

Abstract
Recent studies have characterized the extensive somatic alterations that arise during cancer. However, the somatic evolution of a tumor may be significantly affected by inherited polymorphisms carried in the germline. Here, we analyze genomic data for 5954 tumors to reveal and systematically validate 412 genetic interactions between germline polymorphisms and major somatic events, including tumor formation in specific tissues and alteration of specific cancer genes. Among germline-somatic interactions, we find germline variants in RBFOX1 that increase incidence of SF3B1 somatic mutation by eight-fold via functional alterations in RNA splicing. Similarly, 19p13.3 variants are associated with a four-fold increased likelihood of somatic mutations in PTEN. In support of this association, we find that PTEN knock-down sensitizes the MTOR pathway to high expression of the 19p13.3 gene GNA11. Finally, we observe that stratifying patients by germline polymorphisms exposes distinct somatic mutation landscapes, implicating new cancer genes. This study creates a validated resource of inherited variants that govern where and how cancer develops, opening avenues for prevention research.
Keywords: tumor evolution, germline-somatic interactions, cancer predisposition
Introduction
Cancer is a complex genetic disease influenced by both inherited variants in germline DNA and somatic alterations acquired during formation of the tumor (1,2). Recently, large multi-center efforts such as The Cancer Genome Atlas (TCGA) (3) and the International Cancer Genome Consortium (ICGC) have performed a series of detailed analyses of the somatic alterations affecting tumor genomes (4). These studies have focused primarily on identifying recurrent somatic alterations within and across cancer types, uncovering immense heterogeneity within and between tumors (2).
Prior to tumor genome sequencing, many genes that play a role in cancer were discovered through studies of the germline (5). Linkage studies in families with inherited, typically childhood, cancers identified rare germline mutations in genes related to DNA damage repair, RAS signaling or PIK3 signaling (2,6). In contrast to childhood cancers, adult tumors have largely been considered ‘sporadic’; however, mounting evidence points to a potentially substantial influence from the gremline (7). Studies of homogenous populations in Scandinavia identified a genetic component underlying common adulthood cancers (8,9); more recently, many loci have been implicated through genome-wide association studies (GWAS) in studies of various types of cancer (10-13). Large-scale systematic sequencing of over 4000 tumors (12 cancer types) from TCGA found rare germline truncations in 114 cancer-susceptibility-associated genes, ranging in frequency from 4% (AML) to 19% (ovarian cancer), including BRCA1, BRCA2, FANCM, and MSH6, which are associated with increased somatic mutation frequencies (14).
Other evidence has emerged that germline variants and somatic events can be intricately linked. For example, a recent analysis of rare germline variants and somatic mutations in ovarian cancer highlighted novel ovarian cancer genes and pathways (15). Recent reports have also associated specific haplotypes with JAK2 V617F mutations in myoproliferative neoplasms (16) and with EGFR exon 19 microdeletions in non-small cell lung cancer (17). Germline variation was also found to influence gene expression in breast tumors (18,19). Together, the variants identified thus far are estimated to explain at most 20% of the likely germline contribution to cancer (11), suggesting the existence of many as yet uncharacterized genetic determinants.
Here, we integrate germline genotypes with somatic changes from TCGA to obtain a pan-cancer view of how common inherited variation can prime the later progression of tumors. We seek to identify genetic associations that explain two major classes of somatic events: 1) the tissue site where the tumor develops, and 2) which specific cancer genes are mutated. This analysis identifies an array of germline polymorphisms that increase or decrease the risk for these somatic events, some of which were previously known but most of which are new discoveries.
Results
Structure of germline variation in The Cancer Genome Atlas
We obtained common germline variants and somatic tumor mutations for 6,908 patients from the TCGA Research Network (20). We restricted our analysis to germline variants present at minor allele frequencies of 1% or higher in this patient group, resulting in 706,538 autosomal single nucleotide polymorphisms (SNPs) organized into ∼1.6 million haplotypes (Fig. 1A). Both SNPs and haplotypes were studied, as the two marker types have complementary power to detect common and rare disease-associated variants, respectively (21-23). Examination of patient SNP profiles showed evidence of population substructure (24), even among the majority of patients who self-identified as European ancestry (Fig. S1A). Of these patients, we retained 4,165 who also clustered tightly with Europeans from the HapMap III reference population (25) (discovery cohort, Fig. 1B). For later replication of our findings, we used a validation cohort consisting of 1,789 additional TCGA patients for which full data (tissue type and somatic alterations) became available only after the start of our study (Fig. S1B, S1C).
Fig. 1.
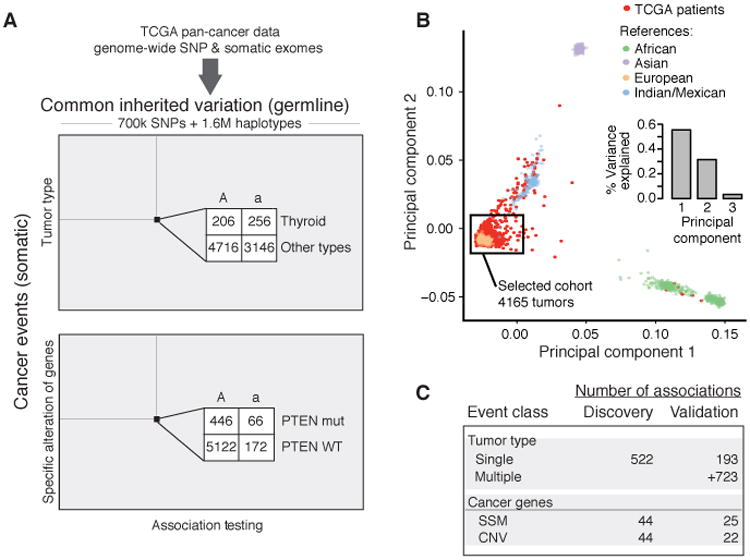
Study design and data. A) 2.3 million germline markers comprising 700K SNPs and 1.6 million multi-SNP haplotypes were tested for association with primary tumor type and somatic mutation status of 138 known cancer genes. B) Principal Components Analysis of TCGA European ancestry samples with HapMap III was used to evaluate population substructure. The first two principal components explain 87% of the variation in genotype among samples. A black box frames the 4165 samples used for the discovery cohort. C) Summary of association results from the discovery phase (P < 10-5) along with the subset of these observed at an FDR < 0.5 in the validation phase. Counts are provided for each class of somatic event. Markers detected at and FDR < 0.50 or lower are also reported in Supplementary Tables 3 and 5. SSM – subtle somatic mutation, CNV – copy number variant.
Associations between germline and tumor site of origin
We first sought to identify germline markers associated with the site at which the tumor develops, i.e. incidence of tumors of a particular tissue type. Previous studies have focused on individual tumor types in isolation, thus it remains unclear to what extent cancer-associated alleles are general versus site specific. All tumors of a particular type were compared to all other types pooled; separate comparisons were performed for each of the 22 TCGA tumor types. SNP or haplotype markers were selected as initial candidates if their p-values of association were less than the “suggestive” threshold commonly used in GWAS (26): 1×10-5 by Fisher's Exact Test, corrected for the number of tumor types assessed. Different tumor types were collected at different times by TCGA; to minimize false positive associations due to such batch effects, we removed markers displaying strong association with batch or plate (Fig. S2) and used genomic control to adjust for inflated p-values (Fig. S3). In cases in which multiple tumor types were each weakly associated with a genotype, these were combined for testing for stronger association as a group. All candidate associations were then tested in the validation cohort, and empirical false discovery rates were estimated using matched numbers of random associations. By this procedure we identified 916 markers of potential interest, 395 of which could be replicated at an empirical FDR < 0.25 (Fig. 1C, 2A; Table S1). Additional markers at each locus were imputed to provide a more complete view of the association. While all TCGA patients have been diagnosed with some type of cancer, these germline markers provide predictive information about where the tumor develops.
Fig. 2.

