO while reducing its systemic toxicity[10–13].
This review aims to summarize the advances that have been made till date regarding therapeutic applications of ATO drug with an emphasis on the ongoing involvement of nanotechnology in the current ATO therapy in treating various malignancies.
Arsenic trioxide (ATO) history: rise, decline and resurgence
ATO presents a rich history in medicinal applications dating back to more than 2400 years[14]. It was used in traditional Chinese medicine to treat syphilis, psoriasis and rheumatism[2]. Other applications were also reported with ATO being effective as an anti-spasmodic, anti-pyretic, anti-periodic, caustic and sedative[14]. However, the awareness of its toxicity began to rise when people realized that long-term chronic exposure to ATO could cause hyperpigmentation of the skin, cancers of the lung, kidney, bladder and prostate along with rare cases of neuropathy, encephalopathy, leucopenia and diabetes[6]. Consequently, by the turn of the twentieth century, its medicinal use started to decline due to its acquired reputation of being a toxic substance and a carcinogen.
ATO resurged as a therapeutic compound in 1990s following an initial study at Harbin Medical University where its anti-leukemic properties were identified in treating APL[14]. A group of physicians at Shanghai Second Medical University continued the study and remarkable results were observed from the clinical trial with patients of relapsed and refractory APL after very low doses of intravenous ATO were administered[15]. It was a major breakthrough in treating APL relapsed patients as the mortality rate with routine treatment i.e. all-trans retinoic acid (ATRA) and anthracycline based chemotherapy was 20-30% and bone marrow transplantation was the only option[14]. The findings, which were later successfully replicated in United States, led to the approval of ATO (TrisenoxTM) by FDA as a frontline drug for relapsed and refractory APL in 2000[6].
Although some successes of ATO therapy have also been observed in other hematological cancers, the therapeutic dosage administered is relatively low, and hence the commonly reported side effects are low grade and reversible after ceasing ATO therapy. However, the same kind of clinical success has not been translated for solid cancers in humans despite ongoing clinical trials to investigate arsenic's efficacy on a variety of solid cancers[15] (http://www.clinicaltrials.gov). This is primarily due to the rapid clearance of ATO and its metabolites from the blood by RES. Compromised therapeutic dose of ATO reaches the tumor site, and, therefore, an increased dosage is required which might, in turn, lead to systemic toxicities and long-term memory damage and hormonal imbalance[8, 16]. Therefore, there is a crucial need for analyzing various possible approaches of increasing the bioavailability of ATO without raising its dosage.
Therapeutic strategies in using ATO alone or in combination
Anti-cancer activity of free ATO has been tested on a variety of solid tumor cell lines with positive experimental results but limited clinical success. Zhao and coworkers[17] administered direct intra-tumoral injections of ATO to human esophageal carcinoma xenografts in mice and investigated ATO distribution and its efficacy within the tumor. Tumour growth inhibition of 13.56%, 62.37% and 76.92% was observed with administration of 1, 5 and 10 mg of ATO respectively without appreciable systemic side effects. These results provided encouraging evidence for potential clinical utility of direct intra-tumoral injections of ATO as one form of treatment for solid cancers. However, there is an escalating need to investigate other approaches where ATO can be delivered selectively to its site of action and to penetrate inside the tumor without being toxic to surrounding healthy tissue. Some of the approaches that have been tested or proposed are outlined below ( Fig. 1 ).
Fig.1.
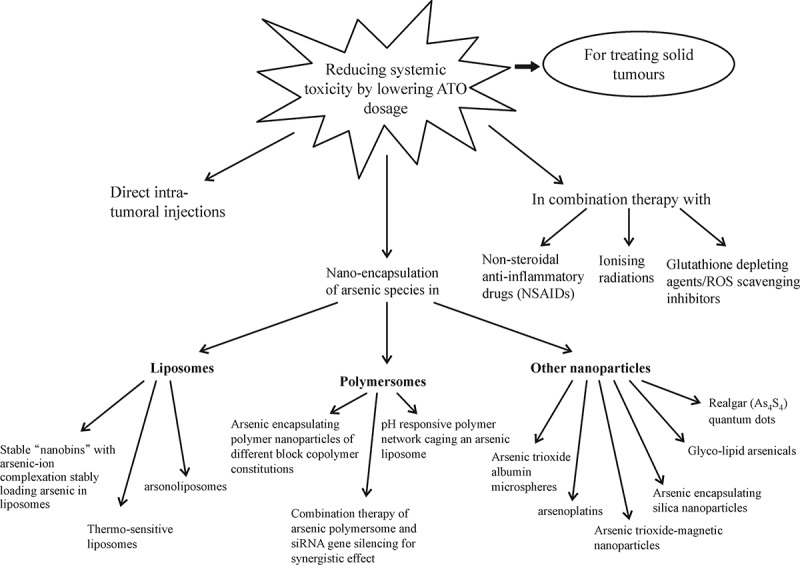
Key therapeutic strategies employed in the attempt to increase the efficacy of ATO as a drug in treating solid tumors without escalating its dosage[ 9, 11– 12, 18– 39].
Glutathione depleting agents
One of the key targets of ATO is glutathione (GSH), a tripeptide molecule which protects the cells from oxidative stress. It is involved in a number of bio-reductive reactions and in the detoxification of xenobiotics[11]. Glutathione titrates intracellular arsenic by forming a complex As(GS) 3[11]. This interaction is exothermic and spontaneous at pH 7.4 and temperature 37°C (∆G= -8.8 kcal/mol). The formation of this complex is preceded by its export out of the cell via multidrug resistance export proteins (MRP1-10) found in cells[8]. Hence, substances which deplete glutathione can sensitize the cells to ATO anti-tumor activity by preventing its export out of the cells. To verify the importance of modulation of glutathione levels, in one experiment, glutathione content was increased by treating the cells with N -acetylcysteine. The anti-tumor activity of ATO was subsequently observed to be suppressed[12]. This makes it evident that simultaneously depleting intracellular glutathione levels might be an effective strategy to intensify ATO action.
