

Abstract
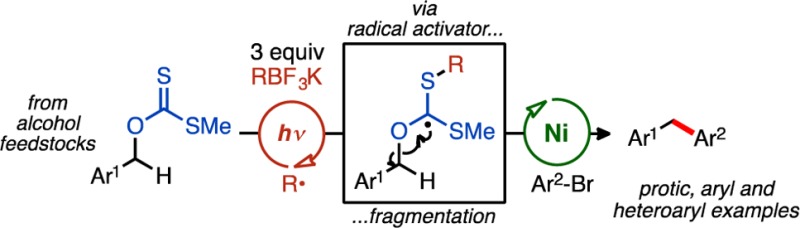
Alkyl xanthate esters are perhaps best known for their use in deoxygenation chemistry. However, their use in cross-coupling chemistry has not been productive, which is due, in part, to inadequate xanthate activation strategies. Herein, we report the use of O-benzyl xanthate esters, readily derived from alcohols, as radical pronucleophiles in Csp3–Csp2 cross-couplings under Ni/photoredox dual catalysis. Xanthate (C–O) cleavage is found to be reliant on photogenerated (sec-butyl) radical activators to form new carbon-centered radicals primed for nickel-catalyzed cross-couplings. Mechanistic experiments support the fact that the key radical components are formed independently, and relative rates are carefully orchestrated, such that no cross reactivity is observed.
Keywords: alkyl xanthate esters, cross-coupling, selective radical generation, carbon-centered radicals, metal-catalyzed reactions
Traditional two-electron, metal-catalyzed Csp3–Csp2 cross-coupling reactions provide efficient and selective access to architecturally rich molecules. However, challenges remain, with regard to accessing reliable and abundant alkyl nucleophiles for this class of coupling transformations.1 In particular, reactive alkylmetallic reagents (RMgX, RZnX, RLi, etc.) have generally limited functional group tolerance,2 although advances in overcoming these barriers have been realized.3 More functional group-tolerant reagents, such as organoboron and organosilicon reagents, often lack the reactivity needed to overcome a rate-limiting transmetalation for effective cross-coupling.
The recent advent of Ni/photoredox dual catalysis by this group and others4 has surfaced as a complementary and mild alternative to traditional two-electron Csp3–Csp2 cross-coupling strategies, taking advantage of a relatively low-barrier interception of alkyl radicals by nickel catalysts,5 ultimately accessing high-valent Ni(III) intermediates. To date, substrate and functional group tolerance, under this single-electron cross-coupling paradigm, has been exceptional.6 In hopes of further expanding this synthetic toolbox and chemical space, novel radical precursors derived from abundant feedstock molecules (e.g., alkyl aldehydes7 and alkylsilanes8) have been targeted that are compatible under this dual catalytic manifold.
Alcohols remain one of the most abundant, naturally occurring organic synthons, and they carry with them enormous molecular diversity. Within cross-coupling contexts, select sp3-hybridized alcohol derivatives have been implemented as carbon electrophiles in low-valent, nickel-catalyzed reactions because of their inherent C–O bond polarization,9 but have rarely functioned as precursors to nucleophilic entities.10 A compelling alternative to C–O bond activation would be the ability to form carbon-centered radicals that might be parlayed toward new C–C bond-forming reactions. Although the strong C–O σ bond (bond dissociation energy (BDE) of ∼96 kcal/mol) presents operational challenges, this strategy, if broadly applied, may offer advantages, particularly in accessing more available and diverse coupling partners.
O-Alkyl thiocarbonate (xanthate) esters (R1OCSSR2) are attractive, bench-stable reagents in their ability to generate carbon radicals under Barton–McCombie-type conditions and are readily prepared in one step from a variety of alcohol feedstocks.11 Under these processes, O-alkyl xanthates are traditionally fragmented with an excess of a reducing agent (e.g., HSnBu3) and substoichiometric amounts of radical initiator (AIBN, peroxide, etc.) to prolong the radical chain event (e.g., Figure 1). A deoxygenation reaction employing specially tailored O-thiocarbamates (Ered(average) ≈ −1.55 V vs SCE) was recently reported, and these derivatives undergo a photocatalyzed, reductive single electron transfer (SET).12
Figure 1.
