

Abstract
Hypoxia triggers a wide range of protective responses in mammalian cells, which are mediated through transcriptional and post-translational mechanisms. Redox signaling in cells by reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) occurs through the reversible oxidation of cysteine thiol groups, resulting in structural modifications that can change protein function profoundly. Mitochondria are an important source of ROS generation, and studies reveal that superoxide generation by the electron transport chain increases during hypoxia. Other sources of ROS, such as the NAD(P)H oxidases, may also generate oxidant signals in hypoxia. This review considers the growing body of work indicating that increased ROS signals during hypoxia are responsible for regulating the activation of protective mechanisms in diverse cell types.
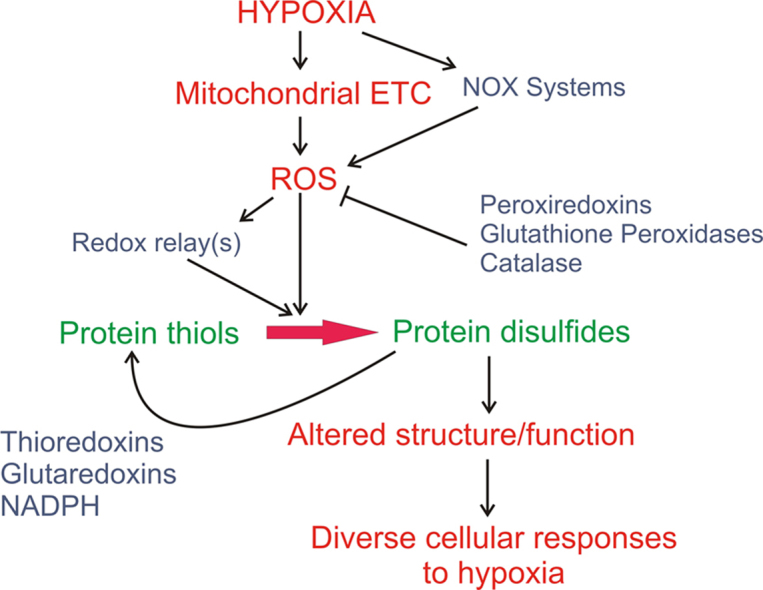
Graphical abstract
1. Introduction
Mammalian cells utilize a wide range of biochemical pathways and systems as they seek to maintain homeostasis, repair or replace damaged cellular components, carry out organ-specific functions, and respond to environmental signals or stresses. Regulation of these pathways is achieved through a variety of cell signaling systems, including protein post-translational changes such as phosphorylation, acetylation, nitrosation, ubiquitinylation and other modifications, as well as signaling mechanisms regulated by Ca2+. Another class of intracellular signaling occurs through reduction-oxidation (redox)-dependent reactions, and cells utilize an extensive array of systems that convey information and control through chemical redox modification of proteins, lipids, and in some cases, DNA.
The essence of redox biochemistry involves the transfer of electrons or reducing equivalents between biomolecules. For example, in intermediary metabolism, metabolic intermediates are oxidized by dehydrogenases that remove electrons during glycolysis or the Krebs cycle; these are then transferred to the mitochondrial electron transport chain by nicotinamide adenine dinucleotide (NADH), flavin adenine dinucleotide (FADH2) or flavin mononucleotide (FMNH2) carriers. Redox reactions are also involved in cell signaling pathways; these frequently involve the generation of reactive oxygen (ROS) or nitrogen (RNS) species that interact with target molecules resulting in an alteration in the biological function of those targets [1]. ROS and RNS can engage in redox reactions, potentially acting as either electron donors or as acceptors. At high concentrations, ROS or RNS can induce oxidant stress that disrupts cell functions or induces tissue damage, such as the oxidative burst observed during ischemia-reperfusion injury. However, a large body of data indicates that lower concentrations of ROS or RNS participate in redox reactions involved in normal physiological cell processes. The regulation of this redox signaling is achieved by controlling the rate of generation ROS or RNS, the location of that generation, the scavenging of the reactive molecules, and by controlling the expression or activity of enzymes that reverse the effects of the oxidant on their molecular targets.
It is important to note that interaction between cellular redox systems generally is mediated by enzymes rather than by spontaneous chemical interaction. For example, although hydrogen peroxide (H2O2, a ROS molecule) and NADH (an electron carrier) can both engage in redox reactions, their mutual interaction is not biologically important because no enzyme system facilitates such a direct interaction. Hence, a high concentration of NADH is not useful for scavenging H2O2, and high levels of H2O2 do not oxidize NADH. By contrast, enzymes such as glutathione reductase can couple the oxidation of nicotinamide adenine dinucleotide phosphate (NADPH) to the reduction of oxidized glutathione. As reduced glutathione (GSH) is an important component of the cell's antioxidant machinery, this allows NADPH to function as an important cofactor in the maintenance of cellular thiol redox balance and ROS scavenging. Another important principle of redox biology is that the cellular concentration of oxidants (say, superoxide or H2O2) can vary widely depending on cellular activity, the degree of stress, and the ability of the cell to maintain homeostasis during the redox stress. Redox signaling or stress can span a wide spectrum, ranging from very low levels of oxidation in quiescent cells to potentially lethal levels in cells exposed to high O2 concentrations (e.g., >50% O2)or subjected to ischemia-reperfusion stress. At high concentrations, oxidants such as H2O2 may react with targets such as protein thiol groups that would not be affected at lower concentrations. Finally, the concept of “cellular redox status” started as the simplistic notion that cells function as homogenous redox systems. It is now recognized that redox conditions are regulated independently in different subcellular compartments, with localized control of ROS generation and compartment-specific expression of antioxidant enzyme systems. And in some cases, redox is regulated in localized subdomains within those compartments. For example, protein thiol redox status in the cytosol is normally highly reduced, yet localized oxidant signals may be generated near the plasma membrane when extracellular growth factor ligand binding leads to activation of ROS-generating enzyme systems coupled to the mitogen receptor. By contrast, the endoplasmic reticulum (ER) maintains a highly oxidizing environment needed for proper protein folding. The nucleus normally maintains a relatively reduced environment, as thiol oxidant stress can interfere with zinc finger domains that are involved in protein-DNA interactions. Hence, recognition of the compartment being assessed experimentally is important for interpreting the measurements obtained in studies of redox biology and signaling.
2. Signaling by ROS in mammalian cells
The principal ROS species produced in cells are superoxide (O2-) and H2O2. Superoxide is a free radical generated when molecular oxygen acquires a single unpaired electron. Superoxide can be generated enzymatically by members of the NAD(P)H oxidase family, or non-specifically through the interaction of O2 with flavin groups, iron-sulfur centers, heme groups, or non-heme iron (see below) [2]. As an anion, superoxide cannot traverse lipid membranes in the absence of anion channels. Once generated, superoxide can affect enzyme function by disrupting iron-sulfur centers, or interacting with heme- and non-heme cofactors. Important physiological roles have been identified for superoxide signaling in mammalian cells. For example, the oxidation status of the [4Fe4S] iron sulfur cluster in human DNA primase can be modified by superoxide. The redox status acts as a reversible on/off switch to regulate its DNA binding, indicating an important role for redox regulation of DNA replication [3].
