

Abstract
Membrane proteins, due to their roles as cell receptors and signaling mediators, make prime candidates for drug targets. The computational analysis of protein-ligand binding affinities has been widely employed as a tool in rational drug design efforts. Although efficient implicit solvent-based methods for modeling globular protein-ligand binding have been around for many years, the extension of such methods to membrane protein-ligand binding is still in its infancy. In this study, we extended the widely used Amber/MMPBSA method to model membrane protein-ligand systems, and we used it to analyze protein-ligand binding for the human purinergic platelet receptor (P2Y12R), a prominent drug target in the inhibition of platelet aggregation for the prevention of myocardial infarction and stroke. The binding affinities, computed by the Amber/MMPBSA method using standard parameters, correlate well with experiment. A detailed investigation of these parameters was conducted to assess their impact on the accuracy of the method. These analyses show the importance of properly treating the non-polar solvation interactions and the electrostatic polarization in the binding of nucleotide agonists and non-nucleotide antagonists to P2Y12R. Based on the crystal structures and the experimental conditions in the binding assay, we further hypothesized that the nucleotide agonists lose their bound magnesium ion upon binding to P2Y12R, and our computational study supports this hypothesis. Ultimately, this work illustrates the value of computational analysis in the interpretation of experimental binding reactions.
Graphical abstract
Introduction
Membrane proteins provide a range of important functions as cell receptors, signaling proteins, transmembrane channels, and more. Their roles as receptors and signaling proteins make them particularly relevant as candidates for drug targets. However, the study of membrane proteins is more complicated than the study of globular proteins. Particularly, the presence of the membrane complicates structural studies, both experimentally and computationally. The presence of the membrane makes it more difficult to employ experimental techniques such as NMR and X-ray crystallography. For instance, the signal from the membrane must be disentangled from that of the protein when using NMR, and membrane proteins are notoriously difficult to crystallize. For computational studies, modeling of the membrane becomes an important consideration.
An active area of computational studies of proteins is the prediction of protein-ligand binding affinities. The Amber 161 and AmberTools 162 suites currently provide the capability of performing such calculations for globular proteins via the widely used MMPBSA module3-8. The consideration of solvation effects in these computational approaches is quite important. It is relatively common knowledge that solvent-solute interactions provide the primary driving force for producing and maintaining the properly folded structures of proteins9-11.
Inclusion of the solvent into a computational model or simulation can generally be classified into one of two different categories: explicit and implicit solvation. In explicit solvation, each atom or molecule of the solvent is modeled individually. While this is generally agreed to be the most accurate method, one is often not interested in the properties of the solvent itself, but rather in the behavior it induces upon the solute. Unfortunately, accurately capturing statistically meaningful characteristics requires sampling either from ensembles of trajectories or from a single very long trajectory. Implicit solvents provide an attractive alternative wherein the effects of the solvent are modeled as a continuum10-26. While the fine-grained details of individual solvent-solute particle interactions are lost, the relevant statistically averaged effects may still be captured when a properly parameterized and transferrable model is used. In addition, since the individual solvent molecules are no longer modeled directly, there are far fewer particles to simulate which reduces the sampling challenges in molecular simulations.
In the case of membrane proteins, the membrane must also be included when modeling solvation effects27-33. In general, the molecules that make up a lipid membrane are much more complex than water or other small organic solvents, and this increases the computational expense of their inclusion. Thus, there has been much effort put into the development and testing of implicit membrane solvent models27-33. Implicit membranes have appeared in several recent computational studies as they can assist in finding the proper native fold of a membrane protein for structural studies and calculations34-36. Implementation of an implicit membrane is currently available in packages such as APBS32, Delphi33, 37, and both the Amber 161 and AmberTools 162 suites. With the implementation of an implicit membrane model into the Amber/PBSA program38-42, the implicit membrane model can be more readily interfaced with the existing MMPBSA framework3-8.
One of the key features to consider in implicit solvent models is the modeling of electrostatic interactions. This is most readily accomplished by employing the Poisson-Boltzmann equation (PBE)43-60. In cases where the electric field is weak and the ion concentration is relatively low (a few hundred millimolar or less), this equation may be approximated as the linear PBE:
| (1) |
where . Here ν denotes the solvent, and I = z2c represents the ionic strength of the solution. The PBE-based solvent models have many biological applications. For example, they have been applied to the prediction of pKa values for ionizable groups in biomolecules61-65, solvation free energies66, 67, binding free energies68-73, and protein folding and design74-83. Even in its simplified linear form, solving the PBE is a non-trivial endeavor. Due to its complexity, there is no general closed form solution; a numerical solution must be sought with the exception of very simplified geometries27, 38-40, 42, 48, 59, 60, 84-115. A semi-analytical Generalized Born (GB) equation was also developed to approximate the PBE solution and is quite popular in biomolecular applications.
In order to apply the PBE or GB frameworks to implicit membrane solvent models, an additional dielectric region must be added (see FIGURE 1). The appropriate dielectric constant of the membrane region is generally thought to be quite low relative to the bulk solvent dielectric constant that is typically set to be between 60 (mimicking SPC water models) and 80 (typical for TIP3P models)116-119. Various dielectric constant profiles were explored during the development of GB implicit membrane models116, 118, 120, and it was demonstrated that a simple two dielectric constant model can reproduce electrostatic free energies relatively well by modeling the membrane as a slab like region with a uniform dielectric constant of about 2. Models with 3 or more layers have been shown to improve the accuracy of the results; however, beyond three layers, the improvement was shown to be marginal118.
Figure 1.
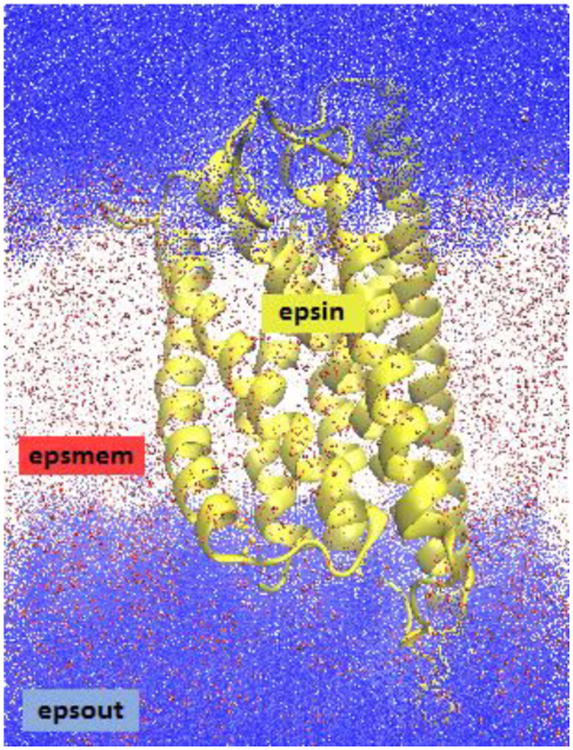
Dielectric constant regions in the P2Y12R system. The protein/ligand is shown in yellow (dielectric: epsin), the implicit membrane is shown in red (dielectric: epsmem), and the surrounding implicit water solvent is shown in blue (dielectric: epsout).
A complete implementation of an implicit membrane under the Amber/PBSA program38-42 requires implementation of appropriate membrane to protein non-polar solvation free energy terms121. While development of these terms is still underway, it is not expected to impact binding free energy calculations for protein-ligand systems in which the binding pocket is sequestered away from the membrane in the protein interior. Thus, such systems make good candidates for testing the electrostatic free energy calculations provided by the current implementation of the implicit membrane model within the Amber/PBSA program38-42.
While three-dimensional structures for globular proteins are quite abundant, such data is less prevalent for membrane proteins. To validate the Amber/PBSA program, and the MMPBSA framework for membrane protein applications, it is necessary to find a protein for which experimental binding affinities and structures of the associated protein-ligand complexes are both available. Recently, a study on the human purinergic platelet receptor (P2Y12R) was reported122, 123, providing crystal structures of the receptor bound to three different ligands and experimental measurements of dissociation constants for the wild type, several select mutants, and two additional ligands. Several antithrombotic drugs target P2Y12R to inhibit platelet aggregation for the purpose of preventing myocardial infarction and stroke. However, limitations of these drugs have motivated the development of a new generation of P2Y12R inhibitors124, 125. Thus, computational modeling, based on the latest structural and functional data, will further facilitate developmental efforts aimed in this direction.
This work documents both the development of an MMPBSA algorithm for membrane protein-ligand binding applications and the validation of the algorithm using P2Y12R complexes with nucleotide agonists and non-nucleotide antagonists. We analyze the sensitivity and the quality of predicted binding affinities with respect to several key polar and non-polar components in the MMPBSA framework. In addition, we also analyze the role of magnesium ions in the binding of nucleotide agonists to P2Y12R. Our results show the application of protein-ligand binding affinity prediction methods towards interpreting experimental binding affinities.
Methods
Preparation of the P2Y12R complex structures
Three separate crystal structures of P2Y12R122, 123, two complexed with agonist ligands: 2-methylthio-adenosine-5′-triphosphate and 2-methylthio-adenosine-5′-diphosphate (2MeSATP and 2MeSADP respectively) and one with an antagonist ligand: AZD-1283 (AZD), were downloaded from the protein databank. As was noted in the corresponding literature122, 123, each of the crystal structures contained several sequence gaps for which no structure could be resolved. The program Modeller126 was used to generate initial structures for these gaps. These homology models were then merged into the crystal structures using the Multi-SEQ127 module in the program VMD128. The single point mutation D294N was modeled in the Amber/LEAP program1, 2, 129.