Radical cleavage of xanthate esters and application under Ni/photoredox dual catalysis.
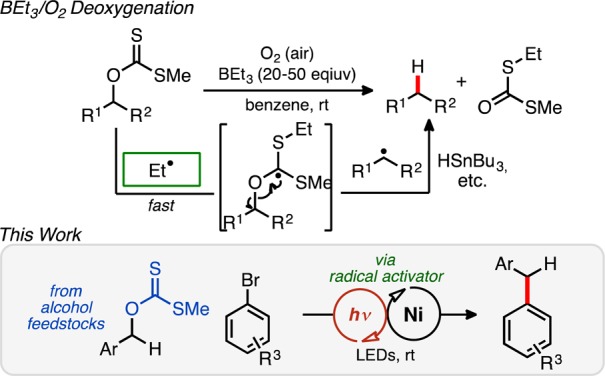
Inspirational to this work, an alternative approach to the classic deoxygenation using Bu3SnH and AIBN, for example, employs BEt3 and O2 as the initiator under otherwise relatively mild reaction conditions (benzene, H2O, room temperature).13 The generated ethyl radical reacts with the xanthate ester, initiating a C–O bond scission event (Figure 1) to produce a new carbon-centered radical that may be rapidly quenched by a hydrogen source, furnishing the reduced adduct.14 Since these pioneering studies, ethyl radical initiators (via BEt3/O2) have been widely adopted in various synthetic capacities,15 although experimental challenges are common.16
We envisioned using carbon radical activators, generated mildly via SET photo-oxidation, to fragment suitable xanthate esters in cooperation with Ni catalysis, allowing access to new Csp3–Csp2 cross-coupled structures. This method would constitute an umpolung of reactivity, because alcohol derivatives in cross-coupling protocols are typically electrophilic (e.g., OMs, OMe), especially under Ni catalysis.9 To our knowledge, O-alkyl xanthate esters have never been employed as either electrophiles or nucleophiles in cross-coupling chemistry.17 In a broader sense, this approach may also validate photo-oxidizable radical precursors as alternatives to pyrophoric BEt3/O2 systems.
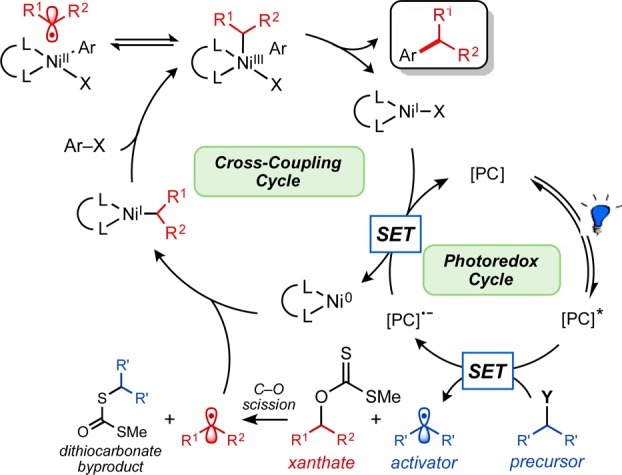
Conceptually, an excited-state photocatalyst ([PC]*; see Scheme 1) may be sufficiently oxidizing to generate an alkyl radical activator (blue) from an established radical precursor (e.g., Y = BF3K, CO2M, or ammonium silicate). This alkyl radical activator could rapidly react with the thiocarbonyl of the xanthate pronucleophile, initializing C–O bond fragmentation. With subsequent xanthate cleavage and extrusion of dithiocarbonate, the alcohol-derived carbon radical (red), may enter the Ni coupling cycle, adding to Ni(0) as delineated in our previous studies.18 Following Ni(I) oxidative addition with the requisite aryl halide, a Ni(III) intermediate may be reached, producing the desired alkylated arene via reductive elimination. A SET event from the reduced form of the photocatalyst may regenerate Ni(0) from the Ni(I) species to close the intertwined dual catalytic cycles (see Scheme 1).