Gartenhaus et al. [13] reported ascorbic acid to be one such compound which depletes intracellular glutathione levels. Ascorbic acid further auto-oxidises to release H 2O 2 which might enhance pro-apoptotic effects of arsenic. L -Buthioninesulphoximine (BSO) is another drug which inhibits a critical step in glutathione synthesis by inhibiting the rate limiting enzyme γ-glutamyl cysteine synthetase along with generating reactive oxygen species (ROS). This sensitizes the cells to ATO treatment and was reported to enhance drug's efficacy in inducing apoptosis on a number of cell lines arising from the cervix, bladder, prostrate, kidney, colon, pancreas and breast cancers[18]. Further biochemical studies indicated that this combination treatment blocks the H 2O 2 scavenging system in the cells by interfering with glutathione along with catalase and glutathione oxidase enzymes. Therefore, this combination of an ROS producing drug and ROS removing inhibitor has the potential to offer selective and efficient anti-cancer treatment[18].
Ionising radiation
When ATO is used in conjunction with ionising radiation, radiation response was reported to be intensified in tumor cells. ATO acts as a radiosensitizer to tumor cells by increasing their susceptibility toward apoptosis. Combined with a low-dose irradiation, the dual treatment has been reported to hold a promise for effective cancer therapy. Kang and Lee[9] observed synergistic effect when human cervical cancer cells were treated with ATO along with ionising radiation. The treatment caused regression in tumor growth by increasing ROS, destabilizing the mitochondrial membrane and activating caspases in cancer cells.
Non-steroidal anti-inflammatory drugs (NSAIDs)
These drugs are known to enhance the susceptibility of cancer cells toward various further anti-cancer treatments such as biologic treatment, chemotherapy and radiotherapy. One such drug sulindac has been reported to sensitize the cells due to its apoptosis inducing and anti-angiogenic properties[40]. It increases the radiation sensitivity of cancer cells by interfering with tumor neovascularisation[40]. Sulindac also exhibits synergistic augmentation of cytotoxicity when used alongside paclitaxel[41]. Similarly, when sulindac was used alongside ATO on human lung cancer NCI-H157 cell line, it enhanced caspase activation via generation of ROS[19]. This synergistic action allowed much lower concentration of ATO to be effective in carrying out its anti-cancer properties.
Another NSAID drug, indomethacin, is a known inhibitor of cyclooxygenase-2 (COX-2), whose expression is significantly increased in a variety of cancers[19]. Hence, by acting as a COX-2 enzyme inhibitor, indomethacin exerts its cytotoxic effects on tumor cells. When it was used in combination with ATO, very low doses of both drugs achieve significant cytotoxicity[42]. The synergism in their anti-cancer action was clearly evident as either drug was not as toxic when it was administered at the same concentration alone[42].
Encapsulation of ATO as a single agent or co-encapsulated with another drug
The advent of nanotechnology has already changed the landscape of drug delivery. Improving the bioavailability of poorly water soluble drugs, selectively delivering the drug in a cell or tissue specific manner, co-delivering two or more drugs, transporting the drug to intracellular compartments/site of action and visualizing the site of delivery using imaging modalities are just few of the promises that nanotechnology holds for drug delivery( Fig. 2 ). ATO, along with few other organic arsenicals, has already been encapsulated in nanoparticles and their efficacy is summarized and evaluated in this review ( Table 1 ).
Fig.2.
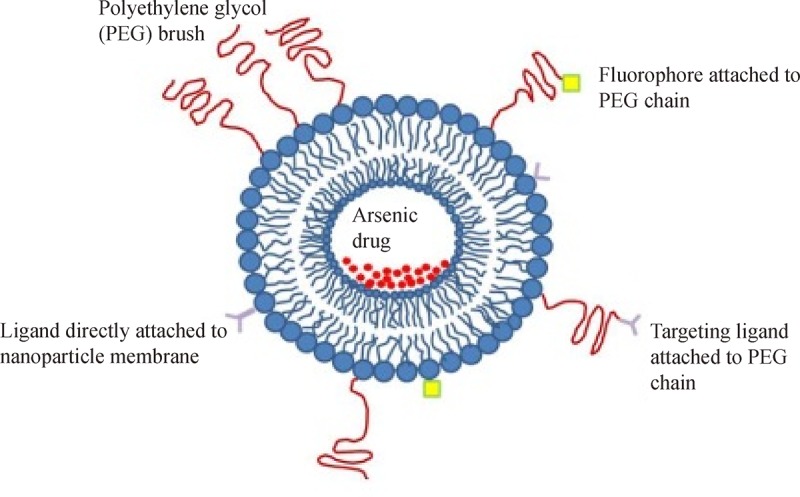
Implementation of nanotechnology for ATO drug delivery.
Tab.1.
Applications of delivering ATO into cancer cells using nanotechnology.