In addition to the ligands obtained from the three P2Y12R crystal structures, two additional antagonist ligands, with reported binding affinity data122, were docked to the receptor from the P2Y12R-AZD crystal structure. The structure for Ticagrelor (TIQ) was extracted from a previously published crystal structure130 (PDB ID: 5ALB). The structure for PSB-0739 (PSB) was generated using MarvinSketch131 to produce a 2D structure file which was subsequently converted into a 3D structure file using the OpenEye toolkits132. The structures were independently docked to the receptor using AutoDock Vina/SMINA133.
The ligands in the three P2Y12R complex structures (2MeSATP, 2MeSADP, and AZD) were extracted to individual structure files for parameterization. Two additional ligands, PSB and TIQ, were obtained from the docking analysis documented above. The two agonists are simple derivatives of ATP and ADP, so their parameters were obtained from the literature134 except for the atomic charges for the 2-methylthio-adenine group. For the rest of the ligands, the Amber/ANTECHAMBER135 program was used to generate force field parameters.
Preparation of the lipid membrane model
P2Y12R is found embedded within platelet outer membranes. The membrane environment for P2Y12R was modeled with an explicit all-atom model for the molecular dynamics (MD) simulation and with an implicit continuum model for the post-processing binding affinity calculation. Construction of the explicit all-atom membrane model was accomplished using the CHARMM-GUI membrane builder web server136. The membrane was constructed with a POPC, POPS, and POPE ratio of 3:2:3 and a cholesterol to lipid ratio of 2:5137. Sphingomyelin lipids were not included since their force field parameters were not yet available.
The aqueous phase of the P2Y12R membrane protein-ligand system was modeled using an explicit all-atom approach with the TIP3P model along with sufficient potassium and chloride ions to mimic a roughly 150 millimolar KCl concentration. The constructed membrane protein-ligand system was loaded into the LEAP1, 2, 129 program for the generation of simulation force field topology and coordinate files.
MD simulation protocol
Each system was first minimized using 500 steps of steepest descent followed by 500 steps of conjugate gradient optimization. All residues taken directly from the crystal structures were held fixed. Residues generated from homology modeling were left unrestrained, along with all solvent molecules including membrane lipids, water molecules, and ions. After minimization, heating was performed in two phases. In the first phase, systems were brought up to 100 K over 2500 time steps (5 ps) under the NVT condition using a Langevin thermostat with a collision frequency of 1.0 per ps. This was followed by heating from 100 K to 303 K over 100 ps under the NPT condition with anisotropic pressure scaling using the Berendsen barostat with a pressure relaxation constant of 2.0 ps and a target pressure of 1.0 atm. In both cases, a cutoff radius of 10 Å was used when computing non-bonded interactions. All these preparation simulations were performed using the MPI parallelized SANDER program from the Amber 16 suite1, 2, 129.
After the initial heating was completed, it was necessary to equilibrate the membrane density prior to fully flexible MD runs. The MD runs utilized Amber's GPU accelerated PMEMD program that does not allow frequent updating of the box size1, 2, 129. Density equilibration was performed over 10 identical 500 ps NPT simulations at a constant temperature of 303 K. As with heating and minimization, portions of the protein substructure containing coordinates from the crystal structure were held fixed. Afterwards, all restraints were removed, and a 20 ns equilibration run was performed to fully relax the system. Finally, a 10 ns production run was used for the MMPBSA calculation, which was found to be sufficient to achieve the averaging required for free energy calculations (SI Figure S1). In order to take full advantage of the GPU accelerated code, a Monte-Carlo thermostat was employed instead of the Berendsen thermostat used during the previous simulation phases.
Binding free energy calculations
Binding free energies were computed using the SANDER/PBSA module in the Amber 2016 release1, 2, 129. The production run trajectory was post processed with CPPTRAJ138 in order to remove the solvent, membrane, and counter ions from the receptor-ligand complex. 1000 frames, taken at equal intervals over the 10 ns production run, were processed using SANDER/PBSA to compute molecular mechanics potential energies and solvation free energies.
The binding free energy for the protein-ligand complex was computed as the difference between the complex free energy and the sum of the receptor and ligand free energies as shown schematically in FIGURE 2 for the membrane protein-ligand system. The SANDER/PBSA calculations were conducted either inside the implicit membrane/water solvent or the pure implicit water solvent according to the following thermodynamic cycle:
Figure 2.
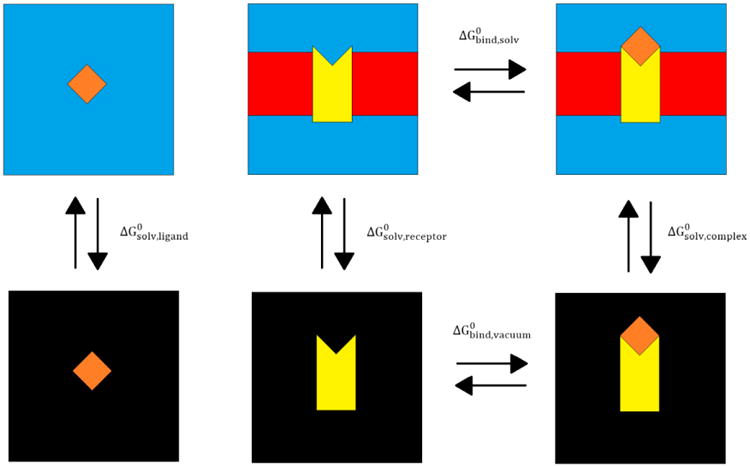
Thermodynamic cycle of the MMPBSA method for a membrane protein-ligand system. The membrane protein is depicted in yellow, the ligand is depicted in orange, the implicit membrane is shown in red, the water solvent is shown with blue, and the vacuum is shown with black. ΔG0 values are labelled for the various transitions from one state to another.
| (2) |
The solvation free energies were calculated using
| (3) |
where the electrostatic terms, , were calculated using the linearized PBE solver as implemented in PBSA38-42. The non-polar solvation terms, , were calculated using either the classical model or the modern model as documented previously139. It is worth pointing out that FIGURE 2 shows that the ligand-binding site of the protein is in the aqueous phase. This implies that the standard non-polar solvent models, optimized for solvation in water, are reasonable approximations. However, it is not unusual to observe binding cavities buried deep within the lipid bilayer. These binding reactions will require a recalibration of the non-polar solvent model, and this scenario will be explored in a future study.
The calculated binding free energies were then compared against the experimental results122. The experimental dissociation constants, Kd, were converted to appropriate binding free energies using the formula:
| (4) |
where R is the gas constant, and T is the temperature.
Estimation of the free energy penalty upon the loss of Mg2+
It is well known that both ATP and ADP have a tendency to form a complex with a magnesium ion in solution140-142. However, in the crystal structures for 2MeSATP and 2MeSADP bound to P2Y12R, magnesium ions were notably absent. Nevertheless, the experimental binding affinity data for 2MeSATP and 2MeSADP bound to P2Y12R was obtained in the presence of 10mM MgCl2122. When the experimental binding affinity measurements were carried out, the presence of magnesium ions in solution may have imposed an energetic penalty on the ligands 2MeSATP and 2MeSADP; if either compound was bound to a magnesium ion in solution, the bound magnesium ion would have to be removed before the ligand could bind to the P2Y12R active site in the same manner that was observed in the crystal structure.
In order to estimate the magnitude of this penalty, we modeled the removal of the bound magnesium ion from both 2MeSATP and 2MeSADP in solution as a binding affinity calculation. The overall reaction is given as:
| (5) |
where AXP = ATP or ADP depending on the ligand in question, and ΔG5 is the overall free energy change for this reaction. We can separate the removal of Mg2+ and the binding of the ligand to P2Y12R into two steps:
| (6) |
| (7) |
with the free energy change for equation (6) given as ΔG6 , and the free energy change for equation (7) given as ΔG7 . Following Hess's Law, we can sum equations (6) and (7) to recover equation (5), and similarly, we can obtain the overall binding free energy as:
| (8) |
To carry out the free energy calculation for equation (6), the 2MeSATP and 2MeSADP ligands were isolated from their respective complex structures. The bound magnesium ion was placed above and in between neighboring phosphate oxygen atoms on the ligand using CHIMERA143. The MD simulation, and MMPBSA binding free energy calculation, was carried out as described previously.
Additional computational details
In each PBSA calculation, a grid spacing of 0.5 Å was used with a grid to solute dimension ratio (fillratio) of 1.5. The geometric multigrid solver option was employed with a convergence threshold of 1.0 × 10-3, and electrostatic focusing was turned off. All PB calculations were conducted using the periodic boundary condition in the PBSA program38-42 in the Amber 2016 suite1, 2, 129.
The solvation system physical constants were set up as follows. The membrane was modeled as a solid slab of 40 Å. The water relative dielectric constant (epsout) was fixed at 80.0. The water phase ionic strength (istrng) was set to be 150 mM. The lower dielectric region within the molecular solutes was defined using the classical solvent excluded surface model with a water solvent probe of 1.4 Å and a membrane solvent probe of 2.3 Å, which is used to account for the larger effective size of a lipid molecule when compared to a water molecule. Further details for extending the classical solvent excluded surface model to membrane systems will be discussed in a separate publication. The default weighted harmonic averaging was employed to assign dielectric constants for boundary grid edges to reduce grid dependence. Charges and radii were assigned using the same parameters as the simulation topology files.
The accuracy of the computed binding free energies was assessed by comparing the RMSD of the calculated versus the experimental values. The Pearson correlation coefficient (R), the slope, and the associated p-value of the linear regression were also analyzed. These were computed for both the absolute binding free energies (ΔG) and the relative binding free energies (ΔΔG) for two different P2Y12 receptors (wild type and mutant D294N) and five different ligands (2MeSATP, 2MeSADP, PSB, TIQ, and AZD).