Scheme 1. Proposed Ni/Photoredox Dual Catalytic Cross-Coupling of O-Alkyl Xanthates Initiated by a Radical Activator.
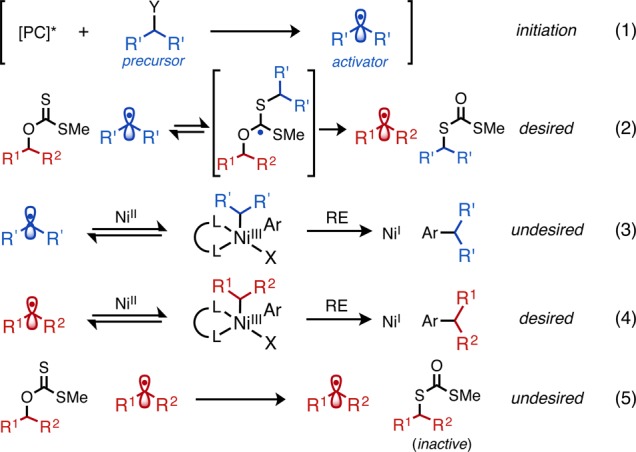
There were several evident challenges with the proposed reaction design. Radical hetero(homo)dimerization would result from multiple radicals simultaneously present, which is a challenge in multicomponent radical reactions. In addition, thiols and sulfides are well-known metal complexing agents, potentially inhibiting reaction progression through nickel sequestration.19 Perhaps most critically, the two radical components must have differentiable reactivity. The nucleophilic radical activator (blue) should be designed to react with the thiocarbonyl electrophile in preference to the nickel catalyst (e.g., eq 2 is desired over eq 3; see Scheme 2) to effect, first, the irreversible C–O cleavage. The stoichiometric concentration of xanthate was anticipated to assist in this selectivity. The xanthate-derived radical (red) must then react smoothly with the nickel catalyst (eq 4 is desired over eq 5; see Scheme 2) with little to no cross reactivity. The potential advantage of using photoredox catalysis in this scenario is the ability to produce precise, substoichiometric quantities of reactive radical activator (<2 mol %), in contrast to radical chain processes, where the concentration of radicals is inherently capricious when using O2 as the initiator.16
Scheme 2. Potential Competing Transformations of Radical Components and Ni/Xanthate Affinity.
Studies commenced with a wide variety of O-alkyl xanthate esters, including unactivated secondary/primary alkyl xanthates and activated secondary benzyl xanthates, with aryl bromide 2. We observed promising reactivity with O-benzyl xanthate ester 1a, alongside the bench-stable [Ni(dtbbpy)(H2O)4]Cl2 (4, 5 mol %) precatalyst.7 Alkylammonium silicates,6g,8 alkyltrifluoroborates,4c,6a−6c and metal carboxylates4d,20 have been effective radical precursors, following SET oxidation under photoredox conditions,21 and all were examined for their efficacy in xanthate (1a) C–O bond scission (Table 1; see the Supporting Information for complete studies). In these preliminary studies, low-molecular-weight alkyltrifluoroborates (labeled as A–E in Table 1) proved to be the more efficacious radical initiators, using the photosensitizer [Ir{dFCF3ppy}2(bpy)]PF6 (5), with sec-BuBF3K (A), furnishing 3a in 94% yield (see entry 1 in Table 1) with no undesired radical couplings detected at room temperature. Cyclopropyltrifluoroborate (C) afforded no product, likely because of its greater s-character and higher oxidation potential.6a Organic and inorganic bases were found to be a hindrance to reaction progression under various conditions examined. Control experiments revealed that light-emitting diodes (LEDs), iridium, and nickel catalysts were all necessary components for this transformation (see entries 2–4 in Table 1). Three equivalents of the alkyltrifluoroborate is deemed necessary, and all sec-BuBF3K is consumed in the reaction, as determined by 19F NMR monitoring (see the Supporting Information). Comprehensively quantifying the fate of the reagent could not be fully ascertained. Regardless, the use of the pyrophoric BEt3 reagent may be replaced with a bench-stable solid.