Germline variants influencing primary tumor type. A) Ideogram of all loci associated with a single tumor type (blue triangles). Red triangles indicate an association with the specific somatic alteration of a cancer gene. B) Manhattan (LocusZoom) plot (78) displaying markers at 8q24.13 associated with incidence of breast cancer. Markers are colored according to linkage disequilibrium (r2 values) derived from the 1000 Genomes European samples. C) Markers at 8q24.13 also associated with age of diagnosis with breast cancer. A/A indicates individuals homozygous for the major allele and A/a, a/a indicate individuals with one or more copies of the minor allele. D) Quantile-quantile plot showing the observed p-values of association (versus random expectation) for 557 loci associated with cancer risk in previous studies. The substantial elevation above the diagonal (red) indicates support for many of these previous loci in the present TCGA analysis. E) Manhattan plot displaying markers at 9q22.23 associated with thyroid carcinoma and genes encoded within that region. Colored genes were found to have altered expression in Thyroid tumors in the presence of the minor allele. F) Mean expression of genes highlighted in panel (E) versus the number of minor alleles. Bars show standard error on mean estimates.
As an example, markers at locus 8q24.13 were identified for their specific and significant association with breast cancer (P < 2×10-10, Fig. 2B), which was reinforced by a second finding of association with age of breast cancer onset (Fig. 2C). While this region had not previously been implicated in cancer predisposition, it had been reported to harbor frequent somatic amplifications in breast cancer cell lines (27). In addition, breast tumors overexpressing genes in this region are associated with shorter time to recurrence (28-31). Among the many other loci associated with tumor type, we identified a SNP at 13q14.2 that falls into an intron of the tumor suppressor RB1 and is associated with multiple tumor types, and a SNP at 11q22.3, a region encoding the cancer genes DDX10 and ATM, that is associated with breast cancer (Table S1). Epigenetic silencing of DDX10 was recently reported in ovarian cancer (32), and rare germline coding variants in ATM have previously been reported in familial breast cancer (33).
To examine the correspondence of this TCGA analysis with previous GWAS of cancer, we analyzed 557 cancer-associated SNPs recorded in the NHGRI GWAS Catalog (26). Most of these previous SNPs had been identified based on an increase in risk for developing cancer as compared to non-diseased controls, whereas the SNPs identified in our study are based on an increased prevalence of tumors of one particular tissue as compared to other tissues. Despite this difference, the previously published markers of cancer risk had substantial correspondence to the markers associated with tumor site, with the strongest signal seen for approximately 15 markers (Fig. 2D, Fig. S4A-S4D, Table S2). The loci validated by both types of study can be prioritized as a set of clearly reproducible cancer risk factors that are also specific to tissue of origin.
Even where the new associations recapitulate a previously reported cancer locus, new insights may be gained, as illustrated for the thyroid cancer locus 9q22.23 (34,35) (Fig. 2E). SNP rs1867277 at this locus, in the promoter region of the FOXE1 transcription factor, was previously reported to be the cause of association; the minor allele was shown to recruit the USF1/USF2 transcription factors, resulting in allele-specific FOXE1 transcription (35). In TCGA, we observed allele-dependent expression of FOXE1, but also of three nearby genes (Fig. 2F): c9orf156 (NAP1), involved in nucleosome assembly with the potential to widely alter gene regulation(36); TRIM14, which may regulate the expression of multiple cancer genes (37); and CORO2A, which de-represses expression of Toll-like receptors to initiate the inflammatory response (38). In TCGA, FOXE1 expression decreased with an increasing number of minor alleles at this locus, whereas Landa et al. reported increased FOXE1 expression (35) (Fig. 2F). This discrepancy in FOXE1 expression levels may arise from performing the analysis in patient tumors, whereas the previous report was based on experiments in cancer cell lines, and in part because risk in this region is driven by at least two distinct haplotypes that may affect the expression of different genes (39,40). Interestingly, targeted overexpression of FOXE1 was not in itself capable of causing thyroid cancer in mice (41), suggesting other genes in this region may play a role. Thus, re-analysis of this locus using genotype and mRNA expression from the TCGA suggests that its role in thyroid cancer may be more complex than previously appreciated.
Associations between the germline and somatic alteration of specific genes
We next sought to identify associations between inherited germline variants and the occurrence of somatic mutations in particular cancer genes. We hypothesized that germline background could generate a context in which a loss or gain of function event in a particular gene could be advantageous to a tumor. As loss or gain of function is commonly achieved through DNA mutation or amplification/deletion, we considered both somatic mutation and somatic copy number changes in our analysis. We used a previously published set of 138 cancer genes which had been compiled based on a strong signal of positive selection for subtle somatic mutations (SSMs, i.e. point mutations, insertions or deletions) or CNVs in tumors (2). Such evidence of positive selection suggests that the alterations affecting these genes are likely to be enriched for functional drivers. Indeed, genes under positive selection in cancer exhibit a bias toward mutations predicted to interfere with protein activity (42,43). Because these genes play a central role in many cancers, we expected that germline loci associated with their alteration status in tumors would provide insight into specific biological contexts that influence which cancer genes most effectively promote tumor growth and survival. For the most frequently mutated genes in this set, such as TP53 which is altered in approximately 35% of TCGA tumors, we estimated that we had 80% statistical power to detect changes in mutation rate of ≥ 1.8-fold (Fig. S5A). For genes near the median mutation frequency in this set, such as BRCA1 which is altered in ∼3% of tumors, we were powered to detect changes in mutation rate of ≥ 3.0-fold.
Associating testing identified a total of 62 associations between germline markers and specific cancer genes, of which 17 were found to replicate in the validation cohort at an empirical FDR < 25%, and of which 35 were deemed of potential interest (empirical FDR < 50%; Fig. 1C; Table S3). These 35 associations covered 28 germline loci and 20 cancer genes. Somatic alteration rates for cancer genes tended to increase with the number of minor alleles at the germline locus, with the largest increases coinciding with the least frequent germline alleles (Fig. 3A-C). The effect sizes were quite large, corresponding to increases in mutation frequency of 1.8- to 14.8-fold (Fig. 3A), and were well correlated between the discovery and validation cohorts (r=0.43, P < 0.01; Fig. S5B). Such large effects linked to common SNPs are particularly striking in comparison to typical GWAS, where effect sizes tend to fall beneath 2-fold (13). For example, a haplotype on 15q22.2 resulted in a 14-fold increased chance of acquiring a CNV affecting GNAQ.
Fig. 3.
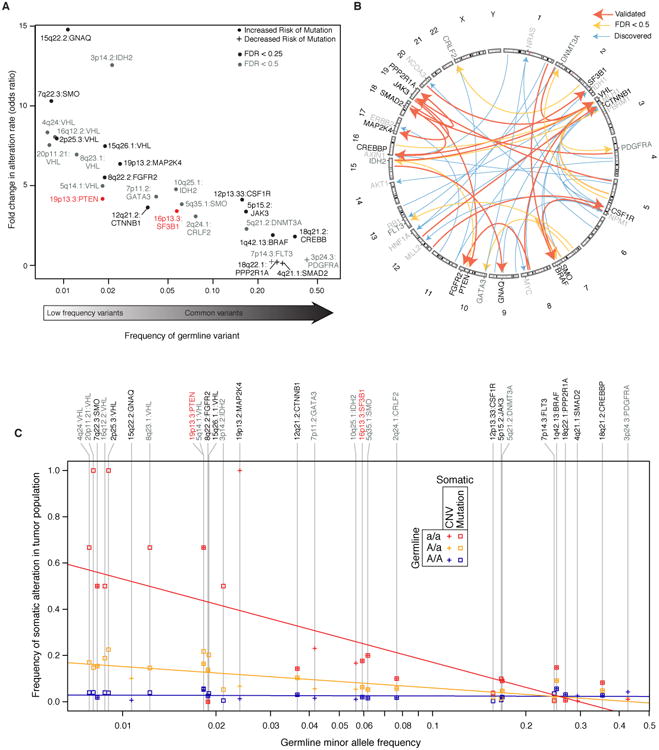
Germline interactions with somatic alteration of specific cancer genes. A) Overview of all potentially interesting (FDR < 0.5; dark grey labels) and validated (FDR < 0.25; black labels) associations of this class, displayed according to the effect size (increase in alteration rate, y-axis) versus the frequency of the germline minor allele (x-axis). We see large effects (from 2-14 fold changes in alteration rate) and an inverse relationship between the magnitude of this effect and the minor allele frequency. Validated loci associated with PTEN mutation and SF3B1 mutation (red) are highlighted in the main text and subsequent figures. B) A Circos plot (80) depicting germline-somatic interactions discovered (blue arrows) and replicated in the validation cohort (orange arrows for FDR < 0.5 and red arrows for FDR < 0.25). C) For each somatically altered gene in (A), the alteration rate is plotted separately for patients with each associated genotype (homozygous major allele, AA; heterozygous, Aa; homozygous minor allele, aa) as a function of the minor allele frequency. Regression lines show the trends for each genotype: homozygous minor allele (red), heterozygous minor allele (orange) and homozygous major allele (green).