Results and Discussion
Impact of the non-polar solvation model
While electrostatic interactions play a major role in PBE-based implicit solvent models, various non-polar interactions, such as cavity surface tension and dispersion must also be accounted for. The PBSA module of Amber currently provides two options for computing non-polar solvation energy terms. The first method (inp1) uses a linear function of the solvent accessible surface area/volume144. The second, more sophisticated, method (inp2) decomposes the non-polar contribution into separate cavity and dispersion terms for better transferrable modeling of the non-polar solvation effects121. To test the relative effectiveness of these two methods, two sets of computations were run using inp1 or inp2 with all other parameters set at their optimal values.
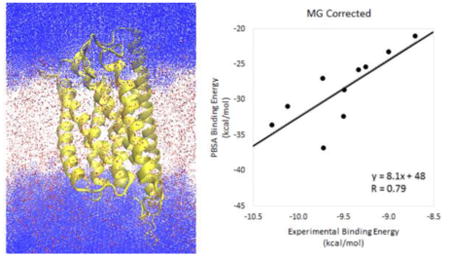
FIGURE 3 shows the values of the Pearson correlation coefficient when inp1 or inp2 are used in the analysis. Additional metrics to assess the agreement are provided in TABLE 1. Using the classical method, inp1, the RMSDs are 59 and 8 kcal/mol (for ΔG and ΔΔG respectively) with the magnesium correction, and 62 and 11 kcal/mol without the magnesium correction. The modern method, inp2, yields RMSDs of 19 and 6 kcal/mol with the magnesium correction, and 23 and 10 kcal/mol without. The lower RMSD values indicate a better agreement using inp2. The same trend holds for the p-value of the correlation; inp1 gives p-values of 0.13 with the magnesium correction and 0.20 without the correction while inp2 yields p-values of 0.0067 and 0.061 respectively. In addition, the R values for inp1 are 0.51 and 0.44 with and without the magnesium correction respectively while the corresponding R values for inp2 are 0.79 and 0.61. In this case, the higher R values for inp2 indicate a stronger correlation. Taken together, the modern inp2 method clearly yields improved results over the classical inp1 method for our system.
Figure 3.
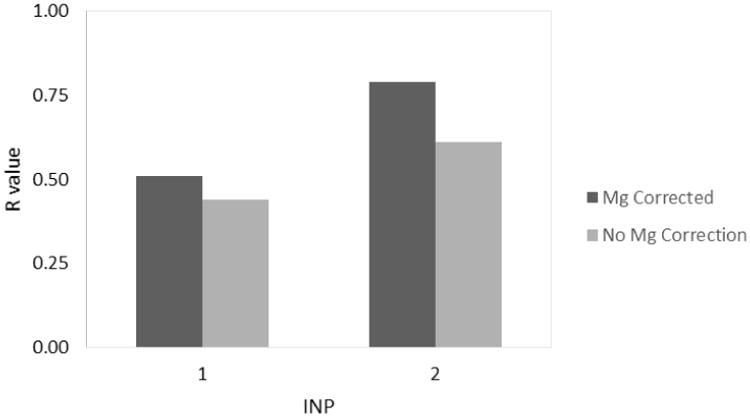
Parameter optimization for the non-polar solvation model, inp (epsin=20, epsmem=4). The R value indicates the correlation for the data set at the given inp value.
Table 1.
The effect of the non-polar solvation model on absolute (ΔG) and relative (ΔΔG) binding affinities. Results that are corrected for the removal of the magnesium ion appear in columns 2-6 while the results that do not take into account the magnesium correction appear in columns 7-11. RMSD values are given in kcal/mol. The raw data used to generate these results is available in Supporting Information (Table S1).
| MG Corrected | No MG Correction | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| INP | RMSD | RMSD(ΔΔG) | Slope | p-value | R | RMSD | RMSD(ΔΔG) | Slope | p-value | R |
| 1 | 59 | 8 | 6.4 | 0.13 | 0.51 | 62 | 11 | 7.7 | 0.20 | 0.44 |
| 2 | 19 | 6 | 8.1 | 6.7E-03 | 0.79 | 23 | 10 | 9.6 | 6.1E-02 | 0.61 |
Impact of the protein dielectric constant
At neutral pH, P2Y12R and all associated ligands except AZD and TIQ are non-neutral in solution. For neutral receptor and ligand systems, the protein dielectric constant is typically assigned to a relatively low value, such as 1 or 2. Charged systems often require a higher dielectric constant to be assigned to the protein in order to compensate for the lack of polarization treatment in typical MMPBSA calculations. To test the effect of the protein dielectric constant upon binding prediction efficacy, a series of calculations was performed with protein dielectric constants of 1, 2, 4, 6, 8, 12, 16, 20, and 24 while all other parameters were held at their optimal values.
FIGURE 4 and TABLE 2 show that a general improvement in the agreement with experiment can be obtained by increasing the protein/solute dielectric constant to about epsin=20. This is indicated by the lowering of the RMSD and p-values and an increase in the value of the correlation coefficient in comparison to these same metrics when measured at nearby epsin values. It is noted that raising the protein dielectric constant has a particularly profound effect on the agreement with heavily charged ligands such as 2MeSATP and 2MeSADP in our system as often observed when modeling binding reactions involving charged ligands/active sites in globular proteins6, 8, 145, 146. The agreement is also expected given that the binding pocket of P2Y12R is still in the water-soluble region of the protein. However, it is worth noting that this finding may not hold for membrane proteins with membrane-accessible binding pockets. High quality structure and affinity data would be necessary to establish a standard practice for such cases.
Figure 4.
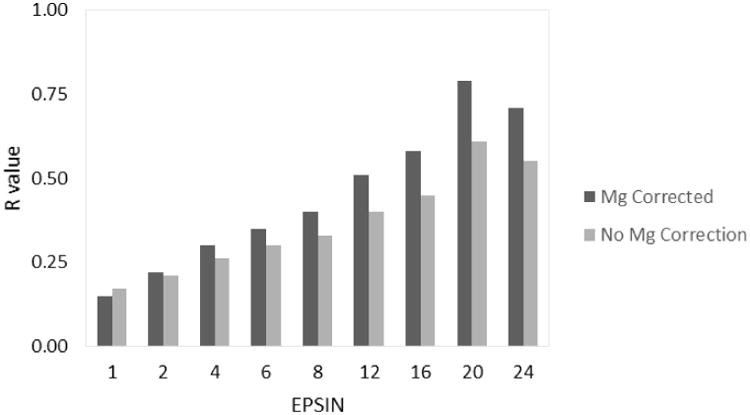
Parameter optimization for the protein dielectric constant, epsin (inp=2, epsmem=4). The R value indicates the correlation for the data set at the given epsin value.
Table 2.
The effect of the protein dielectric constant on absolute (ΔG) and relative (ΔΔG) binding affinities. See TABLE 1 for additional information. RMSD values are given in kcal/mol. The raw data used to generate these results is available in Supporting Information (Table S2).
| MG Corrected | No MG Correction | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EPSIN | RMSD | RMSD(ΔΔG) | Slope | p-value | R | RMSD | RMSD(ΔΔG) | Slope | p-value | R |
| 1 | 87 | 110 | 25 | 0.67 | 0.15 | 120 | 160 | 39 | 0.64 | 0.17 |
| 2 | 53 | 60 | 20 | 0.53 | 0.22 | 78 | 89 | 27 | 0.56 | 0.21 |
| 4 | 37 | 34 | 15 | 0.41 | 0.30 | 50 | 50 | 19 | 0.47 | 0.26 |
| 6 | 31 | 24 | 13 | 0.32 | 0.35 | 40 | 36 | 16 | 0.40 | 0.30 |
| 8 | 27 | 19 | 11 | 0.25 | 0.40 | 35 | 28 | 14 | 0.35 | 0.33 |
| 12 | 23 | 12 | 9.6 | 0.13 | 0.51 | 29 | 19 | 11 | 0.25 | 0.40 |
| 16 | 22 | 10 | 8.5 | 7.9E-02 | 0.58 | 26 | 15 | 10 | 0.19 | 0.45 |
| 20 | 19 | 6 | 8.1 | 6.7E-03 | 0.79 | 23 | 10 | 9.6 | 6.1E-02 | 0.61 |
| 24 | 20 | 7 | 7.4 | 2.1E-02 | 0.71 | 23 | 11 | 8.7 | 0.10 | 0.55 |
Impact of the membrane dielectric constant
Currently, the implicit membrane model implemented in PBSA allows for only a single membrane region. While this may be extended with relative ease in the future, this study focuses on the single membrane dielectric constant protocol. This protocol follows the classical solvent excluded surface definition of molecular surface, in analogy to globular proteins. In doing so, it was reasonable to examine membrane dielectric constants of 1, 2, 4, and 7 with all other parameters set to their optimal values.
From examination of FIGURE 5 and TABLE 3, it is evident that the accuracy of the binding affinity calculations is less sensitive to changes in the membrane dielectric constant in comparison to the differences observed when changing the non-polar solvation model and the protein dielectric constant. Although there is a detectable improvement in the RMSD, p-value, and the correlation coefficient for epsmem=4, other epsmem values are comparable to each other. Again, the finding here may be attributed to the water-accessible nature of the binding pocket in P2Y12R. It is expected that membrane-accessible binding may behave quite sensitively to the membrane dielectric constant.
Figure 5.