In mammalian cells, H2O2 is probably more important than superoxide in terms of the number of known signaling functions. Unlike superoxide, H2O2 can interact with the cysteine mercapto group, causing progressive oxidation to the disulfide, sulfenic, sulfinic, and sulfonic redox states [1]. Disulfide formation represents the lowest degree of oxidation, and is important biologically because it can alter protein function by inducing significant structural changes [4]. Importantly, thiol oxidation is easily reversed by enzymatic mechanisms, making this alteration suitable in terms of a signaling function [5]. Higher states of thiol oxidation can also be important, as in the case of phosphatases that utilize a reactive cysteine at the catalytic site. Attack of that thiol by H2O2 can render the phosphatase inactive, thereby increasing phosphorylation abundance of its target proteins [6], [7]. Lower states of oxidation are easily reversible, but higher states of thiol oxidation may cause prolonged or possibly irreversible inhibition of phosphatases. As phosphatases are important for regulating phosphorylation states of lipids and proteins, this redox-dependent inhibition can have profound effects on cell function.
A conceptual hurdle in understanding how H2O2 functions as a signaling molecule relates to its selectivity and specificity for the intended target proteins. H2O2 is diffusible, and is generated upon dismutation of superoxide either spontaneously or enzymatically by members of the superoxide dismutase (SOD) family of enzymes [8]. It can cross cell membranes via aquaporin channels, and can therefore access multiple subcellular compartments. Given these characteristics, one might ask why H2O2 signaling doesn’t cause accidental oxidation of many non-specific targets along its journey toward the intended target. One answer is that some sources of H2O2 – such as members of the NADPH oxidase (NOX) family – may be anchored to membrane sites close to where redox signaling is needed [9]. For example, NOX isoforms near the plasma membrane are activated by growth factor receptor ligand binding, which may restrict possible targets to those near the inner membrane leaflet [10].
Additional insight into this issue comes from in vitro studies where recombinant proteins are subjected to oxidative attack by addition of exogenous oxidants such as H2O2. For example, the fluorescent protein, roGFP, is a variant of green fluorescent protein (GFP) containing two cysteine residues in the outer surface of the barrel [11], [12]. Oxidation of one cysteine thiol leads to disulfide formation, which alters the fluorescence behavior of the protein [13]. This allows roGFP to act as thiol redox sensor. If purified recombinant roGFP is subjected to oxidant treatment in a test tube, molar concentrations of H2O2 are required to cause oxidation of the protein. By contrast, when expressed in a cell, roGFP begins to oxidize after addition of as little as 10 micromolar H2O2. The difference between in vitro and in vivo sensitivity arises from the involvement of an intermediary protein that mediates a redox relay system. In this case, glutaredoxin, an antioxidant protein in the cell, is easily oxidized by micromolar concentrations of H2O2. Oxidized glutaredoxin then interacts with reduced roGFP to cause its oxidation, yielding reduced glutaredoxin and oxidized roGFP [14]. Had reduced glutaredoxin been added to the in vitro experiment, the recombinant roGFP would have been oxidized by addition of very low concentrations of H2O2. Possibly the first involvement of such a relay described in S. cerevisiae where oxidation of YAP1, a redox-sensitive transcription factor, was found to require the antioxidant enzyme ORP1, an ortholog of glutathione peroxidase-4 [15]. In the absence of ORP1, yeast cells treated with H2O2 fail to activate expression of antioxidant genes under the control of YAP1, whose oxidation is required for activation. In mammalian cells, peroxiredoxin-2 has been shown to participate in a redox relay that leads to STAT3 inactivation in response to H2O2 stress [16]. Thus, specificity of oxidation targets is frequently achieved by transducing the H2O2 signal with a peroxidase that is easily oxidized by low concentrations, and subsequently engages in a redox relay with a small number of highly selected targets that are resistant to non-specific oxidation even at high H2O2 concentrations. These redox intermediaries act in a reverse direction to reduce the target once the H2O2 signal disappears, thereby participating in both activation and inactivation of target proteins. By this mechanism, low signaling concentrations of oxidants such as H2O2 can effectively mediate redox modifications of selective targets, thus avoiding the widespread non-specific oxidation of proteins that would occur with the use of higher concentrations needed to directly oxidize the targets.
3. NAD(P)H oxidases and hypoxia signaling
NAD(P)H oxidases are multimeric membrane-bound complexes that transfer electrons from reduced dinucleotides such as NADH or NADPH to molecular oxygen to produce superoxide or H2O2. In hypoxia, NADPH oxidases may synergize with mitochondria in the production of ROS that are involved in the activation of hypoxia responses [17]. NADPH oxidase consists of multiple subunits, including cytochrome b558, the gp91phox (NOX2), p22phox, cytosolic p47phox and p67phox proteins, along with regulatory proteins such as p40, rac-1, rac-2, and/or rap1A. NAD(P)H-linked oxidase systems have been suggested to function as O2 sensors [18]. Each NOX isoform contains a redox-active subunit and at least one other subunit [18], [19].
While the phagocytic NADPH oxidase activity is regulated by phosphorylation of a NOX2 subunit by protein kinase C, regulation of non-phagocytic NOX isoforms (NOX1, NOX3, NOX4, NOX5, DUOX1 and DUOX2) is less well understood. It is generally assumed that NOX systems could function in O2 sensing based on the assumption that hypoxia should cause a decrease in superoxide production, as O2 is a required substrate for superoxide generation [20]. Presumably, this would result in a redox shift of protein thiols in, for example, membrane voltage-dependent potassium channels (Kv), toward a more reduced state. If these were involved in mediating hypoxia responses, the suggestion was that this response could then activate the downstream response [21]. Of course, to be involved in hypoxia responses the NOX subunits would need to be expressed in the O2-sensitive cells. In that regard, expression has been reported in carotid body glomus cells [22], vascular cells [23], and endothelium [24], all of which are known to be involved in hypoxia responses [25], [26], [27] Direct involvement of these oxidases in hypoxia sensing must be confirmed in genetic deletion models, to determine whether the response to hypoxia is preserved or abolished. Archer et al. tested the hypoxic pulmonary vasoconstrictor response (HPV) in mice carrying a whole-body genetic deletion of the NOX2 subunit gp91phox [28]. Those mice retained their pulmonary vasoconstrictor response to alveolar hypoxia, revealing that the HPV response does not require NOX2. In humans, chronic granulomatous disease is a disorder caused by genetic loss of NOX2 function. While these individuals are vulnerable to infection because of the loss in immune cell antimicrobial function, there is no evidence that oxygen sensing – which is required for embryonic development - is significantly impaired, suggesting that NOX2 is not required for hypoxia sensing by cells. Weissmann et al. measured hypoxic responses in mice with a genetic deletion of the p47phox subunit, and found that the lung vasoconstrictor response was attenuated but still present [29]. Collectively, these results indicate that NOX systems may participate in the hypoxia signaling pathways in the pulmonary circulation but are not be required for triggering the response. However, NOX4 was subsequently show to be dispensable for the development of pulmonary hypertension during chronic hypoxia in rodents [30].
Other NOX systems such as NOX4 have been suggested to function in hypoxia sensing through the generation of superoxide and H2O2 [31]. According to that model, activity of NOX4 is proportional to the local O2 concentration, resulting in a decrease in H2O2 formation under low oxygen conditions. Yet curiously, NOX4 expression is upregulated under hypoxia by the transcription factor HIF-1 [32]. By contrast to the model of Nisimoto et al., other studies suggest that NOX 4 may play a facilitating role in augmenting ROS generation during hypoxia [33], [34]. In either case, although mice with homozygous deletion of the NOX4 gene are vulnerable to heart injury in response to arterial pressure overload, embryonic development, which requires an intact hypoxia sensing response, is preserved. These observations suggest that NOX4 may contribute to redox signaling involved in some, but not all responses to hypoxia.