In what follows, we further investigate validated germline-cancer gene associations at 16p13.3 and 19p13.3 as instructive examples. At each of these loci, one or more genes encoded at the germline locus participates in the same biological pathway as the somatically-mutated cancer gene (Fig 3A,C associations labeled in red).
Intronic SNP in RBFOX1 enables somatic mutation of SF3B1 to affect splicing
In one noteworthy connection between germline and somatic gene mutations, we observed a haplotype at 16p13.3 that was associated with a markedly increased risk of somatic mutation in SF3B1, encoding a component of the U2snRNP spliceosome (Fig. 4A, a striking >8-fold increase in mutation rate under the homozygous minor allele). The most strongly associated haplotype encompassed an enhancer in the fourth intron of RBFOX1 (Fig. 4B), the only gene at this locus. Like SF3B1, RBFOX1 also encodes an RNA-binding protein involved in splicing, which influences the inclusion or exclusion of exons in alternatively spliced isoforms (44). This suggested a model whereby the germline configuration of RNA splicing, linked to variation in RBFOX1 expression, modulates the sensitivity of RNA splicing to subsequent somatic mutations in the U2 spliceosome (Fig. 4C).
Fig. 4.
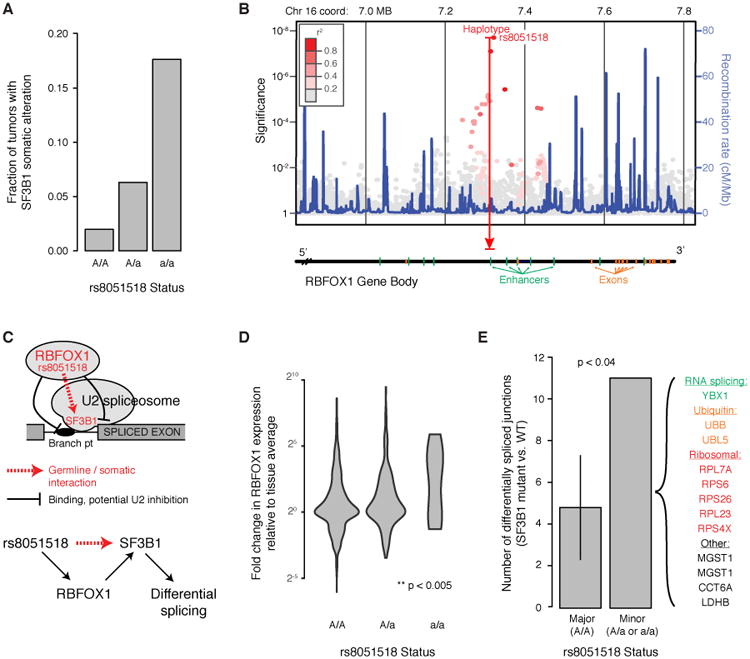
Potentiating SF3B1 mutation through 16p13 germline variation. A) Increase in SF3B1 somatic mutation rate with the rs8051518 minor allele at 16p13. B) Manhattan plot of germline association with SF3B1 mutation rate across this locus, which encodes the single gene RBFOX1. C) Current model by which RBFOX1 functionally interacts with SF3B1 to regulate RNA splicing. D) RBFOX1 increases in mRNA expression in the presence of the rs8051518 minor allele. Analysis is across all TCGA tissues, normalizing for mean expression within each tissue type. E) The number of differentially spliced exon-exon junctions was compared between individuals homozygous for the rs8051518 major allele and those harboring one or more copies of the minor allele. The number of differentially spliced junctions in each group was determined by comparing tumors with WT SF3B1 to tumors with mutant SF3B1. For correct comparison, individuals with the major allele are subsampled so that this cohort is the same size as that of the minor allele (43 individuals, error bar shows ±2σ).
In support of this hypothesis, we first found that the minor allele at this locus tends to substantially increase RBFOX1 expression, especially in homozygotes (Fig. 4D, normalized across tissue types). Next, we investigated how the germline state of RBFOX1, somatic mutation of SF3B1, or the combination of both of these factors impacted RNA splicing patterns in TCGA patients. In patients with the major RBFOX1 allele, SF3B1 mutation had minimal effect on splicing; In contrast, in patients with the minor allele, SF3B1 mutation led to changes in splicing patterns within a number of transcripts (Fig. 4E). Altogether, we identified splice junctions in 11 genes for which there were significant increases or decreases in the fraction of cryptic transcripts, dependent on SF3B1 somatic mutation (Fig. 4E). Such genes include those driving cellular proliferation and metabolism (ribosomal subunits, protein turnover) as well as YBX1, a regulator of alternative splicing. A functional relationship between RBFOX1 germline status and SF3B1 somatic mutation was also supported by statistical modeling, which revealed a significant interaction of these two factors in predicting the overall fraction of cryptic transcripts in a patient (P < 0.04).
19p13.3 allele magnifies effect of PTEN alteration on mTOR signaling
A germline haplotype at locus 19p13.3 was associated with a substantial increase in somatic mutation rate of the PTEN tumor suppressor gene, from approximately 5% for the homozygous major allele to 22% for heterozygotes (Fig. 5A). We noted that two genes at this locus, GNA11 and STK11, function in the PIK3CA/mTOR signaling pathway in which PTEN plays a major repressive role (Fig. 5B). In particular, GNA11 can act as an oncoprotein by activating mTOR signaling (45), whereas STK11 inhibits mTOR activity downstream of PTEN (46,47). The convergence of these three proteins on the mTOR pathway suggested a model in which the minor allele of 19p13.3 affects mTOR signaling, conferring sensitivity of this pathway to later somatic mutation of PTEN.
Fig. 5.
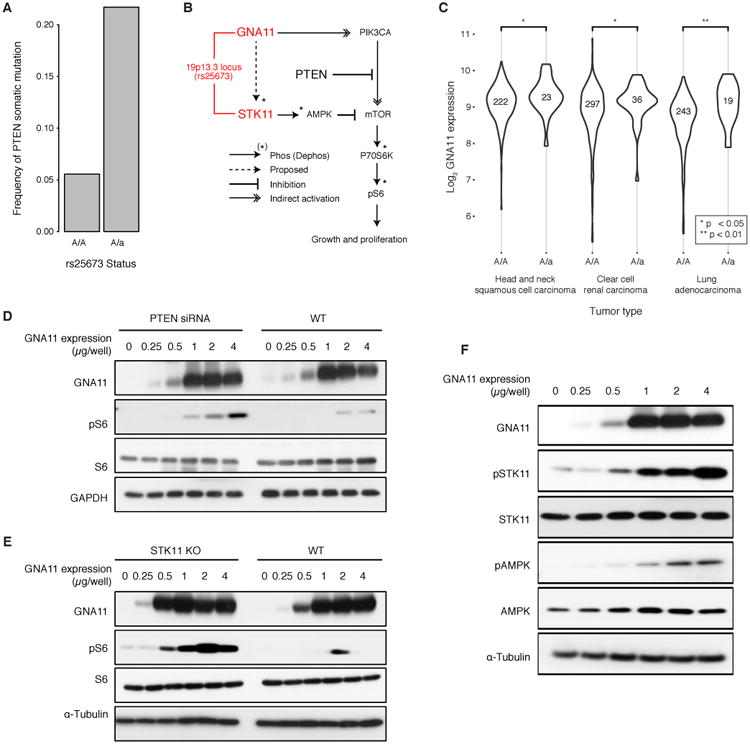
Potentiating PTEN mutation through 19p13 germline variation. A) Increase in PTEN somatic mutation rate depending on the rs25673 minor allele at 19p13. Among the genes encoded at this locus, GNA11 and STK11 function in the mTOR signaling pathway with PTEN. B) Current model in which mTOR signaling, as measured by phospho-S6 (pS6), is activated by GNA11 and repressed by PTEN and STK11. C) GNA11 increases in mRNA expression in the presence of the minor allele in lung adenocarcinoma, renal clear cell carcinoma and head and neck squamous cell carcinoma. D-E) Exogenous control of GNA11 expression regulates mTOR signaling as measured by pS6. The relationship between GNA11 and pS6 is exposed by either D) PTEN knockdown by siRNA or E) STK11 knockout by CRISPR/Cas9. F) Increased expression of GNA11 results in increased phosphorylation of STK11 with concomitant increase in phosphorylated AMPK.
