

Abstract
Over 90% of chondroblastomas contain a heterozygous mutation replacing lysine 36 with methionine (K36M) in the histone H3 variant H3.3. Here, we show that H3K36 methylation is reduced globally in chondroblastomas and in chondrocytes harboring the same genetic mutation due to inhibition of at least two H3K36 methyltransferases, MMSET and SETD2, by the H3.3K36M mutant proteins. Genes with altered expression as well as H3K36 di- and trimethylation in H3.3K36M cells are enriched in cancer pathways. In addition, H3.3K36M chondrocytes exhibit several hallmarks of cancer cells including increased ability to form colonies, resistance to apoptosis and defects in differentiation. Thus, H3.3K36M proteins reprogram H3K36 methylation landscape and contribute to tumorigenesis in part through altering the expression of cancer-associated genes.
Chondroblastomas are locally recurrent primary bone tumors (1). Recently, it has been reported that one allele of the H3F3B gene, one of two genes encoding histone H3 variant H3.3 (2, 3), is frequently mutated in chondroblastoma (4). In addition, global reductions of H3K36me2 and H3K36me3 of endogenous histone H3 in mammalian cells exogenously expressing H3.3K36M mutant protein were observed (5, 6). However, it is largely unknown how the mutant proteins exert their effects on H3K36 methylation of endogenous histones and how the H3.3K36M mutation promotes tumorigenesis of this poorly studied tumor.
We analyzed the levels of H3K36me2/me3 in three primary human chondroblastomas harboring the H3.3K36M mutation and in three giant cell tumors with H3.3G34W mutation (Table S1) using H3K36me2- and H3K36me3-specific antibodies (fig. S1). H3K36me2/me3 were globally reduced in each chondroblastoma specimen, but not in giant cell tumors or normal bone tissues (Fig. 1A).
Figure 1. H3K36 methylation is reduced in tumors and cells containing the H3.3K36M mutation.
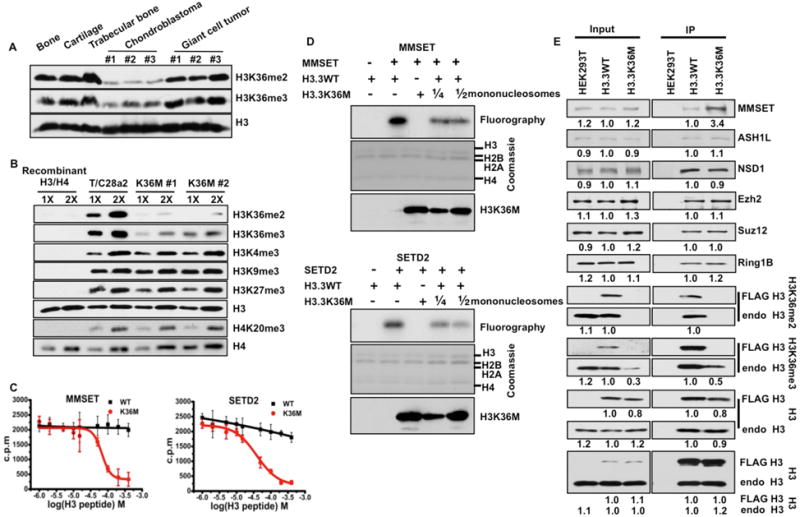
(A–B) H3K36me2/me3 levels are reduced in chondroblastoma tumor samples (A) and in chondrocyte cell lines containing the H3.3K36M mutation (B). (C) The enzymatic activity of the H3K36 methyltransferases MMSET and SETD2 was measured using H3.3-containing recombinant mononucleosomes in the presence of increasing amounts of H3.3K36M peptide or its corresponding H3.3 peptide. Data are mean ± SD (N=3). (D) The H3.3K36M mononucleosomes inhibit the enzymatic activities of MMSET (top panel) and SETD2 (bottom panel) in vitro. (E) MMSET is enriched in H3.3K36M-containing mononucleosomes compared to H3.3 WT nucleosomes. Mononucleosomes were purified from HEK293T cells (negative control), HEK293T cells expressing FLAG-tagged WT H3.3 or H3.3K36M mutant. Proteins from Input and immunoprecipitated (IP) samples were analyzed by Western blotting using the indicated antibodies. The intensity of each blot was quantified, with blots of both input and IP in wild type cells set as 1.
