

SUMMARY
The histone chaperone HIRA is involved in depositing histone variant H3.3 into distinct genic regions including promoters, enhancers and gene bodies. However, how HIRA deposits H3.3 to these regions remains elusive. Through an shRNA screening, we identified single-stranded DNA binding protein RPA as a regulator of the deposition of newly synthesized H3.3 into chromatin. We show that RPA physically interacts with HIRA to form RPA-HIRA-H3.3 complexes, and co-localizes with HIRA and H3.3 at gene promoters and enhancers. Depletion of RPA1, the largest subunit of the RPA complex, dramatically reduces both HIRA association with chromatin and the deposition of newly synthesized H3.3 at promoters and enhancers and leads to altered transcription at gene promoters. These results support a model whereby RPA, best known for its role in DNA replication and repair, recruits HIRA to promoters and enhancers and regulates deposition of newly synthesized H3.3 to these regulatory elements for gene regulation.
eTOC Blurb
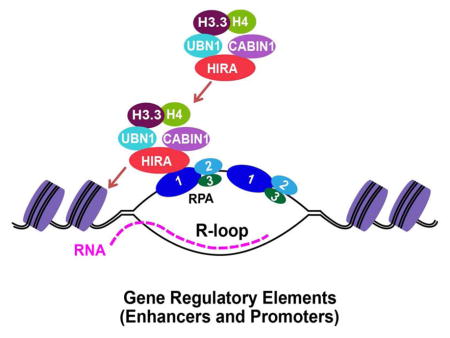
RPA is a DNA binding protein involved in DNA replication and repair. Zhang et al show that RPA is also involved in nucleosome assembly of histone H3 variant H3.3. RPA recruits histone chaperone HIRA, which deposits newly synthesized H3.3 into gene regulatory elements.
INTRODUCTION
Chromatin, a highly organized complex of DNA, RNA and proteins in eukaryotic cells, encodes epigenetic information and maintains genome integrity. Histones, the main protein components of chromatin, package and organize DNA into the structural and fundamental unit of chromatin, the nucleosome. The nucleosome core particle consists of 147 bp DNA wrapped around a histone octamer comprising of one histone (H3-H4)2 tetramer and two histone H2A-H2B dimers. In addition to canonical histones, histone variants, a group of proteins that adopt similar structural fold as core histones, also play an important role in marking chromatin into distinct functional domains and regulating diverse cellular processes including chromosome segregation and gene expression (Burgess and Zhang, 2013; Gurard-Levin et al., 2014; Henikoff and Ahmad, 2005; Szenker et al., 2011). For instance, Canonical H3.1/H3.2 histones are assembled into nucleosomes in a replication-coupled process, whereas histone H3 variant H3.3, differing from H3.1/H3.2 by only four or five amino acid residues, is assembled into nucleosomes in a replication-independent process. H3.3 is enriched at actively transcribed genes, promoters and regulatory elements of developmentally regulated genes as well as at pericentric heterochromatin, and is required for the proper establishment of H3 lysine 27 methylation patterns at promoters of developmentally regulated genes, for silencing of endogenous retroviral elements in mouse embryonic stem cells (Banaszynski et al., 2013; Elsasser et al., 2015), and for marking damaged chromatin and facilitating transcription recovery after genotoxic stress (Adam et al., 2013). Finally, recent studies have revealed missense mutations at K27 and G34 of H3.3 in over 60% pediatric high-grade gliomas as well as high-frequency (over 90%) mutations at K36 and G34 of H3.3 in bone tumors (Behjati et al., 2013; Schwartzentruber et al., 2012; Wu et al., 2012). Thus, H3.3 has diverse functions including gene transcription and silencing, and alterations of which could promote human diseases including cancer
The diverse functions of H3.3 are most likely linked to how it is deposited into chromatin at specific genomic regions. Key regulators of nucleosome assembly of H3.3 are various histone chaperones, such as the HIRA complex and death domain containing protein (DAXX). The HIRA complex, comprised of HIRA, UBN1 and CABIN1, aids in the assembly of H3.3-H4 into nucleosomes at gene regulatory elements including promoters, enhancers, and gene bodies of both active and repressed genes (Goldberg et al., 2010; Ray-Gallet et al., 2002; Tagami et al., 2004). Moreover, HIRA is involved in H3.3 deposition during sperm nucleus decondensation upon fertilization and meiotic sex chromosome inactivation in mouse development (Loppin et al., 2005; van der Heijden et al., 2007). DAXX, which forms a complex with the chromatin re-modeling protein ATRX, is responsible for H3.3 incorporation at telomeres and pericentric heterochromatin and endogenous retroviral element silencing in embryonic stem cells (Drane et al., 2010; Goldberg et al., 2010). Both DAXX and UBN1 recognize A87 and G90, which are unique for H3.3 (Daniel Ricketts et al., 2015; Elsasser et al., 2012; Liu et al., 2012). These results suggest that HIRA and DAXX assemble H3.3 at distinct genomic regions through interactions with additional proteins. Supporting this idea, the SWI/SNF family chromatin remodeler CHD1 was shown to physically associate with HIRA to mediate H3.3 incorporation into chromatin (Konev et al., 2007), and HIRA interacts with RNA polymerase II complex, which may regulate HIRA-mediated deposition of H3.3 throughout gene bodies. However, it remains poorly understood how HIRA-mediated nucleosome assembly of H3.3 at gene regulatory elements is regulated and what the functional implications of H3.3 deposition at these specific regions are.
Replication Protein A (RPA) is a highly conserved heterotrimeric single-stranded DNA (ssDNA) binding protein complex consisting of RPA1, RPA2, and RPA3. RPA is best known for its role in DNA replication and repair (Wold, 1997). Recently, RPA was also implicated in the regulation of transcription in several organisms through associating with transcription factors or RNA polymerase II (Fujimoto et al., 2012; Sikorski et al., 2011). Here we found through an shRNA screening that RPA is required for efficient deposition of newly synthesized H3.3 onto chromatin. RPA forms a complex with HIRA and H3.3 and co-localizes with H3.3 and HIRA to gene regulatory elements. Depletion of RPA results in reduced localization of HIRA and H3.3 to these regions and an altered pattern of nascent transcription at gene promoters. Together, our studies support a model whereby RPA binds to gene regulatory elements and promotes HIRA-mediated nucleosome assembly of H3.3.