In support of this hypothesis, we observed that mRNA expression of GNA11 was higher in the presence of the minor 19p13.3 allele in a majority of tumors types examined (P < 0.05, one-sided Mann Whitney Test with multiple testing correction) (Fig. 5C). To investigate the impact of changing GNA11 expression, we next placed transcription of GNA11 under exogenous control in HEK293T cells and measured the relationship between GNA11 expression level and mTOR signaling activity. Increases in GNA11 mRNA led to corresponding increases in mTOR signaling in wild-type cells; however, this effect was greatly magnified by PTEN knockdown (Fig. 5D). Interestingly, we observed that STK11 loss of function (mutation or deletion) was significantly more likely in the presence of a GNA11 gain of function event (mutation or amplification) (OR: 2.87, P < 1×10-12, Fisher's Exact Test). Based on this result, we anticipated that STK11 might also interfere with GNA11 activation of mTOR signaling, since like PTEN it serves as a repressor of mTOR signaling. Indeed, we observed that the increase in mTOR signaling due to GNA11 was greatly magnified under STK11 knockout (Fig. 5E), just as we had observed for PTEN knockdown. Although further work is needed, these findings lend support to a model by which alterations at locus 19p13.3 increase the activity of GNA11, which in turn, increases the selective advantage provided by PTEN inactivating mutations during tumorigenesis.
Given this particular sensitivity of GNA11-driven mTOR signaling to STK11 knockout, we hypothesized that GNA11 may also act upstream of STK11. Indeed, GNA11 expression was sufficient to increase the level of STK11 protein phosphorylation with a concomitant increase in phosphorylation of AMPK (Fig. 5F), a direct target of STK11 (Fig. 5B) (48). These findings suggest that GNA11 increases mTOR function while indirectly stimulating AMPK-based inhibition of mTOR via STK11 (Fig. 5B dotted line).
Tumor classification by germline identifies new gene mutation landscapes
Since we had identified 28 germline loci that increase the occurrence of somatic alterations in well-known cancer genes, we hypothesized that these same loci might influence mutation rates of other genes not previously linked to cancer. To explore this idea, we used MutSigCV (49) to identify all genes mutated more frequently than expected when grouping TCGA patients in the discovery cohort not by tissue, as has been performed successfully many times in the past (49), but according to the state of their germline at each of the 28 loci. This analysis identified 20 additional genes that had a significantly higher somatic mutation rate than expected when analyzed in a specific germline context (Fig. 6). Some genes had an elevated mutation rate in the presence of the minor allele (Fig. 6 purple blocks), while others were only mutated with the homozygous major allele (Fig. 6 green blocks). An elevated mutation rate relative to the background expectation is a signature of positive selection; thus this approach has the potential to identify genes under positive selection in cancer on a particular genetic background.
Fig. 6.
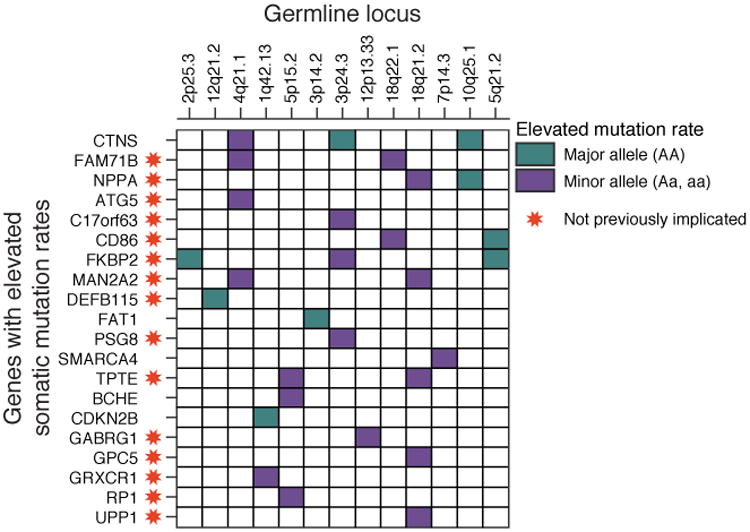
Comprehensive screen for genes with elevated somatic alteration rates, conditioned on germline minor allele status. MutSigCV analysis identified multiple genes with an elevated mutation rate in the presence of the minor allele at 13 loci that were found to influence the somatic alteration rate of a known cancer gene. Among the genes identified, 15 had not previously been identified as frequently mutated genes in cancer (red stars).
Of the 20 genes identified by MutSigCV, fifteen had not previously been identified as frequently mutated in any TCGA or ICGC cancer genome study. Evidence suggests that several of these genes could be relevant to carcinogenesis: for instance, CD86 plays a role in the early T-Cell response (50), TPTE encodes a phosphatase with very high sequence similarity to the known tumor suppressor PTEN(51), and DEFB115 is often co-mutated with protein kinase C isozymes PRKCG, PRKCH and PRKCQ which have been implicated as tumor suppressors (52). Thus mutation analysis based on cohorts defined by genotype, rather than tissue, can provide a powerful strategy to identify novel genes in cancer.
Discussion
Thus far, most studies of the cancer genome have been concerned with understanding the somatic mutations or transcriptional changes that arise during tumor progression. Recently, however, there has been a growing focus on the role of inherited variation in adult cancer, with a view towards next-generation risk assessment and prevention (53). Here, we have described a genome-wide analysis of germline-somatic interactions, based on the availability of germline genotypes and somatic phenotypes for most TCGA patients. We found evidence that genetic background can influence the somatic evolution of a tumor in at least two ways: in determining the site of tumorigenesis; and by modifying the likelihood of acquiring mutations in specific cancer genes. The ability to analyze matched genotypes, somatic genome alterations, and mRNA expression profiles enabled us to not only identify germline-somatic genome linkages but also to investigate which genes at a given locus are impacted transcriptionally and thus may mediate the effects of germline variants. Moreover, by grouping tumor samples according to the state of inherited genetic variants, it was possible to discover recurrent somatic mutations that were not identifiable by any previous cancer analysis, highlighting novel cancer gene candidates.
Collectively, this resource of germline-somatic interactions in cancer will generate many testable hypotheses about the molecular mechanisms underlying adulthood cancer risk. Interactions that are most readily interpreted are those for which genes at the germline locus function in the same biological pathway as the gene that is somatically altered. For example, a locus associated with PTEN mutation encoded two cancer genes in the same pathway, GNA11 and STK11; all three of these genes regulate growth signaling via mTOR (Fig. 5). We were able to show experimentally that both STK11 and PTEN interfere with GNA11 activation of mTOR, suggesting that germline variants increasing the activity of GNA11 could increase the selective advantage of PTEN mutations during tumor progression. Another locus associated with SF3B1 mutation encoded RBFOX1; both genes regulate alternative RNA splicing (Fig. 4). Further investigations showed that SF3B1 mutation is associated with significant differences in cryptic RNA splicing, exclusively in individuals with the minor allele. We expect that further analysis of the many additional germline-somatic interactions reported in this resource will provide clues about the underlying molecular relationships that promote cancer.