| Delivery vehicle | Co-encapsulated agent | Targeting ligand | Type of cancer tested | Treatment response | References |
| Liposomes | Manganese (Mn2+) | F(ab')2 fragments of PGN635, human monoclonal phosphotidyl serine (PS)-targeting antibody | Glioblastoma multiform (GBM) | Encapsulated As3+-Mn2+ dissociates to release As3+ as active drug and Mn2+ as T1 contrast agent for MRI imaging. | [10] |
| Liposomes (Nanobins) | Transition metal ion (Ni, Cu, Zn, Co) | NA | Lymphoma; Breast cancer; SU-DHL-4 human lymphoma cell lines | Liposomal ATO is less harmful to the ovarian functions of female patients. | [20–22] |
| Liposomes | Transition metal ion (Ni, Cu, Zn, Co) | Urokinase plasminogen activator antibody | Ovarian cancer | Uptake of targeted liposomes by cancer cells is 4-fold higher than non-targeted ones. | [23] |
| Liposomes | NA | NA | NA | Liposomal ATO is more effective than free form in reducing the oncogene level (HPV-16) in HPV positive cervical cancers and inducing apoptosis | [24] |
| Liposomes (thermosensitive) | Transition metal ion (Ni, Cu, Zn, Co) | NA | NA | Drug release achieved by a small but rapid increase in temperature ( 4°C) | [25] |
| Liposomes | Transition metal ion (Ni, Cu, Zn, Co) | Folic acid | FR positive human nasopharyngeal and cervical cancer cell lines | Enhanced anti-cancer activity observed with the targeted liposomes | [26] |
| Liposomes (incorporating arsenonolipid) | NA | NA | Three malignant cell lines (HL-60, C6, and GH3) | Induced morphological changes and inhibited survival of cancer cell lines while being non-toxic to control cells. | [27]] |
| Polymersomes (PEGylated PLGA nanoparticles) | NA | NA | Murine peritoneal macrophages | Uptake by macrophages reduced indicating longer circulation half-life of nanoparticles | [28] |
| Polymersomes | NA | anti-CD44v6 single chain variable fragment | Pancreatic cancer cell line, PANC-1 | Targeted nanoparticles display enhanced accumulation in tumor site and successfully induce apoptosis. | [29] |
| Polymersomes with thiol groups for loading of phenylarsine oxide (PAO) utilizing thiol-arsenic chemistry | NA | NA | Breast cancer, MDA-MB-435 cell line | The polymeric system displays similar toxicity as free PAO on the cell line tested. | [[30]] |
| Nanoparticles formed by linking sugar-lipoaminoacid to PAO | NA | NA | Breast cancer MCF-7 cell lines and HT-29 colon cancer cell lines | Inhibition of cell proliferation and apoptosis in MCF-7 but not in HT-29 cell lines showing high selectivity within these arsenicals. | [31] |
| pH responsive polymer network surrounding ATO encapsulating liposome | Ni2+ cation | NA | Human cervical HeLa cell lines | Simultaneous release of both Ni2+ and As3+, with the former diminishing anti-oxidants levels aiding the apoptotic effect of As3+. | [32] |
| Liposomes (thermosensitive) | Mn0.5Zn0.5Fe2O4magnetic nanoparticles | NA | MDA-MB-231 breast cancer cell line | The magnetic nanoparticles serve as the source of hyperthermia causing the liposome to destabilize at its phase transition temperature and release As3+ to induce apoptosis. | [33] |
| Nanoparticles (Arsenic platinum complex) | Pt2+ | NA | Colon HCT-116 and glioblastoma U-87 cell line | Combination of cisplatin and ATO proves more effective than either of the drug. | [34] |
| Magnetite doped mesoporous silica nanoparticles (MSN) | Drug camptothecin in the pores of MSN along with ATO on its surface using thiol binding | NA | Pancreatic cancer cells (BxPC-3) | Displays a synergistic effect and magnetite superparamagnetic crystals can be used as a contrast agent for MRI | [35] |
| Polymersome encapsulating arsenic | Co-treatment with si-RNA polyplex | NA | Human pancreatic cancer | Synergistic effect with si-RNA silencing KRAS oncogene and As3+ inducing apoptosis | [36] |
| Fe3O4 magnetic nanoparticles complexed with As2O3 | Iron oxide nanoparticles | NA | Human cervical cancer cell line (HeLa) | Inhibit cancer-related proteins expression when combined with hyperthermia | [37] |
| Liposomes | Platinum | Folic acid | Folic acid positive tumor cell line | Enhanced uptake and apoptosis induction of targeted liposomes | [26] |
| ATO nanoparticles | Coated with chitosan and 2,3-dimercaptosuccinic acid | NA | Human androgen dependent and independent prostate cancer cell lines | Coated nanoparticles are more stable and less toxic than bare As2O3 nanoparticles. | [38] |
Liposome encapsulation
Liposomes are lipid nanoparticles encapsulating a hydrophilic interior with a hydrophobic membrane. They are formed via self-assembly when certain phospholipids are hydrated in water[44]. The hydrophobic membrane can entrap strongly lipophilic drugs whereas the hydrophilic interiors can encapsulate water soluble molecules like ATO. Zhao et al .[17] prepared liposomes encapsulating ATO and intravenously injected them to investigate their therapeutic effect on C6 gliomas established in rat brains. Liposomal preparation led to a 5-fold increase in arsenic concentration in rat brains as compared to ATO solutions, inducing apoptosis and inhibiting tumor angiogenesis by interfering with the expression of vascular endothelial growth factor (VEGF) with fewer side effects[17]. However, the liposomes formulated were stable for a maximum of three days, which limited their therapeutic use.
Nanobins
ATO forms arsenous acid, As (OH) 3, when dissolved in water at physiologic pH[8]. The latter is able to rapidly cross the lipid membranes; thereby, the liposomes formulated with ATO encapsulated are highly unstable with a stability life span of around 3 days. The issue of such "leaky liposomes" was solved when Chen et al .[26] developed a novel method to stably encapsulate ATO in liposomes utilizing a metal-ion gradient loading mechanism. Arsenic complexes with transition metal ions such as NiII, CoII, and PtII and forms a precipitate. Utilizing this property, liposomes formed are first loaded with acetate solution of a transition metal ion, followed by loading of ATO to the liposomes formulated[22]. Efflux of protonated acetic acid drives ATO to get efficiently encapsulated within the liposomes where it stably precipitates with the transition metal ion in the form of a solid, crystalline substance[8].
Loading of ATO via this mechanism was found to be very efficient and the lipid layer surrounding the drug attenuates its toxicity around 4 times compared with the free drug. These liposomes exhibit a pH dependency by releasing the arsenic payload on encountering lower pH in either acidic tumor milieu or the endosomes after being taken up by the cells. Ahn et al .[21] tested the efficacy of these nanobins for orthotopic model of triple negative breast cancer. A visible shrinkage of tumor was observed in rats which were subjected to nanobin treatment. This could be attributed to the prolonged pharmacokinetics due to enhanced drug exposure to the tumor caused by better penetration of liposomes along with a reduced clearance of encapsulated arsenic within PEGylated liposomes. The shelf life of nanobins was greater than 6 months, probably due to stability provided by direct arsenic-metal bond[8].