We used the CRISPR/Cas9 system (7) to introduce the H3.3K36M mutation into one H3F3B allele (fig. S2) of T/C28a2 cells, which are immortalized human chondrocytes (8). The levels of H3K36me1/me2/me3 were reduced in two independent mutant cell lines as compared to parental T/C28a2 cells (Fig. 1B and fig. S3), with a more dramatic reduction of H3K36me2 than H3K36me3 based on analysis by mass spectrometry (Fig. S3A–B). No apparent changes were observed in H3K4me3, H3K9me3, H3K27me3 or H4K20me3 (Fig. 1B). These results demonstrate that the global reduction of H3K36 methylation in tumor tissues is due to the expression of H3.3K36M mutant proteins.
To understand how H3.3K36M mutant proteins globally reduce H3K36 methylation, we first tested the ability of a H3.3K36M peptide to inhibit the enzymatic activities of four human H3K36 lysine methyltransferases, SETD2, ASH1L, MMSET/WHSC1 and NSD1, which catalyze H3K36me1/me2/me3 (9–11). The purified catalytic domains of each enzyme exhibited methyltransferase activities against H3.3-containing mononucleosomes. The H3.3K36M peptide inhibited the activities of MMSET (IC50=67 μM) and SETD2 (IC50=39 μM) in a dose-dependent manner, compared to the wild type H3 peptide (Fig. 1C and fig. S4A). Moreover, H3.3K36M-containing mononucleosomes also inhibited the enzymatic activities of MMSET and SETD2 (Fig. 1D). In contrast, neither the H3.3K36M peptide nor H3.3K36M mononucleosomes exhibited inhibitory effect on the activities of ASH1L and NSD1 in vitro (fig. S4). We also observed that MMSET, but not ASH1L, NSD1, or two subunits of the H3K27 methyltransferase complex PRC2 (Ezh2 and Suz12) was enriched in the H3.3K36M-containing mononucleosomes compared to wild type H3.3-containing mononucleosomes (Fig. 1E). Finally, in a peptide pull-down assay, MMSET and SETD2 bound to the H3.3K36M peptide more efficiently under higher salt conditions than the corresponding normal H3 peptide (fig. S5A). These results indicate that H3.3K36M mutant protein inhibits at least two mammalian H3K36 methyltransferases, MMSET and SETD2.
Chromatin immunoprecipitation coupled with next-generation sequencing (ChIP-seq) (16) identified 29,250 and 13, 694 H3K36me2 peaks in T/C28a2 cells and H3.3K36M lines, respectively (Fig. 2A–B). H3K36me2 peaks present only in H3.3K36M cells were significantly enriched at promoters, gene bodies, and TES ±2Kbp (p < 0.01) but showed significant depletion at intergenic regions (p < 0.01) compared to H3K36me2 peaks in T/C28a2 cells (Fig. 2A–C). On average, the levels of H3K36me2 in intergenic regions (Fig. 2D) and in gene bodies (Fig. 2E) were reduced in each of the H3.3K36M mutant lines compared to wild type T/C28a2 cells. The reduction of H3K36me2 in four selected genes and two intergenic regions was confirmed using ChIP-PCR (Fig. 2F).
Figure 2. H3.3K36M reprograms chromatin-bound H3K36me2 and H3K36me3.