RESULTS
The RPA Protein Complex Is Required for the Deposition of Newly Synthesized H3.3
To identify regulators that assist in the deposition of newly synthesized H3.3, we performed an shRNA screen to identify genes that when depleted regulate H3.3 deposition. To monitor deposition of newly synthesized H3.3, we utilized a HeLa cell line stably expressing H3.3 tagged with SNAP (Ray-Gallet et al., 2011). The SNAP polypeptide is a mutated form of O6-guanine nucleotide alkyltransferase that covalently reacts with benzyl-guanine (BG). Briefly, after cells were infected with lentivirus expressing shRNAs for 72 h, cells were incubated with cell-permeable non-fluorescent substrates to block the SNAP polypeptide on pre-existing H3.3. After washing away non-fluorescent blocking reagents for 12 hours, cells were incubated with fluorescent substrates (TMR) to label newly synthesized H3.3-SNAP. Newly synthesized H3.3-SNAP was analyzed either by fluorescence intensity (IF) of in individual cells or a chromatin fractionation assay (CFA) that measures H3.3-SNAP on chromatin (Figure 1A). Depletion of HIRA, a histone chaperone known to be involved in the deposition of newly synthesized H3.3, led to a dramatic reduction of the deposition of newly synthesized H3.3 in both assays (Figure S1A–C), suggesting that the H3.3-SNAP approach could be used to identify the factors involved in de novo deposition of newly synthesized H3.3.
Figure 1. The RPA Complex Is Required for the Deposition of Newly Synthesized H3.3.

(A–B) Schematic outline and workflow for identifying genes involved in the deposition of newly synthesized H3.3 using the H3.3-SNAP tag. The experimental outline was described in A and candidate genes (red oval) after each screen procedure and assays used in each procedure (blue box) were outlined in B. A detailed screen procedure was described in Supplemental Information. CFA: chromatin fractionation assays. (C) Depletion of 5 final candidate genes affects the deposition of newly synthesized H3.3 using CFA. The relative SNAP intensity in NT cells (100) as well as in cells depleted with each candidate gene was calculated and reported as the average of two independent experiments. (D–E) Depletion of each subunit of the RPA complex affects the deposition of newly synthesized H3.3 using CFA assays. Each RPA subunit was depleted using two independent shRNAs and deposition of newly synthesized H3.3 was monitored by CFA. (D) The fluorescence intensity of chromatin-bound new H3.3-SNAP (top panel) was detected using a Typhoon FLA 7000, and total proteins (middle panel) were visualized by IRDye Blue Protein Stain. RPA1 and RPA2 levels were analyzed by Western blot with Tubulin as controls (two bottom panels). (E) Image J was used to quantify the SNAP fluorescence intensity and the relative SNAP intensity over total protein levels in NT and RPA depleted cells was reported as mean ±SD of three independent experiments (* p < 0.05). (F–G) Expression of shRNA-resistant RPA1 rescues H3.3 deposition defect in RPA1 depleted cells. (F) Two different amounts (1X and 4X) of shRNA-resistant wild type RPA1 were expressed for 48 hours. RPA1 was then depleted using shRNA (shRNA1) and the deposition of newly synthesized H3.3 was monitored by CFA assays described above. (G) The relative SNAP intensity over total proteins from three independent experiments were calculated (Mean±SD, * p < 0.05).
We screened a custom shRNA library that contained 1,189 shRNAs (about 5 shRNAs per gene) targeting 246 known chromatin regulators and major signaling pathways (Table S1) (Zuber et al., 2011). During the initial screen process using pooled shRNAs targeting each gene, 81 candidate genes were identified that when depleted led to a reduction or increase in the IF of newly synthesized H3.3-SNAP by at least 30% compared to cells infected with lentivirus expressing non-targeting (NT) shRNA (Table S2). We then repeated the screen targeting these 81 genes. In addition, we analyzed the expression of H3.3-SNAP and HIRA, and quantified global nascent transcription levels by 5-ethynyl uridine (EU) staining after the depletion of each candidate gene. 21 candidates were identified that only affected H3.3 deposition while having no apparent effects on the expression of H3.3-SNAP and HIRA proteins and on global transcription (Table S2). To further validate these 21 candidates, we knocked down the expression of each gene using at a minimum 5 individual shRNAs targeting each gene and identified 14 candidate genes that when depleted by at least two individual shRNAs impaired H3.3 deposition. These 14 genes were further tested for H3.3 deposition using CFA assays and 5 candidate genes (CHD2, G9a, ING2, RPA3 and USP51) were found to affect H3.3 deposition in both IF and CFA assays (Figure 1B–C).
RPA3 is a subunit of ssDNA binding protein RPA involved in DNA replication and DNA repair. The effect of RPA3 depletion on H3.3 deposition was unexpected. Therefore, we first analyzed whether RPA1 and RPA2, two other subunits of the RPA complex, are required for the deposition of newly synthesized H3.3. Depletion of RPA1, RPA2 or RPA3, each with two independent shRNAs, resulted in a marked reduction of newly synthesized H3.3 deposition compared to NT control cells by both the CFA (Figure 1D–E) and IF assays (Figure S1D–E). Moreover, depletion of each RPA subunit individually did not affect the expression of the three subunits of the HIRA complex (HIRA, UBN1 and CABIN1) as detected by Western blot or RT-PCR analysis (Figure S1F–G), or by measuring total levels of H3.3-SNAP proteins (Figure S1H). Finally, expression of a shRNA-resistant RPA1 transgene rescued the H3.3 deposition defects in RPA1 depleted cells (Figure 1F–G). Taken together, these results strongly indicate that the RPA complex is important for the efficient deposition of newly synthesized H3.3.
RPA Regulates H3.3 Deposition in the G1 Phase of the Cell Cycle
Depletion of RPA leads to defects in DNA replication, DNA repair and cell cycle progression (Haring et al., 2008). To discern whether the effect of RPA1 depletion on H3.3 deposition was linked to defects in DNA replication/repair and cell cycle, we first analyzed whether RPA was required for the H3.3 deposition in the G1 phase of the cell cycle. We observed that H3.3 deposition/exchange was higher in cells arrested in the G1 phase by Mimosine than unsynchronized cells. Importantly, depletion of RPA1 also led to reduced H3.3 deposition in the G1 phase cells (Figure S2A–D). In addition, we observed that depletion of ORC3, a subunit of the Origin Recognition Complex involved in DNA initiation of replication (Li and Stillman, 2012), while also leading to defects in cell cycle progression and spontaneous DNA damage (Figure S2E–G), had no apparent effect on H3.3 deposition (Figure S2H–I). Thus, it is unlikely that the effect of RPA1 depletion on H3.3 deposition is the secondary effect arising from defects in DNA replication, DNA repair or cell cycle progression in RPA1 depleted cells.
