

Abstract
Background
Sickle cell disease (SCD) is one of the commonest severe monogenic disorders in the world, due to the inheritance of two abnormal haemoglobin (beta globin) genes. SCD can cause severe pain, significant end‐organ damage, pulmonary complications, and premature death. Silent cerebral infarcts are the commonest neurological complication in children and probably adults with SCD. Silent cerebral infarcts also affect academic performance, increase cognitive deficits and may lower intelligence quotient.
Objectives
To assess the effectiveness of interventions to reduce or prevent silent cerebral infarcts in people with SCD.
Search methods
We searched for relevant trials in the Cochrane Library, MEDLINE (from 1946), Embase (from 1974), the Transfusion Evidence Library (from 1980), and ongoing trial databases; all searches current to 19 September 2016. We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group Trials Register: 06 October 2016.
Selection criteria
Randomised controlled trials comparing interventions to prevent silent cerebral infarcts in people with SCD. There were no restrictions by outcomes examined, language or publication status.
Data collection and analysis
We used standard Cochrane methodological procedures.
Main results
We included five trials (660 children or adolescents) published between 1998 and 2016. Four of the five trials were terminated early. The vast majority of participants had the haemoglobin (Hb)SS form of SCD. One trial focused on preventing silent cerebral infarcts or stroke; three trials were for primary stroke prevention and one trial dealt with secondary stroke prevention.
Three trials compared the use of regular long‐term red blood cell transfusions to standard care. Two of these trials included children with no previous long‐term transfusions: one in children with normal transcranial doppler (TCD) velocities; and one in children with abnormal TCD velocities. The third trial included children and adolescents on long‐term transfusion.
Two trials compared the drug hydroxyurea and phlebotomy to long‐term transfusions and iron chelation therapy: one in primary prevention (children), and one in secondary prevention (children and adolescents).
The quality of the evidence was moderate to very low across different outcomes according to GRADE methodology. This was due to trials being at high risk of bias because they were unblinded; indirectness (available evidence was only for children with HbSS); and imprecise outcome estimates.
Long‐term red blood cell transfusions versus standard care
Children with no previous long‐term transfusions and higher risk of stroke (abnormal TCD velocities or previous history of silent cerebral infarcts)
Long‐term red blood cell transfusions may reduce the incidence of silent cerebral infarcts in children with abnormal TCD velocities, risk ratio (RR) 0.11 (95% confidence interval (CI) 0.02 to 0.86) (one trial, 124 participants, low‐quality evidence); but make little or no difference to the incidence of silent cerebral infarcts in children with previous silent cerebral infarcts on magnetic resonance imaging and normal or conditional TCDs, RR 0.70 (95% CI 0.23 to 2.13) (one trial, 196 participants, low‐quality evidence).
No deaths were reported in either trial.
Long‐term red blood cell transfusions may reduce the incidence of: acute chest syndrome, RR 0.24 (95% CI 0.12 to 0.49) (two trials, 326 participants, low‐quality evidence); and painful crisis, RR 0.63 (95% CI 0.42 to 0.95) (two trials, 326 participants, low‐quality evidence); and probably reduces the incidence of clinical stroke, RR 0.12 (95% CI 0.03 to 0.49) (two trials, 326 participants, moderate‐quality evidence).
Long‐term red blood cell transfusions may improve quality of life in children with previous silent cerebral infarcts (difference estimate ‐0.54; 95% confidence interval ‐0.92 to ‐0.17; one trial; 166 participants), but may have no effect on cognitive function (least squares means: 1.7, 95% CI ‐1.1 to 4.4) (one trial, 166 participants, low‐quality evidence).
Transfusions continued versus transfusions halted: children and adolescents with normalised TCD velocities (79 participants; one trial)
Continuing red blood cell transfusions may reduce the incidence of silent cerebral infarcts, RR 0.29 (95% CI 0.09 to 0.97 (low‐quality evidence).
We are very uncertain whether continuing red blood cell transfusions has any effect on all‐cause mortality, Peto odds ratio (OR) 8.00 (95% CI 0.16 to 404.12); or clinical stroke, RR 0.22 (95% CI 0.01 to 4.35) (very low‐quality evidence).
The trial did not report: comparative numbers for SCD‐related adverse events; quality of life; or cognitive function.
Hydroxyurea and phlebotomy versus transfusions and chelation
Primary prevention, children (121 participants; one trial)
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on: silent cerebral infarcts (no infarcts); all‐cause mortality (no deaths); risk of stroke (no strokes); or SCD‐related complications, RR 1.52 (95% CI 0.58 to 4.02) (very low‐quality evidence).
Secondary prevention, children and adolescents with a history of stroke (133 participants; one trial)
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on: silent cerebral infarcts, Peto OR 7.28 (95% CI 0.14 to 366.91); all‐cause mortality, Peto OR 1.02 (95%CI 0.06 to 16.41); or clinical stroke, RR 14.78 (95% CI 0.86 to 253.66) (very low‐quality evidence).
Switching to hydroxyurea and phlebotomy may increase the risk of SCD‐related complications, RR 3.10 (95% CI 1.42 to 6.75) (low‐quality evidence).
Neither trial reported on quality of life or cognitive function.
Authors' conclusions
We identified no trials for preventing silent cerebral infarcts in adults, or in children who do not have HbSS SCD.
Long‐term red blood cell transfusions may reduce the incidence of silent cerebral infarcts in children with abnormal TCD velocities, but may have little or no effect on children with normal TCD velocities. In children who are at higher risk of stroke and have not had previous long‐term transfusions, long‐term red blood cell transfusions probably reduce the risk of stroke, and other SCD‐related complications (acute chest syndrome and painful crises).
In children and adolescents at high risk of stroke whose TCD velocities have normalised, continuing red blood cell transfusions may reduce the risk of silent cerebral infarcts. No treatment duration threshold has been established for stopping transfusions.
Switching to hydroxyurea with phlebotomy may increase the risk of silent cerebral infarcts and SCD‐related serious adverse events in secondary stroke prevention.
All other evidence in this review is of very low‐quality.
Keywords: Adolescent; Child; Humans; Erythrocyte Transfusion; Phlebotomy; Phlebotomy/adverse effects; Anemia, Sickle Cell; Anemia, Sickle Cell/complications; Antisickling Agents; Antisickling Agents/adverse effects; Antisickling Agents/therapeutic use; Brain Infarction; Brain Infarction/etiology; Brain Infarction/prevention & control; Cause of Death; Cognition; Cognition/physiology; Hydroxyurea; Hydroxyurea/adverse effects; Hydroxyurea/therapeutic use; Primary Prevention; Primary Prevention/methods; Quality of Life; Randomized Controlled Trials as Topic; Secondary Prevention; Secondary Prevention/methods; Stroke; Stroke/prevention & control
Interventions to prevent silent strokes in people with sickle cell disease
Review question
We wanted to determine if there were any safe and effective interventions that prevent silent strokes (also known as silent cerebral infarcts) in people with sickle cell disease (SCD).
Background
SCD is a serious inherited blood disorder where the red blood cells, which carry oxygen around the body, develop abnormally. Normal red blood cells are flexible and disc‐shaped, but in sickle cell disease they can become rigid and crescent shaped. Sickled cells are not only less flexible than healthy red blood cells, they are also stickier. This can lead to the blockage of blood vessels, resulting in tissue and organ damage and episodes of severe pain. The abnormal blood cells are more fragile and break apart, which leads to fewer red blood cells, known as anaemia. Sickled red blood cells can block blood flow in vessels in the brain, leading to a silent stroke.
Silent strokes are common, occurring in up to 39% of people with SCD by 18 years of age. Two tests have been used to identify children at higher risk of having a first stroke. Transcranial doppler ultrasonography (TCD) measures the speed of blood flowing through arteries in the brain. Children with a high blood flow have an increased risk of stroke. Whereas magnetic resonance imaging (MRI) takes images of the brain to see if there are any small areas of damage called silent strokes. Children with silent strokes have an increased risk of clinical stroke.
Treatments that have been considered for preventing silent strokes include long‐term red blood cell transfusions, the drug hydroxyurea and stem cell transplantation.
Trial characteristics
Evidence is current to 19 September 2016. We found five randomised controlled trials which enrolled a total of 660 participants. Three trials compared blood transfusions to no blood transfusions and two trials compared blood transfusion to hydroxyurea. Trials were published between 1998 and 2016 and included children and sometimes adolescents; the majority had one form of SCD (HbSS). No trials included stem cell transplantation.
All trials received government funding.
Key Results
In children with abnormal TCD velocities, red blood cell transfusions may decrease the risk of silent strokes, but have little or no effect in children with normal TCD velocities.
In children at higher risk of stroke (abnormal TCD velocities or previous silent stroke), red blood cell transfusions probably reduce the risk of clinical stroke; may reduce the risk of acute chest syndrome and painful crisis; but we are very uncertain whether they have any effect on the risk of death.
In children with normal TCD velocities and previous silent stroke, red blood cell transfusions may improve quality of life, but make little or no difference to IQ.
In children and adolescents who have had at least 12 months of regular red blood cell transfusions to prevent a stroke, continuing red blood cell transfusions may reduce the risk of silent stroke, but we are very uncertain whether they have any effect on the risk of death or clinical stroke.
For children on long‐term red blood cell transfusions with iron chelation (treatment to remove excess iron) who have not had a stroke, we are very uncertain whether switching to hydroxyurea with phlebotomy (withdrawing blood to reduce excess iron) has any effect on the risk of a silent stroke, clinical stroke, death, or SCD‐related complications.
For children and adolescents on long‐term red blood cell transfusions and iron chelation who have had a clinical stroke, we are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on the risk of silent stroke or death. Switching to hydroxyurea and phlebotomy may increase the risk of SCD‐related complications.
Quality of the evidence
In children at higher risk of stroke who have not had previous long‐term transfusions, there is moderate‐quality evidence that long‐term red blood cell transfusions reduce the risk of stroke. The quality of evidence was rated as low to very‐low for the rest of the outcomes including risk of silent cerebral infarcts due to trials being at high risk of bias and because there were a small number of trials and a small number of participants included in the trials.
Summary of findings
Summary of findings for the main comparison.
Long‐term RBC transfusion compared to standard care for prevention of SCI in people with SCD and normal TCD velocities
| Long‐term RBC transfusion compared to standard care for prevention of SCI in people with SCD and normal TCD velocities | ||||||
| Patient or population: prevention of SCI in people with SCD and normal TCD velocities Setting: outpatients Intervention: long‐term RBC transfusion Comparison: standard care | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with standard care | Risk with long‐term RBC transfusion | |||||
| Proportion of participants developing new or progressive SCI lesions | Trial population | RR 0.70 (0.23 to 2.13) | 196 (1 RCT) | ⊕⊕⊝⊝ LOW 1 2 | ||
| 72 per 1000 | 51 per 1000 (17 to 154) | |||||
| All‐cause mortality | No deaths occurred in either trial arm | ‐ | 196 (1 RCT) | ⊕⊕⊝⊝ LOW 1 3 | ||
| SCD ‐ related SAEs ‐ ACS | Trial population | RR 0.20 (0.08 to 0.51) | 196 (1 RCT) | ⊕⊕⊝⊝ LOW 1 4 | ||
| 247 per 1000 | 49 per 1000 (20 to 126) | |||||
| SCD‐related SAEs ‐ pain crisis | Trial population | RR 0.56 (0.40 to 0.78) | 196 (1 RCT) | ⊕⊕⊝⊝ LOW 1 4 | ||
| 577 per 1000 | 323 per 1000 (231 to 450) | |||||
| Clinical stroke | Trial population | RR 0.14 (0.02 to 1.12) | 196 (1 RCT) | ⊕⊕⊝⊝ LOW 1 2 | ||
| 72 per 1000 | 10 per 1000 (1 to 81) | |||||
| Cognitive function assessed with: WASI IQ score | Least square mean 1.7 (SE 95% CI ‐1.1 to 4.4) | ‐ | 196 (1 RCT) | ⊕⊕⊝⊝ LOW 1 4 | Author reported data from SIT 2014 | |
| Quality of life assessed with: Child Health Questionnaire Parent Form 50 | Least square mean 1.7 (SE 95% CI ‐1.1 to 4.4) Diference estimate ‐0.54 (‐0.92 to ‐0.17) | ‐ | 196 (1 RCT) | ⊕⊕⊝⊝ LOW 1 4 | Author reported data from SIT 2014 | |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ACS: acute chest syndrome; CI: confidence interval; RR: risk ratio; RBC: red blood cell; SAEs: serious adverse events; SCD: sickle cell disease; SCI: silent cerebral infarcts; SE: standard error; TCD: transcranial doppler | ||||||
| GRADE Working Group grades of evidence High‐quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate‐quality: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low‐quality: our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low‐quality: we have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 We downgraded the quality of evidence by 1 due to indirectness. Only children with HbSS or HbSβº thalassaemia included. If this review was only considering the quality of evidence for children with HbSS the quality of evidence would not have been downgraded for indirectness.