Some frequently altered cancer genes, such as TP53 and PIK3CA, were not found to be influenced by common germline variants. This result is somewhat puzzling since, due to their frequent mutations in TCGA, our analysis was highly powered to find germline associations with these genes (Fig. S5A). One explanation is that the mutation of these genes is so critical to cancer that it provides a selective advantage to tumor cells regardless of germline background. Another possibility is that germline interactions take place with specific mutation sites within the gene, but not with the gene as a whole, as was previously reported for germline control of JAK2 mutation (16).
An important question is to what extent germline variants, including the ones identified here, can impact precision medicine. Thus far, translating cancer GWAS to the clinic has been challenged by the generally small effect sizes of risk variants identified (13). Here, among individuals with cancer, we identified germline alleles that had very large effects on the progression of later somatic events (e.g. a 15q22.2 allele that increases somatic alteration of GNAQ by >10-fold, Fig. 3A). Moreover, a number of the reported associations were with minor germline alleles that are quite common in the human population (e.g. markers at 10p15.1 or 18q21.2 at frequencies >30%, Fig. 3B), or involve cancer genes with very high somatic mutation frequencies, such as PTEN which is mutated in >60% of some cancer types (54). Such interactions may be of particular interest for their ability to stratify cancer patients, although the true utility of these large effect sizes in a multigenic disease setting remains to be determined. Future studies of germline associations with other clinically important somatic phenotypes, such as site of metastasis, measures of aggressiveness, or therapeutic response, may also reveal large networks of informative interactions. Ultimately, the influence of many germline variants on specific somatic changes in tumors suggests it might be possible to anticipate key events during tumor development, enabling a preventative rather than reactionary approach to therapy.
Materials and Methods
Two phase study
Data were obtained from the TCGA and divided into a discovery and a validation cohort based on availability of all data types before or after 05/2014. Distribution of tumor types in each cohort is shown in Fig. S1C. Assignment of each sample to discovery or validation cohorts is provided in Table S4. We acquired genotype, clinical, copy number and somatic mutation data for all available samples. Human genome assembly GRCh37/UCSC hg19 coordinates were used for all genomic data.
Genotypes
Normal (non-tumor) level 2 genotype calls generated from Affymetrix SNP6.0 array intensities using the BirdSuite software (55) were retrieved from the TCGA data matrix. In these files, each SNP was annotated with an allele count (0=AA, 1=AB, 2=BB, -1=missing) and a confidence score between 0 and 1. Genotypes with a score larger than 0.1 (corresponding to an error rate > 10%) were set to missing and the data were reformatted with PLINK v1.9 (56).
Somatic Phenotypes
Information on tumor type was available for all samples. Exome-wide profiles of somatic DNA mutations and genome-wide CNVs were accessed from the Broad Firehose Analysis Pipeline (57) (April 16, 2014 release) (Fig. S1B). Somatic mutations for 1099 validation samples were obtained using MuTect (58) and Somatic Indel Detector from GATK release 2.2-2 (59).
In addition to TCGA CNV calls, we used the PennCNV (v1.0.3) Affymetrix pipeline to call CNVs using default parameters (60). The Affymetrix Power Tools software package was used to generate signal intensity data from raw CEL files. PennCNV was used to split the signal intensity files by individual, generate CNV calls, merge adjacent CNVs, and annotate CNV calls with UCSC hg19 knownGene annotation.
For each patient, a gene was considered mutated if a DNA mutations mapped to a protein sequence change with SNPEffect4.0 (61) (GRCh37.31, canonical transcripts only). A gene was considered CNV altered if it received a GISTIC2.0 (62) thresholded score of -2 or 2 (0 or 4+ with PennAffyCV) corresponding to homozygous deletion or high level amplification similar to previous studies (63). SSMs, CNVs and their union were each recoded as a binary vector for every tumor, denoting the presence or absence of an event for each of 138 cancer genes.
CNV and somatic mutation rates were used as covariates for statistical modeling. Somatic mutation rate was modeled by the nonsynonymous mutations per MB reported in the Firehose MutSig analysis (49) and CNV rate was modeled as the number of CNVs per sample, approximated by the number times each sample appeared in the focal_input.seg.txt file from the Firehose GISTIC2.0 analysis (62). In each case, values could be assigned to most tumor samples (Fig. S1B).
TCGA Discovery Phase
We discarded 322 SNPs with probe names that did not match the hg19 UCSC genome browser Affymetrix track (track: SNP/CNV Arrays, table:snpArrayAffy6). Allele counts were converted to alleles using the definitions in metadata distributed with Birdsuite and negative strand genotypes were flipped to the positive strand using PLINK.
European ancestry samples were identified using the TCGA metadata (Fig. S1A). Genotypes were filtered with PLINK to remove SNPs with call rate < 95%, SNPs with minor allele frequency (MAF) < 1% and individuals with genotype coverage < 95%. Additional samples were dropped due to ambiguous or conflicting gender assignment and unexpectedly high or low rates of heterozygosity. SNPs not in Hardy Weinberg Equilibrium (p < 10-5) and non-autosomal SNPs were discarded. Batch effects associated with processing groups of samples together (plate effects) can lead to bias in estimates of allele frequencies between groups of samples (64). Thus we discarded 10436 additional SNPs demonstrating strong plate associations (p < 10-8). SNPs associated with > 4 plates were retained. After filtering, 706538 out of 906600 SNPs remained. Post association testing, any tumor type associated markers with plate or batch associations (p < 10-4) were excluded from further analysis (Fig. S2).
TCGA Validation Phase
Affymetrix SNP6.0 genotypes for 1789 validation samples were filtered for call rate, coverage and minor allele frequency. SNPs not present in the discovery set were removed and SNPs missing from the discovery set were imputed using PLINK. Validation set genotypes were phased using Beagle v3.3.2, and haplotypes markers were assigned based on agreement of phased SNP sequences with best associated SNP sequence for each discovery set haplotype as determined by the cluster2hap utility and as previously described (65).
Population Stratification
To further control for population substructure, we performed principal component analysis with the combined TCGA and HapMap Phase III populations. We discarded 107 samples that did not cluster closely with HapMap III European populations. HapMap Phase III genotypes (25) were obtained from the NCBI HapMap ftp site and lifted to hg19 using the liftOver utility (66). Genotypes were merged and reduced to a set of independent SNPs by linkage-based filtering using PLINK. The reduced set of 33724 independent SNPs was used to calculate pairwise identity-by-state (IBS) between all individuals. After performing PCA on the IBS matrix, we removed 10 TCGA individuals clustering with Masai and Yoruban samples and 97 individuals clustering more closely with individuals from the Mexican and Gujarati populations.
Haplotype Inference
Haplotypes were inferred using Beagle software v3.3.2 (67), and encoded as binary markers using the psuedomarkers.jar java utility (parameters: edgecount=60 and othercount=10), resulting in 1598830 haplotype markers.
Power Calculations
Power to detect association with gene alteration status was estimated using the Genetic Power Calculator (68) via the ldDesign R package (69). Empirical estimates of case-control ratio were modeled by the number of TCGA samples harboring alterations in each gene. Allele frequency and prevalence were set to 0.01. D' was set to 1 and the same frequency was used for the marker and quantitative trait locus, simulating the best-case scenario where the causal SNP is genotyped.
Discovery Phase Testing
When testing for marker association with tumor type, samples were partitioned using a one versus rest strategy. For marker association with somatic alteration status at each of the 138 cancer genes, samples were partitioned into two groups based on the presence or absence of a somatic alteration (Fig. 1A).