Most recently, in our laboratories, Wang and colleagues[24] have also established that treatment with liposomal ATO is distinctively effective for HPV infected cervical cancer cells, by reducing the expression of HPV-18 E6 oncogene and inducing apoptosis more effectively than free ATO.
When acetate solution of aqua-cisplatin was employed for arsenic complexation, the liposomes formed had both arsenic and platinum species sequestered inside. Cisplatin (cis-diamine dichloroplatinumII) is widely used to treat a variety of solid tumors such as bladder, ovarian, and testicular carcinomas[44]. On being intracellularly hydrolysed, it yields aqua cisplatin, which induces apoptotic cell death in cancer cells by creating intra and inter-DNA strands crosslinks[34]. This method hence provides an efficient co-encapsulation strategy to load both ATO and platinum drugs within the liposomal aqueous core. To further improve anti-cancer activity of the resulting nanobin, Chen et al .[26] conducted a study using folic acid as a targeting ligand to introduce ATO into malignant epithelial cancer cell lines which overexpressed folic acid receptors (FR). The efficacy of such "targeted" liposomes was analyzed on FR positive KB (human nasopharyngeal) cell line and cellular uptake of ATO via FR mediated endocytosis was observed to be much more than FR negative MCF-7 (breast cancer) cell line. Moreover, the targeted liposomal uptake was 3-6 times higher than untargeted ATO liposomes[26].
The targeting via nano-carriers opens a new horizon for selectively delivering the drug to specific cancer cells. Zhang et al .[23] utilized the overexpression of urokinase system in ovarian epithelial cells and consequently conjugated urokinase plasminogen activator (uPA) antibodies to nanobins. The resulting targeted liposomes were reported to exhibit more than 4-fold increase in their uptake by the ovarian cancer cells than their non-targeted counterparts. They also inhibited tumor cell growth both in vitro and in vivo and induced apoptosis along with downregulating stem cell marker expression. Hence, utilizing specific cancer cell surface characteristics and identifying suitable ligands to be attached to ATO encapsulating liposomes are essential in enhancing the efficacy of ATO therapy.
Thermosensitive liposomes
Mild hyperthermia has frequently been employed as a stimulus to release the drug encapsulated either in the aqueous core or in the lipid bilayer of the liposome[45]. Local mild heating can be applied directly within a narrow temperature range. Moreover, it renders the cells susceptible toward oxidative stress caused by ATO treatment. Needham et al .[46] observed that inclusion of small fraction of lysolipids in double tail lipid liposomes causes the drug to be released quickly at phase transition temperature T m. Winter et al .[25] described a liposomal formulation for encapsulation of ATO which efficiently released the drug on being heated from 27°C to 35°C. This was achieved by incorporating lysolipid such as monopalmitoylphosphotidylcholine (MPPC) in liposomes composed of dipalmitoylphosphotidylcholine (DPPC). Ten mol % of MPPC enabled a very rapid release of encapsulated arsenic within the first hour of temperature change. ATO loading was accomplished using the same principle as described in the nanobin above.
Arsonoliposomes
Arsonoliposomes are nanosized liposomes constituted from arsonolipids, which are analogs of phosphonolipids where arsenic replaces the phosphorus in the lipid polar head group[47]. This type of nanoparticles is designed to combine the anti-cancer properties of arsenic with the enhanced efficacy and reduced toxicity of liposomes. Arsonolipids, when employed on their own, do not possess anti-cancer activity adequate enough to be of therapeutic value[48]. However, on being incorporated in liposomes, they demonstrate increased toxicity to cancer cells while being less toxic to normal cells. Phosphotidyl choline (PC)-based arsonoliposomes suffered from rapid clearance from blood circulation after in vivo administration, along with their aggregating and fusing into larger particles. However, the PEGylation of arsonoliposomes was able to enhance their bioavailability and pharmacokinetic profile and was shown to be effective against brain glioma, pituitary and leukemia cell lines[27].
Polymersome encapsulation
Polymersomes are nanoparticles constituted of block copolymers that are organized in a bilayer, similar to liposomes, with the hydrophobic polymer wall surrounding an aqueous core[44]. PEGylated PLGA nanoparticles were synthesized as drug delivery vehicles for ATO. Loading the nanoparticles with ATO was achieved by slightly modified spontaneous emulsification solvent diffusion (SESD) method after transforming the amorphous ATO into cubic crystal form and thereby, increasing its solubility in organic solvents[28].
Qian et al .[29] synthesized PEG-PDLLA vesicles from amphiphilic diblock copolymer poly ( D, L -lactide) and encapsulated arsenite ion in the nanoparticles' core. To further improve the specificity, they conjugated an antibody fragment, i.e. anti-CD44v6 single chain variable fragment onto the vesicles which demonstrated high specificity and affinity toward membrane antigen CD44v6 overexpressed on a number of epithelium derived cancer cells. The formulated nanoparticles were then tested against human pancreatic cancer cell line PANC-1 and were found to be very efficient in delivering the payload within the tumor site in comparison to the non-targeted ones.
A polymeric micelle system has been developed recently by Zhang et al .[30] utilizing a biodegradable block copolymer. Free thiol groups on the hydrophobic block of the polymer backbone were used to conjugate an even more toxic form of trivalent arsenic species compared to ATO, i.e. phenylarsine oxide (PAO). 65% of free thiol groups were reported to conjugate with PAO whose release from the formulated nanoparticles was triggered by the presence of glutathione, which perhaps acted as a competing agent for thiol group. PAO encapsulation further served to improve micellar stability and prevented micellar dissociation in water or in the presence of detergents like SDS[17].