2 We downgraded the quality of evidence by 1 due to imprecision. The estimate has wide confidence intervals that include clinically relevant benefit and harm
3 We downgraded the quality of evidence by 1 due to imprecision. Rare event, no deaths occurred
4 We downgraded the quality of evidence by 1 due to high risk of bias. Performance bias, unblinded outcome assessment and high cross‐over rates
Summary of findings 2.
Long‐term RBC transfusion compared to standard care for prevention of SCI in people with SCD and abnormal TCD velocities
| Long‐term RBC transfusion compared to standard care for prevention of SCI in people with SCD and abnormal TCD velocities | ||||||
| Patient or population: prevention of SCI in people with SCD and abnormal TCD velocities Setting: outpatients Intervention: long‐term RBC transfusion Comparison: standard care | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with standard care | Risk with long‐term RBC transfusion | |||||
| Proportion of participants developing new or progressive SCI lesions | Trial population | RR 0.11 (0.02 to 0.86) | 124 (1 RCT) | ⊕⊕⊝⊝ LOW 1 2 | ||
| 159 per 1000 | 18 per 1000 (3 to 137) | |||||
| All‐cause mortality | No deaths occurred in either trial arm | ‐ | 130 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 2 3 4 | ||
| SCD ‐ related SAEs ‐ ACS | Trial population | RR 0.30 (0.11 to 0.87) | 130 (1 RCT) | ⊕⊕⊝⊝ LOW 2 3 | ||
| 209 per 1000 | 63 per 1000 (23 to 182) | |||||
| SCD‐related SAEs ‐ pain crisis | Trial population | RR 0.90 (0.44 to 1.86) | 130 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 2 3 5 | ||
| 197 per 1000 | 177 per 1000 (87 to 366) | |||||
| Clinical stroke | Trial population | RR 0.10 (0.01 to 0.73) | 130 (1 RCT) | ⊕⊕⊝⊝ LOW 1 2 | ||
| 164 per 1000 | 16 per 1000 (2 to 120) | |||||
| Cognitive function ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Quality of life ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ACS: acute chest syndrome; CI: confidence interval; RR: risk ratio; RCT: randomised controlled trial; SAEs: serious adverse events; SCD: sickle cell disease; SCI: silent cerebral infarcts; TCD: transcranial doppler | ||||||
| GRADE Working Group grades of evidence High‐quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate‐quality: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low‐quality: our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low‐quality: we have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 We downgraded the quality of evidence by 1 due to risk of bias. The analysis was not intention‐to‐treat
2 We downgraded the quality of evidence by 1 due to indirectness. Only children with HbSS or HbSβº thalassaemia included. If this review was only considering the quality of evidence for children with HbSS the quality of evidence would not have been downgraded for indirectness.
3 We downgraded the quality of evidence by 1 due to risk of bias. Some outcome assessment not blinded and the trial was stopped early
4 We downgraded the quality of evidence by 1 due to imprecision. Rare event. No deaths occurred
5 We downgraded the quality of evidence by 1 due to imprecision. The estimate has wide CIs that include clinically relevant benefit and harm
Summary of findings 3.
Long‐term RBC transfusions compared to standard care for SCD‐related SAEs and clinical stroke in people with SCD at risk of stroke and no previous long‐term RBC transfusions
| Long‐term RBC transfusions compared to standard care for SCD‐related SAEs and clinical stroke in people with SCD at risk of stroke and no previous long‐term RBC transfusions | ||||||
| Patient or population: SCD‐related SAEs and clinical stroke in people with SCD at risk of stroke and no previous long‐term RBC transfusions Setting: outpatient Intervention: long‐term RBC transfusions Comparison: standard care | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with standard care | Risk with long‐term RBC transfusions | |||||
| SCD ‐ related SAEs ‐ ACS | Trial population | RR 0.24 (0.12 to 0.49) | 326 (2 RCTs) | ⊕⊕⊝⊝ LOW 1 2 | ||
| 232 per 1000 | 56 per 1000 (28 to 114) | |||||
| SCD‐related SAEs ‐ pain crisis | Trial population | RR 0.63 (0.42 to 0.95) | 326 (2 RCTs) | ⊕⊕⊝⊝ LOW 1 2 | ||
| 421 per 1000 | 265 per 1000 (177 to 400) | |||||
| Clinical stroke | Trial population | RR 0.12 (0.03 to 0.49) | 326 (2 RCTs) | ⊕⊕⊕⊝ MODERATE 1 | ||
| 110 per 1000 | 13 per 1,000 (3 to 54) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ACS: acute chest syndrome: CI: confidence interval; RR: risk ratio; RBC: red blood cells; SAEs: serious adverse events; SCD: sickle cell disease | ||||||
| GRADE Working Group grades of evidence High‐quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate‐quality: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low‐quality: our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low‐quality: we have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
a Data for the other outcomes for this SoF table are presented separately in Table 1 (normal TCD velocities) and Table 2 (abnormal TCD velocities) We combined the data for these specific comparisons as they are objective outcomes and the status of TCD velocities does not impact these outcomes.
1 We downgraded the quality of the evidence by 1 due to risk of bias. Unblinded trial and cross‐overs, and imbalance between loss to follow‐up between trial arms
2 We downgraded the quality of the evidence by 1 due to indirectness (only children with HbSS or HbSβº thalassaemia included in studies). If this review was only considering the quality of evidence for children with HbSS the quality of evidence would not have been downgraded for indirectness.
Summary of findings 4.
Transfusions continued compared to transfusions halted for prevention of SCI in people with SCD and normalised TCD velocities
| Transfusions continued compared to transfusions halted for prevention of SCI in people with SCD and normalised TCD velocities | ||||||
| Patient or population: prevention of SCI in people with SCD and normalised TCD velocities Setting: outpatients Intervention: transfusions continued Comparison: transfusions halted | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with transfusions halted | Risk with transfusions continued | |||||
| Proportion of participants developing new or progressive SCI lesions | Trial population | RR 0.29 (0.09 to 0.97) | 77 (1 RCT) | ⊕⊕⊝⊝ LOW 1 2 | One patient in each group had no follow‐up MRI | |
| 275 per 1000 | 80 per 1000 (25 to 267) | |||||
| All‐cause mortality | Low** | Peto OR 8.00 (0.16 to 404.12) | 79 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | ||
| 26 per 1000 | 208 per 1000 (4 to 10,507) |
|||||
| High | ||||||
| 33 per 1000 | 264 per 1000 (5 to 13,336) |
|||||
| Clinical stroke | Trial population | RR 0.22 (0.01 to 4.35) | 79 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | ||
| 49 per 1000 | 11 per 1000 (0 to 212) | |||||
| SCD‐related SAEs ‐ ACS | See comment | ‐ | 79 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 4 | No comparative numbers reported | |
| SCD‐related SAEs ‐ pain crisis | See comment | ‐ | 79 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 4 | No comparative numbers reported | |
| Cognitive function ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Quality of life ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ** Mortality risk for control estimates from: Leikin 1989 ACS: acute chest syndrome; CI: confidence interval; RR: risk ratio; SAEs: serious adverse events; SCD: sickle cell disease; SCI: silent cerebral infarcts; TCD: transcranial doppler | ||||||
| GRADE Working Group grades of evidence High‐quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate‐quality: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low‐quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low‐quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
1 We downgraded the quality of evidence by 1 due to risk of bias. There was no description of allocation concealment, there was a risk of performance bias, and the trial was stopped early
2 We downgraded the quality of evidence by 1 due to indirectness. Only children with HbSS or HbSβº thalassaemia included in the trial. If this review was only considering the quality of evidence for children with HbSS the quality of evidence would not have been downgraded for indirectness.
3 We downgraded the quality of evidence by 1 due to imprecision. The estimate has wide CIs that include clinically relevant benefit and harm
4 We downgraded the quality of evidence by 1 due to imprecision. No comparative numbers were provided
Summary of findings 5.
Hydroxyurea compared to RBC transfusions for prevention of SCI in people with SCD who have not had a stroke (primary prevention)
| Hydroxyurea compared to RBC transfusions for prevention of SCI in people with SCD who have not had a stroke (primary prevention) | ||||||
| Patient or population: prevention of SCI in people with SCD who have not had a stroke (primary prevention) Setting: outpatients Intervention: hydroxyurea Comparison: RBC transfusions | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with RBC transfusions | Risk with hydroxyurea | |||||
| Proportion of participants developing new or progressive SCI lesions | No SCIs occurred in either trial arm | ‐ | 121 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | ||
| All‐cause mortality | No deaths occurred in either trial arm | ‐ | (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | ||
| SCD‐related SAEs ‐ ACS | Trial population | RR 2.03 (0.39 to 10.69) | 121 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 3 4 | ||
| 33 per 1000 | 67 per 1000 (13 to 350) | |||||
| SCD‐related SAEs ‐ Pain crisis | Trial population | RR 5.08 (0.61 to 42.23) | 121 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 3 4 | ||
| 16 per 1000 | 83 per 1000 (10 to 692) | |||||
| Clinical stroke | No strokes occurred in either trial arm | ‐ | 121 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | ||
| Cognitive function ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Quality of life ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ACS: Acute chest syndrome; CI: confidence interval; RBC: red blood cells; RR: risk ratio; SAEs: serious adverse events; SCD: sickle cell disease; SCIs: silent cerebral infarcts | ||||||
| GRADE Working Group grades of evidence High‐quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate‐quality: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low‐quality: our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low‐quality: we have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 We downgraded the quality of evidence by 1 due to indirectness. Only children with HbSS or HbSβº thalassaemia included. If this review was only considering the quality of evidence for children with HbSS the quality of evidence would not have been downgraded for indirectness.
2 We downgraded the quality of evidence by 1 due to imprecision. Rare events. No SCIs or strokes occurred
3 We downgraded the quality of evidence by 1 due to risk of bias. Trial was stopped early some outcomes assessed without blinding
4 We downgraded the quality of evidence by 1 due to imprecision. Estimate has wide confidence intervals that include both clinically relevant benefits and harms
Summary of findings 6.