Association testing between all marker-gene and marker-tumor type pairs was performed using a two-sided Fisher's Exact test with PLINK. This test is not affected by class imbalance, which occurs for most phenotypes in this study. Candidate associations were selected based on the “suggestive” GWAS significance threshold, adjusted by the number of phenotypes tested (1×10-5/22 for primary tumor type and 1×10-5/138 for mutation status of cancer genes). P-values were adjusted for inflation using genomic control (Fig. S3, S6). Because allelic tests are biased for SNPs violating Hardy Weinberg Equilibrium, candidate associations were subjected to permutation to obtain empirical p-values; somatic alterations were permuted across samples 1×108 times to generate an empirical null distribution capable of providing a p-value with a resolution of 1×10-8.
Multi-tumor association
To gain power to identify SNPs associated with multiple tumor types, tumors with correlated ORs were tested for association as a group. Specifically, loci receiving at least one weak but insignificant association (P < 0.05) were reevaluated by grouping correlated tumor types. All tumors at the locus with an OR > 1.2 were grouped and a Fisher's Exact Test was performed to determine their combined OR and P-value. This joint P-value was compared to an empirical distribution obtained by permuting tumor type label 1000 times, grouping tumor types with OR > 1.2 and repeating the Fisher's Exact test. Markers were selected for validation if they were among the top 5 most significant tests after permutation, and had a combined OR > 1 and a P-value < 1×10-5.
Controlling for covariates
Firth's penalized logistic regression (R logistf package) (70) was used to control for covariates, including population substructure (1st two principal components PC1 and PC2), individual-specific somatic mutation and/or copy number alteration rates, and gender. For associations with cancer genes, we also controlled for primary tumor type. The Wald test was used to reject the hypothesis that the slope contribution of the genotype in the fitted model was 0 (i.e., that the genotype does not provide information about the phenotype) in the presence of covariates.
Validation
For both gene alteration status and tumor type associations, validation testing was performed using a one-sided Fisher's Exact test consistent with the odds ratio observed in the discovery screen. Candidate associations achieving an empirical false discovery rate (FDR) < 0.5 were considered to be of potential interest are listed in Tables S1 and S3. We considered a marker to have “validated” at an empirical FDR < 0.25. These associations are visualized as a custom track in the UCSC Genome Browser (http://ideker.ucsd.edu/apps/germline/).
Empirical false discovery rate estimation
For each candidate marker-phenotype pair carried forward for validation testing, phenotype labels were permuted 10,000 times. Each of these 10,000 sets of permutated marker-phenotype pairs was then evaluated in the validation cohort. The expected number of false discoveries at a particular significance threshold was estimated as the mean number of marker-phenotype pairs detected at that threshold across the 10,000 permutations. The empirical false discovery rate was estimated as the ratio of false discoveries to the total number of candidates detected at a particular significance threshold. Empirical FDRs were estimated at significance threshold intervals of 0.05.
MutSigCV Analysis
MutSigCV version 1.4 (49) with default parameters and configuration files was used to identify genes with an elevated mutation rate on each germline background. Discovery phase samples were divided into two groups: those with one or more copies of the minor allele, and those with none. MutSigCV was run separately on both groups to identify genes that were mutated at higher frequency than expected according to its gene-specific background mutation model.
Expression Analysis
Expression data (Level 3 normalized data) for all TCGA samples were obtained from the April 16th, 2015 Firehose run (71). To determine a pan-cancer relationship between gene expression and genotype, log2 expression was regressed on minor allele count and tumor type. Significance of effect size was determined by first regressing expression on primary tumor type alone. Variance due to tumor type was removed by summing the expected value over all samples and the residuals after regressing on tumor type. Effect sizes were estimated using a one sided t-test to evaluate the difference in distributions for samples with one or more copies of the minor allele versus samples homozygous for the major allele. T-tests were performed when at least 5 samples were present in both groups.
Alternative Splicing Analysis
TCGA RNASeq data for 95 samples with SF3B1 mutations and 105 controls (Table S5) were processed as previously described (72). The percentage of cryptic reads at each splice site across the genome was calculated for each patient. Only splice sites in the top 85th quartile of coverage were considered for the analyses. Percentages were log transformed and averaged across patients with matching genotypes in order to determine the variation in splicing due to the minor allele and SF3B1 mutation status. Specifically, the number of sites that were differentially spliced between WT and SF3B1 mutant tumors was determined for individuals with one or more copies of the minor allele, and compared to the same statistic for individuals homozygous for the major allele. A site with two-fold change in cryptic splicing was considered differentially spliced. In order to control for the effect of sample size differences when partitioning tumors on germline genotype on identifying differentially spliced junctions, samples with the major allele were down-sampled 1000 times. A two-sample t-test was used to determine whether cryptic splicing occurred at the same frequency for samples with the minor allele and without an SF3B1 mutation as compared to all other samples. To correct for any biases, the observed genotypes were randomly re-assigned 10,000 times and the two-sample t-test re-performed.
Imputation, Locus Annotation and Visualization
A gene was considered to be in cis with a germline marker if it was encoded within 1MB of the marker. Coordinates of promoters, exons and introns were determined from Gencode V19 basic obtained from the UCSC Genome Browser (73). Promoters were assumed to fall within 2KB 5′ of a gene's transcription start site. Enhancer coordinates were obtained from Fantom 5 (74)(robust enhancer set).
Post-association testing, markers were assumed to occur at the same locus if pairwise linkage disequilibrium was sufficiently large (LD; r2 > 0.2) (75). To determine overlap of new markers with existing cancer GWAS SNPs, GWAS SNPs within 1 MB of candidate loci were pulled from the NHGRI GWAS Catalog (downloaded April 2015) (26). Pairwise LD between SNPs at each candidate locus and NHGRI cancer GWAS SNPs was assessed using the SNAP server (76). The NCBI Genome Decoration Page was used for ideogram construction. Published cancer-associated SNPs from the NHGRI GWAS Catalog, and additional SNP markers at loci implicated in this study were imputed using the Michigan Imputation Server (77) (1000 genomes Phase 1v3 reference, Shapeit2). Imputed markers were used for visualizing loci using Locuszoom Software (78). For LocusZoom plot construction, we used default recombination rates, gene locations and NHGRI catalog data. LD values were generated from the 1000 Genomes 2012 European population, and promoter and enhancer regions were determined as described above.
Cell lines
HEK293 cell lines were obtained from the American Type Culture Collection (ATCC) in 2015 and stored in liquid nitrogen vapor. Cells were authenticated using short tandem repeat (STR) fingerprinting within 6 months of freezing. HEK293 cells for the current study were thawed from this stock.
HEK293 control and HEK293 STK11 knockout cell lines were grown and maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin and 250 ng/ml amphotericin B (Sigma Aldrich) at 37°C in humidified air with 5% CO2. Cells were grown to 70-80% confluence prior to re-plating for transfection experiments. STK11 knock out cells were engineered by the CRISPR/Cas9 gene editing system, using the pSpCas9 (BB)-2A-Puro (PX459) V2.0 vector (purchased from Addgene#62988). STK11 knock out was confirmed by western blot (Fig S7A). The corresponding sgRNAs were designed using the CRISPR design website following published protocols (79). Cells were transfected with the CRISPR construct and selected for 3 days using puromycin then replaced by fresh medium to maintain the growth of the cell. Mass culture of cells were confirmed to lack STK11 protein expression by western blotting.
PTEN knock down and GNA11 expression
Twenty-four hours before transfection, HEK 293 cells were plated in 6-well plates at 40% confluence. When indicated, cells were transfected with siRNA targeting PTEN and control siRNA (purchased from Dharmacon, ON-TARGET plus SMART pool) at a final concentration of 10nM, using Turbofect transfection reagent (Thermo Fisher) according to the manufacturer's instructions. The following day, cells were transfected with increasing amounts of pcDNAlll-GNA11 where indicated, to control GNA11 protein expression levels at 0, 0.25, 0.5, 1, 2 and 4 μg/well. Cells lysates were harvested after 24 hours, serum starving for the last 4 hours. Reproducibility of PTEN siRNA knockdown was confirmed by western blot (Fig S7B).