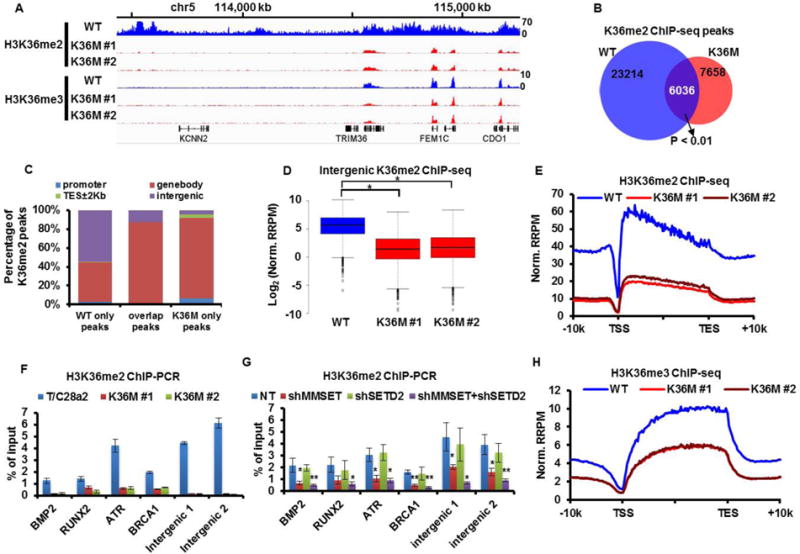
(A) Integrative Genomics Viewer (IGV) tracks showing H3K36me2 and H3K36me3 distribution on chromosome 5 in WT (blue) and two H3.3K36M (K36M) cell lines (red). (B) Venn diagram illustration of H3K36me2 peaks in WT and two H3.3K36M cell lines and their overlap. (C) Genomic distribution of H3K36me2 peaks unique to WT cells (23214), peaks shared by WT and H3.3K36M cells (6036), and peaks unique to H3.3K36M cells (7658). TES: transcription end site. (D–E) Normalized tag distribution profiles of H3K36me2 for intergenic regions (D, *p value < 0.01) and gene bodies (E) in WT and K36M cell lines. RRPM: reference-adjusted reads per million. TSS: transcription starting site (TSS). (F) Analysis of H3K36me2 at four gene loci and two intergenic regions using ChIP-PCR in two H3.3K36M mutant lines. (G) Depletion of MMSET but not SETD2 reduced H3K36me2 at four gene loci and two intergenic regions. Data are mean ± SD (N=3, *p<0.05, **p<0.01) (H) H3K36me3 in gene bodies is reduced in two H3.3K36M chondrocyte lines.
Depletion of SETD2 had no apparent effect on H3K36me2. In contrast, depletion of MMSET alone or in combination with SETD2 resulted in a marked reduction of H3K36me2, but to a lesser extent than cells expressing H3.3K36M based on Western blot analysis (fig. S5B–C). Depletion of SETD2 or MMSET did not affect the expression of three other H3K36 methyltransferases tested (fig. S5D). Finally, H3K36me2 ChIP-PCR and ChIP-seq reveal that depletion of MMSET alone or in combination with SETD2, but not SETD2 alone, led to reduction of H3K36me2 in gene bodies and intergenic regions, but to lesser extent than in H3.3K36M mutant cells (Fig. 2G and fig. S5E–F). Together, these results support the idea that the reduction of H3K36me2 in H3.3K36M cells is mediated, at least in part, through the inhibition of MMSET by the H3.3K36M proteins.