RPA Forms a Complex with HIRA and H3.3
To understand how the RPA complex regulates H3.3 deposition, we determined whether RPA interacts with H3.3 and its two distinct histone chaperones, HIRA and DAXX. Unlike HIRA, depletion of DAXX did not affect the deposition of newly synthesized H3.3 using the H3.3-SNAP staining method (Ray-Gallet et al., 2011)(data not shown), possibly due to the fact that DAXX deposits H3.3 at specific chromatin regions such as telomeric heterochromatin. Therefore, we focused on investigating the potential interactions among RPA, H3.3 and the subunits of the HIRA complex. First, we tested whether each subunit of the RPA complex co-localized with H3.3, HIRA and UBN1 using the LacO-LacI targeting system in the A03_1 cell line, which contains multiple LacO repeats (Liu et al., 2012). We observed that HIRA-EGFP (Figure 2A–B), UBN1-EGFP (Figure 2C–D) and H3.3-EGFP (Figure 2E–F) formed foci that co-localized with each subunit of RPA-mCherry-LacI, whereas HIRA-, UBN1- and H3.3-EGFP did not form foci in cells expressing the mCherry-LacI empty vector (Figure S3A). These results suggest that RPA, HIRA and H3.3 form a complex in cells. To test this idea further, H3.1-Flag or H3.3-Flag was immunoprecipitated from HEK293T cells stably expressing H3.1 or H3.3. In agreement with published reports, DAXX was specifically co-immunoprecipitated with H3.3, but not with H3.1. In contrast, CAF-1 p60 was co-immunoprecipitated with H3.1, but not with H3.3. Under the same conditions, we observed that significantly more RPA1 and RPA2 co-purified with H3.3 than with H3.1 in the presence of ethidium bromide (Figure 2G), which would be expected to disrupt DNA-mediated false-positive interactions (Nguyen and Goodrich, 2006). These results indicate that RPA can interact with H3.3 and the HIRA complex in cells and this interaction is unlikely to be mediated by DNA.
Figure 2. RPA, H3.3 and HIRA Form a Complex.

(A–D) Each subunit of the RPA complex co-localizes with HIRA and UBN1. The A03_1 cell line carrying an array of LacO operators was co-transfected with RPA1-, RPA2-, or RPA3-mCherry-LacI and HIRA- or UBN1-EGFP. Cells were visualized using fluorescence microscopy. Representative images (A, C) and quantification results from two independent experiments (B, D) are shown. More than 50 cells with EGFP and mCherry-LacI were counted in each experiment. (E–F) Each RPA subunit co-localizes with histone H3.3. The experiment was performed as described above. (G) RPA1 and RPA2 were enriched with H3.3 immunoprecipitation (IP) compared to H3.1. H3.1-Flag or H3.3-Flag were IPed from cell lysates with or without ethidium bromide (EB) and proteins in whole cell extracts (Input) and IP samples were analyzed by Western blot. Con: control IP using the cells without H3.1-Flag or H3.3-Flag. (H) Depletion of HIRA results in reduced association of RPA with H3.3. (I) Depletion of RPA does not affect the HIRA-H3.3 interaction.
To understand the interdependence of the interactions among RPA, H3.3 and the HIRA complex, we determined how deletion of HIRA or RPA1 would affect the formation of the RPA-HIRA-H3.3 complex. Depletion of HIRA resulted in reduced association of H3.3 with RPA1 and RPA2. In contrast, depletion of RPA1 had no detectable effects on the association of H3.3 with HIRA (Figure 2H–I), suggesting that the RPA complex associates with H3.3 through its interaction with HIRA.
RPA1 Is the Major Subunit That Binds to the HIRA Complex in Vivo
Both the RPA and HIRA complexes consist of three subunits. We observed that significantly more endogenous HIRA proteins were co-purified with RPA1-EGFP than with RPA2-EGFP, or RPA3-EGFP despite the fact that RPA1-EGFP fusion proteins were expressed at a lower level than either RPA2-EGFP or RPA3-EGFP. Under the same conditions, DAXX was not detectable in the immunoprecipitate (Figure 3A). Similar results were observed when UBN1 was co-expressed with RPA1-, RPA2- or RPA3-EGFP (Figure S3B). These results indicated that RPA1 is likely the main subunit to interact with the HIRA complex.
Figure 3. The HIRA-A Domain Interacts with RPA1-C Domain.

(A) RPA1 interacts with HIRA. RPA1-, RPA2- or RPA3-EGFP was expressed in 293T cells and immunoprecipitated using antibodies against GFP. Proteins were analyzed by Western blot. (B–C) RPA1-C domain interacts with HIRA. (B) Schematic representation of RPA1 deletion mutants. (C) The experiments were performed as described in A. (D–E) RPA1 interacts with the HIRA-A domain. (D) Schematic representation of HIRA deletion mutants. (E) EGFP-tagged HIRA and mutants were expressed in 293T cells in the presence of RPA1-mCherry and were immunoprecipitated using antibodies against GFP. (F–H) The HIRA-A domain interacts with RPA1-C domain in vitro. (F) Schematic representation of purified recombinant RPA or mutant proteins used in (G–H). (G–H) The RPA complex interacts with HIRA-A domain in vitro, mainly through RPA1-C. GST or GST-HIRA-A proteins were used to pull down recombinant RPA complex (G) or RPA mutants (H) after EB treatment. Note that RPA1-C domain alone was not soluble, but formed a soluble complex with RPA2-D and RPA3-E domains (RPA core).
RPA1 contains four oligonucleotide/oligosaccharide-binding (OB) folds, which are also referred to as F, A, B and C DNA binding domains (DBD) (Figure 3B). The RPA1-A and RPA1-B domains are the major ssDNA binding regions, whereas the RPA1-C domain has a lower affinity for ssDNA and is involved in forming the RPA complex core (Fanning et al., 2006). We found RPA1-ABC, -FC and -C domains bound to HIRA (Figure 3C) or UBN1 (Figure S3C), whereas the RPA1-FAB domains did not exhibit detectable association with HIRA or UBN1 (Figure 3C and Figure S3C). These results support the idea that RPA1-C is the major domain mediating the RPA-HIRA interaction.
HIRA-A Domain Interacts with the RPA1-C Domain in Vivo and in Vitro
HIRA consists of three domains: the N-terminal WD40 repeats A domain that binds to UBN1, a central B domain that binds to Asf1a and a C-terminal C domain that binds to CABIN1 (Figure 3D). Three HIRA mutants (HIRA-A,-AB and C domain) were tested for their interactions with RPA1 in vivo. The HIRA-A domain bound to RPA1 as efficiently as wild type HIRA, whereas HIRA-C domain could not bind (Figure 3E). These results indicated that the HIRA-A domain is mainly responsible for the RPA1-HIRA interaction in vivo. Supporting this conclusion, the UBN1-d41-77 mutant that is defective in HIRA binding also exhibited reduced association with RPA1 (Figure S3D–E). Moreover, the HIRA-A domain co-localized with the RPA1-C domain in A03_1 cells (Figure S3F). Using in vitro GST pull-down assays as well as recombinant RPA complexes and the various domains (His-RPA-CDE core, His-RPA2/3-DE and His-RPA1-AB) (Figure 3F and Figure S3G), we found that the HIRA-A domain directly bound to the RPA complex in vitro either in the presence of EB (Figure 3G) or with Micrococcal nuclease treatment to eliminate false positive interactions (Nguyen and Goodrich, 2006) (Figure S3H). Moreover, the HIRA-A domain bound to the RPA-CDE core, less efficiently to RPA1-AB domain, but not to the RPA2/3-DE subcomplex (Figure 3H). These results indicate that the RPA1-C and HIRA-A domains are mainly responsible for the RPA-HIRA interaction, with minor contributions from the RPA1-AB domain.