In addition to the above, a polymeric shell was also employed in caging ATO encapsulating liposome "nanobin" as described previously. The pH responsive polymeric network surrounding the nano-sized liposome allowed the simultaneous release of membrane permeable trivalent arsenic ions along with membrane impermeable Ni cations to which they form a complex. When the pH is reduced to the levels found in late endosomes or tumor acidic milieu, both ions were released simultaneously, unlike nanobins, where nickel ions remained inside the liposome while the arsenic diffused out[49]. This is an attractive strategy because nickel ions have been known to deplete intracellular glutathione and ascorbate levels, thereby rendering the cells even more susceptible toward ATO toxicity[26]. The cytotoxicity of these polymer caged nanobins was tested against HeLa cells and they were found to be more potent in inducing apoptosis than plain liposomes. The triggered release from pH change was due to the collapse of the cross-linked polymeric network which creates pressure points, hence pressing liposomes and consequently destabilizing their lipid bilayer membrane, making them release their payload[49].
Nanoparticle mediated siRNA and arsenic co-therapy
Zeng et al .[36] demonstrated the potential synergistic effect of silencing mutant KRAS gene and arsenic therapy on pancreatic tumor cell lines with excellent results. Mutant KRAS gene plays an important role in cell transformation and cancer development and is known to be activated in a number of pancreatic cancers. Therefore, downregulating it with si-RNA gene therapy becomes an attractive treatment which gave positive results such as inhibition of proliferative, migratory and invasive pancreatic tumor cells[50]. When co-treated with ATO, the apoptotic effect of the latter was substantially improved. Arsenic was encapsulated in poly(ethylene glycol)-block-poly( DL -lactide) vesicles and siRNA-complexed polyplexes were formed from poly(ethylene glycol)-block-poly(L-lysine) polymer. Both of the polymer nanoparticles were simultaneously administered to the human pancreatic cancer cell lines PANC-1 and BXPC-3 and their cytotoxicity was analyzed and substantiated[50].
Other nanoparticles
ATO magnetic nanoparticles
Encapsulating magnetic nanoparticles along with an anti-cancer drug has many advantages. These nanoparticles enable targeting under the influence of magnetic field and improve drug's pharmacokinetics profile in vivo. In addition, these ferromagnetic nanoparticles can also be utilized for inducing hyperthermia in the tumor tissue. Magnetic fluid (fluid comprised of superparamagnetic nanoparticles) absorbs much higher energy than usual materials and transfers all of it to the tumor tissue where temperature may rise to 45°C. This concept, magnetic fluid hyperthermia (MFH), where magnetic nanoparticles are heated by applying alternating magnetic field is gradually becoming an attractive anti-cancer treatment[33]. By behaving as localized points of heat, they may prevent the damage to healthy tissues as is commonly induced in conventional hyperthermia treatment.
In a previous study, Fe3O4 magnetic nanoparticles were synthesized with ATO utilizing co-precipitation and impregnation techniques to form nanosized As2O3-Fe3O4 complexes. These nanoparticles were directly injected into the xenograft cancer tissue of human cervical cancer HeLa cells in nude mice and were found to be internalised by the cancer cells. When combined with MFH, these particles attained a steady rise in temperature ranging from 42° to 65°C which successfully ablated the tissue[37]. In another study, As2O3- MgFe2O4nanoparticles were synthesized via solvent displacement methods and characterized[51].
When such a drug-magnetic nanoparticle complex was co-encapsulated in a thermosensitive liposome, the heat from these magnetic nanoparticles could lead to destabilizing of the liposomal membrane, and the drug could then be released at the specified location. Li et al .[33] successfully used magnetic hyperthermia and thermosensitive liposomes in conjunction by co-encapsulating Mn-Zn ferrite magnetic nanoparticles with ATO in liposomes. Upon exposure to alternating magnetic field, the Mn-Zn ferrite nanoparticles served as the heating source, causing the lipids in liposomal membrane to reach their transition temperatures and thereby release ATO. Their efficacy was tested against human breast cancer MDA-MB 231 cell line and a significant inhibition of cancer cell growth was observed[33].
Song et al. [52] prepared magnetic core-polymer shell drug delivery system encapsulating ATO. The magnetic core employed was a small magnetic nanoparticle like Fe3O4 and the shell was composed of a biocompatible and biodegradable polymer. This ATO drug delivering system has been utilizing double emulsion-solvent evaporation technique by coating the magnetite with the polymer poly lactic acid (PLA). These hybrid nanoparticles had high encapsulation efficiency and were effective in achieving anti-cancer response along with magnetic targeting.
ATO albumin microspheres
Bovine serum albumin microspheres containing ATO were prepared employing chemical crosslink and solidification method[53]. The drug loading experiments exhibited 29% encapsulation efficiency and the release experiments in vitro suggested a slower release of ATO after an initial burst from the microspheres, with a cumulative percentage release close to 95%[54]. When conjugated with cell penetrating peptides, the microspheres could display an increased cellular uptake and consequent intracellular delivery of ATO into the cells. For this purpose, trans-activating transcriptional activator (Tat) peptide was employed and covalently linked to the albumin microspheres for successfully delivering ATO into the bladder cells[55].
ATO loaded silica nanoparticles
Magnetite doped mesoporous silica nanoparticles (MSN) have been recently fabricated where both the internal porous surface and external surface could be utilized for loading different carcinogenic drugs. Moreover, they possessed further attractive features including a large surface area, ease of surface functionalization, biocompatibility and stimuli responsive drug release. Muhammad et al .[34] loaded camptothecin, a poorly hydrophobic drug, into the pores while conjugating ATO onto the surfaces of the designed silica nanoparticles utilizing the interaction between arsenic and sulphydryl groups. Besides, superparamagnetic magnetite nanocrystals tethered onto their surface served two important purposes. They allowed the nanoparticles to deliver drug payload to the site of action and also enabled monitoring of therapeutic response via magnetic resonance imaging (MRI). This dual drug nano-formulation exhibited synergism with very low concentrations of nanoparticles showing cytotoxic effect to cancer cells.