4. Mitochondrial ROS production by the ETC
The mitochondrial electron transport chain (ETC) shuttles electrons obtained during intermediary metabolism (glycolysis and in the Krebs cycle) to O2. Electron transfer steps at complex I, III and IV are coupled to proton translocation across the inner membrane. This creates an electrochemical gradient that is used by complex V to synthesize ATP from ADP and inorganic phosphate. Mitochondria have been known to generate ROS for more than 50 years [35]. Sources of mitochondrial ROS other than the ETC have also been described [36]. The production of ROS by mitochondria was initially viewed as a potentially damaging oxidant process [35], [37]. However, more recent evidence suggests that mitochondria-derived ROS can act as signals that activate both transcriptional and post-translational responses to hypoxia in diverse cell types.
Superoxide can be generated at complex I [38], [39], [40], [41], II [42] or III [43], [44], [45], [46], [47], [48], [49], by the release of electrons from iron-sulfur groups, flavin cofactors, or from ubisemiquinone, a free radical generated in the Q cycle of complex III. Depending on the particular site of generation along the ETC, superoxide can be release into the matrix or the intermembrane space. SOD is expressed in each of these compartments, facilitating H2O2 generation. Complex IV has never been shown to release ROS, largely because it binds O2 with very high affinity while electrons are being sequentially transferred from cytochrome c [50]. After the fourth electron has been transferred, the fully reduced molecule is finally released as H2O.
In the ETC, complexes I and II deliver pairs of electrons to the mobile electron carrier ubiquinone, yielding ubiquinol. Ubiquinol then diffuses in the membrane to reach the Qo site in Complex III, where the two electrons are removed sequentially. The first electron is passed to the Rieske Iron-Sulfur Protein (RISP) in Complex III, which subsequently passes the electron to cytochrome c1, to cytochrome c, and finally to cytochrome oxidase. Removal of the first electron from ubiquinol by RISP generates the transient free radical, ubisemiquinone, at the Qo site. Normally, the second electron is rapidly removed by the b cytochromes, thereby returning ubiquinone to the membrane pool. However, if the removal of the unpaired electron from ubisemiquinone is delayed, superoxide may be generated when the electron is instead captured by O2. The lifetime of the ubisemiquinone radical at complex III represents a potential mechanism for controlling ROS generation at the Qo site. Interventions that prolong the lifetime of ubisemiquinone lead to a marked increase in superoxide formation at the Qo site. The toxin antimycin A, for example, prevents removal of the electron from ubisemiquinone and causes a large increase in superoxide generation at the Qo site [43].
Mitochondrial toxins can either increase or decrease ROS generation, depending on where they act. For example, rotenone acts to block the downstream end of complex I, which can lead to increased superoxide production from that complex as electrons (derived from NADH oxidation) remain on flavin or iron-sulfur groups and have nowhere else to go [38]. At the same time, rotenone would tend to decrease ROS production from complex III because it would inhibit the influx of electrons from complex I. Myxothiazol inhibits the influx of electrons into complex III, which blocks the formation of superoxide by that complex. However, by causing a backup of electrons in complex I and II, myxothiazol has the potential to augment ROS generation from those earlier sites. Antimycin A inhibits the binding of ubiquinone at the downstream side of complex III, and thereby prolongs the lifetime of ubisemiquinone at the entry site. This augments ROS generation by complex III, and may also increase ROS generation by complexes I and II because of the electron backup caused by loss of compex III function [39]. Hence, understanding how disruptions of the ETC may affect ROS generation is complex.
5. ROS generation and redox signaling by the mitochondrial ETC in hypoxia
Hypoxia stimulates oxidant production by the ETC, with studies implicating complexes I and III [51], [52]. This increase is paradoxical, in the sense that the concentration of O2, a substrate for superoxide generation, is lessened [53]. The ROS arising from the electron transport chain transit through the intermembrane space to reach the cytosol where they function as redox second messengers. Using a redox sensor such as roGFP expressed in the cytosol, one can detect O2-dependent increases in the oxidation signal as the cells are subjected to progressively more severe levels of hypoxia (Fig. 1).
Fig. 1.

Cytosolic oxidant stress in pulmonary arterial smooth muscle cells, as assessed using roGFP to detect changes in thiol oxidation. Superfusion with buffer equilibrated to different O2 tensions reveals that hypoxia results in increased oxidant signaling, with the greatest increases at the lowest O2 levels.
While mitochondrial oxidants can certainly contribute to tissue oxidant damage [35], [37] in conditions such as ischemia-reperfusion injury [54], [55], [56], [57], [58], [59] or other disorders [60], [61], a growing body of work reveals that mitochondrial oxidants can also function as redox signals in response to cellular stresses [62] including hypoxia [63], [64]. Changes in mitochondrial redox status could conceivably be a mechanism for altering the generation of ROS, as backup of electrons along the ETC could favor the generation of superoxide even if the PO2 were decreased. The redox signal resulting from the generation of ROS production could then alter cysteine thiol redox status, either directly or via a redox relay system as described above.
The first demonstration that mitochondrial ROS participate in transcription factor activation was reported by Chandel et al., who found that hypoxia-induced ROS signals from mitochondria trigger the activation of hypoxic transcriptional responses through the inhibition of HIF prolyl hydroxylase (PHD2), a negative regulator of HIF-α stability [63], [65]. While the specific pathways linking hypoxia-induced increases in ROS to HIF prolyl hydroxylase are still not fully understood, one possible mechanism relates to the interaction of ROS with the Fe2+ associated with PHD2 [66]. Exogenous oxidants have been shown to inhibit asparaginyl hydroxylase (Factor Inhibiting HIF, FIH); this is a related enzyme that functions as a negative regulator of HIF transcriptional activity under normoxic conditions [67]. In the regulation of HIF signaling, ROS signals could inhibit PHD2 and FIH directly [67], or instead act by regulating post-translational modifications of PHD2 and FIH that inhibit their functions allosterically.
A growing body of data indicates that hypoxia increases ROS generation by complex III, possibly by increasing the ubisemiquinone lifetime at that site. Superoxide generated inside the inner membrane should be ejected into the intermembrane space (IMS) by the strong electrical field within the membrane; the ROS signal can then migrate from the IMS to the cytosol [2]. Genetic studies implicating complex III were reported by Guzy et al., who assessed ROS production during hypoxia using a FRET-based redox sensor to assess thiol oxidation [68]. They observed increases in oxidation of the probe during hypoxia, which were attenuated when expression of the Rieske Iron-Sulfur Protein (RISP) was suppressed using RNA interference. Deletion of RISP prevents ROS generation at complex III and also abolishes electron transport and oxygen consumption by the cell. Mansfield and colleagues also studied ROS production in embryonic cells from mice lacking cytochrome c [69], which is also required for ETC function and oxygen consumption. In those cells, the absence of cytochrome c caused the RISP protein in complex III to remain reduced, thereby preventing the generation of ubisemiquinone at the Qo site of complex III. Deletion of RISP or cytochrome c blocked the generation of an oxidant signal during hypoxia, and inhibited the stabilization of HIF-1α. However, these cells were still able to stabilize HIF-α in response to PHD2 pharmacological inhibition by dimethyloxaloylglycine (DMOG), indicating that mitochondria were responsible for detection of hypoxia. Thus, electron transfer through complex III is critical for the redox signaling of hypoxia in cells.