Similar numbers of H3K36me3 ChIP-seq peaks were detected in gene bodies from T/C28a2 cells and H3.3K36M mutant cells (fig. S6A). However, the amount of H3K36me3 throughout gene bodies was reduced in each of the two H3.3K36M mutant lines compared to T/C28a2 cells (Fig. 2H). Furthermore, over 60% of genes with reduced H3K36me3 also exhibited reduced H3K36me2 (fig. S6B). The reduction of H3K36me3 correlates with reduction of H3K36me2 (fig. S6C), suggesting that both H3K36me2 and H3K36me3 are altered to a similar degree within gene bodies of a large fraction of genes. ChIP-PCR confirmed the reduction of H3K36me3 in gene bodies of four selected genes in two H3.3K36M mutant lines (fig. S6D). Finally, depletion of SETD2 and MMSET reduced H3K36me3 in gene bodies, with MMSET depletion having a lesser effect than SETD2 depletion (fig. S5B–C and fig. S6E–F). These results indicate that reduced H3K36me3 in gene bodies of H3.3K36M mutant chondrocytes is mainly due to SETD2 inhibition although MMSET inhibition may also contribute.
We also analyzed H3K36me2 and H3K36me3 levels in primary chondroblastomas using ChIP-seq. The H3K36me2 and H3K36me3 chromatin occupancy in gene bodies was reduced in two chondroblastoma samples analyzed (fig. S7A–B). Gene set enrichment analysis (GSEA) (12) indicated that genes with reduced H3K36me2 and H3K36me3 in gene bodies in chondrocyte cell lines were also enriched in the corresponding gene sets from tumor samples (fig. S7C–D). These results indicate that H3.3K36M mutant proteins have similar effects on the reduction of H3K36me2 and H3K36me3 in gene bodies in both primary chondroblastoma samples and chondrocyte cell lines containing the same H3.3K36M mutation.
We determined how H3.3K36M mutant proteins associate with chromatin using H3.3K36M-specific antibodies (fig. S8A) and three different assays (Western blot, immunofluorescence (IF) and ChIP-seq). The majority of H3.3K36M mutant proteins were detected on chromatin similarly to wild type H3 based on chromatin fractionation and IF assays (fig. S8B and C). H3.3K36M ChIP-seq identified 6162 overlapping peaks between two H3.3K36M mutant lines (Fig. 3A). H3.3K36M peaks were significantly enriched at promoter, gene bodies, and TES ±2Kbp (p < 0.01), but exhibited significantly depletion (p < 0.01) at intergenic regions compare to randomly shuffled peaks with the same length of H3.3K36M peaks (Fig. 3B). Two of these peaks were confirmed using ChIP-PCR (Fig. 3C). The levels of H3.3K36M mutant proteins correlated with gene expression levels (Fig. 3D). These results indicate that chromatin localization of H3.3K36M proteins is similar to that of wild type H3.3 observed in other cell lines (13, 14). Furthermore, the levels of H3K36M mutant proteins were higher within genes with reduced H3K36me2 or H3K36me3 than those without changes in H3K36me2/me3 (Fig. 3E and fig. S9A). Conversely, the levels of H3K36me2 and H3K36me3 were lower within H3.3K36M peak regions than in the surrounding regions (Fig. 3F and fig. S9B–C). The inverse relationship between the levels of H3.3K36M mutant proteins and H3K36me2/me3 on chromatin supports the idea that the reduction of H3K36me2/me3 is due, at least in part, to the inhibition of MMSET and SETD2 by H3.3K36M mutant proteins incorporated into the chromatin.
Figure 3. H3K36M levels in chromatin are inversely correlated with H3K36me2/me3.