Table 1. Deviations from Standard Reaction Conditions Employing Xanthate 1a under Ni/Photoredox Dual Catalysisa.

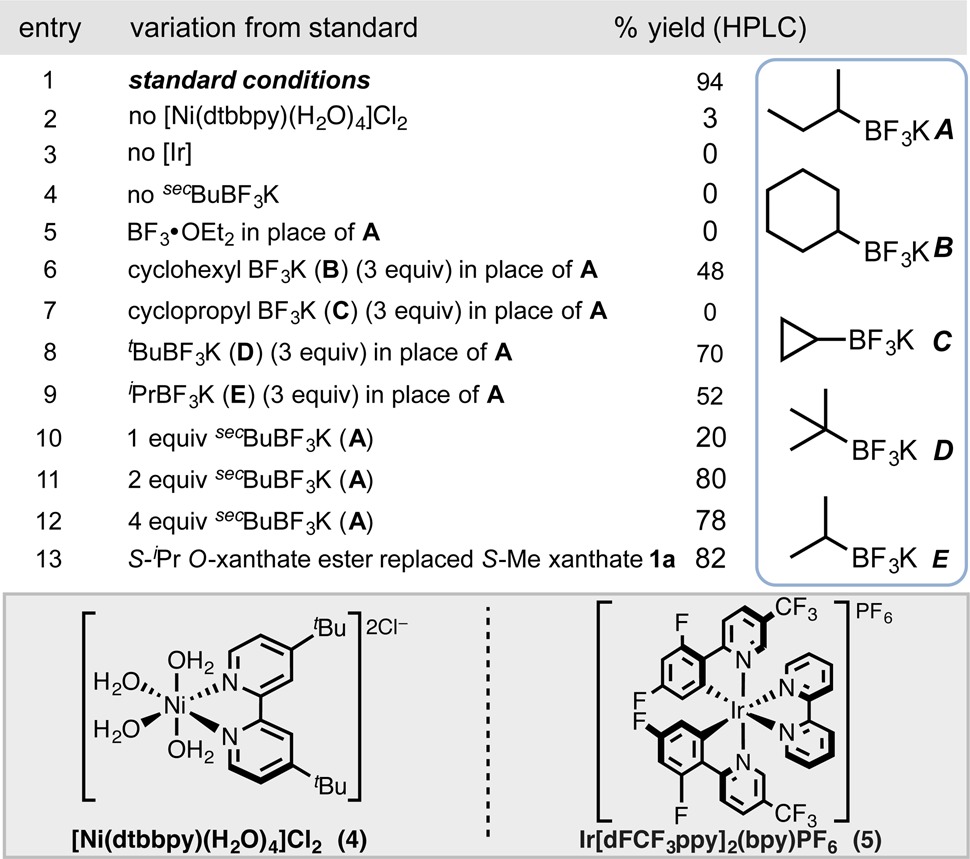
Reactions performed on 0.1 mmol scale for 48 h, unless otherwise noted. HPLC yields reported as compared to internal standards. See the Supporting Information for additional details.
With satisfactory conditions in hand to promote the coupling of benzyl O-xanthate esters, an array of xanthates (prepared using base, CS2, MeI, and used without purification) were examined with bromide 2 (see Scheme 3). Electron-rich and electron-deficient O-benzyl xanthates are similarly well-tolerated under the conditions. Oxygenated arenes (3b, 3c, 3j), including free phenol (3h), are perhaps most compatible under the developed dual catalytic conditions. Heteroaromatic, methanol-derived xanthates are easily prepared and employed, yielding pyridines (3g), furans (3j), and thiophenes (3l). These base-free coupling conditions also tolerate protic functional groups (3f, 3h), where many analogous, benzyl-derived pronucleophiles (e.g., BnBF3K or BnZnCl, derived from benzyl halides), in contrast, would be difficult to prepare directly in their unprotected form. Secondary benzyl and unactivated alkyl xanthates (Chugaev elimination product could be observed)22 were not well-tolerated in these studies, even upon mild heating, and often resulted in a mixture of products.