Further work implicating complex III in oxidant signal generation during hypoxia was reported by Orr et al., who performed a screen to identify small molecules that inhibit superoxide generation by complex III without affecting electron transport or mitochondrial oxidative phosphorylation (OXPHOS). The compounds they identified were found to inhibit cellular responses to hypoxia, including HIF-1α stabilization [6].
A critical test of the mitochondrial ROS hypothesis requires an ability to assess oxidant status in subcellular compartments. Studies by Waypa et al. addressed this by targeting expression of roGFPs, described above, to different subcellular compartments [70]. Experimentally, roGFP is expressed in a cell and is excited with light at two different wavelengths (405 and 488 nm). Emitted light at 525 nm then provides a ratiometric assessment of its oxidation that is independent of protein levels or excitation intensity. Redox status of the protein is reversible, so the sensor can be calibrated by applying chemical reducing and oxidizing agents. This is accomplished at the end of the experiment by adding dithiothreitol (to reduce thiols) followed by aldrithiol (to oxidize thiols). The ratios corresponding to maximum reduction and maximum oxidation so obtained are then used to calculate the redox status during the study (Fig. 2) [71]. This is analogous to the use of ratiometric probes used to assess intracellular Ca2+.
Fig. 2.

Use of redox-sensitive proteins to assess thiol redox status in live cells. Thiol oxidation and reduction produce reciprocal changes in emission when excited at two different wavelengths. Ratiometric measurements in the cells can be calibrated at the end of the experiment using chemical oxidizing (aldrithiol) and reducing (dithiothreitol) agents, yielding a percent oxidation.
Limiting expression of the protein to specific subcellular compartments is accomplished using targeting sequences. For example, roGFP expression can be directed to the mitochondrial matrix by appending the mitochondrial targeting sequence from cytochrome oxidase subunit IV [70]. To express the sensor in the intermembrane space, it was appended to the carboxy terminus of glycerol phosphate dehydrogenase (GPD), a protein embedded in the inner membrane whose lipophobic carboxy tail resides in the intermembrane space. Electron micrographs of immunogold particles directed at the protein confirm the expected subcellular localization [70].
Pulmonary arterial smooth muscle cells are intrinsically O2-sensitive and respond to hypoxia by contracting in a Ca2+-dependent manner. Waypa et al. examined redox responses to hypoxia in the cytosol, IMS, and matrix compartments using the targeted roGFP sensors. Differences in baseline oxidation were noted during normoxia, with oxidation in the matrix > IMS > cytosol. During acute hypoxia, oxidation increased in the cytosol and in the IMS [72]. However, oxidation in the matrix decreased during acute hypoxia. These findings reveal several important points. First, significant differences in basal redox status are evident in different subcellular sites. Second, hypoxia increases thiol oxidation in the IMS and cytosol, consistent with a release of superoxide from the inner membrane. Important differences in the redox response to hypoxia occur across subcellular compartments, as some undergo oxidation and others do not. What effect does this redox response have on the increase in Ca2+ during hypoxia? They found that ROS scavengers such as over-expression of catalase or the SOD-catalase mimetic EUK134 [73] prevented the increase in cytosolic Ca2+, revealing that increases in ROS act in a signal transduction system that triggers the response to hypoxia in these cells [74], [75], [76].
In studies using mice with conditional deletion of RISP, Waypa et al. found that genetic deletion in smooth muscle cells abolished hypoxia-induced increases in roGFP oxidation in the cytosol and mitochondrial IMS of pulmonary artery smooth muscle cells, while it abolished the acute hypoxia-induced increases in cytosolic Ca2+ in the same cell type [72]. However, addition of exogenous H2O2 elicited increases in cytosolic Ca2+, indicating that hypoxia-induced ROS signals are required to trigger the increases in Ca2+ that mediate vasoconstriction. Indeed, adult mice with smooth muscle-specific deletion of RISP showed an abrogation of the normal hypoxia-induced increase in pulmonary arterial pressure, as estimated from right ventricular systolic pressure measurements. Collectively these observations identify an important role for mitochondrial oxidant signals in regulating the pulmonary vascular response to hypoxia.
Complex I is a potential source of superoxide generation, principally at flavin-linked sites (FMN) and iron-sulfur centers [77]. ROS generation by complex I is highly dependent on the mitochondrial membrane potential, and conditions that hyperpolarize mitochondria can augment superoxide production via reverse electron flux from the ubiquinone pool into the complex [78]. In terms of hypoxia responses, ROS generation from complex I has been implicated in signaling within the carotid body, as detailed in the following section.
6. Redox regulation of hypoxic responses in diverse tissues
The carotid body is an organelle located at the bifurcation of the common carotid artery and is known to function as a sensor of arterial blood O2 tension. Type I (glomus) cells likely function as the sensory elements of this organ, based on their expression of K+ channels that become rapidly inactivated during acute hypoxia [79]. A key mechanistic question relates to how the potassium channels are regulated by O2 tension, as there is little evidence that the channels themselves are being directly regulated by interaction with O2. A study by Fernandez-Aguera et al. provided evidence that redox signals from the mitochondria may contribute to this regulation [52]. Using mice with conditional genetic deletion of Ndufs2, a critical subunit of mitochondrial complex I, they found that knockout of this subunit from catecholaminergic cells – which include carotid body glomus cells– demonstrated a loss of hypoxic sensitivity. This loss of sensitivity was evident in the loss of hypoxia-stimulated minute ventilation in intact animals ventilated with hypoxic gas mixtures, as well as at the level of hypoxia-induced catecholamine release, carotid sinus nerve firing, and intracellular Ca2+ measurements, all of which increase rapidly during superfusion with buffers at low O2 tensions. These findings were consistent with the longstanding observation that rotenone, an inhibitor of complex I function, also inhibits carotid body hypoxic responsiveness. Using patch-clamp recordings, they found that Ndufs2-deficient carotid body cells failed to demonstrate the normal hypoxia-induced decrease in K+ channel conductivity, and appeared instead to be chronically inhibited. Antioxidant treatments lessened this inhibition in the knockout cells, whereas exogenous oxidants in controls cells mimicked hypoxic responses and prevented hypoxic inactivation of potassium channels. The authors concluded that hypoxia inhibits K+ channels through an ROS-dependent process; loss of complex I function led to a loss of hypoxia-induced increases in ROS, although basal ROS signaling seemed to have increased. These findings are interesting because they support the idea that mitochondrial ROS generation increases - rather than decreases - during hypoxia [53].
In view of previous studies implicating complex III in hypoxia responsiveness, how can we reconcile the apparently independent roles of complex I and III in redox-regulated hypoxia sensing? One possibility is that they actually represent the same mechanism. Loss of ETC function at complex I will limit electron flux into complex III, so ROS signals coming from III will be attenuated by the lack of an electron source. Loss of complex III activity will similarly block electron flux through complex I, although that complex will still have an ample source of electrons as NADH concentrations increase. If complex III increases its generation of ROS to signal the onset of hypoxia, this response would be lost upon inactivation of electron flow through complex I (by Ndufs2 knockout or rotenone) or by loss of the Rieske Iron-Sulfur center in complex III (which prevents ROS generation). Future experiments testing the role of RISP deletion in carotid body cells on carotid body hypoxia responsiveness would be needed to test this hypothesis. Similarly, studies of Ndufs2 knockouts in pulmonary blood vessels are needed to test whether the loss of complex I function phenotypically mimics the effects of complex III inactivation.