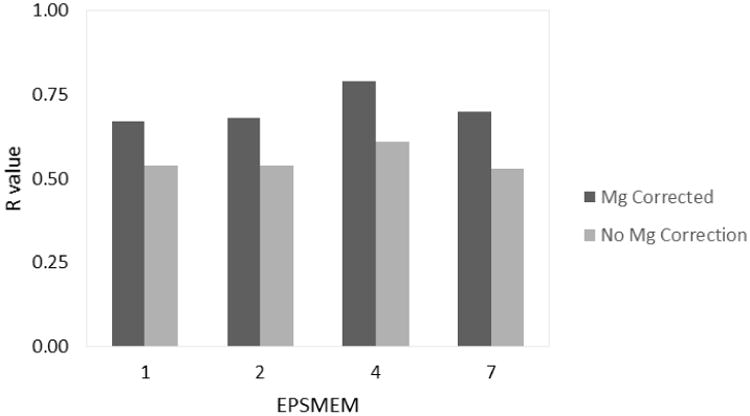
Parameter optimization for the membrane dielectric constant, epsmem (inp=2, epsin=20). The R value indicates the correlation for the data set at the given epsmem value.
Table 3.
The effect of the membrane dielectric constant on absolute (ΔG) and relative (ΔΔG) binding affinities. See TABLE 1 for additional information. The raw data used to generate these results is available in Supporting Information (Table S1).
| MG Corrected | No MG Correction | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EPSMEM | RMSD | RMSD(ΔΔG) | Slope | p-value | R | RMSD | RMSD(ΔΔG) | Slope | p-value | R |
| 1 | 23 | 10 | 11 | 3.4E-02 | 0.67 | 27 | 15 | 12 | 0.10 | 0.54 |
| 2 | 22 | 9 | 9.5 | 3.1E-02 | 0.68 | 26 | 14 | 11 | 0.11 | 0.54 |
| 4 | 19 | 6 | 8.1 | 6.7E-03 | 0.79 | 23 | 10 | 9.6 | 6.1E-02 | 0.61 |
| 7 | 20 | 7 | 7.6 | 2.4E-02 | 0.70 | 24 | 11 | 9.1 | 0.12 | 0.53 |
Effect of the magnesium correction
Finally, we address the effect of modeling explicit binding of the two nucleotide agonists to the magnesium ion. FIGURES 3-5 and TABLES 1-3 show that modeling the removal of a magnesium ion, as described in the Methods section, leads to much higher R values, lower p values, and lower RMSD values compared to the uncorrected data across the board. The effect of the magnesium correction can be seen most clearly in FIGURES 6 & 7 as the data points for 2MeSATP and 2MeSADP rise in free energy to improve the agreement with the linear trend established by the other less charged or neutral ligands. FIGURE 6 compares the absolute binding free energies (ΔG) between our calculated MMPBSA values and the experimental results while FIGURE 7 provides an analogous comparison using the relative binding free energies (ΔΔG). The large systematic bias inherent in MMPBSA binding affinity calculations manifests itself as both a large slope and a large y-intercept in FIGURE 6. Using the relative free energy values in FIGURE 7 removes the large y-intercept while keeping the slope and overall correlation the same. It is clear that the rather large error in the uncorrected set is due to the binding data obtained from 2MeSATP and 2MeSADP. When the correction is applied, the agreement improves specifically because it counteracts the extremely favorable electrostatic interactions with these two ligands in the standard MMPBSA calculation. Note that this is so even when a very high apparent protein dielectric constant is used.
Figure 6.
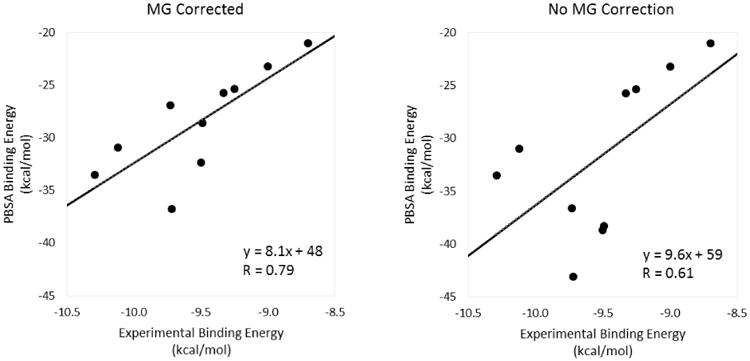
Absolute binding free energy (ΔG) correlation plots (inp=2, epsin=20, and epsmem=4). The plot that was corrected for the removal of the magnesium ion appears on the left while the plot that did not take into account the magnesium correction appears on the right.
Figure 7.
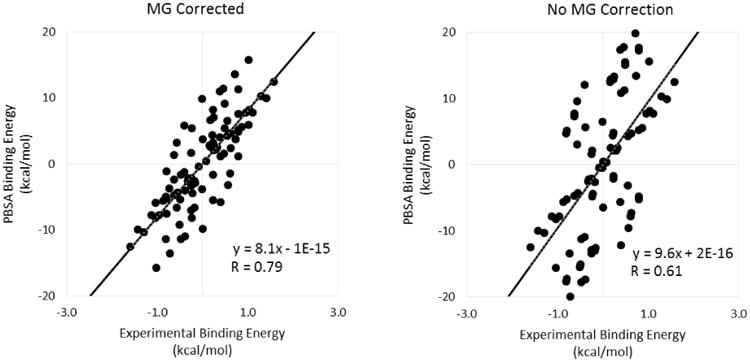
Relative binding free energy (ΔΔG) correlation plots (inp=2, epsin=20, and epsmem=4). The plot that was corrected for the removal of the magnesium ion appears on the left while the plot that did not take into account the magnesium correction appears on the right.
Our hypothesis that the bound magnesium ion is lost upon 2MeSATP and 2MeSADP binding to P2Y12R is based on inspection of the complex crystal structures, and the fact that the binding assay was conducted in the presence of 10mM MgCl2122. This is supported by our computational modeling of the binding reactions, which utilizes standard setups of the widely used MMPBSA method. It should be noted that our binding affinity modeling was conducted without the normal mode entropy analysis. This analysis usually does not contribute favorably to the overall agreement with experiment due to the approximation used6, 8, 145. Additionally, it is possible that binding-induced conformational changes cannot be fully taken into account by the widely used single-trajectory approach. The more extensive multi-trajectory approach, or more high-end free energy simulation methods with enhanced sampling, will be explored in the future.
Nevertheless, additional structural analysis shows that P2Y12R is indeed an interesting case. FIGURE 8 compares the electrostatic potential at the binding site for P2Y12R bound to 2MeSATP123 (FIGURE 8A, PDB ID: 4PY0) with two other proteins which have a magnesium ion bound to ATP in their binding sites: G protein-coupled receptor kinase 1, GRK1147 (FIGURE 8B, PDB ID: 3C4X), and Flagella-related protein H, FlaH148 (FIGURE 8C, PDB ID: 4YDS). It can be seen in FIGURES 8B and 8C that a negatively charged region (red) is present in both GRK1 and FlaH to stabilize the magnesium ion in the binding site. However, no comparable region appears that would stabilize a magnesium ion in P2Y12R as FIGURE 8A shows that the ligand binding site for P2Y12R is electropositive (blue). The electropositive nature of the P2Y12R active site might explain why the P2Y12R crystal structures, with bound 2MeSATP or 2MeSADP, were obtained without a magnesium ion to begin with.
Figure 8.
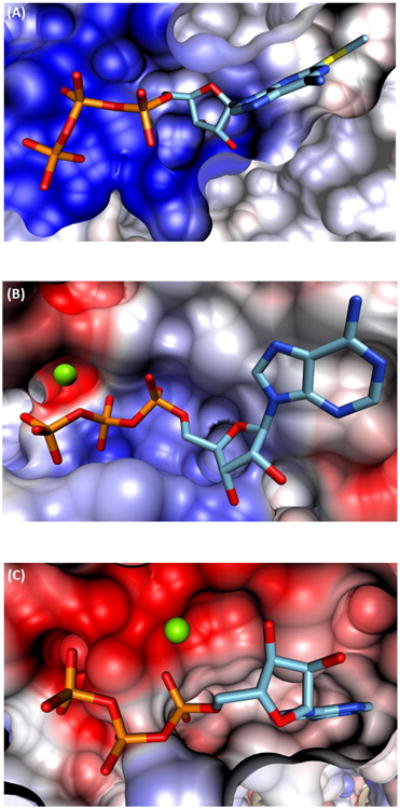
Cross-sectional comparison of the ATP binding site for A) P2Y12R, B) GRK1, and C) FlaH. Blue and red coloring reflects a net positive or negative charge, carbon bonds are shown in cyan, phosphorus bonds are depicted in orange, and the magnesium ions are shown in green.
On the other hand, it is also reasonable to question whether the binding poses observed in the crystal structures are the active forms in the binding assay. Indeed, the authors of the crystal structure study speculated that their P2Y12R-2MeSADP structure might be in an “agonist-bound inactive state” based on the positions of certain helices in comparison with other structures.123 However, the binding poses observed in the crystal structure active sites do make physical sense in that the electrostatic interactions between the highly negatively charged ligands and the many positively charged side chains in the P2Y12R active site are favorable for binding. Absence of additional reactants or additional experimental measurements (i.e. NMR), it is difficult to justify a mechanism that contradicts the binding poses observed in the existing high quality structural data.
Concluding Remarks and Future Directions
In this study, the widely used Amber/MMPBSA procedure was extended to model membrane protein-ligand binding affinities; in particular, it was used to model the binding of several ligands to the human purinergic platelet receptor, P2Y12R. A good agreement with experimental binding affinities was observed. A detailed investigation of simulation parameters was conducted to assess their impact on the accuracy of the MMPBSA results. The reported optimization procedure also illustrated the various details that should be considered while applying the MMPBSA method in studies of other membrane protein systems.