Hydroxyurea compared to RBC transfusion for prevention of SCI in people with SCD who had a stroke (secondary prevention)
| Hydroxyurea compared to RBC transfusion for prevention of SCI in people with SCD who had a stroke (secondary prevention) | ||||||
| Patient or population: prevention of SCI in people with SCD who had a stroke (secondary prevention) Setting: outpatients Intervention: hydroxyurea Comparison: RBC transfusion | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with RBC transfusion | Risk with hydroxyurea | |||||
| Proportion of participants developing new or progressive SCI lesions | Moderate | Peto OR 7.28 (0.14 to 366.91) | 133 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | ||
| 72 per 1000 | 524 per 1000 (10 to 26,418) | |||||
| All‐cause mortality | Low** | OR 1.02 (0.06 to 16.41) | 133 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | ||
| 26 per 1000 | 208 per 1000 (4 to 10,507) |
|||||
| High | ||||||
| 33 per 1000 | 264 per 1000 (5 to 13,336) |
|||||
| SCD‐related SAEs ‐ ACS | Trial population | RR 0.33 (0.04 to 3.08) | 133 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | ||
| 45 per 1000 | 15 per 1000 (2 to 140) | |||||
| SCD‐related SAEs ‐ Pain crisis | Trial population | RR 3.15 (1.23 to 8.11) | 133 (1 RCT) | ⊕⊕⊝⊝ LOW 1 2 | ||
| 76 per 1000 | 239 per 1000 (93 to 614) | |||||
| Clinical stroke | Moderate | RR 14.78 (0.86 to 253.66) | 133 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | ||
| 72 per 1000 | 1000 per 1000 (62 to 1000) | |||||
| Cognitive function ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Quality of life ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). **Mortality risk for control estimates from: Leikin 1989 CI: confidence interval; RR: risk ratio; OR: odds ratio | ||||||
| GRADE Working Group grades of evidence High‐quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate‐quality: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low‐quality: our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low‐quality: we have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 We downgraded the quality of the evidence by 1 due to risk of bias. Trial was not blinded and was stopped early
2 We downgraded the quality of the evidence by 1 due to indirectness. Only children with HbSS and HbSβº thalassaemia were included. If this review was only considering the quality of evidence for children with HbSS the quality of evidence would not have been downgraded for indirectness.
3 We downgraded the quality of the evidence by 1 due to imprecision. The estimate has wide confidence intervals that include clinically relevant benefit or harm or both
Background
Description of the condition
Sickle cell disease (SCD) is a genetic haemoglobin disorder, which can cause severe pain, significant end‐organ damage, pulmonary complications, and premature death (Chakravorty 2015). It is one of the most common severe monogenic disorders in the world, due to the inheritance of two abnormal haemoglobin (beta globin) genes (Rees 2010). Populations originating from sub‐Saharan Africa, South and Central America, the Caribbean, the Middle East, India and parts of the Mediterranean are predominantly affected. Reductions in infant and child mortality and increasing migration from highly affected countries have made this a worldwide problem (Piel 2012). Over 12,500 people in the UK and 100,000 in the USA suffer from the disease (NICE 2010; Pleasants 2014). A recent study estimated that approximately 305,800 babies were born with SCD in 2010, of which two thirds were born in Africa, and this could increase to approximately 404,200 by 2050 (Piel 2012).
The term 'sickle cell disease' refers to all genotypes that cause the clinical syndrome. There are three main types of SCD. Sickle cell anaemia is the most common form of the disease (up to 70% of cases of SCD in people of African origin) and is due to the inheritance of two beta globin S (βS) alleles (haemoglobin (Hb)SS). The second most common genotype (up to 30% of cases in people of African origin) is haemoglobin SC disease (HbSC disease) it is due to the co‐inheritance of the βS and βC alleles and tends to be a more moderate form of the disease. The third major type of SCD occurs when βS is inherited with a β‐thalassaemia allele, causing HbS/β‐thalassaemia (Rees 2010). People who have inherited a thalassaemia null mutation (HbSߺ) have a disease that is clinically indistinguishable from sickle cell anaemia, whereas people with HbSβ+ thalassaemia have a milder disorder. In high‐income countries, people with SCD are expected to live into their 40s, 50s and beyond, whereas in low‐income countries (including some African nations) it is estimated that between 50% to 90% of children born with HbSS die before their fifth birthday (Gravitz 2014; Grosse 2011).
In SCD under conditions of low oxygen levels, acidity and cellular dehydration, the HbS molecules polymerise and lead to membrane damage that distorts the red blood cells (RBC) which take on the appearance of sickle‐shaped cells. The main determinant of disease severity is the rate and extent of this HbS polymerisation (Rees 2010). This is exemplified by the co‐inheritance of genetic factors that affect the intracellular HbS or fetal haemoglobin concentration, e.g. the protective effects of co‐inherited α‐thalassaemia (Rumaney 2014; Steinberg 2012) or hereditary persistence of fetal haemoglobin (Akinsheye 2011; Steinberg 2012). Sickling of RBC results in two main events: the blockage of blood flow resulting in organ and tissue ischaemia; and haemolytic anaemia (Sparkenbaugh 2013). Both of these processes are thought to lead to increased inflammation and an increased tendency to develop a clot (Frenette 2007; Rees 2010). Reduced blood flow is mediated via a dynamic interaction between RBC containing sticky HbS, the vessel wall, and white cells (Rees 2010). Sickle RBC also have a shorter lifespan of 10 to 12 days, versus 120 days for normal RBC, due to intravascular and extravascular haemolysis, leading to anaemia (Kato 2006a). Chronic intravascular haemolysis leads to a reduced nitric oxide level within the blood; nitric oxide is sequestered by free haemoglobin (Hb), which over time favours endothelial dysfunction and the development of pulmonary hypertension (Kato 2006a; Kato 2006b).
The causes of cerebral infarcts in SCD are unclear and several of the mechanisms listed above may converge leading to vessel wall damage, and the narrowing and occlusion of cerebral blood vessels.
Silent cerebral infarcts
Silent cerebral infarcts (SCI) are the commonest neurological complication in children, and possibly in adults (DeBaun 2016). These are defined as the presence of abnormalities on a magnetic resonance imaging (MRI) scan consistent with cerebral infarction (T‐2 weighted and FLAIR imaging) without a clinical history or abnormalities on physical examination that are consistent with a previous stroke (DeBaun 2012). Some studies specify MRI lesions have to be at least 3 mm in diameter in children (Casella 2010), whereas in adults a more restrictive definition is sometimes used which includes a lesion measuring at least 5 mm on MRI (DeBaun 2012).
The occurrence of SCI in children with SCD increases the risk for stroke, and new or enlarged SCI. It also affects academic performance, increases cognitive deficits and may lower intelligence quotient (IQ) compared either to children with SCD who have normal MRI scans or with siblings without SCD (DeBaun 2012;DeBaun 2014).
The lack of longitudinal studies has made it difficult to define the natural history and prevalence of SCI in both children and adults. The 'Cooperative Study of Sickle Cell Disease' (CSSCD) cohort estimated a prevalence of 22% in children with HbSS aged six to 19 years; a French study reported a cumulative risk of SCI of 19% by eight years of age; 32% by 14 years of age; and 39% by 18 years of age (Bernaudin 2015a; DeBaun 2016). In children in Kuwait, SCI is much more uncommon, with one study estimating a prevalence of only 3% (Adekile 2002). This may be explained by the fact that most people with SCD in the Arabian peninsula have persistently elevated Hb F levels, even as adults (Marouf 2003). It is still unclear as to when SCI first occurs in young children and if incidence rates change in adolescence (Bernaudin 2011; DeBaun 2012). Knowledge of the prevalence of SCI in adults with SCD is limited by the small number of studies and the small number of participants in studies, nevertheless it appears that up to one third of adults with SCD may develop SCI. Although more common in people with HbSS disease, SCI are also identified in people with HbSC disease (5% to 31%) and in people with HbSβ (3% to 38%) (DeBaun 2012). While SCI are uncommon in children in Kuwait, they are common in adults, with one study estimating a prevalence of 20% (Marouf 2003). These studies suggest no plateau in incidence and thus the resulting need for trials to study both primary and secondary prevention (Bernaudin 2011; DeBaun 2012).
Relatively little is known about the causes of SCI and the optimal preventive therapy. However, the consistent finding that anaemia is strongly associated with SCI suggests that cerebral haemodynamic insufficiency (demand for oxygen exceeds supply) is a central component (DeBaun 2012). This is consistent with that fact that the majority of SCI are confined to deep white matter which suggests hypoperfusion or hypoxic events (Bernaudin 2015a; van der Land 2016).
SCI may or may not be associated with increased transcranial doppler (TCD) velocities (tests that measure the speed of blood flow through the brain's blood vessels (either the internal carotid artery or the middle cerebral artery) by ultrasound). The TCD velocities are classed as normal (less than 170 cm per second); conditional (170 cm to less than 200 cm per second); or abnormal (at least 200 cm per second) (Adams 1998).
Risk factors for people with SCD developing SCI include lower haemoglobin levels, lower fetal haemoglobin, internal carotid artery stenosis, elevated systolic blood pressure (SBP) and a history of seizures (Bernaudin 2015a; DeBaun 2012; van der Land 2016) .
In high‐income countries randomised controlled trials (RCTs) (STOP, STOP II) have demonstrated that regular blood transfusion therapy (typically monthly) prevents strokes in children with SCD and high transcranial doppler (TCD) velocities (Abboud 2011; Adams 1998).
Description of the intervention
RBC transfusions
Chronic RBC transfusions, either given as simple or exchange transfusions, form part of the management of a number of SCD complications such as the primary prevention of strokes in children with abnormal TCD velocities (Adam 2008) or the prevention of further chest crises in people with recurrent episodes (Howard 2015). Blood transfusion can be given by simple, top‐up transfusions to increase the number of normal RBC or by exchange transfusion in which the HbS is replaced by healthy RBC with a goal of reducing HbS to below 30% (Kanter 2013).
Children with elevated TCD velocities or previously elevated TCD velocities that have normalised developed fewer new SCI lesions with regular blood transfusions (Abboud 2011; Pegelow 2001).
In people with SCD, RBC transfusions have reduced sickle cell‐related complications and improved quality of life, but are not without potentially serious complications. The benefits of transfusion therapy must be balanced against risks including infections, iron overload, acute or delayed haemolytic transfusion reactions, and increased complexity of compatibility testing (Chou 2013a; Chou 2013b; Porter 2013; Scheunemann 2010; Ubesie 2012).
Hydroxyurea (hydroxycarbamide)
Hydroxyurea has been in use since the 1980s and shown in clinical trials to be beneficial for SCD in reducing vaso‐occlusive crises, chest crises and in improving survival (Field 2014). Hydroxyurea is currently the only approved therapeutic drug for the treatment of sickle cell anaemia (for adults with severe vaso‐occlusive episodes of pain or acute chest syndrome) and its use has become widespread in both children and adults with SCD. Hydroxyurea significantly decreases haemolytic rate and improves the degree of baseline anaemia, which suggests that it could also decrease the rate of SCI (Bernaudin 2015a).
Haematopoietic stem cell transplantation
Haematopoietic (blood forming) stem cell transplantation (HSCT) is the only known treatment for SCD that reduces or eliminates the sickling of RBC. Allogeneic (from a relative (matched or mismatched) or matched unrelated donor) haematopoietic stem cells from bone marrow, peripheral blood, or umbilical cord blood are transplanted to produce partial or total correction of the sickle haemoglobin phenotype (Oringanje 2016). Risks are associated with both myeloablative conditioning (preparative regimen prior to the transplant) and the allogeneic stem cells. Risks include death, infertility and gonadal failure, development of secondary malignancies, graft versus host disease (GVHD) (an immune reaction of donor cells against recipient tissues), post‐transplant immunological and neurological complications, and failure of the transplant (recurrence of SCD). HSCT has been used mostly in children under 16 years of age and there is limited evidence for its use in adults who may be at a higher risk of death (Oringanje 2016).