In multiple cell types including cultured endothelial cells, Hernansanz-Agustin and colleagues found that acute hypoxia (1–2% O2) led to an increase in the oxidation of dihydroethidium, a chemical probe that can be oxidized by superoxide [80]. A parallel burst in H2O2 was detected using carboxydichlorofluoricein diacetate (CDCFDA), suggesting that the superoxide is converted to peroxide in the cells. A mitochondria-targeted dihydroethidium probe also detected an oxidant burst during hypoxia, which was lost in ρ0 cells that lack a functional mitochondrial ETC. Those findings implicate mitochondria as a source of redox signaling during hypoxia. Finally, that study reported that HIF-1α stabilization was lost in ρ0 cells, whereas treatment with the superoxide dismutase mimetic Tiron tended to increase HIF-1α stabilization and CDCFDA oxidation, suggesting that HIF-1α stabilization is positively regulated by H2O2 rather than by superoxide.
In a recent study, Briggs et al. examined HIF regulation in triple-negative breast cancer cells, and found that these cells secrete glutamate to the extracellular space [81]. There, it acts in an autocrine/paracrine manner to inhibit the xCT glutamate-cystine transporter, a major antiporter mediating cellular uptake of cystine, which is then reduced to cysteine as needed for the maintenance of redox balance. When inhibited, cellular levels of cysteine decline and PHD2 (EglN1 in their paper) becomes oxidized and inactivated, leading to constitutive normoxic stabilization of HIF-1α. Curiously, they did not find evidence of increased ROS in response to xCT inhibition or growth in cystine-free media, leading them to conclude that EglN1 is a sensor of cysteine. However, other groups have reported that inhibition of the cysteine/glutamate antiporter in tumor cells leads to rapid induction of oxidant stress leading to ferroptosis, an iron-dependent cell death pathway [82]. Moreover, the inactivation of EglN1 occurred through thiol oxidation, which is normally regulated by NADPH-dependent antioxidant systems. Further studies are needed to reconcile these differences, and to determine how EglN1 redox status could be regulated by cysteine, independently of ROS, in the study of Briggs et al.
7. Mitochondrial ROS signals regulate AMPK response to hypoxia
Redox signals from mitochondria in hypoxic cells also regulate post-translational responses including the activation of AMP kinase (AMPK). In lung alveolar epithelial cells, hypoxia suppresses vectoral sodium transport from the alveoli to the interstitium, a response that is important for fluid removal from the lung airspace [83]. The mechanism involves ROS-triggered internalization of the Na+/K+-ATPase, a transporter on the basolateral membrane of the epithelium. That response is mediated by AMPK activation, which triggers PKC-zeta translocation to the plasma membrane by phosphorylating it at Thr410. Gusarova et al. found that catalase overexpression, or depletion of mitochondrial DNA (ρ0 cells), blocked the ability of alveolar epithelial cells to internalize the Na+/K+ ATPase during hypoxia [84]. In addition, Emerling et al. reported that AMPK phosphorylation during hypoxia failed to develop in ρ0 cells, and that exogenous antioxidants prevented AMPK activation in hypoxia [85]. Gusarova and colleagues subsequently reported that the activation of AMPK during hypoxia is triggered by extracellular Ca2+ entry through Ca2+ release-activated Ca2+ (CRAC) channels [86]. Thus, hypoxia-induced ROS signals lead to release of intracellular calcium stores and activation of CRAC channels that amplify the Ca2+ signal, thereby activating AMPK via Ca2+/calmodulin-dependent kinase kinase beta (CaMMKβ) mechanism. The AMPK activates PKC-ζ which then triggers internalization of the Na+/K+-ATPase. Collectively these studies suggest that mitochondrial redox signals regulate AMPK activation during hypoxia. A summary of these responses in relation to thiol redox status has been reported [87].
AMPK is also activated during bioenergetics crises, when ATP levels decrease and ADP and AMP accumulate. However, the protective effects of AMPK cannot prevent a bioenergetic crisis if it only becomes activated after the problem has developed, as defined by a decrease in ATP and increase in AMP or ADP. The possibility that AMPK could be activated by physiological hypoxia, in anticipation of a bioenergetic crisis, is therefore an attractive notion [88]. Hypoxia-induced mitochondrial redox signals, which are activated during moderate levels of hypoxia, could provide such a mechanism.
Studies reveal that ROS signals during hypoxia can indeed activate AMPK in diverse cell types, through a Ca2+-dependent mechanism [86], [88]. During hypoxia, ROS signals initiate release of intracellular calcium from the endoplasmic reticulum (ER). The resulting decrease in ER Ca2+ leads to the oligomerization of stromal interaction molecule 1 (STIM1), the ER calcium sensor, which organizes the calcium release-activated calcium (CRAC) channels at sites where ER and plasma membranes associate. CaMKKβ is a calcium-sensitive kinase that can activate AMPK in the absence of a bioenergetic deficiency [89]. Knockdown of CaMKKβ abolishes the AMPK response to hypoxia, indicating that hypoxia can trigger AMPK activation without increased [AMP] through ROS-dependent CRAC channel activation, leading to increases in cytosolic Ca2+ that activate the AMPK upstream kinase CaMKKβ [88].
8. Summary
Redox signaling is an important mechanism that contributes to cellular homeostasis in both normal and tumor cells, under both normoxic and hypoxic conditions. Mitochondria and other NAD(P)H systems participate in redox signaling through their ability to generate ROS; these signals are locally regulated and often act on the intended thiol targets through intermediary redox shuttling systems. Opposing this oxidant signaling, systems including thioredoxins, glutaredoxins, glutathione peroxidases and peroxiredoxins can reverse thiol oxidation of proteins. Subcellular compartments regulate thiol redox status independently, and contain their own independent antioxidant protein systems. Although seemingly counter-intuitive, mitochondrial augment the release of ROS signals to the cytosol during hypoxia, leading to the activation of diverse protective systems through transcriptional and post-translational mechanisms. Future studies are required to more fully understand the roles of redox signaling in hypoxia, and to understand how these signals are regulated spatially and temporally in the cell.
Acknowledgments
This work was supported by NIH grants HL35440 and HL122062 (P.T.S.) and a Parker B. Francis Fellowship (K.A.S).