Testing of the non-polar solvent model indicates that the modern dispersion/cavity method (inp2), which separately models dispersion and hydrophobic interactions, yields improved agreement with experiment all around as compared with the classical, but simpler, linear response model (inp1). These results suggest that the modern approach is preferred when performing binding free energy calculations for membrane protein-ligand systems, or at least it is preferred for ligand binding cavities that are fully exposed to water, an observation that is consistent with studies of globular proteins8.
An investigation of protein dielectric constants led to an optimized value of 20. This is due to the highly charged and partially exposed nature of the active site of P2Y12R, and this is also consistent with many previous studies of globular proteins6, 8, 145, 146. The introduction of a higher protein/solute dielectric constant is a reasonable, but crude, treatment of the screening effect of electrostatic interactions due to polarization of electronic, orientational, and solvent-exchange origins. The screening effect reduces otherwise very favorable electrostatic interactions, rendering these interactions comparable to hydrophobic interactions in most biochemical systems.
A similar investigation of membrane dielectric constants led to an optimized value of 4. This is within the 1-4 range that is indicative of highly hydrophobic regions of membranes, and it agrees well with other values reported in the literature30, 117, 119, 149. It is worth noting that an epsmem value of 1, 2, or even 7 can be used without sacrificing much accuracy as the effect of changing the membrane dielectric constant is more subtle in comparison to changes in the protein dielectric constant. This result seems reasonable for our system. An active site exposed on the protein surface should be somewhat separated from the hydrophobic, non-polar region in the surrounding lipid bilayer, and therefore, the effect of the membrane dielectric constant on the binding free energy should be more modest. Finally, it is worth noting that the magnesium correction had no influence on the trends observed in the analysis of simulation parameters. If the uncorrected data is analyzed on its own in FIGURES 3-5 and TABLES 1-3, we see the same general trends that we see when we analyze the corrected data on its own.
We hypothesized that the bound magnesium ion is lost when 2MeSATP or 2MeSADP bind to P2Y12R. This was based on inspection of the complex crystal structures, and the fact that the binding assay was conducted in the presence of 10mM MgCl2122. This is supported by our modeling study of the binding reactions, which uses standard setups from a widely employed method. Additional structural analysis shows that P2Y12R is indeed an interesting case. The electropositive nature of its active site might explain why the P2Y12R complex structures were obtained without magnesium ions. Nevertheless, the approach we have taken above is just one possible way to resolve the anomaly in the 2MeSATP and 2MeSADP binding affinities. Another possibility is that the bound magnesium ion may remain in the P2Y12R binding site upon 2MeSATP and 2MeSADP binding. This is akin to reducing the net charges of the two highly charged ligands. However, in this situation the remaining charges of the ligands are still very high, and the electrostatic interactions remain highly favorable, even when a high apparent protein dielectric constant is used, leading to poor overall agreement with experiment (data not shown). Of course, this possibility also contradicts the magnesium-free binding of 2MeSATP and 2MeSADP observed in the P2Y12R crystal structures.
Supplementary Material
Acknowledgments
This work is supported in part by the NIH (GM093040 & GM079383).
Footnotes
Supporting Information: Two tables of raw data that were used to produce Tables 1-3 and Figures 6 and 7, and a plot showing the convergence of ΔG values over the 10 ns of the production run
References
- 1.Case DA, Betz RM, Botello-Smith W, Cerutti DS, T E Cheatham I, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, et al. Amber 16. University of California, San Francisco; San Francisco, CA: 2016. [Google Scholar]
- 2.Case DA, Betz RM, Botello-Smith W, Cerutti DS, T E Cheatham I, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, et al. AmberTools 16. University of California, San Francisco; San Francsico, CA: 2016. [Google Scholar]
- 3.Srinivasan J, Cheatham TE, Cieplak P, Kollman PA, Case DA. Continuum Solvent Studies of the Stability of DNA, RNA, and Phosphoramidate - DNA Helices. J Am Chem Soc. 1998;120:9401–9409. [Google Scholar]
- 4.Kollman PA, Massova I, Reyes C, Kuhn B, Huo SH, Chong L, Lee M, Lee T, Duan Y, Wang W, et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 5.Gohlke H, Case DA. Converging Free Energy Estimates: MM-PB(GB)SA Studies on the Protein-Protein Complex Ras-Raf. J Comput Chem. 2004;25:238–250. doi: 10.1002/jcc.10379. [DOI] [PubMed] [Google Scholar]
- 6.Yang TY, Wu JC, Yan CL, Wang YF, Luo R, Gonzales MB, Dalby KN, Ren PY. Virtual Screening Using Molecular Simulations. Proteins: Struct, Funct, Bioinf. 2011;79:1940–1951. doi: 10.1002/prot.23018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J Chem Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 8.Wang C, Nguyen PH, Pham K, Huynh D, Le TBN, Wang H, Ren P, Luo R. Calculating Protein–Ligand Binding Affinities With MMPBSA: Method and Error Analysis. J Comput Chem. 2016;37:2436–2446. doi: 10.1002/jcc.24467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perutz MF. Electrostatic Effects in Proteins. Science. 1978;201:1187–1191. doi: 10.1126/science.694508. [DOI] [PubMed] [Google Scholar]
- 10.Davis ME, Mccammon JA. Electrostatics in Biomolecular Structure and Dynamics. Chem Rev. 1990;90:509–521. [Google Scholar]
- 11.Honig B, Nicholls A. Classical Electrostatics in Biology and Chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 12.Honig B, Sharp K, Yang AS. Macroscopic Models Of Aqueous-Solutions - Biological And Chemical Applications. J Phys Chem. 1993;97:1101–1109. [Google Scholar]
- 13.Beglov D, Roux B. Solvation of Complex Molecules in a Polar Liquid: An Integral Equation Theory. J Chem Phys. 1996;104:8678–8689. [Google Scholar]
- 14.Cramer CJ, Truhlar DG. Implicit Solvation Models: Equilibria, Structure, Spectra, and Dynamics. Chem Rev. 1999;99:2161–2200. doi: 10.1021/cr960149m. [DOI] [PubMed] [Google Scholar]
- 15.Bashford D, Case DA. Generalized Born Models of Macromolecular Solvation Effects. Annu Rev Phys Chem. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 16.Baker NA. Improving Implicit Solvent Simulations: A Poisson-Centric View. Curr Opin Struct Biol. 2005;15:137–143. doi: 10.1016/j.sbi.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 17.Chen JH, Im WP, Brooks CL. Balancing Solvation and Intramolecular Interactions: Toward a Consistent Generalized Born Force Field. J Am Chem Soc. 2006;128:3728–3736. doi: 10.1021/ja057216r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feig M, Chocholousova J, Tanizaki S. Extending the Horizon: Towards the Efficient Modeling of Large Biomolecular Complexes in Atomic Detail. Theor Chem Acc. 2006;116:194–205. [Google Scholar]
- 19.Koehl P. Electrostatics Calculations: Latest Methodological Advances. Curr Opin Struct Biol. 2006;16:142–151. doi: 10.1016/j.sbi.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Im W, Chen JH, Brooks CL. In: Advances in Protein Chemistry. Baldwin R, Baker DJ, editors. Vol. 72. Academic Press; 2006. pp. 173–198. [DOI] [PubMed] [Google Scholar]
- 21.Lu BZ, Zhou YC, Holst MJ, McCammon JA. Recent Progress in Numerical Methods for the Poisson-Boltzmann Equation in Biophysical Applications. Commun Comput Phys. 2008;3:973–1009. [Google Scholar]
- 22.Wang J, Tan CH, Tan YH, Lu Q, Luo R. Poisson-Boltzmann Solvents in Molecular Dynamics Simulations. Commun Comput Phys. 2008;3:1010–1031. [Google Scholar]
- 23.Altman MD, Bardhan JP, White JK, Tidor B. Accurate Solution of Multi-Region Continuum Biomolecule Electrostatic Problems Using the Linearized Poisson-Boltzmann Equation with Curved Boundary Elements. J Comput Chem. 2009;30:132–153. doi: 10.1002/jcc.21027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai Q, Wang J, Hsieh MJ, Ye X, Luo R. In: Annual Reports in Computational Chemistry. Ralph AW, editor. Vol. 8. Elsevier; 2012. pp. 149–162. [Google Scholar]
- 25.Xiao L, Wang C, Luo R. Recent Progress in Adapting Poisson–Boltzmann Methods to Molecular Simulations. J Theor Comput Chem. 2014;13:1430001. [Google Scholar]
- 26.Botello-Smith WM, Cai Q, Luo R. Biological Applications of Classical Electrostatics Methods. J Theor Comput Chem. 2014;13:1440008. [Google Scholar]
- 27.Forsten KE, Kozack RE, Lauffenburger DA, Subramaniam S. Numerical-Solution of the Nonlinear Poisson-Boltzmann Equation for a Membrane-Electrolyte System. J Phys Chem. 1994;98:5580–5586. [Google Scholar]