How the intervention might work
RBC transfusion
The mechanisms for the reduction in stroke risk from chronic transfusion are not known (DeBaun 2006). However, a reduction in cells containing high amounts of HbS or an increase in Hb level could have beneficial effects on cerebral blood vessels or interactions between RBC and endothelial cells (Adams 1998).
Children with the lowest baseline haemoglobin levels have higher odds of SCI than do those with the highest haemoglobin levels (DeBaun 2012). Also, an acute reduction in haemoglobin level (less than 55 g/L) is associated with an increase in new‐onset SCI, whether or not the child has SCD (Dowling 2012).
Transfusion does have an immediate haemodynamic effect measured by the reduction of middle cerebral artery velocity (Venketasubramanian 1994).
The STOP trial has shown that RBC transfusions are effective for preventing stroke in children with elevated TCD velocities. It is also conceivable that RBC transfusions may prevent further SCI injury, even though the microvascular pathology of SCI is different from the involvement of the larger vessels (internal carotid and medium sized vessels) in stroke (DeBaun 2014).
Hydroxyurea
In preliminary studies hydroxyurea was substituted successfully for chronic transfusion for preventing secondary strokes. In a cohort study from 1992 to 2010, participants with severe baseline anaemia treated with hydroxyurea had a reduction in SCI from 37.1% in a previous cohort (1988 to 2007) to 32.4% by age 14 years (Bernaudin 2015a). Preliminary data from single‐arm trials also suggest that hydroxyurea may be of benefit for the secondary prevention of SCI (Bernaudin 2011). Hydroxyurea is known to modestly increase the level of HbF via a range of mechanisms, including epigenetic modifications (Pule 2015). In RCTs on the use of hydroxyurea in SCD, it was found to increase total Hb and HbF levels and reduce vaso‐occlusive crises; however, its benefit could not be solely attributed to the rise in HbF, with likely other mechanisms including effects on platelet count, white count, and RBC adhesion to endothelium (Charache 1995; Wang 2011). Hydroxyurea also decreases intravascular haemolysis which may ameliorate nitric oxide sequestration.
HSCT
Allogeneic HSCT is the only curative treatment for SCD and a potential option for the primary or secondary prevention of SCI (Bernaudin 2007). In 36 HSCT transplant participants with a history of stroke, two had a recurrence post‐transplantation, one participant experienced a transient ischaemic attack (TIA), and one had a severe cerebrovascular disorder with Moya‐Moya disease and experienced fatal intracranial haemorrhage. After a median follow‐up of six years, the risk of stroke recurrence was 5.6%; however, no strokes or silent ischaemic lesions occurred in participants with successful engraftment (Bernaudin 2007). In a study carried out in the USA (eight participants with evidence of SCI before transplantation and who also had post‐transplant studies) lesions were stable in three participants and four participants had lesions that decreased in size by brain MRI. There were no clinical strokes after transplantation in this group (Walters 2010).
In HSCT, high doses of chemotherapy are used to destroy an individual's own stem cells, which are then replaced with stem cells from a donor who is unaffected by SCD. The blood of transplant recipients contains normal RBC produced by the donated stem cells. Stem cell recipients typically need to take immunosuppressants for months to a few years. These medications can cause serious side effects.
Why it is important to do this review
SCI are the most common neurological injury in children and can occur in up to a third of adults with SCD. People with SCI have an increased risk for stroke and lower academic performance. There are no evidence‐based strategies currently established for the primary prevention of SCI (DeBaun 2016). The effectiveness of either RBC transfusion, hydroxyurea or HSCT is unclear in secondary prevention of SCI. It is important for clinicians and people with SCD to understand what treatments are most effective based on the quality of evidence for both primary and secondary prevention in order to manage and reduce the serious sequelae of SCI.
Objectives
To assess the effectiveness of RBC transfusions and hydroxyurea alone or in combination and HSCT to reduce or prevent SCI in people with SCD.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs).
Types of participants
Participants with homozygous SCD (SS), sickle beta thalassaemia (Sβ and Sβ+) and sickle haemoglobin C disease (SC) of all ages and both sexes, with or without evidence of SCI.
Types of interventions
RBC transfusions versus standard care
HSCT versus standard care
Hydroxyurea versus standard care or placebo
RBC transfusions versus HSCT
RBC transfusions versus hydroxyurea
Hydroxyurea versus HSCT
RBC transfusions combined with hydroxyurea versus standard care or hydroxyurea or RBC transfusions or HSCT alone
Types of outcome measures
Primary outcomes
Proportion of participants developing new or progressive SCI lesions on MRI
All‐cause mortality
Serious adverse events (SAEs) associated with different therapies or SCD
We will categorise the proportion of participants developing new or progressive MRI lesions, all cause mortality, and SAEs according to short‐, medium‐, and long‐term outcomes. We will report the exact definition of these time frames over time periods that are common to as many trials as possible (e.g. five years and under, six to 10 years, over 10 years).
Secondary outcomes
Clinical stroke (according to short‐, medium‐, and long‐term outcomes)
Cognitive function as assessed by validated scales (such as Wechsler scales) from baseline and at various time intervals as reported in trials (at least six months)
Quality of life as assessed by validated scales (at least six months)
Any adverse events associated with different therapies
Search methods for identification of studies
We searched for all relevant published and unpublished trials without restrictions on language, year or publication status
Electronic searches
We identified trials from the Cochrane Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register using the terms: (sickle cell OR (haemoglobinopathies AND general)) AND stroke.
The Haemoglobinopathies Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (Clinical Trials) (updated each new issue of the Cochrane Library) and weekly searches of MEDLINE. Unpublished work is identified by searching the abstract books of five major conferences: the European Haematology Association conference; the American Society of Hematology conference; the British Society for Haematology Annual Scientific Meeting; the Caribbean Health Research Council Meetings; and the National Sickle Cell Disease Program Annual Meeting. For full details of all searching activities for the register, please see the relevant section of the Cochrane Cystic Fibrosis and Genetic Disorders Group's website.
Date of the most recent search of the Cochrane Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register: 06 October 2016.
In addition to this we searched the following databases for RCTs on 19 September 2016.
The Cochrane Library (CENTRAL, DARE, HTA, NHSEED) – 19 September 2016 (www.cochranelibrary.com) (Appendix 1)
MEDLINE (OvidSP, Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE Daily and Ovid MEDLINE, 1946 to 19 September 2016) (Appendix 2)
PubMed (Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, for recent records not yet added to MEDLINE) (www.ncbi.nlm.nih.gov/sites/entrez) (Appendix 3)
Embase (OvidSP, 19 September 2016) (Appendix 4)
CINAHL (EBSCOHost, 1937 to 19 September 2016) (Appendix 5)
Transfusion Evidence Library (1950 to 19 September 2016) (www.transfusionevidencelibrary.com) (Appendix 6)
LILACS (1982 to 19 September 2016) (lilacs.bvsalud.org/en/) (Appendix 7)
Web of Science (Conference Proceedings Citation Index‐ Science (CPCI‐S) ‐ 1990 to 19 September 2016) (Appendix 8).
We also searched the following trial databases for ongoing trials on 19 September 2016.
ClinicalTrials.gov (clinicaltrials.gov/) (Appendix 9)
WHO International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/) (Appendix 10)
We combined searches in MEDLINE and Embase with RCT filters based on the recommended sensitivity‐maximising Cochrane RCT search filters, as detailed in the Cochrane Handbook for Systematic Reviews of Interventions (Lefebvre 2011), and in CINAHL with an RCT filter based on the Scottish Intercollegiate Guidelines Network's (SIGN) RCT filter (www.sign.ac.uk/methodology/filters.html).
Searching other resources
We handsearched reference lists of included trials in order to identify further relevant trials. We contacted the lead authors of the included trials to identify any unpublished material, missing data or information regarding any ongoing trials.
Data collection and analysis
Selection of studies
We selected trials according to chapter seven of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a; Higgins 2011b). Two review authors (LE, PF) independently screened all electronically‐derived citations and abstracts of papers identified by the search strategy for relevance. At this stage we excluded trials that were clearly irrelevant based on the abstract. Two independent review authors (LE, PF) formally assessed the full texts of all potentially relevant trials for eligibility against the criteria outlined above. We requested additional information from trial authors as necessary. The two review authors discussed the results of trial selection and resolved our discrepancies. In the event that this was not possible, we would have referred the decision of eligibility to a third review author (MA). We reported the results of trial selection using a PRISMA flow diagram (Moher 2009).
Data extraction and management
Two review authors (LE, PF) independently extracted data for all trials according to Cochrane guidelines (Higgins 2011a). We resolved disagreements between the review authors by consensus. The review authors were not blinded to names of authors, institutions, journals, or the outcomes of the trials. We used Covidence to extract data and to assess the risk of bias for all included trials (Covidence 2015). We used the available tables in the Review Manager software to present extracted data on trial characteristics (Review Manager 2014).
We extracted the following data.
General information
Review author's name; date of data extraction; study ID; first author of trial; author's contact address (if available); citation of paper; objectives of the trial.
Trial details
Trial design; location; setting; sample size; power calculation; treatment allocation; inclusion and exclusion criteria; reasons for exclusion; comparability of groups; length of follow‐up; stratification; stopping rules described; statistical analysis; results; conclusion; and funding.
Characteristics of participants
Age; gender; total number recruited; total number randomised; total number analysed; types of underlying disease; proportion of participants with lesions; TCD velocities; lost to follow‐up numbers; dropouts (percentage in each arm) with reasons; protocol violations; previous treatments; current treatment; prognostic factors; haemoglobin S levels; SCD complications.
Interventions
Experimental and control interventions; method of RBC transfusion (simple, partial or full exchange transfusion); type of RBC transfusion (intermittent or chronic); dose and duration of hydroxycarbamide therapy; HSCT (relative, other donor, cord blood, bone marrow, peripheral blood, extent of HLA matching, preparative regimen); standard care.
Outcomes measured
Proportion of participants developing new or enlarged lesions; mortality (all cause); SAEs related to treatments or sickle lung disease; clinical stroke; cognitive function; quality of life.
We used full‐text versions, clinical trial registration information, and abstracts to extract data. We extracted data using one form only for trials reported in more than one publication. Where sources did not provide sufficient information, we contacted authors and trial groups for additional details.
Assessment of risk of bias in included studies
We performed an assessment of all RCTs using the Cochrane 'risk of bias’ tool according to the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c). Two review authors (LE, PF) worked independently to assess each element of potential bias listed below as 'high', 'low' or 'unclear' risk of bias. We reported a brief description of the judgement statements upon which we assessed potential bias in the 'Characteristics of included studies' table. We ensured that a consensus on the degree of risk of bias was met through comparison of the review authors' statements and if necessary, we would have consulted a third review author (MA). We used Cochrane's tool for assessing the risk of bias and included the following domains.
Selection bias (random sequence generation and allocation concealment)
Performance bias (blinding of participants and personnel)
Detection bias (blinding of outcome assessment)
Attrition bias (incomplete outcome data)
Reporting bias (selective reporting)
Other bias
Measures of treatment effect
Where data allowed, we performed quantitative assessments using the Review Manager software (RevMan 2014).