RPA1 and RPA2 Co-localize with the HIRA Complex and H3.3 at Gene Regulatory Elements
To gain further insights into the function of RPA in regulation of H3.3 deposition, we performed chromatin immunoprecipitation coupled with next-generation sequencing (ChIP-seq) of endogenous RPA1 and RPA2 in HeLa cells. Metagene analysis of RPA1 and RPA2 ChIP-seq results indicated that both RPA1 and RPA2 were enriched at promoter regions, defined as −2kb to +1kb of TSS (transcription start site), with more RPA1 and RPA2 at the promoter regions of highly transcribed genes than lowly expressed genes (Figure 4A and 4B). RPA2 ChIP followed by quantitative real-time PCR confirmed RPA2 enrichments at three selected promoter regions compared to corresponding flanking regions (Figure 4C). Using MACS peak calling programs (Liu, 2014), we identified 4904 RPA1 peaks and 4360 RPA2 peaks (Figure S4A). Over 30% of RPA1 and RPA2 peaks overlapped significantly. The relatively low overlap is likely due, in part, to low enrichment of RPA1 and RPA2 ChIP and apparently noisy RPA1 and RPA2 ChIP-seq peaks (Figure 4D and Figure S4D). However, calculation of RPA2 ChIP-seq read density surrounding RPA1 ChIP-seq peaks indicated that RPA2 co-localized with RPA1 and this pattern was reproducible (Figure S4B).
Figure 4. RPA Co-localizes with HIRA and H3.3 at gene regulatory elements.

(A–C) RPA1 and RPA2 are enriched around transcription start sites (TSS) of highly transcribed genes. Normalized reads density plots (RPKM: Reads Per Kilobase per Million mapped reads) of RPA1 (A) and RPA2 (B) ChIP-seq signals from 5 Kb upstream of TSS to 5 Kb downstream of transcription end sites (TES). Genes were split into five groups based on their expression levels in HeLa cells, with 0–20% representing the genes with the highest expression. (C) ChIP-qPCR validation of RPA2 enrichment at TSS regions compared to corresponding flanking regions. Results are average of two independent experiments. (D–E) RPA1 and RPA2 co-localize with H3.3 and H3.3 chaperones genome-wide. (D) Integrative Genomics Viewer (IGV) tracks showing the distributions of RPA1, RPA2, HIRA, UBN1, and H3.3 at the RHOBTB3 gene locus. (E) Normalized tag density plots of RPA1, RPA2, HIRA, UBN1, ASF1a, and H3.3 ChIP-seq signals in a 4 Kb window centered on a composite of the RPA1 and RPA2 overlapping peaks. (F) Unsupervised clustering identifies three distinct clusters of RPA1 and RPA2 overlapping peaks. Promoter is defined as 2 Kb upstream and 1 Kb downstream of TSS. ChIP-seq results for HIRA, UBN1, ASF1a, and H3.3 from published work (Pchelintsev et al., 2013) were used in analysis of D–F. ChIP-seq results for histone marks and transcription factors are from the ENCODE project (http://www.genome.gov/encode/).
Because of the low enrichment of RPA1 and RPA2 ChIP-seq peaks, we utilized 1536 RPA1 and RPA2 overlapping peaks for all following analysis. First, 457 and 461 of the 1536 peaks were localized at gene promoters and intergenic enhancers, respectively, indicating a significant enrichment of RPA at these gene regulatory elements including promoters and enhancers (Figure S4C).
To explore whether RPA co-localizes with the HIRA complex and H3.3, we calculated the ChIP-seq read density of HIRA, UBN1, ASF1a, and H3.3 in the regions surrounding the 1536 RPA1/RPA2 overlapping peaks using published datasets (Pchelintsev et al., 2013). We observed that RPA1 peaks co-localized with RPA2, HIRA, UBN1 and ASF1a peaks in HeLa cells (Figure 4E). Interestingly, H3.3 ChIP-seq peaks, but not that of HIRA, UBN1 or exhibited a bifurcated distribution pattern surrounding the center of RPA1/RPA2 peaks (Figure 4E, Figure S4E–H).
To further delineate the properties of RPA ChIP-seq peaks, we performed unsupervised clustering of the 1536 peaks according to their overlap with peaks for HIRA, UBN1, ASF1a and H3.3, and histone marks associated with enhancers (H3K4me1 and H3K27ac), promoters (H3K4me3) and transcriptional silencing (H3K9me3 and H3K27me3) as well as selected transcription factors obtained from publicly available databases in HeLa cells (Pchelintsev et al., 2013). This analysis showed that RPA1 and RPA2, like HIRA and H3.3, did not co-occupy with the repressive histone marks (H3K27me3 and H3K9me3). Moreover, the 1536 peaks, while all co-localizing with HIRA, UBN1, ASF1a and H3.3, occurred in three distinct clusters (Cluster 1, 2 and 3, Figure 4F).
Cluster 1 was enriched with H3K4me1/H3K4me2, H3K27ac, H2AZ, p300, a subset of specific transcription factors and DNase hypersensitive site (DNaseHS), but exhibited very low overlap with RNA polymerase II, H3K4me3 and promoters. These chromatin features suggest that this class of RPA1/RPA2 peaks co-localizes with active enhancers.
Cluster 2 showed enrichment of H3K4me1/H3K4me2, but less overlap with p300, H3K27ac and DNaseHS compared to Cluster 1. Therefore, it is likely that these RPA binding regions represent poised enhancer elements. Cluster 3 exhibited a strong overlap with gene promoters, RNA polymerase II, H2AZ, H3K4me3, H3K27ac and DNaseHS, (Figure 4F). These results are consistent with the idea that the RPA complex, together with HIRA and H3.3, are enriched at promoter and enhancer regions in HeLa cells.