Scheme 3. O-Benzyl Xanthate and Aryl Halide Scope.
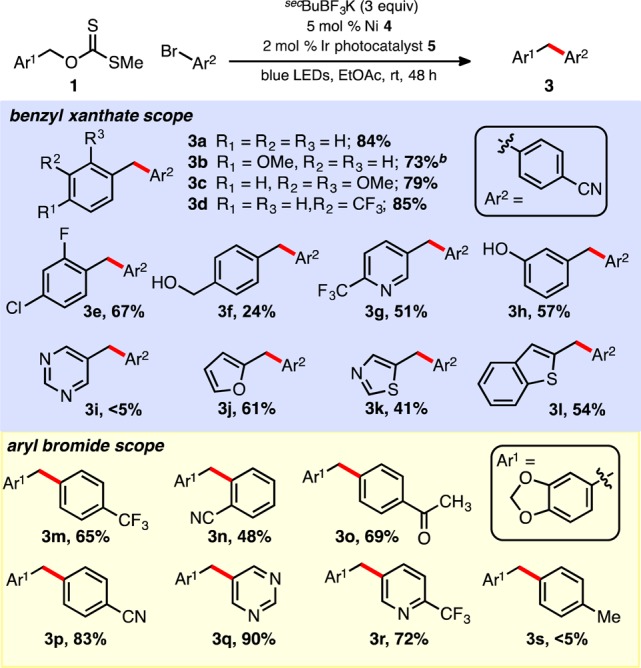
Conditions are as described in Table 1 (entry 1). 0.5 mmol scale reactions; isolated yields are reported. bTwo equivalents of secBuBF3K employed.
Numerous aryl bromides were examined next, alongside an O-benzyl xanthate ester under the optimized conditions (Scheme 3). Electron-deficient aryl bromides and heteroaryl bromides are quite compatible, although electron-rich (e.g., 3s) and even electron-neutral bromides are largely not as robust. Functional handles such as ketones (3o) and nitriles (3n, 3p) are also well tolerated. Pyridines (3r) and a pyrimidine (3q) can be prepared in moderate to excellent yields (72%–90%).
To showcase the potential utility of benzyl alcohol-derived pronucleophiles, we explored a bidirectional synthesis of 9 from 4-bromobenzyl alcohol (7), using sequential Ni/photoredox dual catalyzed reactions (Scheme 4). Bicycloheptylsilicate 6, which is a secondary alkyl radical precursor,8 was coupled efficiently with 7 under mild conditions, yielding intermediate 8. Benzyl alcohol 8 underwent xanthate activation, and following workup and solvent exchange, reacted smoothly with 3-bromopyrimidine to furnish the final dialkylated product 9 in 62% yield over the two-step sequence.
Scheme 4. Rapid, Bidirectional Coupling Affords 9 under Ni/Photoredox Dual Catalysis.
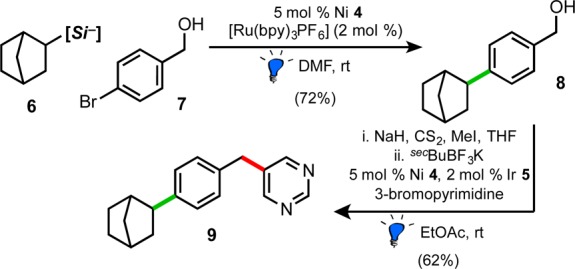
Isolated yields. [Si–] = bis(catechol) ammonium silicate.