Arsenoplatins
Arsenoplatins are small molecule complexes of aqueous form of ATO linked chemically to Pt(II). They are formed when cisplatin, an inorganic drug, is heated with ATO in an acetonitrile mixture for three days[34]. The rationale behind their synthesis is that cisplatin, cis-diamminedichloroplatinum(II), widely used to treat ovarian, head, neck, bladder and testicular cancers, shows synergistic activity when used alongside with ATO. Both of them induce apoptosis in cancer cells albeit with different mechanisms, cisplatin by causing inter and intra-DNA crosslinking and arsenic by the mechanisms listed above. Miodragović et al. [34] reported the formation of arsenoplatins, which are stable in solution, display superior anti-cancer activity and also overcome drug resistance which is a major limitation of platinum drugs. The As-Pt core is suitable for ligand substitution reactions and its efficacy has been demonstrated for a variety of cisplatin-resistant cancer cell lines, such as cisplatin resistant ovarian, colon and glioblastoma cell lines.
Others
Wimmer et al. [31] developed a glycol lipid arsenical and reported its anti-proliferative effects on breast cancer cells MCF-7. This compound was formed by chemically attaching lipoamino acid to sugar. The former increased the lipophilicity of the compound and augmented arsenic's toxic effects. In addition, realgar (As 4S4) quantum dots have been produced recently both as a colloidal dispersion and as a hydrogel; the latter is pH responsive and both forms have been reported to possess superior anti-tumor properties[8]. Most recently, chitosan, a well-known surface modifier, has been reported to be used for coating ATO nanoparticles. The resulting nanoparticles were biocompatible and had an improved stability and toxicity profile against prostate cancer[38]. Additionally, these nanoparticles induced cell cycle arrest by p21 WAF/CIP1 expression via epigenetic remodelling in androgen dependent and independent cell lines[56].
Conclusion
Despite an increasing awareness of toxicity arising from arsenic compounds, ATO has established itself as an effective, broad spectrum anti-cancer drug. Following its successful establishment as the front line treatment for APL patients, researchers and clinicians have been working toward improving its effectiveness in treatment and reducing its side effects for solid cancers. Its poor bioavailability due to rapid clearance from blood by RES limits the therapeutic dose of arsenic reaching the tumor site. Therefore, using nanotechnology to facilitate ATO delivery into solid tumors becomes the focus in this field. Up to date, immense progress has been made in delivering ATO compounds specifically to the cancer cells. ATO along with other arsenic compounds has successfully been encapsulated in liposomes, polymersomes and other nanoparticles. In addition, ligands unique to the receptors overexpressed on specific cancer cells have been conjugated to these nanoparticles. Hence, it is promising that delivering ATO using nanotechnology would be employed as an anti-cancer drug option in treating a variety of solid cancers with better efficacy and much less toxicity.
Contributor Information
Anam Akhtar, Department of Natural Sciences, School of Science and Technology, Middlesex University, London NW4 4BT, UK..
Scarlet Xiaoyan Wang, Department of Natural Sciences, School of Science and Technology, Middlesex University, London NW4 4BT, UK..
Lucy Ghali, Department of Natural Sciences, School of Science and Technology, Middlesex University, London NW4 4BT, UK..
Celia Bell, Department of Natural Sciences, School of Science and Technology, Middlesex University, London NW4 4BT, UK..
Xuesong Wen, Department of Natural Sciences, School of Science and Technology, Middlesex University, London NW4 4BT, UK..
References
- 1. Shen S, Li XF, Cullen WR, et al. Arsenic binding to proteins[J]. Chem Rev, 2013, 113(10): 7769–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miller WHJr, Schipper HM, Lee JS, et al. Mechanisms of action of arsenic trioxide[J]. Cancer Res, 2002, 62(14): 3893–3903. [PubMed] [Google Scholar]
- 3. Wang S, Wu X, Tan M, et al. Fighting fire with fire: poisonous Chinese herbal medicine for cancer therapy[J]. J Ethnopharmacol, 2012, 140(1): 33–45. [DOI] [PubMed] [Google Scholar]
- 4. Evens AM, Tallman MS, Gartenhaus RB. The potential of arsenic trioxide in the treatment of malignant disease: past, present, and future[J]. Leuk Res, 2004, 28(9): 891–900. [DOI] [PubMed] [Google Scholar]
- 5. Shen ZX, Chen GQ, Ni JH, et al. , and the Clinical Efficacy and Pharmacokinetics in Relapsed Patients. Use of arsenic trioxide (As 2O 3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients[J]. Blood, 1997, 89(9): 3354–3360. [PubMed] [Google Scholar]
- 6. Antman KH. Introduction: the history of arsenic trioxide in cancer therapy[J]. Oncologist, 2001, 6(2Suppl 2): 1–2. [DOI] [PubMed] [Google Scholar]
- 7. Zhang P, Wang SY, Hu XH. Arsenic trioxide treated 72 cases of acute promyelocytic leukemia[J]. Chinese Journal of Hematology, 1996, 17(1): 58–62. [Google Scholar]
- 8. Swindell EP, Hankins PL, Chen H, et al. Anticancer activity of small-molecule and nanoparticulate arsenic(III) complexes[J]. InorgChem, 2013, 52(21): 12292–12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kang YH, Lee SJ. Role of p38 MAPK and JNK in enhanced cervical cancer cell killing by the combination of arsenic trioxide and ionizing radiation[J]. Oncol Rep, 2008, 20(3): 637–643. [PubMed] [Google Scholar]