Depletion of RPA1 Results in Reduced Association of HIRA with Chromatin and Impairs H3.3 Deposition at Promoters and Enhancers
We next hypothesized that RPA is involved in targeting/recruiting HIRA to gene regulatory elements, which in turn regulates the deposition/exchange of newly synthesized H3.3 at these regions. To test this hypothesis, we first determined whether depletion of RPA1 affected HIRA chromatin localization by performing HIRA ChIP-seq in RPA1-depleted HeLa cells as well as in HIRA-depleted cells as a control. To analyze the effect of RPA depletion on HIRA localization, we calculated the average normalized read density of HIRA ChIP-seq peaks in RPA1-depleted cells surrounding the HIRA and RPA1 overlapping peaks obtained in control cells. As shown in Figure 5A, the average read density surrounding HIRA and RPA overlapping peaks was dramatically reduced in HIRA- and RPA1- deleted cells compared with control cell (Figure 5A). Box-plot representation of read densities of individual peaks also indicated that RPA1 depletion, like HIRA depletion, led to reduced binding of HIRA to chromatin (Figure 5B). Reduced HIRA binding at a representative gene promoter (DDX58) was shown in Figure 5C. The reduction of HIRA at two selected TSS gene loci was confirmed by HIRA ChIP-PCR (Figure 5D). These results indicate that RPA1 is required for efficient association of HIRA with chromatin.
Figure 5. Depletion of RPA1 Results in Reduced Association of HIRA with Chromatin and Impairs Deposition of Newly Synthesized H3.3.

(A–C) Depletion of RPA1 results in reduced association of HIRA with chromatin. (A) ChIP-seq density profiles of endogenous HIRA in NT and HIRA- or RPA1-deleted HeLa cells. 4-Kb regions centered on HIRA and RPA1 overlapping peaks were analyzed. (B) Box plot of HIRA ChIP-seq read densities of HIRA peaks in NT and HIRA- or RPA1-depleted HeLa cells. Student t-test was applied to calculate p-values between NT and shHIRA or NT and shRPA1 (* p < 0.01). (C) An example of HIRA ChIP-seq read density distribution at the DDX58 gene locus in NT, HIRA- or RPA1-depleted cells. (D) The effect of RPA1 and HIRA depletion on HIRA binding at two TSSs was analyzed using ChIP-qPCR. (E–H) Depletion of RPA1 impairs deposition of newly synthesized H3.3 at selected promoters and enhancers. (E) Diagram illustrating the experimental procedures detecting deposition of newly synthesized H3.3. (F) HIRA and RPA1 knockdown was analyzed by Western blot. (G–H) Analysis of deposition of new H3.3 at four selected promoters (G) and three selected enhancers (H) by ChIP-qPCR. The primer sequences were in Table S3 and ChIP-seq peaks were shown in Figure S4D–G and Figure S5A–C. The results in D, G and H were from three independent experiments (mean ± SD) (* p < 0.05).
To analyze the effect of RPA1 depletion on new H3.3 deposition at gene regulatory regions, we depleted RPA1 or HIRA (as a control) in HeLa cells, transiently expressed H3.3-HA for 24 hours and then performed H3.3-HA ChIP (Figure 5E–F). Analysis of H3.3-HA ChIP DNA using primers amplifying 4 different TSSs and three different enhancer elements (Figure S4D–H and S5A–C) indicate that deposition of newly synthesized H3.3 at the four TSS sites and the three enhancer sites was reduced after depletion of RPA1 or HIRA (Figure 5G–H). These results support the idea that RPA1 is required for efficient deposition of H3.3 at gene regulatory elements, possibly through promoting association of HIRA with these regions.
The Ability of RPA1 to Bind ssDNA Is Required for H3.3 Deposition
RPA is an ssDNA binding protein. The RPA1-CM mutant (containing mutations at 6 charged residues in the RPA-A domain) is unable to bind ssDNA, whereas the RPA-aroA mutant (F238A and F269A) shows a reduced affinity for ssDNA (Haring et al., 2008). We, therefore, tested whether expression of these two mutants could rescue the H3.3 deposition defects by RPA1 depletion. At the similar expression level as exogenous wild type RPA1, the RPA1-aroA mutant exhibited significantly reduced ability to rescue H3.3 deposition defects in RPA1 depleted cells compared to wild type RPA1. While the RPA1-CM mutant was also unable to rescue the H3.3 deposition defects, the expression of the RPA1-CM mutant was lower than exogenous wild type RPA1, possibly due to its instability (Figure S5D–E). Nonetheless, these results suggest that the ability of RPA1 to bind ssDNA is required for its function in regulating H3.3 deposition.
Formation of RNA-DNA hybrid (R-loop structure), which leads to generation of ssDNA, is frequently detected at promoters (Sanz et al., 2016). We next tested whether the R-loop structure is required for H3.3 deposition at gene regulatory elements. Overexpression of RNase H1, which cleaves the RNA in an R-loop, led to reduced H3.3 deposition at 5 out of 7 foci tested (Figure S5F–G). These results are consistent with the idea that RPA binds to ssDNA, possibly resulting from R-loop formation at gene regulatory elements, and promotes the deposition of H3.3 at these regions.
RPA1 and HIRA Regulate Transcription Including Nascent Divergent Transcription of a Common Set of Genes
Since RPA1 and HIRA are co-localized to gene regulatory regions including promoters and enhancers, we sought to determine whether RPA1 and HIRA are required for transcription at these regulatory elements. To assess nascent RNA synthesis in intact cells, we used the Bru-seq technique. Briefly, after HIRA- and RPA1-depletion, cells were pulsed with 2 mM bromouridine (Bru) for 30 min. Cells were then lysed, total RNA isolated and nascent Bru-containing RNA captured using anti-BrdU antibodies conjugated to magnetic beads as previously described (Paulsen et al., 2014; Paulsen et al., 2013). As shown in Figure 6A–B, depletion of HIRA led to a change in the transcription of 2946 sense transcripts at promoters and 1546 divergent transcripts using the cutoff p-value of 1x10−5. Interestingly, there was little overlap between altered sense and divergent transcripts, suggesting that HIRA depletion affects transcription in either the sense or divergent direction. Similar results were observed in RPA1-depleted cells (Figure 6B). Importantly, over 43% sense and divergent transcripts affected by RPA1 depletion overlapped with sense and divergent transcripts altered by HIRA depletion (Figure 6B). These results provide additional support to the idea that RPA and HIRA affect transcription of a common set of genes, and that HIRA and RPA1 regulate the directionality of transcription at gene promoter regions.
Figure 6. RPA1 and HIRA Regulate Nascent Divergent Transcription.