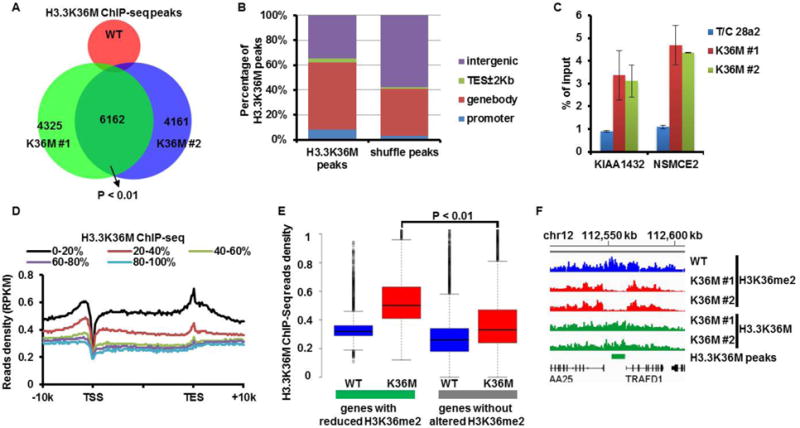
(A) Venn diagram showing H3K36M ChIP-seq peaks identified in WT and two H3K36M cell lines. (B) Most of H3.3K36M ChIP-seq peaks are in gene bodies and intergenic regions. (C) Validation of H3.3K36M peaks by ChIP-PCR in H3.3K36M mutant lines. (D) The normalized read density (RPKM: Reads Per Kilobase per Million mapped reads) of H3K36M ChIP-seq signals from 10Kb upstream of TSS to 10Kb downstream of TES. Genes were split into five groups, with 0–20% representing genes with the highest expression. (E) H3K36M ChIP-seq read densities in H3.3K36M cell lines at two groups of genes: genes with or without significantly reduced H3K36me2. (F) Genome browser track examples of the occupancy profiles of H3K36me2 and H3.3K36M in chromosome 12. Green bar: H3.3K36M peak.
Gene expression analysis using RNA-seq indicated that the expression of 567 and 799 genes was elevated and reduced, respectively, in two H3.3K36M mutant cell lines compared to parental T/C28a2 cells (fig. S10A). In addition, the expression of intergenic regions with reduced H3K36me2 was lower in H3.3K36M cells than in wild type cells (fig. S10B). GSEA analysis indicated that genes with reduced expression in H3.3K36M mutant chondrocyte lines were significantly enriched among genes with reduced expression in chondroblastoma samples, whereas genes with increased expression were not enriched (fig. S10C–D). A lack of correlation of genes with increased expression between H3.3K36M mutant lines and chondroblastoma samples was likely due to heterogeneity of chondroblastoma samples (Table S1). The correlation of genes with reduced expression between the H3.3K36M mutant chondrocyte lines and chondroblastoma datasets argue for common mechanisms linked to H3.3K36M expression and the loss of H3K36me2/me3. Indeed, incorporation of H3.3K36M mutant proteins into nucleosomes occurs immediately prior to or simultaneously with the reduction of H3K36me2 and H3K36me3, and alters gene expression based on analysis of a limited number of gene loci (fig. S11A–B). Finally, we observed that genes with reduced H3K36me2 or H3K36me3 within gene bodies exhibited a significant correlation with changes in gene expression in both cell lines (Fig. 4A) and chondroblastoma samples (fig. S11C). These results suggest that H3K36me2 and H3K36me3 levels are associated with changes in gene expression in both cell lines and tumors. It is known that H3K36me2/me3 antagonizes H3K27me3 (15, 16), raising the possibility that changes in expression of some genes in H3.3K36M mutant cells may also be linked to deregulation of H3K27me3-repressed genes.
Figure 4. H3.3K36M alters the H3K36 methylation landscape and expression of genes linked to carcinogenesis.