Western blotting
Immunodetection was carried out using antibodies from Cell Signaling Technology against PTEN, total STK11, Phospho-STK11 (pSTK11; Ser428), ribosomal protein S6, phospho-S6 (pS6; Ser240/244), total AMPK and phospho-AMPKα (pAMPKα; Thr172). GAPDH and α-tubulin were included as a loading control. GNA11 (D-17) was detected using a primary antibody from Santa Cruz Biotechnology (sc-394). Secondary horseradish peroxidase-linked goat anti-rabbit IgG antibodies were obtained from Southern Biotech.
Supplementary Material
Statement of Significance.
This study systematically identifies germline variants that directly impact tumor evolution, either by dramatically increasing alteration frequency of specific cancer genes or by influencing the site where a tumor develops.
Acknowledgments
We thank all members of the Ideker Lab, as well as Cherie Ng, Kelly Frazer, Erin Smith, and Richard Kolodner for scientific feedback and discussion. The results presented here are in whole or part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
Financial Support: This work was supported by NIH grants DP5-OD017937 to H.C. and U24 CA184427 to T.I., as well as the Cancer Cell Map Initiative supported by the Fred Luddy Family Foundation.
Footnotes
COI Disclosure Statement: The authors declare no potential conflicts of interest.
Author Contributions: Project conceived by T.I. and H.C. Data acquisition, processing and analysis by H.C. CNV calling by K.F. MutSig analysis by M.H. Expression analysis by H.C., A.G., R.M., J.J. Alt splicing analysis by C.B, R.M., E.V.N, Y.S., E.W., G.Y. mTOR pathway experiments by X.W., S.G. Figures by H.C., R.M. and T.I. Manuscript written by T.I., H.C., R.M., S.L. and J.K.
References
- 1.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–24. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collins FS, Barker AD. Mapping the cancer genome Pinpointing the genes involved in cancer will help chart a new course across the complex landscape of human malignancies. Sci Am. 2007;296(3):50–7. [PubMed] [Google Scholar]
- 4.Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, et al. International network of cancer genome projects. Nature. 2010;464(7291):993–8. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hofree M, Carter H, Kreisberg J, Bandyopadhyay S, Mischel P, Friend S et al. Challenges in identifying cancer genes by analysis of exome sequencing data. Volume In Press: Nature Communications. 2016 doi: 10.1038/ncomms12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene. 2004;23(38):6445–70. doi: 10.1038/sj.onc.1207714. [DOI] [PubMed] [Google Scholar]
- 7.Lu Y, Ek WE, Whiteman D, Vaughan TL, Spurdle AB, Easton DF, et al. Most common ‘sporadic’ cancers have a significant germline genetic component. Hum Mol Genet. 2014;23(22):6112–8. doi: 10.1093/hmg/ddu312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Czene K, Lichtenstein P, Hemminki K. Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish Family-Cancer Database. Int J Cancer. 2002;99(2):260–6. doi: 10.1002/ijc.10332. [DOI] [PubMed] [Google Scholar]
- 9.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 10.Chang CQ, Yesupriya A, Rowell JL, Pimentel CB, Clyne M, Gwinn M, et al. A systematic review of cancer GWAS and candidate gene meta-analyses reveals limited overlap but similar effect sizes. Eur J Hum Genet. 2014;22(3):402–8. doi: 10.1038/ejhg.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galvan A, Ioannidis JP, Dragani TA. Beyond genome-wide association studies: genetic heterogeneity and individual predisposition to cancer. Trends Genet. 2010;26(3):132–41. doi: 10.1016/j.tig.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hosking FJ, Dobbins SE, Houlston RS. Genome-wide association studies for detecting cancer susceptibility. Br Med Bull. 2011;97:27–46. doi: 10.1093/bmb/ldq038. [DOI] [PubMed] [Google Scholar]
- 13.Varghese JS, Easton DF. Genome-wide association studies in common cancers--what have we learnt? Curr Opin Genet Dev. 2010;20(3):201–9. doi: 10.1016/j.gde.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 14.Lu C, Xie M, Wendl MC, Wang J, McLellan MD, Leiserson MD, et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat Commun. 2015;6:10086. doi: 10.1038/ncomms10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanchi KL, Johnson KJ, Lu C, McLellan MD, Leiserson MD, Wendl MC, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun. 2014;5:3156. doi: 10.1038/ncomms4156. doi:1038/ncomms4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campbell PJ. Somatic and germline genetics at the JAK2 locus. Nat Genet. 2009;41(4):385–6. doi: 10.1038/ng0409-385. [DOI] [PubMed] [Google Scholar]
- 17.Liu W, He L, Ramirez J, Krishnaswamy S, Kanteti R, Wang YC, et al. Functional EGFR germline polymorphisms may confer risk for EGFR somatic mutations in non-small cell lung cancer, with a predominant effect on exon 19 microdeletions. Cancer Res. 2011;71(7):2423–7. doi: 10.1158/0008-5472.CAN-10-2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Q, Seo JH, Stranger B, McKenna A, Pe'er I, Laframboise T, et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell. 2013;152(3):633–41. doi: 10.1016/j.cell.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen QR, Hu Y, Yan C, Buetow K, Meerzaman D. Systematic genetic analysis identifies Cis-eQTL target genes associated with glioblastoma patient survival. PLoS One. 2014;9(8):e105393. doi: 10.1371/journal.pone.0105393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The Cancer Genome Atlas Research Network. http://cancergenome.nih.gov/
- 21.de Bakker PI, Yelensky R, Pe'er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37(11):1217–23. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 22.Sham PC, Rijsdijk FV, Knight J, Makoff A, North B, Curtis D. Haplotype association analysis of discrete and continuous traits using mixture of regression models. Behav Genet. 2004;34(2):207–14. doi: 10.1023/B:BEGE.0000013734.39266.a3. [DOI] [PubMed] [Google Scholar]
- 23.Shim H, Chun H, Engelman CD, Payseur BA. Genome-wide association studies using single-nucleotide polymorphisms versus haplotypes: an empirical comparison with data from the North American Rheumatoid Arthritis Consortium. BMC Proc. 2009;3(7):S35. doi: 10.1186/1753-6561-3-s7-s35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marchini J, Cardon LR, Phillips MS, Donnelly P. The effects of human population structure on large genetic association studies. Nat Genet. 2004;36(5):512–7. doi: 10.1038/ng1337. [DOI] [PubMed] [Google Scholar]
- 25.Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA, Peltonen L, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467(7311):52–8. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42(Database issue):D1001–6. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saito S, Morita K, Hirano T. High frequency of common DNA copy number abnormalities detected by bacterial artificial chromosome array comparative genomic hybridization in 24 breast cancer cell lines. Hum Cell. 2009;22(1):1–10. doi: 10.1111/j.1749-0774.2008.00061.x. [DOI] [PubMed] [Google Scholar]
- 28.Auvinen P, Rilla K, Tumelius R, Tammi M, Sironen R, Soini Y, et al. Hyaluronan synthases (HAS1-3) in stromal and malignant cells correlate with breast cancer grade and predict patient survival. Breast Cancer Res Treat. 2014;143(2):277–86. doi: 10.1007/s10549-013-2804-7. [DOI] [PubMed] [Google Scholar]
- 29.Cipriano R, Miskimen KL, Bryson BL, Foy CR, Bartel CA, Jackson MW. Conserved oncogenic behavior of the FAM83 family regulates MAPK signaling in human cancer. Mol Cancer Res. 2014;12(8):1156–65. doi: 10.1158/1541-7786.MCR-13-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalashnikova EV, Revenko AS, Gemo AT, Andrews NP, Tepper CG, Zou JX, et al. ANCCA/ATAD2 overexpression identifies breast cancer patients with poor prognosis, acting to drive proliferation and survival of triple-negative cells through control of B-Myb and EZH2. Cancer Res. 2010;70(22):9402–12. doi: 10.1158/0008-5472.CAN-10-1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klopfleisch R, Gruber AD. Derlin-1 and stanniocalcin-1 are differentially regulated in metastasizing canine mammary adenocarcinomas. J Comp Pathol. 2009;141(2-3):113–20. doi: 10.1016/j.jcpa.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 32.Gai M, Bo Q, Qi L. Epigenetic down-regulated DDX10 promotes cell proliferation through Akt/NF-κB pathway in ovarian cancer. Biochemical and biophysical research communications. 2016;469(4):1000–5. doi: 10.1016/j.bbrc.2015.12.069. [DOI] [PubMed] [Google Scholar]