(A–B) Depletion of HIRA and RPA1 results in altered transcription at the promoter regions. After HIRA- and RPA1-depletion, nascent transcripts were analyzed using Bru-Seq. Top panel in (A) illustrates our approach to assess strand-specific transcription at promoters using Bru-seq. First, the sense and divergent reads of Bru-seq at ±2 Kb of TSS were counted. DESeq (Love et al., 2014) was used to calculate the p value. Bottom panels are volcano plots of altered sense or divergent transcripts at the promoter regions after the deletion of HIRA or RPA1. The Y-axis represents the value of -log10 (p-value) calculated by DEseq (Love et al., 2014), and the X-axis showing the Log2 fold changes of the corresponding transcripts. (B) Four-set Venn diagram separation of significantly changed sense and divergent transcripts after deletion of HIRA or RPA1 in HeLa cells. Datasets were obtained from Figure 6A with the cutoff p value of 10−5, and plotted by VennPlex (Cai et al., 2013). (C–D) HIRA, RPA and H3.3 are enriched at TSS regions with altered sense transcripts after HIRA (C) and RPA1 (D) depletion. (C) Normalized read density plots (TSS±5Kb) from HIRA and H3.3 ChIP-seq at two groups of TSSs with or without changes in sense transcripts at the promoter regions after HIRA knockdown. (D) Normalized read density plots (TSS±5Kb) from RPA1 and H3.3 ChIP-seq at TSSs with or without changes at sense transcripts after RPA1 knockdown.
To explore why depletion of HIRA and RPA1 affected the expression of nascent transcripts, we compared levels of HIRA, RPA1, and H3.3 at TSS region of transcripts altered after the deletion of HIRA or RPA1 to those unaltered transcripts. HIRA and H3.3 levels at gene promoter regions of altered sense transcripts (Figure 6C) and divergent transcripts (Figure S6A) after HIRA deletion were higher than those of unaffected transcripts. Similarly, RPA1 and H3.3 levels at sense transcripts (Figure 6D) and divergent transcripts (Figure S6B) altered in RPA1 depleted cells were higher than those without change. These results are consistent with the idea that RPA1 and HIRA regulate H3.3 deposition into the chromatin of a select set of gene promoter regions and thereby regulate the directionality and the rate of transcription initiation of these genes.
DISCUSSION
In this study, we show that RPA, an ssDNA binding protein best known for its role in DNA replication and DNA repair, interacts with HIRA and co-localizes with HIRA and H3.3 to promoter and enhancer elements. Depletion of RPA results in a reduced association of HIRA to chromatin, impairs the deposition of H3.3 at these gene regulatory elements, and alters the pattern of sense and divergent transcription from promoters. These studies reveal a mechanism whereby RPA and HIRA regulate the deposition of newly synthesized H3.3 at gene regulatory elements and directionality of transcription at gene promoters.
The RPA Complex Regulates Deposition of Newly synthesized H3.3 at Gene Regulatory Elements
The histone variant H3.3 is deposited at distinct chromatin regions including telomeric heterochromatin, gene bodies, enhancers and promoters (Chen et al., 2013; Goldberg et al., 2010). HIRA is the classic histone chaperone depositing H3.3 at genic regions including gene bodies, promoters and enhancers (Pchelintsev et al., 2013). It is proposed that the ability of HIRA to bind RNA polymerase II is responsible for recruiting HIRA for nucleosome assembly of H3.3 at gene bodies (Ray-Gallet et al., 2011). However, it remains unclear how HIRA is recruited to gene regulatory elements. We have provided several lines of evidence supporting the idea that RPA binds HIRA and regulates HIRA-mediated H3.3 deposition at gene regulatory elements. First, we show that depletion of each RPA subunit individually using shRNA compromises H3.3 deposition, suggesting that the RPA complex is required for optimal deposition of newly synthesized H3.3 histones into chromatin. Second, RPA interacts with HIRA in vitro and in vivo and forms a complex that also contains H3.3 in vivo. Third, RPA1 and RPA2 co-localize with HIRA and H3.3 and are enriched at promoters and enhancers genome wide. Finally, depletion of RPA1 reduces HIRA chromatin localization globally and compromises H3.3 deposition at promoters and enhancers. These results support a model whereby RPA binds to gene regulatory elements and facilitates the association of HIRA to these regions, which in turn regulates deposition of newly synthesized H3.3.
How is RPA is recruited to promoters and enhancers? Nucleosomes at promoters and gene regulatory elements are very dynamic, most likely due to the presence of histone variants H3.3 and H2AZ (Jin and Felsenfeld, 2007). Moreover, divergent transcription and eRNAs are frequently detected at promoters and enhancers, respectively (Wu and Sharp, 2013). These RNA transcripts generated at promoters and enhancers may promote the formation of R-loop, a structure that contains a DNA: RNA hybrid and a displaced ssDNA. Therefore, it is possible that RPA binds the displaced ssDNA in these R-loops at gene regulatory regions. Supporting this idea, we show that the ability of RPA1 to bind ssDNA is important for its role in H3.3 deposition. Overexpression of RNase H1, which disrupts the R loop structure, affects H3.3 deposition at selected enhancers and promoters. Interestingly, R-loops are enriched at promoters (Sanz et al., 2016). Recently, it was reported that RPA physically interacts with transcription factor HSF1 and assists its access to the HSP70 promoter by recruiting the FACT complex (Fujimoto et al., 2012). Therefore, it is also possible that RPA is recruited to gene promoters and enhancers through protein-protein interactions. Supporting this possibility, we observed that RPA1 and RPA2 ChIP peaks are enriched with one very similar motif that represents the consensus binding sequence for three transcription factors (ZN652, OTX1, GSC2, data not shown). Regardless of the specific mechanism responsible for recruiting RPA to the gene regulatory elements, we suggest that the RPA-HIRA-H3.3 axis promotes nucleosome formation on double stranded DNA because it has been shown that nucleosome assembly occurs preferentially on dsDNA (Almouzni et al., 1990).
RPA and HIRA Regulate the Directionality of Transcription at Promoter Regions
When nascent transcripts at gene promoters were analyzed using Bru-seq, we found that a specific set of genes showed altered generation of nascent transcripts in either the sense or divergent directions following HIRA- or RPA1-depletion. Over 40% of these transcripts showed an overlap between HIRA and RPA1 depletion. These results indicate that RPA and HIRA regulate gene expression through a common mechanism at these genes, most likely by regulating H3.3 deposition/exchange at these regions. Strikingly, depletion of HIRA or RPA1 affects transcription in either the sense or divergent direction from the promoter. This suggests that deposition of H3.3 by HIRA and RPA1 into a promoter region regulates the directionality of transcription initiation. In budding yeast, three subunits of HIRA complex were also found to affect directionality of transcription at gene promoters through a genome wide screen (Marquardt et al., 2014). We suggest that HIRA- and RPA1-mediated H3.3 deposition and directionality of transcription at gene promoters represent a conserved mechanism by which cells can fine-tune the regulation of gene expression.