Additional experiments aided elucidation of the mechanistic nuances of this reaction, and data collected generally support the proposed dual-catalyzed mechanism. Remarkably, the undesired sec-butyl arene cross-coupled product 10 was never observed throughout these studies when xanthate was present (Figure 2). However, in the absence of the xanthate ester under otherwise standard reaction conditions, full conversion to 10 can be observed,6a,24 critically demonstrating the rapid nature of sec-butyl radical addition to the xanthate thiocarbonyl and the ability to control reaction outcomes based on relative rates as initially designed (Figure 2a). Furthermore, homodimerized radical activator 11 can be observed in the reaction,23 in addition to the dialkyl dithiocarbonate 12 byproduct that is generated, validating the notion that sec-butyl radical is key to reaction initiation. Studies also revealed that external addition of dialkyl dithiocarbonate 12 (0.7 equiv) to the reaction decreased the reaction rate, presumably through nickel coordination.25
Figure 2.
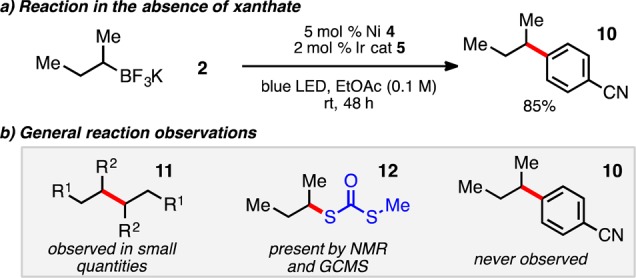
Mechanistic studies and observations.
In conclusion, we present the first example of O-benzyl xanthate ester pronucleophiles derived from abundant benzyl alcohols in Csp3–Csp2 cross-coupling reactions under photoredox/Ni dual catalysis, which is reliant on the unique generation of radical activators to promote xanthate cleavage. The mild reaction conditions employ low-molecular-weight, commercially available potassium alkyltrifluoroborates as radical precursors for xanthate C–O bond scission under base-free conditions. The relative rates of two distinct radical components are judiciously controlled in the reaction, capitalizing on the inherent selectivity of photoredox catalysis. The development of a versatile new functional group other than halide derivatives adds an important new dimension to Csp3–Csp2 cross-coupling chemistry.
Acknowledgments
Dr. Charles Ross, III (University of Pennsylvania), is acknowledged for collection of HRMS data; Sigma–Aldrich is acknowledged for the kind donation of IrCl3; and Dr. Álvaro Gutiérrez-Bonet is acknowledged for the preparation of [Ni(dtbbpy)(H2O)4]Cl2. David Primer (University of Pennsylvania) and John Tellis (Genentech) are thanked for insightful contributing discussions. Dr. Simon Lang (University of Pennsylvania) is thanked for additional data collection.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.7b00772.
Experimental details and spectral data (PDF)
We thank NIGMS (No. R01 GM113878) for financial support of this research.
The authors declare no competing financial interest.
Supplementary Material
References
- Hartwig J. F.Organotransition Metal Chemistry: From Bonding to Catalysis; University Science: Sausalito, CA, 2010. [Google Scholar]
- Rudolph A.; Lautens M. Angew. Chem., Int. Ed. 2009, 48, 2656. 10.1002/anie.200803611. [DOI] [PubMed] [Google Scholar]
- a Benischke A. D.; Knoll I.; Rérat A.; Gosmini C.; Knochel P. Chem. Commun. 2016, 52, 3171. 10.1039/C5CC10272C. [DOI] [PubMed] [Google Scholar]; b Toriyama F.; Cornella J.; Wimmer L.; Chen T.-G.; Dixon D. D.; Creech G.; Baran P. S. J. Am. Chem. Soc. 2016, 138, 11132. 10.1021/jacs.6b07172. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a review, see:; c Haas D.; Hammann J. M.; Greiner R.; Knochel P. ACS Catal. 2016, 6, 1540.and references therein 10.1021/acscatal.5b02718. [DOI] [Google Scholar]