- 28.Spassov VZ, Yan L, Szalma S. Introducing an Implicit Membrane in Generalized Born/Solvent Accessibility Continuum Solvent Models. J Phys Chem B. 2002;106:8726–8738. [Google Scholar]
- 29.Im W, Feigh M, Brooks CL., III An Implicit Membrane Generalized Born Theory for the Study of Structure, Stability, and Interactions of Membrane Proteins. Biophys J. 2003;85:2900–2918. doi: 10.1016/S0006-3495(03)74712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanizaki S, Feig M. A Generalized Born Formalism for Heterogeneous Dielectric Environments: Application to the Implicit Modeling of Biological Membranes. J Chem Phys. 2005;122:124706. doi: 10.1063/1.1865992. [DOI] [PubMed] [Google Scholar]
- 31.Tanizaki S, Feig M. Molecular Dynamics Simulations of Large Integral Membrane Proteins with an Implicit Membrane Model. J Phys Chem B. 2006;110:548–556. doi: 10.1021/jp054694f. [DOI] [PubMed] [Google Scholar]
- 32.Callenberg KM, Choudhary OP, de Forest GL, Gohara DW, Baker NA, Grabe M. APBSmem: A Graphical Interface for Electrostatic Calculations at the Membrane. PloS One. 2010;5 doi: 10.1371/journal.pone.0012722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Li C, Sarkar S, Zhang J, Witham S, Zhang Z, Wang L, Smith N, Petukh M, Alexov E. DelPhi: A Comprehensive Suite for DelPhi Software and Associated Resources. BMC Biophysics. 2012;5:9. doi: 10.1186/2046-1682-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schafer NP, Truong HH, Otzen DE, Lindorff-Larsen K, Wolynes PG. Topological Constraints and Modular Structure in the Folding and Functional Motions of GlpG, An Intramembrane Protease. Proc Natl Acad Sci U S A. 2016;113:2098–2103. doi: 10.1073/pnas.1524027113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marassi FM, Ding Y, Schwieters CD, Tian Y, Yao Y. Backbone Structure of Yersinia Pestis Ail Determined in Micelles by NMR-Restrained Simulated Annealing with Implicit Membrane Solvation. J Biomol NMR. 2015;63:59–65. doi: 10.1007/s10858-015-9963-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bajda M, Filipek S. Study of Early Stages of Amyloid Aβ13-23 Formation Using Molecular Dynamics Simulation in Implicit Environments. Comput Biol Chem. 2015;56:13–18. doi: 10.1016/j.compbiolchem.2015.02.014. [DOI] [PubMed] [Google Scholar]
- 37.Li C, Li L, Zhang J, Alexov E. Highly Efficient and Exact Method for Parallelization of Grid - Based Algorithms and Its Implementation in DelPhi. J Comput Chem. 2012;33:1960–1966. doi: 10.1002/jcc.23033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang J, Cai Q, Li ZL, Zhao HK, Luo R. Achieving Energy Conservation in Poisson-Boltzmann Molecular Dynamics: Accuracy and Precision with Finite-Difference Algorithms. Chem Phys Lett. 2009;468:112–118. doi: 10.1016/j.cplett.2008.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Luo R. Assessment of Linear Finite-Difference Poisson-Boltzmann Solvers. J Comput Chem. 2010;31:1689–1698. doi: 10.1002/jcc.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cai Q, Hsieh MJ, Wang J, Luo R. Performance of Nonlinear Finite-Difference Poisson-Boltzmann Solvers. J Chem Theory Comput. 2010;6:203–211. doi: 10.1021/ct900381r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Cai Q, Xiang Y, Luo R. Reducing Grid Dependence in Finite-Difference Poisson-Boltzmann Calculations. J Chem Theory Comput. 2012;8:2741–2751. doi: 10.1021/ct300341d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Botello-Smith WM, Luo R. Applications of MMPBSA to Membrane Proteins I: Efficient Numerical Solutions of Periodic Poisson-Boltzmann Equation. J Chem Inf Model. 2015;55:2187–2199. doi: 10.1021/acs.jcim.5b00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warwicker J, Watson HC. Calculation of the Electric-Potential in the Active-Site Cleft Due to Alpha-Helix Dipoles. J Mol Biol. 1982;157:671–679. doi: 10.1016/0022-2836(82)90505-8. [DOI] [PubMed] [Google Scholar]
- 44.Bashford D, Karplus M. Pkas Of Ionizable Groups In Proteins - Atomic Detail From A Continuum Electrostatic Model. Biochemistry. 1990;29:10219–10225. doi: 10.1021/bi00496a010. [DOI] [PubMed] [Google Scholar]
- 45.Jeancharles A, Nicholls A, Sharp K, Honig B, Tempczyk A, Hendrickson TF, Still WC. Electrostatic Contributions To Solvation Energies - Comparison Of Free-Energy Perturbation And Continuum Calculations. J Am Chem Soc. 1991;113:1454–1455. [Google Scholar]
- 46.Gilson MK. Theory Of Electrostatic Interactions In Macromolecules. Curr Opin Struct Biol. 1995;5:216–223. doi: 10.1016/0959-440x(95)80079-4. [DOI] [PubMed] [Google Scholar]
- 47.Edinger SR, Cortis C, Shenkin PS, Friesner RA. Solvation Free Energies of Peptides: Comparison of Approximate Continuum Solvation Models with Accurate Solution of the Poisson-Boltzmann Equation. J Phys Chem B. 1997;101:1190–1197. [Google Scholar]
- 48.Luo R, David L, Gilson MK. Accelerated Poisson-Boltzmann Calculations for Static and Dynamic Systems. J Comput Chem. 2002;23:1244–1253. doi: 10.1002/jcc.10120. [DOI] [PubMed] [Google Scholar]
- 49.Lu Q, Luo R. A Poisson-Boltzmann Dynamics Method with Nonperiodic Boundary Condition. J Chem Phys. 2003;119:11035–11047. [Google Scholar]
- 50.Tan C, Yang L, Luo R. How Well Does Poisson-Boltzmann Implicit Solvent Agree With Explicit Solvent? A Quantitative Analysis. J Phys Chem B. 2006;110:18680–18687. doi: 10.1021/jp063479b. [DOI] [PubMed] [Google Scholar]
- 51.Cai Q, Wang J, Zhao HK, Luo R. On Removal of Charge Singularity in Poisson-Boltzmann Equation. J Chem Phys. 2009;130:145101. doi: 10.1063/1.3099708. [DOI] [PubMed] [Google Scholar]
- 52.Ye X, Cai Q, Yang W, Luo R. Roles of Boundary Conditions in DNA Simulations: Analysis of Ion Distributions with the Finite-Difference Poisson-Boltzmann Method. Biophys J. 2009;97:554–562. doi: 10.1016/j.bpj.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ye X, Wang J, Luo R. A Revised Density Function for Molecular Surface Calculation in Continuum Solvent Models. J Chem Theory Comput. 2010;6:1157–1169. doi: 10.1021/ct900318u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luo R, Moult J, Gilson MK. Dielectric Screening Treatment of Electrostatic Solvation. J Phys Chem B. 1997;101:11226–11236. [Google Scholar]
- 55.Wang J, Tan C, Chanco E, Luo R. Quantitative Analysis of Poisson-Boltzmann Implicit Solvent in Molecular Dynamics. Phys Chem Chem Phys. 2010;12:1194–1202. doi: 10.1039/b917775b. [DOI] [PubMed] [Google Scholar]
- 56.Hsieh MJ, Luo R. Exploring a Coarse-Grained Distributive Strategy for Finite-Difference Poisson-Boltzmann Calculations. J Mol Model. 2011;17:1985–1996. doi: 10.1007/s00894-010-0904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cai Q, Ye X, Wang J, Luo R. On-the-Fly Numerical Surface Integration for Finite-Difference Poisson-Boltzmann Methods. J Chem Theory Comput. 2011;7:3608–3619. doi: 10.1021/ct200389p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Botello-Smith WM, Liu X, Cai Q, Li Z, Zhao H, Luo R. Numerical Poisson-Boltzmann Model for Continuum Membrane Systems. Chem Phys Lett. 2013;555:274–281. doi: 10.1016/j.cplett.2012.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu X, Wang C, Wang J, Li Z, Zhao H, Luo R. Exploring a Charge-Central Strategy in the Solution of Poisson's Equation for Biomolecular Applications. Phys Chem Chem Phys. 2013;15:129–141. doi: 10.1039/c2cp41894k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang C, Wang J, Cai Q, Li ZL, Zhao H, Luo R. Exploring High Accuracy Poisson-Boltzmann Methods for Biomolecular Simulations. Comput Theor Chem. 2013;1024:34–44. doi: 10.1016/j.comptc.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luo R, Head MS, Moult J, Gilson MK. pK(a) Shifts in Small Molecules and HIV Protease: Electrostatics and Conformation. J Am Chem Soc. 1998;120:6138–6146. [Google Scholar]
- 62.Georgescu RE, Alexov EG, Gunner MR. Combining Conformational Flexibility and Continuum Electrostatics for Calculating pK(a)s in Proteins. Biophys J. 2002;83:1731–1748. doi: 10.1016/S0006-3495(02)73940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nielsen JE, McCammon JA. On the Evaluation and Optimization of Protein X-ray Structures for pKa Calculations. Protein Sci. 2003;12:313–326. doi: 10.1110/ps.0229903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Warwicker J. Improved pK(a) Calculations Through Flexibility Based Sampling of a Water-Dominated Interaction Scheme. Protein Sci. 2004;13:2793–2805. doi: 10.1110/ps.04785604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tang CL, Alexov E, Pyle AM, Honig B. Calculation of pK(a)s in RNA: On the Structural Origins and Functional Roles of Protonated Nucleotides. J Mol Biol. 2007;366:1475–1496. doi: 10.1016/j.jmb.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 66.Shivakumar D, Deng YQ, Roux B. Computations of Absolute Solvation Free Energies of Small Molecules Using Explicit and Implicit Solvent Model. J Chem Theory Comput. 2009;5:919–930. doi: 10.1021/ct800445x. [DOI] [PubMed] [Google Scholar]