RPA and HIRA bind gene regulatory regions of thousands of genes. However, depletion of HIRA and RPA affect transcription a few hundred genes based on RNA-seq analysis (data not shown). This apparent paradox likely arises from compensatory mechanisms that regulate H3.3 deposition and gene expression. For instance, Drosophia cells lacking both copies of H3.3 genes are viable and the expression of less than 400 genes in male animals were affected, likely due to compensation from increased expression of H3.1 (Sakai et al., 2009). Moreover, it has been shown that Xnp, the homolog for human ATRX, and HIRA work in parallel for deposition of H3.3 at nucleosome free regions in Drosophila (Schneiderman et al., 2012). Thus, while H3.3 and HIRA are essential for mouse development (Szenker et al., 2011), compensatory pathways may offset gene expression changes caused by depletion of HIRA and H3.3 in cell lines.
In addition to gene transcription, H3.3 is important for proper establishment of H3 lysine 27 methylation patterns at promoters of developmentally regulated genes and silencing of endogenous retroviral elements in mouse embryonic stem cells (Banaszynski et al., 2013; Elsasser et al., 2015). Furthermore, H3.3 marks damaged chromatin and facilitates transcription recovery after genotoxic stress (Adam et al., 2013), and is required for maintaining DNA replication fork progression after UV damage (Frey et al., 2014). It would be interesting in future experiments to determine whether the RPA-HIRA-H3.3 axis regulates any of these important processes and how this deposition mechanism is restricted during normal DNA replication where nucleosome assembly of H3.1 dominates.
EXPERIMENTAL PROCEDURES
Assays for Monitoring Deposition of Newly Synthesized H3.3 Using the SNAP Tag
Deposition of newly synthesized H3.3 using the SNAP Tag was performed as described previously (Ray-Gallet et al., 2011). Briefly, 10 mM SNAP blocking reagent (New England Biolabs) was added to the medium at 37°C for 30 min to quench old H3.3-SNAP. Cells were then washed with PBS three times and incubated in the medium for another 30 min, followed by three more washes using PBS. Fresh medium was added and the cells incubated for 12 h. Then 2 mM TMR was added to the medium for 20 min at 37 °C. Cells were first pre-extracted with 0.5% Triton X-100 buffer and fixed in 3% paraformaldehyde. A fluorescence microscope (100× objective) was used to record fluorescence images and Image J was used to quantify the SNAP fluorescence intensity. For each experiment, more than 100 cells were counted. Alternatively, after the TMR staining, the cells were collected, washed with PBS and subjected to the chromatin fractionation assay following the described procedure (Mendez and Stillman, 2000). Briefly, cells were extracted with 0.5% Triton X-100 extraction buffer on ice for 5 min followed by high-speed centrifugation. The chromatin pellet was washed with PBS and then boiled in 1X SDS sample buffer. Proteins in chromatin pellet were separated by SDS-PAGE and SNAP-tagged H3.3 was detected on a Typhoon 7900, and the total proteins were visualized by IRDye Blue Protein Stain and used for loading controls. Image J was used to quantify the SNAP fluorescence intensity and total protein signals.
Bromouridine Labeling, Isolation of Bru-RNA and Strand-Specific cDNA Library Preparation
Bromouridine labeling, isolation of Bru-RNA and strand specific cDNA library preparation were performed as described previously (Paulsen et al., 2014; Paulsen et al., 2013). Briefly, bromouridine (Aldrich) was added to the media of HeLa cell to a final concentration of 2 mM at 37° C for 30 min. Cells were then directly lysed by Trizol and total RNA isolated. The Bru-labeled RNA was captured from the total RNA by incubation with anti-BrdU antibodies (BD Biosciences) conjugated to magnetic beads under gentle agitation at room temperature for 1 h. Isolated Bru-labeled RNA was then used to prepare strand-specific DNA libraries using the Illumina Tru Seq Kit (Illumina) according to the manufacturer’s instructions with modifications previously described (Paulsen et al., 2014).
Reagents and other experimental procedures including immunoprecipitation, GST-pull down assays and RNA-seq, ChIP-seq and BrU-seq were described in Supplemental Information.
Supplementary Material
Highlights.
RPA regulates the deposition of newly synthesized H3.3 independent of cell cycle
RPA interacts with HIRA directly and forms a RPA-HIRA-H3.3 complex
RPA co-localizes with HIRA and H3.3 at gene regulatory elements
RPA and HIRA regulate transcriptional directionality at promoters
Acknowledgments
We are grateful to Mark Alexandrow providing the A03_1 cell line, to Bruce Stillman and Peter Adams for antibodies, to Gloria E.O. Borgstahl and Guohong Li for plasmids and Michelle T. Paulsen for technical assistance with Bru-seq. This work is supported by NIH grants (GM118015 and CA157489 to ZZ, HG006786 to ML, and CA086862 to MSW) and by the Epigenomics Program of Mayo Clinic Center for Individualized Medicine.
Footnotes
ACCESSION NUMBERS
The high-throughput sequencing datasets have been deposited in Gene Expression Omnibus (GEO) under the accession number GSE76661.