- Seminal report:; a Kalyani D.; McMurtrey K. B.; Neufeldt S. R.; Sanford M. S. J. Am. Chem. Soc. 2011, 133, 18566. 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ye Y.; Sanford M. S. J. Am. Chem. Soc. 2012, 134, 9034. 10.1021/ja301553c. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zuo Z. W.; Ahneman D. T.; Chu L. L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345, 437. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Examples with transition metals other than nickel have been reported:; a Sahoo B.; Hopkinson M. N.; Glorius F. J. Am. Chem. Soc. 2013, 135, 5505. 10.1021/ja400311h. [DOI] [PubMed] [Google Scholar]; b Hopkinson M. N.; Sahoo B.; Li J.-L.; Glorius F. Chem.—Eur. J. 2014, 20, 3874. 10.1002/chem.201304823. [DOI] [PubMed] [Google Scholar]
- a Primer D. N.; Karakaya I.; Tellis J. C.; Molander G. A. J. Am. Chem. Soc. 2015, 137, 2195. 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Karakaya I.; Primer D. N.; Molander G. A. Org. Lett. 2015, 17, 3294. 10.1021/acs.orglett.5b01463. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yamashita Y.; Tellis J. C.; Molander G. A. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 12026. 10.1073/pnas.1509715112. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tellis J. C.; Amani J.; Molander G. A. Org. Lett. 2016, 18, 2994. 10.1021/acs.orglett.6b01357. [DOI] [PubMed] [Google Scholar]; e Karimi-Nami R.; Tellis J. C.; Molander G. A. Org. Lett. 2016, 18, 2572. 10.1021/acs.orglett.6b00911. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Patel N. R.; Molander G. A. J. Org. Chem. 2016, 81, 7271. 10.1021/acs.joc.6b00800. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Vara B. A.; Jouffroy M.; Molander G. A. Chem. Sci. 2017, 8, 530. 10.1039/C6SC03236B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez-Bonet A.; Tellis J. C.; Matsui J. K.; Vara B. A.; Molander G. A. ACS Catal. 2016, 6, 8004. 10.1021/acscatal.6b02786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouffroy M.; Primer D. N.; Molander G. A. J. Am. Chem. Soc. 2016, 138, 475. 10.1021/jacs.5b10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kuwano R.; Kondo Y. Org. Lett. 2004, 6, 3545. 10.1021/ol048540e. [DOI] [PubMed] [Google Scholar]; b Guan B. T.; Xiang S. K.; Wang B. Q.; Sun Z. P.; Wang Y.; Zhao K. Q.; Shi Z. J. J. Am. Chem. Soc. 2008, 130, 3268. 10.1021/ja710944j. [DOI] [PubMed] [Google Scholar]; For enantioselective and stereospecific variants, see:; c Taylor B. L. H.; Swift E. C.; Waetzig J. D.; Jarvo E. R. J. Am. Chem. Soc. 2011, 133, 389. 10.1021/ja108547u. [DOI] [PubMed] [Google Scholar]; d Harris M. R.; Hanna L. E.; Greene M. A.; Moore C. E.; Jarvo E. R. J. Am. Chem. Soc. 2013, 135, 3303. 10.1021/ja311783k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- While this research was underway, a Csp3–Csp2 cross-coupling of redox-active, alkyl metal oxalates (radical pronucleophiles) was reported with various alcohol partners:Zhang X.; MacMillan D. W. C. J. Am. Chem. Soc. 2016, 138, 13862. 10.1021/jacs.6b09533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For pioneering reports, see:; a Barton D. H. R.; McCombie S. W. J. Chem. Soc., Perkin Trans. 1 1975, 1574. 10.1039/p19750001574. [DOI] [Google Scholar]; b Barrett A. G. M.; Prokopiou P. A.; Barton D. H. R. J. Chem. Soc., Perkin Trans. 1 1981, 1510. 10.1039/p19810001510. [DOI] [Google Scholar]; c Barton D. H. R.; Crich D.; Löbberding A.; Zard S. Z. Tetrahedron 1986, 42, 2329. 10.1016/S0040-4020(01)90614-3. [DOI] [Google Scholar]; d For a review on thiocarbonyls in radical chemistry, see:Crich D.; Quintero L. Chem. Rev. 1989, 89, 1413. 10.1021/cr00097a001. [DOI] [Google Scholar]