- 67.Nicholls A, Mobley DL, Guthrie JP, Chodera JD, Bayly CI, Cooper MD, Pande VS. Predicting Small-Molecule Solvation Free Energies: An Informal Blind Test for Computational Chemistry. J Med Chem. 2008;51:769–779. doi: 10.1021/jm070549+. [DOI] [PubMed] [Google Scholar]
- 68.Swanson JMJ, Henchman RH, McCammon JA. Revisiting Free Energy Calculations: A Theoretical Connection to MM/PBSA and Direct Calculation of the Association Free Energy. Biophys J. 2004;86:67–74. doi: 10.1016/S0006-3495(04)74084-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bertonati C, Honig B, Alexov E. Poisson-Boltzmann Calculations of Nonspecific Salt Effects on Protein-Protein Binding Free Energies. Biophys J. 2007;92:1891–1899. doi: 10.1529/biophysj.106.092122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brice AR, Dominy BN. Analyzing the Robustness of the MM/PBSA Free Energy Calculation Method: Application to DNA Conformational Transitions. J Comput Chem. 2011;32:1431–1440. doi: 10.1002/jcc.21727. [DOI] [PubMed] [Google Scholar]
- 71.Luo R, Gilson HSR, Potter MJ, Gilson MK. The Physical Basis of Nucleic Acid Base Stacking in Water. Biophys J. 2001;80:140–148. doi: 10.1016/S0006-3495(01)76001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.David L, Luo R, Head MS, Gilson MK. Computational Study of KNI-272, a Potent Inhibitor of HIV-1 Protease: On the Mechanism of Preorganization. J Phys Chem B. 1999;103:1031–1044. [Google Scholar]
- 73.Luo R, Gilson MK. Synthetic Adenine Receptors: Direct Calculation of Binding Affinity and Entropy. J Am Chem Soc. 2000;122:2934–2937. [Google Scholar]
- 74.Marshall SA, Vizcarra CL, Mayo SL. One- and Two-Body Decomposable Poisson-Boltzmann Methods for Protein Design Calculations. Protein Sci. 2005;14:1293–1304. doi: 10.1110/ps.041259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hsieh MJ, Luo R. Physical Scoring Function Based on AMBER Force Field and Poisson-Boltzmann Implicit Solvent for Protein Structure Prediction. Proteins: Struct, Funct, Bioinf. 2004;56(3):475–486. doi: 10.1002/prot.20133. [DOI] [PubMed] [Google Scholar]
- 76.Wen EZ, Luo R. Interplay of Secondary Structures and Side-Chain Contacts in the Denatured State of BBA1. J of Chem Phys. 2004;121:2412–2421. doi: 10.1063/1.1768151. [DOI] [PubMed] [Google Scholar]
- 77.Wen EZ, Hsieh MJ, Kollman PA, Luo R. Enhanced Ab Initio Protein Folding Simulations in Poisson-Boltzmann Molecular Dynamics with Self-Guiding Forces. J Mol Graphics Modell. 2004;22:415–424. doi: 10.1016/j.jmgm.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 78.Lwin TZ, Luo R. Overcoming Entropic Barrier with Coupled Sampling at Dual Resolutions. J Chem Phys. 2005;123:194904. doi: 10.1063/1.2102871. [DOI] [PubMed] [Google Scholar]
- 79.Lwin TZ, Zhou RH, Luo R. Is Poisson-Boltzmann Theory Insufficient for Protein Folding Simulations? J Chem Phys. 2006;124:034906. doi: 10.1063/1.2161202. [DOI] [PubMed] [Google Scholar]
- 80.Lwin TZ, Luo R. Force Field Influences in Beta-Hairpin Folding Simulations. Protein Science. 2006;15:2642–2655. doi: 10.1110/ps.062438006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tan YH, Luo R. Protein Stability Prediction: A Poisson-Boltzmann Approach. J Phys Chem B. 2008;112:1875–1883. doi: 10.1021/jp709660v. [DOI] [PubMed] [Google Scholar]
- 82.Tan Y, Luo R. Structural and Functional Implications of p53 Missense Cancer Mutations. BMC Biophysics. 2009;2:5. doi: 10.1186/1757-5036-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Korman TP, Tan YH, Wong J, Luo R, Tsai SC. Inhibition Kinetics and Emodin Cocrystal Structure of a Type II Polyketide Ketoreductase. Biochemistry. 2008;47:1837–1847. doi: 10.1021/bi7016427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miertus S, Scrocco E, Tomasi J. Electrostatic Interaction of a Solute with a Continuum - a Direct Utilization of Abinitio Molecular Potentials for the Prevision of Solvent Effects. Chem Phys. 1981;55:117–129. [Google Scholar]
- 85.Hoshi H, Sakurai M, Inoue Y, Chujo R. Medium Effects on the Molecular Electronic-Structure 1. the Formulation of a Theory for the Estimation of a Molecular Electronic-Structure Surrounded by an Anisotropic Medium. J Chem Phys. 1987;87:1107–1115. [Google Scholar]
- 86.Zauhar RJ, Morgan RS. The Rigorous Computation of the Molecular Electric-Potential. J Comput Chem. 1988;9:171–187. [Google Scholar]
- 87.Rashin AA. Hydration Phenomena, Classical Electrostatics, and the Boundary Element Method. J Phys Chem. 1990;94:1725–1733. [Google Scholar]
- 88.Yoon BJ, Lenhoff AM. A Boundary Element Method for Molecular Electrostatics with Electrolyte Effects. J Comput Chem. 1990;11:1080–1086. [Google Scholar]
- 89.Juffer AH, Botta EFF, Vankeulen BAM, Vanderploeg A, Berendsen HJC. The Electric-Potential Of A Macromolecule In A Solvent - A Fundamental Approach. J Comput Phys. 1991;97:144–171. [Google Scholar]
- 90.Zhou HX. Boundary-Element Solution of Macromolecular Electorstatic-Interaction Energy Between 2 Proteins. Biophys J. 1993;65:955–963. doi: 10.1016/S0006-3495(93)81094-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bharadwaj R, Windemuth A, Sridharan S, Honig B, Nicholls A. The Fast Multipole Boundary-Element Method for Molecular Electrostatics - an Optimal Approach for Large Systems. J Comput Chem. 1995;16:898–913. [Google Scholar]
- 92.Purisima EO, Nilar SH. A Simple Yet Accurate Boundary-Element Method for Continuum Dielectric Calculations. J Comput Chem. 1995;16:681–689. [Google Scholar]
- 93.Liang J, Subramaniam S. Computation of Molecular Electrostatics with Boundary Element Methods. Biophys J. 1997;73:1830–1841. doi: 10.1016/S0006-3495(97)78213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vorobjev YN, Scheraga HA. A fast Adaptive Multigrid Boundary Element Method for Macromolecular Electrostatic Computations in a Solvent. J Comput Chem. 1997;18:569–583. [Google Scholar]
- 95.Totrov M, Abagyan R. Rapid Boundary Element Solvation Electrostatics Calculations in Folding Simulations: Successful Folding of a 23-Residue Peptide. Biopolymers. 2001;60:124–133. doi: 10.1002/1097-0282(2001)60:2<124::AID-BIP1008>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 96.Boschitsch AH, Fenley MO, Zhou HX. Fast Boundary Element Method for the Linear Poisson-Boltzmann Equation. J Phys Chem B. 2002;106:2741–2754. [Google Scholar]
- 97.Lu BZ, Cheng XL, Huang JF, McCammon JA. Order N algorithm for Computation of Electrostatic Interactions in Biomolecular Systems. Proc Natl Acad Sci U S A. 2006;103:19314–19319. doi: 10.1073/pnas.0605166103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lu B, Cheng X, Huang J, McCammon JA. An Adaptive Fast Multipole Boundary Element Method for Poisson-Boltzmann Electrostatics. J Chem Theory Comput. 2009;5:1692–1699. doi: 10.1021/ct900083k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bajaj C, Chen SC, Rand A. An Efficient Higher-Order Fast Multipole Boundary Element Solution For Poisson-Boltzmann-Based Molecular Electrostatics. SIAM J Sci Comput. 2011;33:826–848. doi: 10.1137/090764645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cortis CM, Friesner RA. Numerical Solution of the Poisson-Boltzmann Equation Using Tetrahedral Finite-Element Meshes. J Comput Chem. 1997;18:1591–1608. [Google Scholar]
- 101.Holst M, Baker N, Wang F. Adaptive Multilevel Finite Element Solution of the Poisson-Boltzmann Equation I. Algorithms and Examples. J Comput Chem. 2000;21:1319–1342. [Google Scholar]
- 102.Baker N, Holst M, Wang F. Adaptive Multilevel Finite Element Solution of the Poisson-Boltzmann Equation II. Refinement at Solvent-Accessible Surfaces in Biomolecular Systems. J Comput Chem. 2000;21:1343–1352. [Google Scholar]
- 103.Shestakov AI, Milovich JL, Noy A. Solution of the Nonlinear Poisson-Boltzmann Equation Using Pseudo-Transient Continuation and the Finite Element Method. J Colloid Interface Sci. 2002;247:62–79. doi: 10.1006/jcis.2001.8033. [DOI] [PubMed] [Google Scholar]
- 104.Chen L, Holst MJ, Xu JC. The Finite Element Approximation of the Nonlinear Poisson-Boltzmann Equation. SIAM J Numer Anal. 2007;45:2298–2320. [Google Scholar]
- 105.Xie D, Zhou S. A New Minimization Protocol for Solving Nonlinear Poisson–Boltzmann Mortar Finite Element Equation. BIT Numer Math. 2007;47:853–871. [Google Scholar]
- 106.Bond SD, Chaudhry JH, Cyr EC, Olson LN. A First-Order System Least-Squares Finite Element Method for the Poisson-Boltzmann Equation. J Comput Chem. 2010;31:1625–1635. doi: 10.1002/jcc.21446. [DOI] [PubMed] [Google Scholar]
- 107.Klapper I, Hagstrom R, Fine R, Sharp K, Honig B. Focusing of Electric Fields in the Active Site of Copper-Zinc Superoxide Dismutase Effects of Ionic Strength and Amino Acid Modification. Proteins: Struct, Funct, Genet. 1986;1:47–59. doi: 10.1002/prot.340010109. [DOI] [PubMed] [Google Scholar]