References
- 1.Brigelius-Flohe R., Flohe L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011;15(8):2335–2381. doi: 10.1089/ars.2010.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sabharwal S.S., Schumacker P.T. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat. Rev. Cancer. 2014;14(11):709–721. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Brien E., Holt M.E., Thompson M.K., Salay L.E., Ehlinger A.C., Chazin W.J., Barton J.K. The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport. Science. 2017;355:6327. doi: 10.1126/science.aag1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Winterbourn C.C. Revisiting the reactions of superoxide with glutathione and other thiols. Arch. Biochem. Biophys. 2016;595:68–71. doi: 10.1016/j.abb.2015.11.028. [DOI] [PubMed] [Google Scholar]
- 5.Winterbourn C.C. Are free radicals involved in thiol-based redox signaling? Free Radic. Biol. Med. 2015;80:164–170. doi: 10.1016/j.freeradbiomed.2014.08.017. [DOI] [PubMed] [Google Scholar]
- 6.Orr A.L., Vargas L., Turk C.N., Baaten J.E., Matzen J.T., Dardov V.J., Attle S.J., Li J., Quackenbush D.C., Goncalves R.L., Perevoshchikova I.V., Petrassi H.M., Meeusen S.L., Ainscow E.K., Brand M.D. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015;11(11):834–836. doi: 10.1038/nchembio.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tonks N.K. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121(5):667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 8.Winterbourn C.C., Hampton M.B. Redox biology: signaling via a peroxiredoxin sensor. Nat. Chem. Biol. 2015;11(1):5–6. doi: 10.1038/nchembio.1722. [DOI] [PubMed] [Google Scholar]
- 9.Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008;275(13):3249–3277. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 10.Bae Y.S., Oh H., Rhee S.G., Yoo Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells. 2011;32(6):491–509. doi: 10.1007/s10059-011-0276-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanson G.T., Aggeler R., Oglesbee D., Cannon M., Capaldi R.A., Tsien R.Y., Remington S.J. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 2004;279(13):13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- 12.Dooley C.T., Dore T.M., Hanson G.T., Jackson W.C., Remington S.J., Tsien R.Y. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J. Biol. Chem. 2004;279(21):22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- 13.Remington S.J. Fluorescent proteins: maturation, photochemistry and photophysics. Curr. Opin. Struct. Biol. 2006;16(6):714–721. doi: 10.1016/j.sbi.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Gutscher M., Sobotta M.C., Wabnitz G.H., Ballikaya S., Meyer A.J., Samstag Y., Dick T.P. Proximity-based protein thiol oxidation by H2O2-scavenging peroxidases. J. Biol. Chem. 2009;284(46):31532–31540. doi: 10.1074/jbc.M109.059246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma L.H., Takanishi C.L., Wood M.J. Molecular mechanism of oxidative stress perception by the Orp1 protein. J. Biol. Chem. 2007;282(43):31429–31436. doi: 10.1074/jbc.M705953200. [DOI] [PubMed] [Google Scholar]
- 16.Sobotta M.C., Liou W., Stocker S., Talwar D., Oehler M., Ruppert T., Scharf A.N., Dick T.P. Peroxiredoxin-2. and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015;11(1):64–70. doi: 10.1038/nchembio.1695. [DOI] [PubMed] [Google Scholar]
- 17.Ward J.P. A twist in the tail: synergism between mitochondria and NADPH oxidase in the hypoxia-induced elevation of reactive oxygen species in pulmonary artery. Free Radic. Biol. Med. 2008;45(9):1220–1222. doi: 10.1016/j.freeradbiomed.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 18.Wolin M.S., Ahmad M., Gao Q., Gupte S.A. Cytosolic NAD(P)H regulation of redox signaling and vascular oxygen sensing. Antioxid. Redox Signal. 2007;9(6):671–678. doi: 10.1089/ars.2007.1559. [DOI] [PubMed] [Google Scholar]
- 19.Muller G., Morawietz H. NAD(P)H oxidase and endothelial dysfunction. Horm. Metab. Res. 2009;41(2):152–158. doi: 10.1055/s-0028-1086023. [DOI] [PubMed] [Google Scholar]
- 20.Archer S.L., Nelson D.P., Weir E.K. Detection of activated O2 species in vitro and in rat lungs by chemiluminescence. J. Appl. Physiol. 1985;67(5):1912–1921. doi: 10.1152/jappl.1989.67.5.1912. [DOI] [PubMed] [Google Scholar]
- 21.Michelakis E.V., Thebaud B., Weir E.K., Archer S.L. Hypoxic pulmonary vasoconstriction: redox regulation of O2-sensitive K+ channels by a mitochondrial O2-sensor in resistance artery smooth muscle cells. J. Mol. Cell. Cardiol. 2004;37(6):1119–1136. doi: 10.1016/j.yjmcc.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 22.Kummer W., Acker H. Immunohistochemical demonstration of four subunits of neutrophil NAD(P)H oxidase in type I cells of carotid body. J. Appl. Physiol. 1995;78(5):1904–1909. doi: 10.1152/jappl.1995.78.5.1904. [DOI] [PubMed] [Google Scholar]
- 23.Griendling K.K., Sorescu D., Ushio-Fukai M. NAD(P)H oxidase - role in cardiovascular biology and disease. Circ. Res. 2000;86(5):494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 24.Babior B.M. The NADPH oxidase of endothelial cells. IUBMB Life. 2000;50(4–5):267–269. doi: 10.1080/713803730. [DOI] [PubMed] [Google Scholar]
- 25.Shimoda L.A., Sham J.S.K., Shimoda T.H., Sylvester J.T. L-type Ca2+ channels, resting [Ca2+]i, and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000;279(5):L884–L894. doi: 10.1152/ajplung.2000.279.5.L884. [DOI] [PubMed] [Google Scholar]
- 26.Liu Q., Sham J.S.K., Shimoda L.A., Sylvester J.T. Hypoxic constriction of porcine distal pulmonary arteries: endothelium and endothelin dependence. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001;280(5):L856–L865. doi: 10.1152/ajplung.2001.280.5.L856. [DOI] [PubMed] [Google Scholar]
- 27.Yuan G., Khan S.A., Luo W., Nanduri J., Semenza G.L., Prabhakar N.R. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J. Cell. Physiol. 2011 doi: 10.1002/jcp.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Archer S.L., Reeve H.L., Michelakis E., Puttagunta L., Waite R., Nelson D.P., Dinauer M.C., Weir E.K. O2 sensing is preserved in mice lacking the gp91 phox subunit of NADPH oxidase. Proc. Natl. Acad. Sci. USA. 1999;96:7944–7949. doi: 10.1073/pnas.96.14.7944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weissmann N., Zeller S., Schafer R.U., Turowski C., Ay M., Quanz K., Ghofrani H.A., Schermuly R.T., Fink L., Seeger W., Grimminger F. Impact of mitochondria and NADPH oxidases on acute and sustained hypoxic pulmonary vasoconstriction. Am. J. Respir. Cell. Mol. Biol. 2006;34(4):505–513. doi: 10.1165/rcmb.2005-0337OC. [DOI] [PubMed] [Google Scholar]