- 33.Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38(8):873–5. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 34.Gudmundsson J, Sulem P, Gudbjartsson DF, Jonasson JG, Sigurdsson A, Bergthorsson JT, et al. Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations. Nat Genet. 2009;41(4):460–4. doi: 10.1038/ng.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landa I, Ruiz-Llorente S, Montero-Conde C, Inglada-Pérez L, Schiavi F, Leskelä S, et al. The variant rs1867277 in FOXE1 gene confers thyroid cancer susceptibility through the recruitment of USF1/USF2 transcription factors. PLoS Genet. 2009;5(9):e1000637. doi: 10.1371/journal.pgen.1000637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andrews AJ, Chen X, Zevin A, Stargell LA, Luger K. The histone chaperone Nap1 promotes nucleosome assembly by eliminating nonnucleosomal histone DNA interactions. Mol Cell. 2010;37(6):834–42. doi: 10.1016/j.molcel.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nenasheva VV, Kovaleva GV, Uryvaev LV, Ionova KS, Dedova AV, Vorkunova GK, et al. Enhanced expression of trim14 gene suppressed Sindbis virus reproduction and modulated the transcription of a large number of genes of innate immunity. Immunol Res. 2015;62(3):255–62. doi: 10.1007/s12026-015-8653-1. [DOI] [PubMed] [Google Scholar]
- 38.Huang W, Ghisletti S, Saijo K, Gandhi M, Aouadi M, Tesz GJ, et al. Coronin 2A mediates actin-dependent de-repression of inflammatory response genes. Nature. 2011;470(7334):414–8. doi: 10.1038/nature09703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tcheandjieu C, Lesueur F, Sanchez M, Baron-Dubourdieu D, Guizard AV, Mulot C, et al. Fine-mapping of two differentiated thyroid carcinoma susceptibility loci at 9q22.33 and 14q13.3 detects novel candidate functional SNPs in Europeans from metropolitan France and Melanesians from New Caledonia. Int J Cancer. 2016;139(3):617–27. doi: 10.1002/ijc.30088. [DOI] [PubMed] [Google Scholar]
- 40.He H, Li W, Liyanarachchi S, Srinivas M, Wang Y, Akagi K, et al. Multiple functional variants in long-range enhancer elements contribute to the risk of SNP rs965513 in thyroid cancer. Proc Natl Acad Sci U S A. 2015;112(19):6128–33. doi: 10.1073/pnas.1506255112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nikitski A, Saenko V, Shimamura M, Nakashima M, Matsuse M, Suzuki K, et al. Targeted Foxe1 Overexpression in Mouse Thyroid Causes the Development of Multinodular Goiter But Does Not Promote Carcinogenesis. Endocrinology. 2016;157(5):2182–95. doi: 10.1210/en.2015-2066. [DOI] [PubMed] [Google Scholar]
- 42.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gonzalez-Perez A, Lopez-Bigas N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012;40(21):e169. doi: 10.1093/nar/gks743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499(7457):172–7. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shoushtari AN, Carvajal RD. GNAQ and GNA11 mutations in uveal melanoma. Melanoma Res. 2014;24(6):525–34. doi: 10.1097/CMR.0000000000000121. [DOI] [PubMed] [Google Scholar]
- 46.Iglesias-Bartolome R, Martin D, Gutkind JS. Exploiting the head and neck cancer oncogenome: widespread PI3K-mTOR pathway alterations and novel molecular targets. Cancer Discov. 2013;3(7):722–5. doi: 10.1158/2159-8290.CD-13-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zbuk KM, Eng C. Hamartomatous polyposis syndromes. Nat Clin Pract Gastroenterol Hepatol. 2007;4(9):492–502. doi: 10.1038/ncpgasthep0902. [DOI] [PubMed] [Google Scholar]
- 48.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9(8):563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Melichar B, Nash MA, Lenzi R, Platsoucas CD, Freedman RS. Expression of costimulatory molecules CD80 and CD86 and their receptors CD28, CTLA-4 on malignant ascites CD3+ tumour-infiltrating lymphocytes (TIL) from patients with ovarian and other types of peritoneal carcinomatosis. Clin Exp Immunol. 2000;119(1):19–27. doi: 10.1046/j.1365-2249.2000.01105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leslie NR, Downes CP. PTEN function: how normal cells control it and tumour cells lose it. Biochem J. 2004;382(Pt 1):1–11. doi: 10.1042/BJ20040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, et al. Cancer-associated protein kinase C mutations reveal kinase's role as tumor suppressor. Cell. 2015;160(3):489–502. doi: 10.1016/j.cell.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marcus PM, Freedman AN, Khoury MJ. Targeted Cancer Screening in Average-Risk Individuals. Am J Prev Med. 2015;49(5):765–71. doi: 10.1016/j.amepre.2015.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008;40(10):1253–60. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Broad Institute TCGA Genome Data Analysis Center: Analysis-ready standardized TCGA data from Broad GDAC Firehose stddata__2014_06_14 run. Broad Institute of MIT and Harvard; 2015. [DOI] [Google Scholar]
- 58.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486(7403):405–9. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17(11):1665–74. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Baets G, Van Durme J, Reumers J, Maurer-Stroh S, Vanhee P, Dopazo J, et al. SNPeffect 4.0: on-line prediction of molecular and structural effects of protein-coding variants. Nucleic Acids Res. 2012;40(Database issue):D935–9. doi: 10.1093/nar/gkr996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12(4):R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45(10):1127–33. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pluzhnikov A, Below JE, Konkashbaev A, Tikhomirov A, Kistner-Griffin E, Roe CA, et al. Spoiling the whole bunch: quality control aimed at preserving the integrity of high-throughput genotyping. Am J Hum Genet. 2010;87(1):123–8. doi: 10.1016/j.ajhg.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang QS, Browning BL, Browning SR. Genome-wide haplotypic testing in a Finnish cohort identifies a novel association with low-density lipoprotein cholesterol. Eur J Hum Genet. 2014 doi: 10.1038/ejhg.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuhn RM, Haussler D, Kent WJ. The UCSC genome browser and associated tools. Brief Bioinform. 2013;14(2):144–61. doi: 10.1093/bib/bbs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81(5):1084–97. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19(1):149–50. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- 69.Ball R, Ball MR. Package ‘ldDesign’. 2013 [Google Scholar]
- 70.Heinze G. A comparative investigation of methods for logistic regression with separated or nearly separated data. Statistics in medicine. 2006;25(24):4216–26. doi: 10.1002/sim.2687. [DOI] [PubMed] [Google Scholar]
- 71.Broad Institute TCGA Genome Data Analysis Center Analysis-ready standardized TCGA data from Broad GDAC Firehose stddata__2015_04_02 run. Broad Institute of MIT and Harvard; 2015. [DOI] [Google Scholar]
- 72.DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, et al. Transcriptome sequencing reveals potential mechanism of cryptic 3' splice site selection in SF3B1-mutated cancers. PLoS Comput Biol. 2015;11(3):e1004105. doi: 10.1371/journal.pcbi.1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32(Database issue):D493–6. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507(7493):455–61. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Freedman ML, Monteiro AN, Gayther SA, Coetzee GA, Risch A, Plass C, et al. Principles for the post-GWAS functional characterization of cancer risk loci. Nat Genet. 2011;43(6):513–8. doi: 10.1038/ng.840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O'Donnell CJ, de Bakker PI. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24(24):2938–9. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284–7. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–7. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Krzywinski MI, Schein JE, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: An information aesthetic for comparative genomics. Genome Research. 2009 doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.