For dichotomous outcomes we recorded the number of events and the total number of participants in both the treatment and control groups. For dichotomous outcomes we reported the pooled risk ratio (RR) with 95% confidence intervals (CI). Where the number of observed events were small (less than 5% of sample per group), and where trials have balanced treatment groups, we reported the Peto odds ratio (OR) with 95% CI (Deeks 2011).
For continuous outcomes we recorded the mean, standard deviation (SD) and total number of participants in both the treatment and control groups. For continuous outcomes using the same scale, we performed analyses using the mean difference (MD) with 95% CIs. If continuous outcomes were reported using different scales we used standardised mean difference (SMD).
Where available, we extracted and reported hazard ratios (HRs) for mortality data. If HRs were not available, we made every effort to estimate as accurately as possible the HR using the available data and a purpose built method based on the Parmar and Tierney approach (Parmar 1998; Tierney 2007).
Where appropriate, we reported the number needed to treat to benefit (NNTB) and the number needed to treat to harm (NNTH) with CIs. If we could not report the available data in any of the formats described above, we performed a narrative report, and when appropriate we presented data in tables.
Unit of analysis issues
We did not expect to encounter unit of analysis issues as cluster randomised trials, cross‐over trials, and multiple observations for the same outcome were not included in this review. Should we have found any trials with these designs, we would have treated these in accordance with the advice given in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011d). If participants would have been randomised more than once we would have contacted the authors of the trial to provide us with data on outcomes associated with the initial randomisation. For trials with multiple treatment groups we would only include subgroups that were considered relevant to the analysis. We tabulated all subgroups in the 'Characteristics of included studies' section. When appropriate, we combined groups to create a single pair‐wise comparison. If this was not possible, we selected the most appropriate pair of interventions and excluded the others (Higgins 2011d).
Dealing with missing data
Where data were identified to be missing or unclear in published literature, we contacted the lead authors of trials (e.g. STOP 2 2005; Vichinsky 2010), for additional data, but no data have been received to date. We recorded the number of participants lost to follow‐up for each trial. Where possible, we analysed data on an intention‐to‐treat (ITT) basis, but if data were insufficient, we presented a per protocol analyses (Higgins 2011b).
Assessment of heterogeneity
If the clinical and methodological characteristics of individual trials were sufficiently homogeneous, we combined the data to perform a meta‐analysis. We assessed the statistical heterogeneity of treatment effects between trials using a Chi² test with a significance level at P < 0.1. We used the I² statistic to quantify the degree of potential heterogeneity and classified it as moderate if I² > 50%, or considerable if I² > 80%. We expected to identify at least moderate clinical and methodological heterogeneity within the included trials, and hence used the random‐effects model throughout. If statistical heterogeneity was considerable, we would not report the overall summary statistic. We would have assessed potential causes of heterogeneity by sensitivity and subgroup analyses (Deeks 2011).
Assessment of reporting biases
We did not explore publication bias as we identified fewer than 10 trials. We would have explored potential publication bias (small trial bias) by generating a funnel plot and using a linear regression test and considered a P value of less than 0.1 as significant for this test (Sterne 2011).
Data synthesis
We performed analyses according to the recommendations in the Cochrane Handbook for Systematic Reviews of Interventions using aggregated data for analysis (Deeks 2011). For statistical analysis, we entered data into the Review Manager software (RevMan 2014). One review author (PF) entered the data and a second (LE) checked for accuracy.
Where meta‐analysis was feasible, and had moderate statistical heterogeneity, we used the random‐effects model for pooling the data. We used the Mantel‐Haenszel method for dichotomous outcomes or Peto method as necessary, and the inverse variance method (and SMDs as necessary) for continuous outcomes. If we had identified heterogeneity over 80%, we would not have performed a meta‐analysis; rather we would have reported a narrative review of the results.
Subgroup analysis and investigation of heterogeneity
In two trials, we performed a subgroup analysis based on TCD velocities, as one trial included participants with normal TCD velocities and the other included participants with abnormal TCD velocities
If adequate data were available, we would have performed subgroup analyses according to Cochrane recommendations on the rest of the outcomes in order to assess the effect on heterogeneity (Deeks 2011).
Age of participant: neonate, child (one to 15 years), adult (16 years and older)
Genotype (homozygous SCD (SS), sickle beta thalassaemia (Sβ0 and Sβ+) and sickle haemoglobin C disease (SC))
TCD velocities (normal (less than 170 cm/s, conditional 170 to less than 200 cm/s, abnormal at least 200 cm/s)
Presence of previous SCI on MRI
Follow‐up duration: longer‐term RCTs (one year or longer) versus shorter term RCTs (less than one year)
Sensitivity analysis
We would have assessed the robustness of our findings by performing the following sensitivity analyses according to Cochrane recommendations if sufficient data were available (Deeks 2011).
Including only those trials with a 'low risk of bias' (e.g. RCTs with methods assessed as low risk for random sequence generation and concealment of treatment allocation).
Including only those trials with less than a 20% dropout rate.
Summary of findings table
We used the GRADE approach to create a 'Summary of findings' table, as suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2011a; Schünemann 2011b). We used the GRADE approach to rate the quality of the evidence as 'high', 'moderate', 'low', or 'very low' using the five GRADE considerations below.
Risk of bias: not serious, serious or very serious
Inconsistency: not serious, serious or very serious
Indirectness: not serious, serious or very serious
Imprecision: not serious, serious or very serious
Publication bias: undetected, likely or very likely
We presented the following outcomes within a summary of findings table for each intervention comparison.
Proportion of participants developing new or progressive MRI lesions
All‐cause mortality
Serious adverse events
Clinical stroke
Cognitive function
Quality of life
We used and specified time periods that are common to as many trials as possible (e.g. five years and under, six to 10 years, over 10 years).
Results
Description of studies
See Characteristics of included studies and Characteristics of excluded studies.
Results of the search
See PRISMA flow diagram (Figure 1).
Figure 1.

Study flow diagram.
In the searches for this review we identified a total of 1016 citations, which were reduced to 671 citations once duplicates were removed. Two review authors (PF, LE) excluded 543 citations on the basis of the abstract, and two authors (PF, LE) reviewed 127 full text articles for relevance. We excluded 12 trials within 23 publications that were not relevant, and identified seven trials within 104 publications.
Included studies
Six trials met the inclusion criteria: five completed trials (660 participants) (SIT 2014; STOP 1998; STOP 2 2005; SWiTCH 2012; TWITCH 2016); and one ongoing trial with an estimated completion date of October 2017 (NCT01389024) (see Characteristics of ongoing studies).
One additional trial is awaiting classification to determine if it meets the inclusion criteria (Vichinsky 2010) (see Characteristics of studies awaiting classification). The primary author has been contacted for the full trial report and results.
Trial design
All five randomised trials were multicentre, ranging from 12 centres (STOP 1998; STOP 2 2005) to 29 centres (SIT 2014).
Four trials were terminated early (STOP 1998; STOP 2 2005; SWiTCH 2012; TWITCH 2016):
STOP 1998 was terminated 16 months early by the trial's data monitoring board when a 92% reduction in incidence of stroke in the transfused group was seen.
STOP 2 2005 was terminated two years early due to safety concerns.
SWiTCH 2012 was stopped early due to futility for the composite primary endpoint.
TWITCH 2016, a non‐inferiority trial, was stopped after the first scheduled interim analysis because non‐inferiority had been demonstrated.
The SIT trial was the only trial that was not stopped before the planned end of recruitment and follow‐up (SIT 2014).
Trial size
The number of participants enrolled in the five trials ranged from 79 (STOP 2 2005) to 196 (SIT 2014). Power calculations were reported in four trials (SIT 2014; STOP 1998; STOP 2 2005; TWITCH 2016), three of these trials were stopped early (STOP 1998; STOP 2 2005; TWITCH 2016). The STOP 2 trial planned to recruit 100 children and the TWiTCH trial planned to recruit 148 children (STOP 2 2005; TWITCH 2016).
Setting
The trials were published between 1998 (STOP 1998) and 2016 (TWITCH 2016). All were multicentre trials (12 to 29 recruitment centres). One trial was conducted in the USA (SWiTCH 2012), three trials in the USA and Canada (STOP 1998; STOP 2 2005; TWITCH 2016); and the SIT trial in the USA, Canada, France and the UK (SIT 2014).
Participant
All trials included participants with HbSS disease and HbSβº thalassaemia. Two trials also included participants with HbS/O Arab disease (SWiTCH 2012; TWITCH 2016). Three trials did not specify the distribution of phenotypes (SIT 2014; STOP 1998; STOP 2 2005). In the SWiTCH trial 100% of participants in the transfusion arm and 99% in the no transfusion arm had the HbSS phenotype (SWiTCH 2012), and in TWiTCH trial 97% in the transfusion arm and 100% in hydroxyurea arm had the HbSS phenotype (TWITCH 2016). In the STOP trial, α‐thalassaemia trait was more common in the RBC transfusion arm: 14 (22%) in the RBC arm and 7 (9%) in the standard care arm (STOP 1998).
All participants in the trials were children and adolescents aged from two to 20 years. Two trials included participants over 16 years (STOP 2 2005; SWiTCH 2012). The ages in the trials ranged from the lowest mean (SD) age of eight years (three years) in the SIT and STOP trials (SIT 2014; STOP 1998) to a mean (SD) high of 13 years (four years) in the SWiTCH trial (SWiTCH 2012).
Participants tended to be equally divided between males and females with the highest participation of males (57%) in the SIT trial and the lowest (39%) in the TWiTCH trial (SIT 2014; TWITCH 2016).
All trials excluded females who were pregnant or people who had HIV. Other inclusion and exclusion criteria varied depending on the objectives of the trial. In the SWiTCH trial, individuals were included if they had an overt clinical stroke after the age 12 months (SWiTCH 2012), whereas in the other four trials individuals were excluded if they had a clinical history of stroke (SIT 2014; STOP 1998; STOP 2 2005; TWITCH 2016).
In the SIT trial, children had to have evidence of at least one SCI confirmed on MRI and normal TCD velocities (SIT 2014). In a further three trials, individuals had to have abnormal TCD velocities prior to any transfusion therapy (greater than or equal to 200 cm/s) (STOP 1998; STOP 2 2005; TWITCH 2016), and in the STOP 2 trial, these abnormal TCD velocities had to have normalised with transfusion therapy (STOP 2 2005). Three trials excluded individuals with a history of seizures (SIT 2014; STOP 1998; STOP 2 2005); two trials excluded individuals with a severe vasculopathy (STOP 2 2005; TWITCH 2016); and one trial excluded children with a previous TIA (TWITCH 2016).
Interventions
All trials had a transfusion arm with the aim of keeping HbS to 30% or less with local discretion as to the type of RBC transfusion administered (simple, manual exchange or automated exchange). Three trials reported using leucocyte‐depleted RBC and blood matched for C, D, E and Kell antigen (SIT 2014; STOP 1998; STOP 2 2005), the remaining two trials did not report the type of blood component used (SWiTCH 2012; TWITCH 2016).
In two trials participants in the transfusion arm also received iron chelation (SWiTCH 2012; TWITCH 2016), in the SIT and STOP 2 trials, participants received iron chelation if required (SIT 2014; STOP 2 2005) and in the remaining trial, participants did not receive iron chelation (STOP 1998).
Three trials compared long‐term transfusion therapy to standard care with no hydroxyurea treatment (SIT 2014; STOP 1998; STOP 2 2005). In two of these trials participants had not had previous long‐term transfusions (SIT 2014; STOP 1998), and in one trial all participants had previous long‐term transfusions to prevent primary stroke (STOP 2 2005). In the STOP 2 trial, the transfusion‐halted arm could receive transfusions to treat SCD complications but if hydroxyurea or regular transfusions were initiated it was considered a cross‐over and data were censored at treatment initiation (STOP 2 2005).