- 30.Veith C., Kraut S., Wilhelm J., Sommer N., Quanz K., Seeger W., Brandes R.P., Weissmann N., Schroder K. NADPH oxidase 4 is not involved in hypoxia-induced pulmonary hypertension. Pulm. Circ. 2016;6(3):397–400. doi: 10.1086/687756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nisimoto Y., Diebold B.A., Cosentino-Gomes D., Lambeth J.D. Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry. 2014;53(31):5111–5120. doi: 10.1021/bi500331y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diebold I., Petry A., Hess J., Gorlach A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell. 2010;21(12):2087–2096. doi: 10.1091/mbc.E09-12-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S., Tabar S.S., Malec V., Eul B.G., Klepetko W., Weissmann N., Grimminger F., Seeger W., Rose F., Hanze J. NOX4 regulates ROS levels under normoxic and hypoxic conditions, triggers proliferation, and inhibits apoptosis in pulmonary artery adventitial fibroblasts. Antioxid. Redox Signal. 2008;10(10):1687–1698. doi: 10.1089/ars.2008.2035. [DOI] [PubMed] [Google Scholar]
- 34.Mittal M., Roth M., Konig P., Hofmann S., Dony E., Goyal P., Selbitz A.C., Schermuly R.T., Ghofrani H.A., Kwapiszewska G., Kummer W., Klepetko W., Hoda M.A., Fink L., Hanze J., Seeger W., Grimminger F., Schmidt H.H., Weissmann N. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007;101(3):258–267. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]
- 35.Jensen P.K. Antimycin-insensitive oxidation of succinate and reduced nicotinamide-adenine dinucleotide in electron-tranport particles. Biochim. Biophys. Acta. 1966;122:157–166. doi: 10.1016/0926-6593(66)90057-9. [DOI] [PubMed] [Google Scholar]
- 36.Brand M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016;100:14–31. doi: 10.1016/j.freeradbiomed.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 37.Boveris A., Oshino N., Chance B. The cellular production of hydrogen peroxide. Biochem. J. 1972;128(3):617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turrens J.F., Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Votyakova T.V., Reynolds I.J. DeltaPsi(m)-dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 2001;79(2):266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- 40.Genova M.L., Ventura B., Giuliano G., Bovina C., Formiggini G., Castelli G.P., Lenaz G. The site of production of superoxide radical in mitochondrial Complex I is not a bound ubisemiquinone but presumably iron-sulfur cluster N2. FEBS Lett. 2001;505(3):364–368. doi: 10.1016/s0014-5793(01)02850-2. [DOI] [PubMed] [Google Scholar]
- 41.Li Y., Trush M.A. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem. Biophys. Res. Commun. 1998;253(2):295–299. doi: 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- 42.Misra H.P., Fridovich I. The univalent reduction of oxygen by reduced flavins and quinones. J. Biol. Chem. 1972;247:188–192. [PubMed] [Google Scholar]
- 43.Turrens J.F., Alexandre A., Lehninger A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 44.Garcia-Ruiz C., Colell A., Morales A., Kaplowitz N., Fernandez-Checa J.C. Role of oxidative stress generated from the mitochondrial electron transport chain and mitochondrial glutathione status in loss of mitochondrial function and activation of transcription factor nuclear factor-kappa B: studies with isolated mitochondria and rat hepatocytes. Mol. Pharmacol. 1995;48(5):825–834. [PubMed] [Google Scholar]
- 45.Kwong L.K., Sohal R.S. Substrate and site specificity of hydrogen peroxide generation in mouse mitochondria. Arch. Biochem. Biophys. 1998;350:118–126. doi: 10.1006/abbi.1997.0489. [DOI] [PubMed] [Google Scholar]
- 46.Garcia-Ruiz C., Colell A., Mari M., Morales A., Fernandez-Checa J.C. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J. Biol. Chem. 1997;272(17):11369–11377. doi: 10.1074/jbc.272.17.11369. [DOI] [PubMed] [Google Scholar]
- 47.Quillet-Mary A., Jaffrezou J.P., Mansat V., Bordier C., Naval J., Laurent G. Implication of mitochondrial hydrogen peroxide generation in ceramide-induced apoptosis. J. Biol. Chem. 1997;272(34):21388–21395. doi: 10.1074/jbc.272.34.21388. [DOI] [PubMed] [Google Scholar]
- 48.Gille L., Nohl H. The ubiquinol/bc1 redox couple regulates mitochondrial oxygen radical formation. Arch. Biochem. Biophys. 2001;388(1):34–38. doi: 10.1006/abbi.2000.2257. [DOI] [PubMed] [Google Scholar]
- 49.Zhang L., Yu L.D., Yu C.A. Generation of superoxide anion by succinate-cytochrome c reductase from bovine heart mitochondria. J. Biol. Chem. 1998;273(51):33972–33976. doi: 10.1074/jbc.273.51.33972. [DOI] [PubMed] [Google Scholar]
- 50.Fabian M., Palmer G. Hydrogen peroxide is not released following reaction of cyanide with several catalytically important derivatives of cytochrome c oxidase. FEBS Lett. 1998;422:1–4. doi: 10.1016/s0014-5793(97)01561-5. [DOI] [PubMed] [Google Scholar]
- 51.Duranteau J., Chandel N.S., Kulisz A., Shao Z., Schumacker P.T. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J. Biol. Chem. 1998;273(19):11619–11624. doi: 10.1074/jbc.273.19.11619. [DOI] [PubMed] [Google Scholar]
- 52.Fernandez-Aguera M.C., Gao L., Gonzalez-Rodriguez P., Pintado C.O., rias-Mayenco I., Garcia-Flores P., Garcia-Perganeda A., Pascual A., Ortega-Saenz P., Lopez-Barneo J. Oxygen sensing by arterial chemoreceptors depends on mitochondrial Complex I signaling. Cell Metab. 2015;22(5):825–837. doi: 10.1016/j.cmet.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 53.Guzy R.D., Schumacker P.T. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006;91(5):807–819. doi: 10.1113/expphysiol.2006.033506. [DOI] [PubMed] [Google Scholar]
- 54.Ambrosio G., Zweier J.L., Duilio C., Kuppusamy P., Santoro G., Elia P.P., Tritto I., Cirillo P., Condorelli M., Chiariello M. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J. Biol. Chem. 1993;268(25):18532–18541. [PubMed] [Google Scholar]
- 55.Becker L.B., Vanden Hoek T.L., Shao Z.H., Li C.Q., Schumacker P.T. Generation of superoxide in cardiomyocytes during ischemia before reperfusion. Am. J. Physiol. 1999;277(6 Pt 2):H2240–H2246. doi: 10.1152/ajpheart.1999.277.6.H2240. [DOI] [PubMed] [Google Scholar]
- 56.Di Lisa F., Menabo R., Canton M., Petronilli V. The role of mitochondria in the salvage and the injury of the ischemic myocardium. Biochim. Biophys. Acta. 1998;1366(1–2):69–78. doi: 10.1016/s0005-2728(98)00121-2. [DOI] [PubMed] [Google Scholar]
- 57.Ferrari R. The role of mitochondria in ischemic heart disease. J. Cardiovasc. Pharmacol. 1996;28(Suppl. 1):S1–S10. doi: 10.1097/00005344-199600003-00002. [DOI] [PubMed] [Google Scholar]
- 58.Turrens J.F., Beconi M., Barilla J., Chavez U.B., McCord J.M. Mitochondrial generation of oxygen radicals during reoxygenation of ischemic tissues. Free Radic. Res. Commun. 1991;12–13(Pt 2):681–689. doi: 10.3109/10715769109145847. [DOI] [PubMed] [Google Scholar]
- 59.Vanden Hoek T.L., Li C., Shao Z., Schumacker P.T., Becker L.B. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J. Mol. Cell. Cardiol. 1997;29:2571–2583. doi: 10.1006/jmcc.1997.0497. [DOI] [PubMed] [Google Scholar]
- 60.Freeman B.A., Crapo J.D. Hyperoxia increases oxygen radical production in rat lungs and lung mitochondria. J. Biol. Chem. 1981;256(21):10986–10992. [PubMed] [Google Scholar]