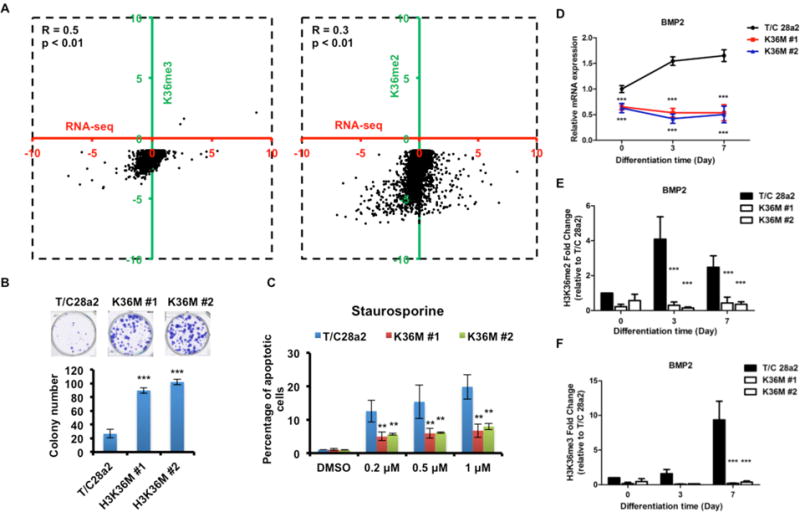
(A) Log2 fold occupancy changes in H3K36me3 (left) and H3K36me2 (right) were calculated for H3.3K36M cells vs. WT cells and plotted relative to corresponding fold changes in mRNA detected by RNA-seq. Genes with significantly changed H3K36me3 or H3K36me2 in gene bodies (p value < 10−5) in both cell lines were chosen for analysis. Each dot represents a single gene. (B) H3.3K36M increases colony formation of T/C 28a2 cells. Top: representative images. (C) Annexin V positive cells were analyzed and quantified by FACS. (D) Expression of BMP2 was analyzed by RT-PCR during differentiation conditions and normalized against GAPDH. (E–F) H3K36me2 and H3K36me3 ChIP assays in cells differentiated for 0, 3 or 7 days. ChIP DNA was analyzed using primers amplifying the BMP2 gene body. Fold enrichment relative to T/C 28a2 cells at the start of differentiation was calculated. Results in B–F represent the mean ± SD (N=3, ** p<0.01 and *** p<0.001).
We performed Ingenuity Pathway Analysis (IPA) using three gene sets with altered occupancy of H3K36me2 (1143 genes) and H3K36me3 (1359 genes) and altered gene expression (598 genes) in both chondrocytes and chondroblastoma tumor samples. Genes associated with “Molecular mechanisms of cancer” IPA canonical pathway was highly enriched in all three datasets (fig. S12A). Several genes assigned to the “Molecular mechanisms of cancer” pathway are known to be involved in DNA repair, differentiation and apoptosis (fig. S12 and Table S2). We investigated whether H3.3K36M mutant chondrocytes display cancer-associated cellular phenotypes including DNA repair in which H3K36me3 has a role (17–19). The H3.3K36M mutations did not affect proliferation of chondrocyte cell lines using MTT assay (fig. S13A), but increased the ability of these cells to form colonies (Fig. 4B). Moreover, H3.3K36M mutant chondrocyte cells were less sensitive to staurosporine-induced apoptosis (Fig. 4C) and formed denser micromasses when placed into chondrocyte differentiation medium (fig. S13B). The effect of H3.3K36M mutation on staurosporine-induced apoptosis could be detected at the same time when H3.3K36M mutant proteins were incorporated into chromatin (fig. S13C). Finally, the H3.3K36M mutant cells were also defective in homologous recombination (HR) (fig. S13D) but had no apparent defects in non-homologous end joining or mismatch repair (fig. S13E–F). Thus, H3.3K36M mutant cells exhibit several cancer-associated cellular phenotypes.
Consistent with differentiation defects, expression of BMP2 and other genes that regulate chondrocyte differentiation (20) were reduced in H3.3K36M cell lines based on analysis of RNA-seq results (fig. S14A). In the micromass assays, BMP signaling is required for hypertrophic chondrocyte differentiation (21). The mRNA levels of BMP2 and SOX9 were reduced in micromass cultures of T/C28a2 cells expressing the H3.3K36M mutant protein compared to the parental cell line (Figs. 4D and fig. S14B). Consistent with the reduction in gene expression, ChIP-PCR results showed that H3K36me2 and H3K36me3 occupancy of BMP2 was reduced during differentiation (Fig. 4E–F). In addition to genes (BMP2 and RUNX2) involved in differentiation, the expression of two genes involved in HR, BRCA1 and ATR, was reduced in H3.3K36M mutant lines as well as in MMSET and SETD2 depleted cells (fig. S14C–D). Thus, the cancer-associated cellular phenotypes observed in H3.3K36M mutant cells are caused at least in part by alterations in H3K36 methylation and expression of key genes that regulate oncogenesis and chondrogenesis.