- 108.Davis ME, McCammon JA. Solving The Finite-Difference Linearized Poisson-Boltzmann Equation - A Comparison Of Relaxation And Conjugate-Gradient Methods. J Comp Chem. 1989;10:386–391. [Google Scholar]
- 109.Nicholls A, Honig B. A Rapid Finite-Difference Algorithm, Utilizing Successive Over-Relaxation To Solve The Poisson-Boltzmann Equation. J Comp Chem. 1991;12:435–445. [Google Scholar]
- 110.Luty BA, Davis ME, McCammon JA. Solving the Finite-Difference Nonlinear Poisson-Boltzmann Equation. J Comput Chem. 1992;13:1114–1118. [Google Scholar]
- 111.Holst M, Saied F. Multigrid Solution of the Poisson-Boltzmann Equation. J Comput Chem. 1993;14:105–113. [Google Scholar]
- 112.Holst MJ, Saied F. Numerical-Solution Of The Nonlinear Poisson-Boltzmann Equation - Developing More Robust And Efficient Methods. J Comput Chem. 1995;16:337–364. [Google Scholar]
- 113.Bashford D. An Object-Oriented Programming Suite for Electrostatic Effects in Biological Molecules. Lect Notes Comput Sci. 1997;1343:233–240. [Google Scholar]
- 114.Im W, Beglov D, Roux B. Continuum Solvation Model: Computation of Electrostatic Forces from Numerical Solutions to the Poisson-Boltzmann Equation. Comput Phys Commun. 1998;111:59–75. [Google Scholar]
- 115.Rocchia W, Alexov E, Honig B. Extending the Applicability of the Nonlinear Poisson-Boltzmann equation: Multiple Dielectric Constants and Multivalent Ions. J Phys Chem B. 2001;105:6507–6514. [Google Scholar]
- 116.Im W, Feig M, Brooks CL. An Implicit Membrane Generalized Born Theory for the Study of Structure, Stability, and Interactions of Membrane Proteins. Biophys J. 2003;85:2900–2918. doi: 10.1016/S0006-3495(03)74712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nymeyer H, Zhou HX. A Method to Determine Dielectric Constants in Nonhomogeneous Systems: Application to Biological Membranes. Biophys J. 2008;94:1185–1193. doi: 10.1529/biophysj.107.117770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tanizaki S, Feig M. A Generalized Born Formalism for Heterogeneous Dielectric Environments: Application to the Implicit Modeling of Biological Membranes. The Journal of chemical physics. 2005;122:124706. doi: 10.1063/1.1865992. [DOI] [PubMed] [Google Scholar]
- 119.Stern HA, Feller SE. Calculation of the Dielectric Permittivity Profile for a Nonuniform System: Application to a Lipid Bilayer Simulation. J Chem Phys. 2003;118:3401. [Google Scholar]
- 120.Ulmschneider MB, Ulmschneider JP, Sansom MS, Di Nola A. A Generalized Born Implicit-Membrane Representation Compared to Experimental Insertion Free Energies. Biophys J. 2007;92:2338–2349. doi: 10.1529/biophysj.106.081810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tan C, Tan YH, Luo R. Implicit Nonpolar Solvent Models. J Phys Chem B. 2007;111:12263–74. doi: 10.1021/jp073399n. [DOI] [PubMed] [Google Scholar]
- 122.Zhang K, Zhang J, Gao ZG, Zhang D, Zhu L, Han GW, Moss SM, Paoletta S, Kiselev E, Lu W. Structure of the Human P2Y12 Receptor in Complex with an Antithrombotic Drug. Nature. 2014;509:115–118. doi: 10.1038/nature13083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang J, Zhang K, Gao ZG, Paoletta S, Zhang D, Han GW, Li T, Ma L, Zhang W, Müller CE, et al. Agonist-Bound Structure of the Human P2Y12 Receptor. Nature. 2014;509:119–122. doi: 10.1038/nature13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Savi P, Zachayus JL, Delesque-Touchard N, Labouret C, Hervé C, Uzabiaga MF, Pereillo JM, Culouscou JM, Bono F, Ferrara P, et al. The Active Metabolite of Clopidogrel Disrupts P2Y12 Receptor Oligomers and Partitions Them Out of Lipid Rafts. Proc Natl Acad Sci U S A. 2006;103:11069–11074. doi: 10.1073/pnas.0510446103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Algaier I, Jakubowski JA, Asai F, Von Kügelgen I. Interaction of the Active Metabolite of Prasugrel, R-138727, with Cysteine 97 and Cysteine 175 of the Human P2Y12 Receptor. J Thromb Haemostasis. 2008;6:1908–1914. doi: 10.1111/j.1538-7836.2008.03136.x. [DOI] [PubMed] [Google Scholar]
- 126.Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinf. 2016;54:5.6.1–5.6.37. doi: 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Roberts E, Eargle J, Wright D, Luthey-Schulten Z. MultiSeq: Unifying Sequence and Structure Data for Evolutionary Analysis. BMC Bioinf. 2006;7:382. doi: 10.1186/1471-2105-7-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Humphrey W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. J Mol Graphics. 1996;14:33–8. 27–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 129.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber Biomolecular Simulation Programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Buchanan A, Newton P, Pehrsson S, Inghardt T, Antonsson T, Svensson P, Sjögren T, Öster L, Janefeldt A, Sandinge AS, et al. Structural and Functional Characterization of a Specific Antidote for Ticagrelor. Blood. 2015;125:3484–3490. doi: 10.1182/blood-2015-01-622928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.software for chemical visualization. ChemAxon; Cambridge, MA: 2016. MarvinSketch, version 16.3.2. [Google Scholar]
- 132.software toolkits for cheminformatics and modeling. OpenEye; Santa Fe, NM: 2016. OpenEye Toolkits. [Google Scholar]
- 133.Koes DR, Baumgartner MP, Camacho CJ. Lessons Learned in Empirical Scoring with Smina From the CSAR 2011 Benchmarking Exercise. J Chem Inf Model. 2013;53:1893–1904. doi: 10.1021/ci300604z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Meagher KL, Redman LT, Carlson HA. Development of Polyphosphate Parameters for Use with the AMBER Force Field. J Comput Chem. 2003;24:1016–1025. doi: 10.1002/jcc.10262. [DOI] [PubMed] [Google Scholar]
- 135.Wang J, Wang W, Kollman PA, Case DA. Antechamber, an Accessory Software Package for Molecular Mechanical Calculations. J Comput Chem. 2005;25:1157–1174. [Google Scholar]
- 136.Jo S, Kim T, Im W. Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. PLoS One. 2007;2:e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Biró E, Akkerman JW, Hoek FJ, Gorter G, Pronk LM, Sturk A, Nieuwland R. The Phospholipid Composition and Cholesterol Content of Platelet-Derived Microparticles: a Comparison with Platelet Membrane Fractions. J Thromb Haemostasis. 2005;3:2754–2763. doi: 10.1111/j.1538-7836.2005.01646.x. [DOI] [PubMed] [Google Scholar]
- 138.Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 139.Tan C, Tan YH, Luo R. Implicit Nonpolar Solvent Models. J Phys Chem B. 2007;111:12263–12274. doi: 10.1021/jp073399n. [DOI] [PubMed] [Google Scholar]
- 140.Liao JC, Sun S, Chandler D, Oster G. The Conformational States of Mg-ATP in Water. Eur Biophys J. 2004;33:29–37. doi: 10.1007/s00249-003-0339-2. [DOI] [PubMed] [Google Scholar]
- 141.Alberty RA. Standard Gibbs Free Energy, Enthalpy, and Entropy Changes as a Function of pH and pMg for Several Reactions Involving Adenosine Phosphates. J Biol Chem. 1969;244:3290–3302. [PubMed] [Google Scholar]
- 142.Gout E, Rébeillé F, Douce R, Bligny R. Interplay of Mg2+, ADP, and ATP in the Cytosol and Mitochondria: Unravelling the Role of Mg2+ in Cell Respiration. Proc Natl Acad Sci U S A. 2014;111:E4560–E4567. doi: 10.1073/pnas.1406251111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--A Visualization System for Exploratory Research and Analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 144.Eisenberg D, McLachlan AD. Solvation Energy in Protein Folding and Binding. Nature. 1986;319:199–203. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- 145.Hou T, Wang J, Li Y, Wang W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J Chem Inf Model. 2011;51:69–82. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Talley K, Ng C, Shoppell M, Kundrotas P, Alexov E. On the Electrostatic Component of Protein-Protein Binding Free Energy. PMC Biophys. 2008;1:1–23. doi: 10.1186/1757-5036-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Singh P, Wang B, Maeda T, Palczewski K, Tesmer JJ. Structures of Rhodopsin Kinase in Different Ligand States Reveal Key Elements Involved in G Protein-Coupled Receptor Kinase Activation. J Biol Chem. 2008;283:14053–14062. doi: 10.1074/jbc.M708974200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chaudhury P, Neiner T, D'Imprima E, Banerjee A, Reindl S, Ghosh A, Arvai AS, Mills DJ, van der Does C, Tainer JA, et al. The Nucleotide-Dependent Interaction of FlaH and FlaI is Essential for Assembly and Function of the Archaellum Motor. Mol Microbiol. 2016;99:674–685. doi: 10.1111/mmi.13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dias RP, Li L, Soares TA, Alexov E. Modeling the Electrostatic Potential of Asymmetric Lipopolysaccharide Membranes: the MEMPOT Algorithm Implemented in DelPhi. J Comput Chem. 2014;35:1418–1429. doi: 10.1002/jcc.23632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.