- 10. Zhang L, Zhang Z, Mason RP, et al. Convertible MRI contrast: Sensing the delivery and release of antigliomanano-drugs[J]. Sci Rep, 2015, 5: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Emadi A, Gore SD. Arsenic trioxide- An old drug rediscovered[J]. Blood Rev, 2010, 24(4-5): 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grad JM, Bahlis NJ, Reis I, et al. Ascorbic acid enhances arsenic trioxide-induced cytotoxicity in multiple myeloma cells[J]. Blood, 2001, 98(3): 805–813. [DOI] [PubMed] [Google Scholar]
- 13. Gartenhaus RB, Prachand SN, Paniaqua M, et al. Arsenic trioxide cytotoxicity in steroid and chemotherapy-resistant myeloma cell lines: enhancement of apoptosis by manipulation of cellular redox state[J]. Clin Cancer Res, 2002, 8(2): 566–572. [PubMed] [Google Scholar]
- 14. Waxman S, Anderson KC. History of the development of arsenic derivatives in cancer therapy[J]. Oncologist, 2001, 6(2Suppl 2): 3–10. [DOI] [PubMed] [Google Scholar]
- 15. Murgo AJ. Clinical trials of arsenic trioxide in hematologic and solid tumors: overview of the National Cancer Institute Cooperative Research and Development Studies[J]. Oncologist, 2001, 6(2Suppl 2): 22–28. [DOI] [PubMed] [Google Scholar]
- 16. Platanias LC. Biological responses to arsenic compounds[J]. J BiolChem, 2009, 284(28): 18583–18587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao S, Zhang X, Zhang J, et al. Intravenous administration of arsenic trioxide encapsulated in liposomes inhibits the growth of C6 gliomas in rat brains[J]. J Chemother, 2008, 20(2): 253–262. [DOI] [PubMed] [Google Scholar]
- 18. Maeda H, Hori S, Ohizumi H, et al. Effective treatment of advanced solid tumors by the combination of arsenic trioxide and L-buthionine-sulfoximine[J]. Cell Death Differ, 2004, 11(7): 737–746. [DOI] [PubMed] [Google Scholar]
- 19. Kim HR, Kim EJ, Yang SH, et al. Combination treatment with arsenic trioxide and sulindac augments their apoptotic potential in lung cancer cells through activation of caspase cascade and mitochondrial dysfunction[J]. Int J Oncol, 2006, 28(6): 1401–1408. [PubMed] [Google Scholar]
- 20. Ahn RW, Barrett SL, Raja MR, et al. Nano-encapsulation of arsenic trioxide enhances efficacy against murine lymphoma model while minimizing its impact on ovarian reserve in vitro and in vivo[J]. PLoS One, 2013, 8(3): e58491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ahn RW, Chen F, Chen H, et al. A novel nanoparticulate formulation of arsenic trioxide with enhanced therapeutic efficacy in a murine model of breast cancer[J]. Clin Cancer Res, 2010, 16(14): 3607–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen H, MacDonald RC, Li S, et al. Lipid encapsulation of arsenic trioxide attenuates cytotoxicity and allows for controlled anticancer drug release[J]. J Am ChemSoc, 2006, 128(41): 13348–13349. [DOI] [PubMed] [Google Scholar]
- 23. Zhang Y, Kenny HA, Swindell EP, et al. Urokinase plasminogen activator system-targeted delivery of nanobins as a novel ovarian cancer therapy[J]. Mol Cancer Ther, 2013, 12(12): 2628–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X, Li D, Ghali L, et al. Therapeutic potential of delivering arsenic trioxide into HPV-infected cervical cancer cells using liposomal nanotechnology[J]. Nanoscale Res Lett, 2016, 11(1): 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winter ND, Murphy RKJ, O'Halloran TV, et al. Development and modeling of arsenic-trioxide-loaded thermosensitive liposomes for anticancer drug delivery[J]. J Liposome Res, 2011, 21(2): 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen H, Ahn R, Van den Bossche J, et al. Folate-mediated intracellular drug delivery increases the anticancer efficacy of nanoparticulate formulation of arsenic trioxide[J]. Mol Cancer Ther, 2009, 8(7): 1955–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gortzi O, Papadimitriou E, Kontoyannis CG, et al. Arsonoliposomes, a novel class of arsenic-containing liposomes: effect of palmitoyl-arsonolipid-containing liposomes on the viability of cancer and normal cells in culture[J]. Pharm Res, 2002, 19(1): 79–86. [DOI] [PubMed] [Google Scholar]
- 28. Wang Z, Liu W, Xu H, et al. Preparation and in vitro Studies of Stealth PEGylated PLGA Nanoparticles as Carriers for Arsenic Trioxide[J]. Chin J ChemEng, 2007, 15(6): 795–801. [Google Scholar]
- 29. Qian C, Wang Y, Chen Y, et al. Suppression of pancreatic tumor growth by targeted arsenic delivery with anti-CD44v6 single chain antibody conjugated nanoparticles[J]. Biomaterials, 2013, 34(26): 6175–6184. [DOI] [PubMed] [Google Scholar]
- 30. Zhang Q, Vakili MR, Li XF, et al. Polymeric micelles for GSH-triggered delivery of arsenic species to cancer cells[J]. Biomaterials, 2014, 35(25): 7088–7100. [DOI] [PubMed] [Google Scholar]
- 31. Wimmer N, Robinson JA, Gopisetty-Venkatta N, et al. Novel Glyco-lipid-arsenicals (III) with Anti-proliferative Effects on MCF-7 Human Breast Cancer Cells[J]. Understanding Biology Using Peptides. Springer New York, 2006, 365–366. [DOI] [PubMed] [Google Scholar]