Hydroxyurea was the comparator in the SWiTCH and TWiTCH trials, which was initiated at 20 mg/kg/day with escalation to a maximum tolerated dose (MTD) with transfusion overlap for four to nine months until MTD was reached (SWiTCH 2012; TWITCH 2016). Once MTD was reached, phlebotomy was commenced with a target of 10 mL/kg of blood removed monthly to reduce iron burden (maximum 500 mL).
There were no randomised trials that had an HSCT therapy comparison.
Outcomes
Outcomes varied across trials depending on the objectives. The primary outcome in the SIT trial was the recurrence of infarct or haemorrhage as determined by neuroimaging (SIT 2014). Secondary outcomes were TIA and changes in cognition.
In both STOP trials, the primary outcomes were cerebral infarction or intracranial haemorrhage and the STOP 2 trial also included reversion to abnormal velocity on TCD (STOP 1998; STOP 2 2005). In both trials secondary outcomes reported were death, and transfusion‐ and SCD‐related adverse events.
In the SWiTCH trial, the primary outcome was a composite primary endpoint of secondary stroke recurrence rate and quantitative liver iron concentration, while non‐stroke neurological events, non‐neurological sickle cell clinical events, quality of life evaluation, and measures of organ function were all secondary outcomes (SWiTCH 2012).
In the TWiTCH trial, the primary outcome was TCD time‐averaged mean velocity on the index side, defined as the cerebral hemisphere with the higher mean arterial velocity at baseline assessment (TWITCH 2016). TCD velocity on the non‐index side, new stroke or non‐stroke neurological events, new brain MRI or MRA lesions, hepatic iron overload, sickle‐related events, status, quality of life, growth, and treatment‐related complications were secondary outcomes.
Source
All five trials received government funding.
Excluded studies
We excluded 12 trials (23 references) primarily because they either reported the wrong outcomes or they were not randomised (see Characteristics of excluded studies).
One trial (12 references) and two ongoing trials were not designed to assess SCI‐related outcomes (BABY HUG 2011; NCT02560935; NCT02675790). These trials did not or are not performing baseline and exit MRIs.
Eight trials were not randomised (Adams 1999, Bernaudin 2015; Bernaudin 2016; Steinberg 2010; NCT01340404; NCT00402480; NCT01801423; NCT00004485).
One reference was a review (Kawadler 2016).
Risk of bias in included studies
Please refer to the figures section of the review for visual representations of the assessments of risk of bias across all trials and for each item in the included trials (Figure 2; Figure 3). See the risk of bias section in the 'Characteristics of included studies' section for further information about the bias identified within the individual trials.
Figure 2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Figure 3.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Random sequence generation (selection bias)
We considered four trials to be at low risk of bias, as randomisation was done centrally by the statistical data co‐ordinating centre or was randomly generated or both (SIT 2014; STOP 1998; STOP 2 2005;TWITCH 2016). We judged the SWiTCH trial to be at unclear risk of bias as the method of randomisation was not adequately reported (SWiTCH 2012).
Allocation concealment (selection bias)
We considered three trials to be at low risk of bias for allocation concealment as assignment was done by a central statistical data centre or allocation was statistically determined or both (SIT 2014; STOP 1998;TWITCH 2016). We considered two trials to be at unclear risk of bias because no description of allocation concealment was provided (STOP 2 2005; SWiTCH 2012).
Blinding
Blinding of participants and personnel (performance bias)
We considered all five trials to be at high risk of performance bias as it is impractical to mask a blood transfusion intervention so all participants and personnel were unblinded.
Blinding of outcome assessment (detection bias)
We considered all five trials to be at low risk of bias for the outcome assessment of SCI, clinical stroke and mortality as these outcomes were either adjudicated by experts masked to treatment assignments or an objective outcome (mortality).
We judged all five trials to be at high risk of bias for all other outcomes as all were unblinded.
Incomplete outcome data
We considered four trials to be at low risk for attrition bias as they all used an intention‐to‐treat analysis and all participants were accounted for in the trials (SIT 2014; STOP 1998; SWiTCH 2012; TWITCH 2016).
We judged the STOP trial to be at an unclear risk of bias for the SCI outcome as only those participants with an MRI at randomisation were included and treatment classification was based on the actual trial experience (STOP 1998).
We judged the STOP 2 trial to be at unclear risk for attrition bias as 17% of participants discontinued or data were censored (STOP 2 2005). However, we considered the trial to be at low risk of bias for stroke, SCI and TIA as only one participant in each group did not have a follow‐up MRI.
Selective reporting
We considered one trial to be at low risk of reporting bias as a protocol was provided and all planned outcomes were reported (SIT 2014). We rated three trials as having an unclear risk for reporting bias (STOP 1998; STOP 2 2005; TWITCH 2016): one trial had no protocol and no prospective trial registration (STOP 1998); one trial did not report any secondary outcomes and it was not clear if all adverse events were reported as some participant data were censored (STOP 2 2005); and the remaining trial did not report some secondary outcomes and it was unclear if these outcomes will be reported in future publications (TWITCH 2016).
We judged one trial to be at high risk for selective reporting bias for outcomes other than SCI, stroke, and mortality, as several secondary outcomes were not reported (i.e. quality of life, growth and development, organ damage, transfusion‐related, chelation‐related and phlebotomy related complications) (SWiTCH 2012).
Other potential sources of bias
We considered all trials to be at unclear risk for other sources of bias. We judged four trials to have unclear risk due to early termination of these trials (STOP 1998; STOP 2 2005; SWiTCH 2012; TWITCH 2016). In addition to this, in the TWiTCH trial, children with severe vasculopathy were excluded during screening, so these children might not be suitable candidates for hydroxyurea and longer follow‐up is required to determine if findings are maintained over time (TWITCH 2016). The SIT trial was considered to have unclear risk because there was a 20% cross‐over rate to either transfusion or hydroxyurea treatment and also because hydroxyurea was started in 17% of participants due to disease severity even though it was part of the exclusion criteria (SIT 2014). In the SWiTCH trial, more participants had moya‐moya in the hydroxyurea arm (11 participants) than the transfusion arm (five participants), it was not known if there was a difference between treatment arms in the number of participants with severe vasculopathy (SWiTCH 2012).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6
RBC transfusion versus standard care
Three trials including 405 participants compared RBC transfusions to standard care and all trials assessed the primary prevention of stroke (SIT 2014; STOP 1998; STOP 2 2005). In the SIT trial, the primary endpoint was the occurrence of any infarct including a new or enlarged SCI (SIT 2014).
Two trials included children who were at higher risk of a primary stroke who did not have previous long‐term RBC transfusions to prevent stroke (SIT 2014; STOP 1998). Children in the SIT trial had evidence of previous SCI on MRI but normal or conditional TCD velocities (SIT 2014). Children in the STOP trial had abnormal TCD velocities and 35% of these children also had SCI on MRI (STOP 1998). Data on the incidence of SCI is extracted from an associated trial publication (Pegelow 2001). SCI was not a primary outcome in this trial, and intention‐to‐treat analysis was not used with treatment classification based on the actual trial experience.
One trial included children and adolescents who were at higher risk of a primary stroke (previous history of abnormal TCD velocities) who had previous long‐term RBC transfusions (at least 30 months) to prevent stroke (STOP 2 2005). The majority of participants in the STOP 2 trial had also participated in the first STOP trial, participants were included if their TCD velocities had normalised (STOP 1998; STOP 2 2005). Data on the incidence of SCI are extracted from an associated trial publication (Abboud 2011).
Participants in the STOP 2 trial are analysed separately under the 'Transfusion continued versus transfusion halted comparison' (STOP 2 2005).
Primary outcomes
1. Proportion of participants developing new or progressive SCI lesions on MRI
Participants with normal TCD velocities
Long‐term RBC transfusions may make little or no difference to the incidence of SCI in children with previous SCI on MRI and normal TCD velocities, RR 0.70 (95% CI 0.23 to 2.13) (one trial, 196 participants, low‐quality evidence) (SIT 2014) (Analysis 1.1; Table 1).
Analysis 1.1.
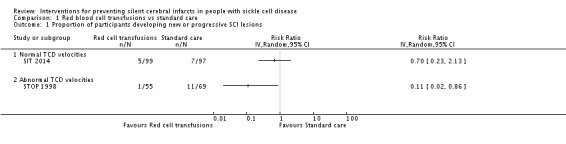
Comparison 1 Red blood cell transfusions vs standard care, Outcome 1 Proportion of participants developing new or progressive SCI lesions.
Participants with abnormal TCD velocities
Long‐term RBC transfusions may reduce the incidence of SCI in children with abnormal TCD velocities, RR 0.11 (95% CI 0.02 to 0.86) (one trial, 124 participants, low‐quality evidence) (STOP 1998) (Analysis 1.1; Table 2).
2. All‐cause mortality
No participants died in the SIT and STOP trials (two trials, 326 participants), therefore, we have no evidence of any difference in mortality rates between the treatments (low to very low‐quality evidence) (SIT 2014; STOP 1998) (Table 1; Table 2).
3. SAEs associated with different therapies or SCD
SCD‐related SAEs ‐ ACS
Participants with normal TCD velocities
Long‐term RBC transfusions may reduce the incidence of ACS in children with previous SCI on MRI and normal TCD velocities, RR 0.20 (95% CI 0.08 to 0.51) (one trial, 196 participants, low‐quality evidence) (SIT 2014) (Analysis 1.2; Table 1).
Analysis 1.2.
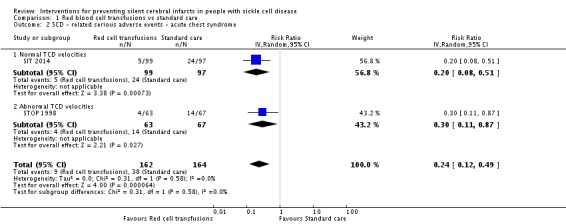
Comparison 1 Red blood cell transfusions vs standard care, Outcome 2 SCD ‐ related serious adverse events ‐ acute chest syndrome.
Participants with abnormal TCD velocities
Long‐term RBC transfusions may reduce the incidence of ACS in children with abnormal TCD velocities, RR 0.30 ( 95% CI 0.11 to 0.87) (one trial, 130 participants, low‐quality evidence) (STOP 1998) (Analysis 1.2; Table 2).
Participants with no previous long‐term RBC transfusions
Long‐term RBC transfusions may reduce the incidence of ACS in children with a higher risk of stroke (abnormal TCD velocities or previous history of SCI), RR 0.24 (95% CI 0.12 to 0.49) (two trials, 326 participants, low‐quality evidence) (SIT 2014; STOP 1998) (Analysis 1.2; Table 3).
SCD‐related SAEs ‐ painful crisis
Participants with normal TCD velocities
Long‐term RBC transfusions may reduce the incidence of painful crisis in children with previous SCI on MRI and normal TCD velocities, RR 0.56 (95% CI 0.40 to 0.78) (one trial, 196 participants, low‐quality evidence) (SIT 2014) (Analysis 1.3; Table 1).
Analysis 1.3.
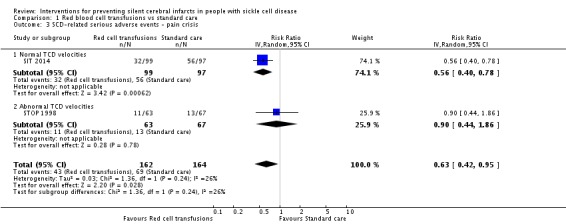
Comparison 1 Red blood cell transfusions vs standard care, Outcome 3 SCD‐related serious adverse events ‐ pain crisis.