AUTHOR CONTRIBUTIONS
H.Z. and Z.Z conceived and designed the experiments. H.Z. performed most of experiments. H.G. performed all of the bioinformatics analysis. H. Z. and Z. W. performed H3.3 SNAP staining screening and all the experiments for revision. J.L. performed RPA1 and RPA2 ChIP. T.O. and M.W. provided key resources and reagents. M.L. performed Bru-seq. Z.Z., H.Z., H.G. and Z.W. wrote the paper. All authors read and edited the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adam S, Polo SE, Almouzni G. Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell. 2013;155:94–106. doi: 10.1016/j.cell.2013.08.029. [DOI] [PubMed] [Google Scholar]
- Almouzni G, Clark DJ, Mechali M, Wolffe AP. Chromatin assembly on replicating DNA in vitro. Nucleic acids research. 1990;18:5767–5774. doi: 10.1093/nar/18.19.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski LA, Wen D, Dewell S, Whitcomb SJ, Lin M, Diaz N, Elsasser SJ, Chapgier A, Goldberg AD, Canaani E, et al. Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells. Cell. 2013;155:107–120. doi: 10.1016/j.cell.2013.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013;45:1479–1482. doi: 10.1038/ng.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RJ, Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol. 2013:14–22. doi: 10.1038/nsmb.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Chen H, Yi T, Daimon CM, Boyle JP, Peers C, Maudsley S, Martin B. VennPlex--a novel Venn diagram program for comparing and visualizing datasets with differentially regulated datapoints. PLoS One. 2013;8:e53388. doi: 10.1371/journal.pone.0053388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Zhao J, Wang Y, Wang M, Long H, Liang D, Huang L, Wen Z, Li W, Li X, et al. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Genes Dev. 2013;27:2109–2124. doi: 10.1101/gad.222174.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel Ricketts M, Frederick B, Hoff H, Tang Y, Schultz DC, Singh Rai T, Grazia Vizioli M, Adams PD, Marmorstein R. Ubinuclein-1 confers histone H3.3-specific-binding by the HIRA histone chaperone complex. Nat Commun. 2015;6:7711. doi: 10.1038/ncomms8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010;24:1253–1265. doi: 10.1101/gad.566910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsasser SJ, Huang H, Lewis PW, Chin JW, Allis CD, Patel DJ. DAXX envelops a histone H3.3-H4 dimer for H3.3-specific recognition. Nature. 2012;491:560–565. doi: 10.1038/nature11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsasser SJ, Noh KM, Diaz N, Allis CD, Banaszynski LA. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature. 2015 doi: 10.1038/nature14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning E, Klimovich V, Nager AR. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic acids research. 2006;34:4126–4137. doi: 10.1093/nar/gkl550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey A, Listovsky T, Guilbaud G, Sarkies P, Sale JE. Histone H3.3 Is Required to Maintain Replication Fork Progression after UV Damage. Curr Biol. 2014;24:2195–2201. doi: 10.1016/j.cub.2014.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Takaki E, Takii R, Tan K, Prakasam R, Hayashida N, Iemura S, Natsume T, Nakai A. RPA assists HSF1 access to nucleosomal DNA by recruiting histone chaperone FACT. Mol Cell. 2012;48:182–194. doi: 10.1016/j.molcel.2012.07.026. [DOI] [PubMed] [Google Scholar]
- Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurard-Levin ZA, Quivy JP, Almouzni G. Histone chaperones: assisting histone traffic and nucleosome dynamics. Annu Rev Biochem. 2014;83:487–517. doi: 10.1146/annurev-biochem-060713-035536. [DOI] [PubMed] [Google Scholar]
- Haring SJ, Mason AC, Binz SK, Wold MS. Cellular functions of human RPA1. Multiple roles of domains in replication, repair, and checkpoints. The Journal of biological chemistry. 2008;283:19095–19111. doi: 10.1074/jbc.M800881200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K. Assembly of variant histones into chromatin. Annu Rev Cell Dev Biol. 2005;21:133–153. doi: 10.1146/annurev.cellbio.21.012704.133518. [DOI] [PubMed] [Google Scholar]
- Jin C, Felsenfeld G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007;21:1519–1529. doi: 10.1101/gad.1547707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konev AY, Tribus M, Park SY, Podhraski V, Lim CY, Emelyanov AV, Vershilova E, Pirrotta V, Kadonaga JT, Lusser A, et al. CHD1 motor protein is required for deposition of histone variant H3.3 into chromatin in vivo. Science. 2007;317:1087–1090. doi: 10.1126/science.1145339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Stillman B. The origin recognition complex: a biochemical and structural view. Sub-cellular biochemistry. 2012;62:37–58. doi: 10.1007/978-94-007-4572-8_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CP, Xiong C, Wang M, Yu Z, Yang N, Chen P, Zhang Z, Li G, Xu RM. Structure of the variant histone H3.3-H4 heterodimer in complex with its chaperone DAXX. Nat Struct Mol Biol. 2012;19:1287–1292. doi: 10.1038/nsmb.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T. Use model-based Analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods Mol Biol. 2014;1150:81–95. doi: 10.1007/978-1-4939-0512-6_4. [DOI] [PubMed] [Google Scholar]
- Loppin B, Bonnefoy E, Anselme C, Laurencon A, Karr TL, Couble P. The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature. 2005;437:1386–1390. doi: 10.1038/nature04059. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt S, Escalante-Chong R, Pho N, Wang J, Churchman LS, Springer M, Buratowski S. A chromatin-based mechanism for limiting divergent noncoding transcription. Cell. 2014;157:1712–1723. doi: 10.1016/j.cell.2014.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602–8612. doi: 10.1128/mcb.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TN, Goodrich JA. Protein-protein interaction assays: eliminating false positive interactions. Nature methods. 2006;3:135–139. doi: 10.1038/nmeth0206-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Magnuson B, Wilson TE, Ljungman M. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods. 2014;67:45–54. doi: 10.1016/j.ymeth.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Tsan YC, Chang CW, Tarrier B, Washburn JG, Lyons R, et al. Coordinated regulation of synthesis and stability of RNA during the acute TNF-induced proinflammatory response. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:2240–2245. doi: 10.1073/pnas.1219192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pchelintsev NA, McBryan T, Rai TS, van Tuyn J, Ray-Gallet D, Almouzni G, Adams PD. Placing the HIRA histone chaperone complex in the chromatin landscape. Cell Rep. 2013;3:1012–1019. doi: 10.1016/j.celrep.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray-Gallet D, Quivy JP, Scamps C, Martini EM, Lipinski M, Almouzni G. HIRA is critical for a nucleosome assembly pathway independent of DNA synthesis. Mol Cell. 2002;9:1091–1100. doi: 10.1016/s1097-2765(02)00526-9. [DOI] [PubMed] [Google Scholar]
- Ray-Gallet D, Woolfe A, Vassias I, Pellentz C, Lacoste N, Puri A, Schultz DC, Pchelintsev NA, Adams PD, Jansen LE, et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol Cell. 2011;44:928–941. doi: 10.1016/j.molcel.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Sakai A, Schwartz BE, Goldstein S, Ahmad K. Transcriptional and developmental functions of the H3.3 histone variant in Drosophila. Curr Biol. 2009;19:1816–1820. doi: 10.1016/j.cub.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz LA, Hartono SR, Lim YW, Steyaert S, Rajpurkar A, Ginno PA, Xu X, Chedin F. Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol Cell. 2016;63:167–178. doi: 10.1016/j.molcel.2016.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneiderman JI, Orsi GA, Hughes KT, Loppin B, Ahmad K. Nucleosome-depleted chromatin gaps recruit assembly factors for the H3.3 histone variant. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:19721–19726. doi: 10.1073/pnas.1206629109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- Sikorski TW, Ficarro SB, Holik J, Kim T, Rando OJ, Marto JA, Buratowski S. Sub1 and RPA associate with RNA polymerase II at different stages of transcription. Mol Cell. 2011;44:397–409. doi: 10.1016/j.molcel.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Res. 2011;21:421–434. doi: 10.1038/cr.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116:51–61. doi: 10.1016/s0092-8674(03)01064-x. [DOI] [PubMed] [Google Scholar]
- van der Heijden GW, Derijck AA, Posfai E, Giele M, Pelczar P, Ramos L, Wansink DG, van der Vlag J, Peters AH, de Boer P. Chromosome-wide nucleosome replacement and H3.3 incorporation during mammalian meiotic sex chromosome inactivation. Nat Genet. 2007;39:251–258. doi: 10.1038/ng1949. [DOI] [PubMed] [Google Scholar]
- Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Sharp PA. Divergent transcription: a driving force for new gene origination? Cell. 2013;155:990–996. doi: 10.1016/j.cell.2013.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.