Our results show that H3.3K36M mutant proteins are incorporated into chromatin in manner similar to wild type H3.3 and inhibit at least MMSET and SETD2 to reduce H3K36 methylation. Moreover, H3.3K36M mutant proteins impact different H3K36 methyltransferases differently, and promote chondroblastoma tumorigenesis likely through alterations of several cancer-related processes including colony formation, apoptosis and chondrocytic differentiation.
Supplementary Material
Acknowledgments
We thank Dr. Goldring for T/C28a2 cells and Dr. Lou for plasmids. We thank Bruce Eckloff and Thomas Wood for sequencing, Benjamin Madden for mass spectrometry analysis. These studies were supported by NIH grants (GM81838 and CA157489 (ZZ), AR68103 (JJW), AR65397 (EWB), AR049069 (AVW), DK58185 (TO) and from the Natural Science Foundation of China (grant No. 31210103914 (RMX)) and by the Epigenomics Program (CIM), Mayo Clinic. ChIP-seq and RNA-seq data sets have been deposited into the Gene Expression Omnibus under accession number GSE7523
References
- 1.Qasem SA, DeYoung BR. Semin Diagn Pathol. 2014;31:10–20. doi: 10.1053/j.semdp.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 2.Talbert PB, Henikoff S. Nat Rev Mol Cell Biol. 2010;11:264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 3.Szenker E, Ray-Gallet D, Almouzni G. Cell Res. 2011;21:421–434. doi: 10.1038/cr.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Behjati S, et al. Nat Genet. 2013;45:1479–1482. doi: 10.1038/ng.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis PW, et al. Science. 2013;340:857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan KM, Han J, Fang D, Gan H, Zhang Z. Cell Cycle. 2013;12:2546–2552. doi: 10.4161/cc.25625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang F, Wen Y, Guo X. Hum Mol Genet. 2014;23:R40–46. doi: 10.1093/hmg/ddu125. [DOI] [PubMed] [Google Scholar]
- 8.Goldring MB, et al. J Clin Invest. 1994;94:2307–2316. doi: 10.1172/JCI117595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuo AJ, et al. Mol Cell. 2011;44:609–620. doi: 10.1016/j.molcel.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner EJ, Carpenter PB. Nat Rev Mol Cell Biol. 2012;13:115–126. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y, et al. J Biol Chem. 2009;284:34283–34295. doi: 10.1074/jbc.M109.034462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subramanian A, et al. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldberg AD, et al. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin C, et al. Nat Genet. 2009;41:941–945. doi: 10.1038/ng.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang GG, Cai L, Pasillas MP, Kamps MP. Nat Cell Biol. 2007;9:804–812. doi: 10.1038/ncb1608. [DOI] [PubMed] [Google Scholar]
- 16.Yuan W, et al. J Biol Chem. 2011;286:7983–7989. doi: 10.1074/jbc.M110.194027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jha DK, Strahl BD. Nat Commun. 2014;5:3965. doi: 10.1038/ncomms4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfister SX, et al. Cell Rep. 2014;7:2006–2018. doi: 10.1016/j.celrep.2014.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li F, et al. Cell. 2013;153:590–600. doi: 10.1016/j.cell.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kozhemyakina E, Lassar AB, Zelzer E. Development. 2015;142:817–831. doi: 10.1242/dev.105536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barna M, Niswander L. Dev Cell. 2007;12:931–941. doi: 10.1016/j.devcel.2007.04.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.