Participants with abnormal TCD velocities
Long‐term RBC transfusions may be no different than standard care in reducing the incidence of painful crisis in children with abnormal TCD velocities, RR 0.90 (95% CI 0.44 to 1.86) (one trial, 130 participants, very low‐quality evidence) (STOP 1998) (Analysis 1.3; Table 2).
Participants with no previous long‐term RBC transfusions
Long‐term RBC transfusions may reduce the incidence of painful crisis in children with a higher risk of stroke (abnormal TCD velocities or previous history of SCI), RR 0.63 (95% CI 0.42 to 0.95) (two trials, 326 participants, low‐quality evidence) (SIT 2014; STOP 1998) (Analysis 1.3; Table 3).
Secondary outcomes
1. Clinical stroke
Participants with normal TCD velocities
Long‐term RBC transfusions may make little or no difference on the incidence of clinical stroke in children with previous SCI on MRI and normal TCD velocities, RR 0.14, 95% CI 0.02 to 1.12) (one trial, 196 participants, low‐quality evidence) (SIT 2014) (Analysis 1.4; Table 1).
Analysis 1.4.
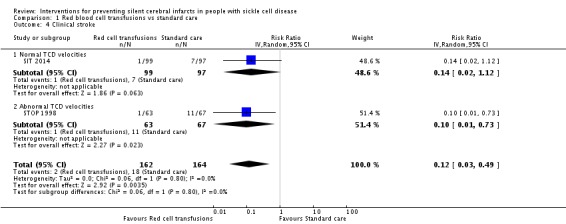
Comparison 1 Red blood cell transfusions vs standard care, Outcome 4 Clinical stroke.
Participants with abnormal TCD velocities
Long‐term RBC transfusions may reduce the incidence of stroke in children with abnormal TCD velocities, RR 0.10 (95% CI 0.01 to 0.73) (one trial, 130 participants, low‐quality evidence) (STOP 1998) (Analysis 1.4; Table 2).
Participants with no previous long‐term RBC transfusion
Compared to children receiving standard care, long‐term RBC transfusions probably reduce the incidence of clinical stroke in children with a higher risk of stroke (abnormal TCD velocities or previous history of SCI), RR 0.12 (95% CI 0.03 to 0.49) (two trials, 326 participants, moderate‐quality evidence) (SIT 2014; STOP 1998) (Analysis 1.4; Table 3).
2. Cognitive function as assessed by validated scales
Participants with normal TCD velocities
The SIT trial reported the difference between trial arms in the Weschler Abbreviated Scale of Intelligence (WASI) in performance, verbal, and full IQ scores (data from baseline to interim and to exit visits were analysed by the trial authors who reported estimated least squares means (LSM) and based on two‐way repeated measures analysis of variance) (SIT 2014).
The SIT trial authors reported that long‐term RBC transfusion may make little or no difference to WASI performance, verbal or full IQ scores in children with SCI (LSM: 1.7, 95% CI ‐1.1 to 4.4) (one trial, 166 participants, low‐quality evidence; analysis performed by the SIT 2014 trial authors) (SIT 2014) (Table 1).
Participants with abnormal TCD velocities
In the STOP trial, at the time of discharge, of the 11 children in the standard care group diagnosed with cerebral infarction, two had a major disability, five had mild to moderate disability, two had symptoms but no disability and one was asymptomatic (STOP 1998). Measures of cognitive function were not reported in the trial, but noted that after 24 months, children receiving transfusion had improved growth (height, weight and body mass index (BMI)) which approached normal, whereas there was no improvement in growth rate in children receiving standard care (STOP 1998).
3. Quality of life as assessed by validated scales
Participants with normal TCD velocities
The SIT trial reported quality of life measured on the Child Health Questionnaire Parent Form 50 (CHQ PF50) completed at baseline and at the occurrence of an overt stroke or at trial exit at 36 months (SIT 2014).
It is not known what the minimal clinically important differences are for the CHQ PF50.
Trial authors stated that quality of life may improve slightly in children receiving long‐term RBC transfusions, difference estimate ‐0.54 (95% CI ‐0.92 to ‐0.17) (one trial, 196 participants, low‐quality evidence) (analysis performed by the SIT trial authors) (SIT 2014) (Table 1).
Participants with abnormal TCD velocities
This outcome was not reported in the STOP trial (STOP 1998).
4. Any adverse events associated with different therapies
Transfusion‐related adverse events ‐ alloimmunisation
Participants with normal TCD velocities
We are very uncertain whether children receiving regular long‐term transfusions have a higher risk for developing alloimmunisations than children receiving standard care, RR 3.16 (95% CI 0.18 to 57.17) (one trial, 121 participants, low‐quality evidence) (SIT 2014) (Analysis 1.5). For data on transfused participants in the SIT trial, the quality of the evidence was low due to high risk of bias and indirectness (SIT 2014).
Analysis 1.5.
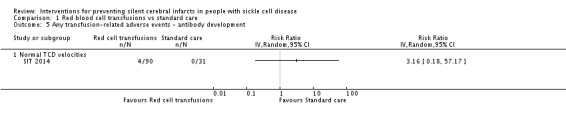
Comparison 1 Red blood cell transfusions vs standard care, Outcome 5 Any transfusion‐related adverse events ‐ antibody development.
Participants with abnormal TCD velocities
In the STOP trial, 10 participants in the transfusion arm developed an alloimmunisation, despite a more rigorous matching protocol than usual (STOP 1998). It was not reported if any participants in the standard care developed any alloimmunisations.
Transfusion‐related adverse events ‐ transfusion reactions
Participants with normal TCD velocities
We are very uncertain whether children receiving regular long‐term transfusions have a higher risk of developing a transfusion reaction than children receiving standard care, RR 5.17 (95% CI 0.71 to 37.52) (one trial, 121 participants, low‐quality evidence) (SIT 2014) (Analysis 1.6). The quality of the evidence was low due to high risk of bias and indirectness.
Analysis 1.6.

Comparison 1 Red blood cell transfusions vs standard care, Outcome 6 Any transfusion‐related adverse events ‐ transfusion reactions.
Participants with abnormal TCD velocities
In the STOP trial, 12 participants in the transfusion arm had 16 mild reactions to blood products or transfusion procedures (STOP 1998). This was not reported for the standard treatment arm.
Iron overload
Participants with normal TCD velocities
Trial authors reported that there was an increased risk of iron overload (measured by serum ferritin greater than 1500 μg/L) in children receiving long‐term RBC transfusions, incidence rate ratio 14.42 (95% CI 5.41 to 875.17) (one trial, 121 participants, low‐quality evidence) (analysis reported in the SIT trial) (SIT 2014) (Table 10). The quality of the evidence was low due to high risk of bias and imprecision.
Table 1.
1 Adverse events per 100 person‐years and incidence rate ratios for transfusion‐related complications
| Outcomes | Studies | Number of participants with at least one event | Adverse events/100 person‐years |
Incidence rate ratioc (95% CI) |
||
| Transfusion | Standard | Transfusion | Standard | |||
| Transfusion reactions | SIT 2014 | 15 out of 90a | 1 out of 31b | 8.85 | 1.66 | 5.33 (1.67 to 23.52) |
| Ferritin > 1500 μg/L | SIT 2014 | 76 out of 90a | 3 out of 31b | 534.70 | 37.07 | 14.42 (5.41 to 875.17) |
CI: confidence interval
a 9 participants who declined transfusion were excluded from the analysis. b 31 participants assigned to observation received one or more transfusions. c The incidence ratio was calculated as the rate of adverse events per 100 person‐years in the transfusion group divided by the rate of adverse events per 100 person‐years in the observation group. The 95% CIs were calculated with the use of the bootstrap method with 10,000 replications
Participants with abnormal TCD velocities
In the STOP trial, iron overload developed faster than anticipated in children receiving transfusion, with mean (SD) serum ferritin rising to 1804 μg/L (773 μg/L) at 12 months and 2509 μg/L (974 μg/L) at 24 months (STOP 1998). This outcome was not reported in the children receiving standard care who had transfusions.
Other sickle cell‐related complications
One trial reported additional SCD‐related adverse events as events per 100 person years and incidence rate ratios (SIT 2014) (Table 11).
Table 2.
2 Adverse events per 100 person years and incidence rate ratios for SCD complications
| Outcomes | Studies | Number of participants with at least one event | Adverse events/100 person‐years |
Incidence Rate Ratioa (95% CI) |
||
| Transfusion | Standard | Transfusion | Standard | |||
| Acute chest syndrome | STOP 1998 | 4 out of 63 | 14 out of 67 | 4.8b | 15.3b | ‐ |
| SIT 2014 | 5 out of 99 | 24 out of 97 | 1.81b | 14.35b | 0.41 (0.20 to 0.75) |
|
| Painful crisis | STOP 1998 | 11 out of 63 | 13 out of 67 | 16.2 | 27.6 | ‐ |
| SIT 2014 | 32 out of 99 | 56 out of 97 | 41.58 | 102.21 | 0.13 (0.04 to 0.28) |
|
| Priapism | SIT 2014 | 1 out of 59 | 7 out of 52 | 0.84 | 6.65 | 0.13 (0.03 to 0.55) |
| Symptomatic avascular necrosis of the hip | SIT 2014 | 1 out of 99 | 6 out of 97 | 0.4 | 2.25 | 0.22 (0.05 to 0.85) |
CI: confidence interval
aThe incidence ratio was calculated as the rate of adverse events per 100 person‐years in the transfusion group divided by the rate of adverse events per 100 person‐years in the observation group. The 95% confidence intervals were calculated with the use of the bootstrap method with 10,000 replications.
bOne child from the standard care group was excluded from these analyses due to a stroke on day 16 of the trial.
Transfusions continued versus transfusions halted
Outcomes are reported from one trial in 79 participants whose initial abnormal TCD velocities have normalised (STOP 2 2005).
Primary outcomes
1. Proportion of participants developing new or progressive SCI lesions on MRI
Continuing long‐term RBC transfusions may reduce the incidence of SCI in children and adolescents whose TCD velocities have normalised, RR 0.29 (95% CI 0.09 to 0.97) (one trial, 77 participants, low‐quality evidence) (STOP 2 2005) (Analysis 2.1; Table 4).
Analysis 2.1.

Comparison 2 Transfusions continued vs transfusions halted, Outcome 1 Proportion of participants developing new or progressive SCI lesions.
2. All‐cause mortality
We are very uncertain whether continuing long‐term RBC transfusions reduces mortality in children and adolescents with normalised TCD velocities.
One participant who was assigned to continue transfusion, died due to complications of ACS, Peto OR 8.00 (95% CI 0.16 to 404.12) (one trial, 79 participants, very low‐quality evidence) (STOP 2 2005) (Analysis 2.2; Table 4).
Analysis 2.2.
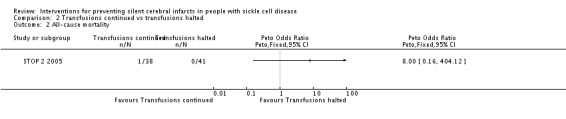
Comparison 2 Transfusions continued vs transfusions halted, Outcome 2 All‐cause mortality.
3. SAEs associated with different therapies or SCD
SCD‐related SAEs ‐ ACS
The authors state that there were fewer incidences of ACS in participants continuing transfusions. In the halted transfusion group, 18 of 41 participants had at least one episode of ACS. Comparative numbers for the transfusion group were not reported (STOP 2 2005).