- 61.Shimada H., Hirai K., Simamura E., Pan J. Mitochondrial NADH-quinone oxidoreductase of the outer membrane is responsible for paraquat cytotoxicity in rat livers. Arch. Biochem. Biophys. 1998;351(1):75–81. doi: 10.1006/abbi.1997.0557. [DOI] [PubMed] [Google Scholar]
- 62.Boveris A., Cadenas E. Mitochondrial production of hydrogen peroxide regulation by nitric oxide and the role of ubisemiquinone. IUBMB Life. 2000;50(4–5):245–250. doi: 10.1080/713803732. [DOI] [PubMed] [Google Scholar]
- 63.Chandel N.S., Maltepe E., Goldwasser E., Mathieu C.E., Simon M.C., Schumacker P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chandel N.S., Schumacker P.T. Cellular oxygen sensing by mitochondria: old questions, new insight. J. Appl. Physiol. 2000;88(5):1880–1889. doi: 10.1152/jappl.2000.88.5.1880. [DOI] [PubMed] [Google Scholar]
- 65.Chandel N.S., McClintock D.S., Feliciano C.E., Wood T.M., Melendez J.A., Rodriguez A.M., Schumacker P.T. Reactive oxygen species generated at mitochondrial Complex III stabilize HIF-1-alpha during hypoxia: a mechanism of O2 sensing. J. Biol. Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 66.Gerald D., Berra E., Frapart Y.M., Chan D.A., Giaccia A.J., Mansuy D., Pouyssegur J., Yaniv M., Mechta-Grigoriou F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118(6):781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 67.Masson N., Singleton R.S., Sekirnik R., Trudgian D.C., Ambrose L.J., Miranda M.X., Tian Y.M., Kessler B.M., Schofield C.J., Ratcliffe P.J. The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep. 2012;13(3):251–257. doi: 10.1038/embor.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guzy R.D., Hoyos B., Robin E., Chen H., Liu L., Mansfield K.D., Simon M.C., Hammerling U., Schumacker P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1(6):401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 69.Mansfield K.D., Guzy R.D., Pan Y., Young R.M., Cash T.P., Schumacker P.T., Simon M.C. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005;1(6):393–399. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Waypa G.B., Marks J.D., Guzy R., Mungai P.T., Schriewer J., Dokic D., Schumacker P.T. Hypoxia Triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ. Res. 2010;106(3):526–535. doi: 10.1161/CIRCRESAHA.109.206334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lohman J.R., Remington S.J. Development of a family of redox-sensitive green fluorescent protein indicators for use in relatively oxidizing subcellular environments. Biochemistry. 2008;47(33):8678–8688. doi: 10.1021/bi800498g. [DOI] [PubMed] [Google Scholar]
- 72.Waypa G.B., Marks J.D., Guzy R.D., Mungai P.T., Schriewer J.M., Dokic D., Ball M.K., Schumacker P.T. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am. J. Respir. Crit. Care Med. 2013;187(4):424–432. doi: 10.1164/rccm.201207-1294OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Desireddi J.R., Farrow K.N., Marks J.D., Waypa G.B., Schumacker P.T. Hypoxia increases ROS signaling and cytosolic Ca(2+) in pulmonary artery smooth muscle cells of mouse lungs slices. Antioxid. Redox Signal. 2010;12(5):595–602. doi: 10.1089/ars.2009.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Waypa G.B., Guzy R., Mungai P.T., Mack M.M., Marks J.D., Roe M.W., Schumacker P.T. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ. Res. 2006;99(9):970–978. doi: 10.1161/01.RES.0000247068.75808.3f. [DOI] [PubMed] [Google Scholar]
- 75.Waypa G.B., Marks J.D., Mack M.M., Boriboun C., Mungai P.T., Schumacker P.T. Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circ. Res. 2002;91(8):719–726. doi: 10.1161/01.res.0000036751.04896.f1. [DOI] [PubMed] [Google Scholar]
- 76.Waypa G.B., Chandel N.S., Schumacker P.T. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ. Res. 2001;88(12):1259–1266. doi: 10.1161/hh1201.091960. [DOI] [PubMed] [Google Scholar]
- 77.Hirst J., King M.S., Pryde K.R. The production of reactive oxygen species by complex I. Biochem. Soc. Trans. 2008;36(Pt 5):976–980. doi: 10.1042/BST0360976. [DOI] [PubMed] [Google Scholar]
- 78.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lopez-Barneo J., Lopez-Lopez J.R., Urena J., Gonzalez C. Chemotransduction in the carotid body: K+ current modulated by PO2 in type I chemoreceptor cells. Science. 1988;241:580–582. doi: 10.1126/science.2456613. [DOI] [PubMed] [Google Scholar]
- 80.Hernansanz-Agustin P., Izquierdo-Alvarez A., Sanchez-Gomez F.J., Ramos E., Villa-Pina T., Lamas S., Bogdanova A., Martinez-Ruiz A. Acute hypoxia produces a superoxide burst in cells. Free Radic. Biol. Med. 2014;71:146–156. doi: 10.1016/j.freeradbiomed.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 81.Briggs K.J., Koivunen P., Cao S., Backus K.M., Olenchock B.A., Patel H., Zhang Q., Signoretti S., Gerfen G.J., Richardson A.L., Witkiewicz A.K., Cravatt B.F., Clardy J., Kaelin W.G., Jr. Paracrine induction of HIF by glutamate in breast cancer: EglN1 Senses Cysteine. Cell. 2016;166(1):126–139. doi: 10.1016/j.cell.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., Morrison B., III, Stockwell B.R. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Matthay M.A., Zemans R.L. The acute respiratory distress syndrome: pathogenesis and treatment. Annu. Rev. Pathol. 2011;6:147–163. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gusarova G.A., Dada L.A., Kelly A.M., Brodie C., Witters L.A., Chandel N.S., Sznajder J.I. Alpha1-AMP-activated protein kinase regulates hypoxia-induced Na,K-ATPase endocytosis via direct phosphorylation of protein kinase C zeta. Mol. Cell. Biol. 2009;29(13):3455–3464. doi: 10.1128/MCB.00054-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Emerling B.M., Weinberg F., Snyder C., Burgess Z., Mutlu G.M., Viollet B., Budinger G.R., Chandel N.S. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic. Biol. Med. 2009;46(10):1386–1391. doi: 10.1016/j.freeradbiomed.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gusarova G.A., Trejo H.E., Dada L.A., Briva A., Welch L.C., Hamanaka R.B., Mutlu G.M., Chandel N.S., Prakriya M., Sznajder J.I. Hypoxia leads to Na,K-ATPase downregulation via Ca(2+) release-activated Ca(2+) channels and AMPK activation. Mol. Cell. Biol. 2011;31(17):3546–3556. doi: 10.1128/MCB.05114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bogdanova A., Petrushanko I.Y., Hernansanz-Agustin P., Martinez-Ruiz A. "Oxygen Sensing" by Na,K-ATPase: these Miraculous Thiols. Front. Physiol. 2016;7:314. doi: 10.3389/fphys.2016.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mungai P.T., Waypa G.B., Jairaman A., Prakriya M., Dokic D., Ball M.K., Schumacker P.T. Hypoxia triggers AMPK activation through ROS-mediated activation of CRAC channels. Mol. Cell. Biol. 2011 doi: 10.1128/MCB.05124-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fogarty S., Hawley S.A., Green K.A., Saner N., Mustard K.J., Hardie D.G. Calmodulin-dependent protein kinase kinase-beta activates AMPK without forming a stable complex: synergistic effects of Ca2+ and AMP. Biochem. J. 2010;426(1):109–118. doi: 10.1042/BJ20091372. [DOI] [PMC free article] [PubMed] [Google Scholar]