- Traditional O-alkyl xanthates have very low reduction potentials, as was confirmed by our CV studies: Eox = +1.75 V. For a report of photoredox-active dithiocarbamates, see:Chenneberg L.; Baralle A.; Daniel M.; Fensterbank L.; Goddard J.-P.; Ollivier C. Adv. Synth. Catal. 2014, 356, 2756. 10.1002/adsc.201400729. [DOI] [Google Scholar]
- Initial reports; see:; a Nozaki K.; Oshima K.; Utimoto K. Tetrahedron Lett. 1988, 29, 6125. 10.1016/S0040-4039(00)82283-2. [DOI] [Google Scholar]; b Barton D. H. R.; Ok J. D.; Jaszberenyi J. C. Tetrahedron Lett. 1990, 31, 3991. 10.1016/S0040-4039(00)94480-0. [DOI] [Google Scholar]; Also see:Spiegel D. A.; Wiberg K. B.; Schacherer L. N.; Medeiros M. R.; Wood J. L. J. Am. Chem. Soc. 2005, 127, 12513. 10.1021/ja052185l. [DOI] [PubMed] [Google Scholar]
- For a report on Et3B/air reaction kinetics and mechanism, see:Jin J.; Newcomb M. J. Org. Chem. 2007, 72, 5098. 10.1021/jo070336s. [DOI] [PubMed] [Google Scholar]
- For a review and references therein, see:; a Ollivier C.; Renaud P. Chem. Rev. 2001, 101, 3415. 10.1021/cr010001p. [DOI] [PubMed] [Google Scholar]; b N-Heterocyclic carbene boryl radicals; see:Ueng S.-H.; Solovyev A.; Yuan X.; Geib S. J.; Fensterbank L.; Lacôte E.; Malacria M.; Newcomb M.; Walton J. C.; Curran D. P. J. Am. Chem. Soc. 2009, 131, 11256. 10.1021/ja904103x. [DOI] [PubMed] [Google Scholar]
- Curran D. P.; McFadden T. R. J. Am. Chem. Soc. 2016, 138, 7741. 10.1021/jacs.6b04014. [DOI] [PubMed] [Google Scholar]
- Fu and coworkers briefly explored xanthate electrophiles; see:Oelke A. J.; Sun J.; Fu G. C. J. Am. Chem. Soc. 2012, 134, 2966. 10.1021/ja300031w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez O.; Tellis J. C.; Primer D. N.; Molander G. A.; Kozlowski M. C. J. Am. Chem. Soc. 2015, 137, 4896. 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyle M. D.; Bartholomew C. H. Catalysts 2015, 5, 145. 10.3390/catal5010145. [DOI] [Google Scholar]
- a Johnston C. P.; Smith R. T.; Allmendinger S.; MacMillan D. W. C. Nature 2016, 536, 322. 10.1038/nature19056. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zuo Z.; Cong H.; Li W.; Choi J.; Fu G. C.; MacMillan D. W. C. J. Am. Chem. Soc. 2016, 138, 1832. 10.1021/jacs.5b13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellis J. C.; Kelly C. B.; Primer D. N.; Jouffroy M.; Patel N. R.; Molander G. A. Acc. Chem. Res. 2016, 49, 1429. 10.1021/acs.accounts.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkeser R. A.; Hazdra J. J. J. Am. Chem. Soc. 1959, 81, 228. 10.1021/ja01510a052. [DOI] [Google Scholar]
- 1,1′-Bi(cyclohexane) dimer can be observed by GCMS when activator B (Table 1) was employed.
- Spectral confirmation of 10 is confirmed:Liu Z.; Dong N.; Xu M.; Sun Z.; Tu T. J. Org. Chem. 2013, 78, 7436. 10.1021/jo400803s. [DOI] [PubMed] [Google Scholar]
- Byproduct 12 was added to otherwise normal reaction conditions and suppressed product formation (see the Supporting Information for a complete study).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.