- 32. Lee SM, Chen H, O'Halloran TV, et al. "Clickable" polymer-caged nanobins as a modular drug delivery platform[J]. J Am ChemSoc, 2009, 131(26): 9311–9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang L, Wang Z, Liu J, et al. Preparation of a new nanosized As 2O 3/Mn 0.5Zn 0.5Fe 2O 4 thermosensitive magnetoliposome and its antitumor effect on MDA_MB_231 cells[J]. J NanosciNanotechnol, 2011, 11(12): 10755–10759. [DOI] [PubMed] [Google Scholar]
- 34. Miodragović ÐU, Quentzel JA, Kurutz JW, et al. Robust structure and reactivity of aqueous arsenous acid-platinum(II) anticancer complexes[J]. AngewChemInt Ed Engl, 2013, 52(41): 10749–10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Muhammad F, Zhao J, Wang N, et al. Lethal drug combination: arsenic loaded multiple drug mesoporous silica for theranostic applications[J]. Colloids Surf B Biointerfaces, 2014, 123: 506–514. [DOI] [PubMed] [Google Scholar]
- 36. Zeng L, Li J, Wang Y, et al. Combination of siRNA-directed Kras oncogene silencing and arsenic-induced apoptosis using a nanomedicine strategy for the effective treatment of pancreatic cancer[J]. Nanomedicine, 2014, 10(2): 463–472. [DOI] [PubMed] [Google Scholar]
- 37. Du Y, Zhang D, Liu H, et al. Thermochemotherapy effect of nanosized As2O3/Fe3O4 complex on experimental mouse tumors and its influence on the expression of CD44v6, VEGF-C and MMP-9[J]. BMC Biotechnol, 2009, 9(1): 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jadhav V, Sachar S, Chandra S, et al. Synthesis and Characterization of Arsenic Trioxide Nanoparticles and Their In Vitro Cytotoxicity Studies on Mouse Fibroblast and Prostate Cancer Cell Lines[J]. J NanosciNanotechnol, 2015, 15: 1–7. 26328301 [Google Scholar]
- 39. Zhao Z, Zhang H, Chi X, et al. Silica nanovehicles endow arsenic trioxide with an ability to effectively treat cancer cells and solid tumours[J]. J Mater Chem B Mater Biol Med, 2014, 2(37): 6313–6323. [DOI] [PubMed] [Google Scholar]
- 40. Milas L. Cyclooxygenase-2 (COX-2) enzyme inhibitors as potential enhancers of tumor radioresponse[J]. SeminRadiatOncol, 2001, 11(4): 290–299. [DOI] [PubMed] [Google Scholar]
- 41. Soriano AF, Helfrich B, Chan DC, et al. Synergistic effects of new chemopreventive agents and conventional cytotoxic agents against human lung cancer cell lines[J]. Cancer Res, 1999, 59(24): 6178–6184. [PubMed] [Google Scholar]
- 42. Mandegary A, Torshabi M, Seyedabadi M, et al. Indomethacin-Enhanced Anticancer Effect of Arsenic Trioxide in A549 Cell Line: Involvement of Apoptosis and Phospho-ERK and p38 MAPK Pathways[J]. BioMed Research International, 2013, 2013: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery[J]. ACS Nano, 2009, 3(1): 16–20. [DOI] [PubMed] [Google Scholar]
- 44. Soussan E, Cassel S, Blanzat M, et al. Drug delivery by soft matter: matrix and vesicular carriers[J]. AngewChemInt Ed Engl, 2009, 48(2): 274–288. [DOI] [PubMed] [Google Scholar]
- 45. Griffin RJ, Monzen H, Williams BW, et al. Arsenic trioxide induces selective tumour vascular damage via oxidative stress and increases thermosensitivity of tumours[J]. Int J Hyperthermia, 2003, 19(6): 575–589. [DOI] [PubMed] [Google Scholar]
- 46. Needham D, Ponce AM, Wright A, et al. Targeted bioavailability of drugs by triggered release from liposomes[J]. Future Lipidol, 2006, 1(1): 25–34. [Google Scholar]
- 47. Koutsopoulos S, Fatouros DG, Ioannou PV, et al. Thermal behavior of novel non-sonicated arsonolipid-containing liposomes[J]. BiophysChem, 2006, 121(2): 150–154. [DOI] [PubMed] [Google Scholar]
- 48. Fatouros DG, Ioannou PV, Antimisiaris SG. Arsonoliposomes: novel nanosized arsenic-containing vesicles for drug delivery[J]. J NanosciNanotechnol, 2006, 6(9-10): 2618–2637. [DOI] [PubMed] [Google Scholar]
- 49. Salnikow K, Donald SP, Bruick RK, et al. Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress[J]. J BiolChem, 2004, 279(39): 40337–40344. [DOI] [PubMed] [Google Scholar]
- 50. Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses[J]. Science, 2008, 321(5897): 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang Guo-Fu, LXH , Zhao Zhe, et al. Characterization and Tissue Pharmacokinetics in Mice of Arsenic Trioxide Magnetic Nanoparticles[J]. Chemical Journal of Chinese Universities, 2010, 5: 010. [Google Scholar]
- 52. Song X, You J, Wang J, et al. Preparation and investigation of arsenic trioxide-loaded polylactic acid/magnetic hybrid nanoparticles[J]. Chem Res Chin Univ, 2014, 30(2): 326–332. [Google Scholar]
- 53. Yang Z, Yang M, Peng J. Evaluation of arsenic trioxide-loaded albumin nanoparticles as carriers: preparation and antitumor efficacy[J]. Drug Dev Ind Pharm, 2008, 34(8): 834–839. [DOI] [PubMed] [Google Scholar]
- 54. Zhou J, Zeng F, Xiang G, et al. Preparation of arsenic trioxide albumin microspheres and its release characteristics in vitro.[Medical Sciences][J]. J HuazhongUnivSciTechnolog Med Sci, 2005, 25(3): 310–312., 319. [DOI] [PubMed] [Google Scholar]
- 55. Zhou J, Wang QH, Liu JH, et al. Effects of Tat peptide on intracellular delivery of arsenic trioxide albumin microspheres[J]. Anticancer Drugs, 2012, 23(3): 303–312. [DOI] [PubMed] [Google Scholar]
- 56. Jadhav V, Ray P, Sachdeva G, et al. Biocompatible arsenic trioxide nanoparticles induce cell cycle arrest by p21(WAF1/CIP1) expression via epigenetic remodeling in LNCaP and PC3 cell lines[J]. Life Sci, 2016, 148: 41–52. [DOI] [PubMed] [Google Scholar]