SCD‐related SAEs ‐ painful crisis
The authors state that there were fewer painful crises in the continuing transfusion arm. Comparative numbers were not reported (STOP 2 2005).
Secondary outcomes
1. Clinical stroke
We are very uncertain whether continuing long‐term RBC transfusions reduces the incidence of clinical stroke in children and adolescents whose TCD velocities have normalised, RR 0.22 (95% CI 0.01 to 4.35) (one trial, 79 participants, very low‐quality evidence). The CIs of the treatment effect estimate are wide due to the low frequency of stroke events in the two treatment arms (STOP 2 2005) (Analysis 2.3; Table 4).
Analysis 2.3.
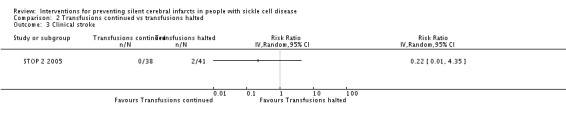
Comparison 2 Transfusions continued vs transfusions halted, Outcome 3 Clinical stroke.
2. Cognitive function as assessed by validated scales
This outcome was not reported.
3. Quality of life as assessed by validated scales
This outcome was not reported.
4. Any adverse events associated with different therapies
Transfusion‐related adverse events ‐ transfusion reactions
Seven participants in the continuing transfusion arm had nine reactions to transfusions. One of the reactions was serious and required hospitalisation. This outcome was not reported for the halted transfusion arm (STOP 2 2005).
Transfusion‐related adverse events ‐ alloimmunisation
One participant who was in the continuing transfusion arm developed an alloimmunisation. It was not reported if any participants in the halted transfusion arm developed any alloimmunisations (STOP 2 2005).
Infections
No cases of hepatitis C were identified among the 68 participants who had serologic testing at the end of the trial.
Hydroxyurea versus RBC transfusions
Two trials are included in this comparison (254 participants) (TWITCH 2016; SWiTCH 2012). One trial assessed primary prevention of stroke (TWITCH 2016) and one trial assessed secondary prevention of stroke (SWiTCH 2012). The SWiTCH trial also compared blood transfusion with iron chelation to hydroxyurea with phlebotomy with a composite primary endpoint allowing for an increased stroke risk, but superiority in iron removal in the hydroxyurea with phlebotomy group. In TWITCH participants also underwent serial phlebotomy to manage iron overload, phlebotomy was not done if haemoglobin concentration was less than 80 g/L.
Data on SCI was extracted from an associated publication (Helton 2014a) for the SWiTCH trial (SWiTCH 2012).
Results from the two trials are not combined.
1. Proportion of participants developing new or progressive SCI lesions on MRI
Primary prevention
No SCI occurred in the TWiTCH trial (one trial, 121 participants, very low‐quality evidence) (TWITCH 2016) (Table 5).
Secondary prevention
We are very uncertain whether switching to hydroxyurea and phlebotomy increases the incidence of SCI in children and adolescents with a history of stroke, Peto OR 7.28 (95% CI 0.14 to 366.91) (one trial, 133 participants, very low‐quality evidence) (SWiTCH 2012) (Analysis 3.1; Table 6).
Analysis 3.1.
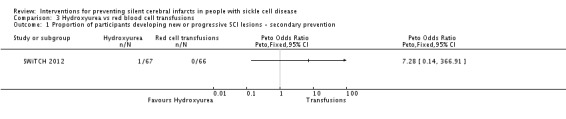
Comparison 3 Hydroxyurea vs red blood cell transfusions, Outcome 1 Proportion of participants developing new or progressive SCI lesions ‐ secondary prevention.
2. All‐cause mortality
Primary prevention
No deaths occurred in the TWiTCH trial (one trial, 121 participants, very low‐quality evidence) (TWITCH 2016) (Table 5).
Secondary prevention
We are very uncertain whether switching to hydroxyurea and phlebotomy reduces mortality compared to RBC transfusions and chelation in children and adolescents with a history of stroke, Peto OR 1.02 (95% CI 0.06 to 16.41) (one trial, 133 participants, very low‐quality evidence) (SWiTCH 2012) (Analysis 3.2; Table 6).
Analysis 3.2.

Comparison 3 Hydroxyurea vs red blood cell transfusions, Outcome 2 All‐cause mortality ‐ secondary prevention.
3. SAEs associated with different therapies or SCD
SCD‐related SAEs ‐ ACS
Primary prevention
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on the risk of ACS compared to continuing to receive RBC transfusions and chelation in children who have a previous history of abnormal TCD velocities and at least 12 months of transfusions, RR 2.03 (95% CI 0.39 to 10.69) (one trial; 121 participants, very low‐quality evidence) (TWITCH 2016) (Analysis 3.3; Table 5).
Analysis 3.3.

Comparison 3 Hydroxyurea vs red blood cell transfusions, Outcome 3 SCD‐related SAEs ‐ Acute chest syndrome ‐ primary prevention.
Secondary prevention
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on the risk of ACS compared to continuing to receive RBC transfusions and chelation in children and adolescents who have a previous history of stroke, RR 0.33 (95% CI 0.04 to 3.08) (one trial, 133 participants, very low‐quality evidence) (SWiTCH 2012) (Analysis 3.4; Table 6).
Analysis 3.4.
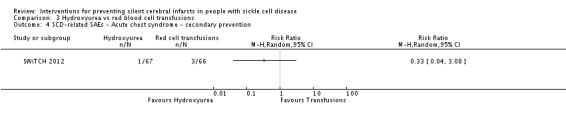
Comparison 3 Hydroxyurea vs red blood cell transfusions, Outcome 4 SCD‐related SAEs ‐ Acute chest syndrome ‐ secondary prevention.
SCD‐related SAEs ‐ painful crisis
Primary prevention
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on the risk of painful crisis compared to continuing to receive RBC transfusions and chelation in children who have a previous history of abnormal TCD velocities and at least 12 months of transfusions, RR 5.08 (95% CI 0.61 to 42.23) (one trial, 121 participants, very low‐quality evidence) (TWITCH 2016) (Analysis 3.5; Table 5).
Analysis 3.5.

Comparison 3 Hydroxyurea vs red blood cell transfusions, Outcome 5 SCD‐related SAEs ‐ Pain crisis ‐ primary prevention.
Secondary prevention
Switching to hydroxyurea and phlebotomy may increase the risk of painful crisis compared to continuing to receive RBC transfusions and chelation in children and adolescents who have a previous history of stroke, RR 3.15 (95% CI 1.23 to 8.11) (one trial, 133 participants, low‐quality evidence) (SWiTCH 2012) (Analysis 3.6; Table 6).
Analysis 3.6.

Comparison 3 Hydroxyurea vs red blood cell transfusions, Outcome 6 SCD‐related SAEs ‐ Pain crisis ‐ secondary prevention.
Total SCD‐related SAEs
Primary prevention
We are very uncertain if switching to hydroxyurea and phlebotomy has any effect on total SAEs in children who have a previous history of abnormal TCD velocities and at least 12 months of transfusions, RR 1.52 (95% CI 0.58 to 4.02) (one trial, 121 participants, very low‐quality evidence) (TWITCH 2016) (Analysis 3.7). The quality of the evidence was very low due to a high risk of bias, imprecision and indirectness.
Analysis 3.7.

Comparison 3 Hydroxyurea vs red blood cell transfusions, Outcome 7 Total SCD‐related SAEs ‐ primary prevention.
Secondary prevention
Switching to hydroxyurea and phlebotomy may increase the risk of SCD related serious adverse events compared to continuing to receive RBC transfusions and chelation in children and adolescents with a history of stroke, RR 3.10 (95% CI 1.42 to 6.75) (one trial, 133 participants, low‐quality evidence) (SWiTCH 2012) (Analysis 3.8). The quality of the evidence was low due to a high risk of bias and indirectness.
Analysis 3.8.
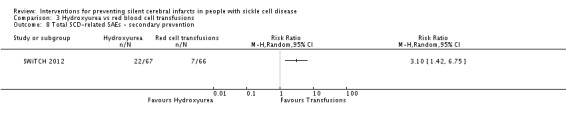
Comparison 3 Hydroxyurea vs red blood cell transfusions, Outcome 8 Total SCD‐related SAEs ‐ secondary prevention.
Secondary outcomes
1. Clinical stroke
Primary prevention
No clinical strokes occurred in the TWiTCH trial (TWITCH 2016). General stroke adjudications were done for 29 possible new neurological events and no child had a positive adjudication for stroke (very low‐quality evidence) (Table 5).
Secondary prevention
We are very uncertain whether switching to hydroxyurea and phlebotomy increase the risk of stroke compared to RBC transfusions and chelation in children and adolescents with a history of stroke, RR 14.78 (95% CI 0.86 to 253.66) (one trial, 133 participants, very low‐quality evidence) (SWiTCH 2012) (Analysis 3.9; Table 6).
Analysis 3.9.
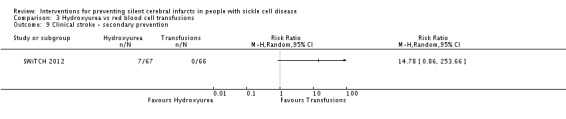
Comparison 3 Hydroxyurea vs red blood cell transfusions, Outcome 9 Clinical stroke ‐ secondary prevention.
Seven participants, all in the hydroxyurea and phlebotomy group, experienced a stroke and all had baseline MRA exams which showed severe vasculopathy, including two with moya‐moya. There were more participants with moya‐moya in the hydroxyurea arm (11 participants) compared to the transfusion arm (five participants), the number of participants with severe vasculopathy in both treatment arms was not reported (SWiTCH 2012).
2. Cognitive function as assessed by validated scales
Primary prevention
Cognitive function is not yet reported in the TWiTCH trial (TWITCH 2016).
Secondary prevention
Measures of cognitive function are not reported in the SWiTCH trial (SWiTCH 2012).
3. Quality of life as assessed by validated scales
Quality of life has not yet been reported in either trial
4. Any adverse events associated with different therapies
Iron overload
Primary prevention
Switching to hydroxyurea and phlebotomy may reduce serum ferritin levels compared to continuing to receive RBC transfusions and chelation in children who have a previous history of abnormal TCD velocities and at least 12 months of transfusions, MD ‐1.40 (95% CI ‐1.94 to ‐0.86) (one trial, 121 participants, low‐quality evidence) (TWITCH 2016) (Analysis 3.10). Quality of evidence was low due to high risk of bias and indirectness.
Analysis 3.10.

Comparison 3 Hydroxyurea vs red blood cell transfusions, Outcome 10 Transfusion‐related complications ‐ serum ferritin ‐ primary prevention.
Switching to hydroxyurea and phlebotomy may have little or no effect on liver iron concentrations compared to continuing to receive RBC transfusions and chelation in children who have a previous history of abnormal TCD velocities and at least 12 months of transfusions, MD ‐1.80 (95% CI ‐5.16 to 1.56) (one trial, 121 participants, low‐quality evidence) (TWITCH 2016) (Analysis 3.11). The quality of the evidence was low due to high risk of bias and indirectness.
Analysis 3.11.
Comparison 3 Hydroxyurea vs red blood cell transfusions, Outcome 11 Transfusion‐related complications ‐ liver iron concentration ‐ primary prevention.
Secondary prevention
Switching to hydroxyurea and phlebotomy may reduce serum ferritin levels compared to continuing to receive RBC transfusions and chelation in children and adolescents with a history of stroke (at the final assessment, median serum ferritin was 1994 μg/L, interquartile range (IQR) 998 to 3475, in the hydroxyurea arm and 4064 μg/L, IQR 2330 to 7126, in the transfusion arm; one trial, 133 participants; low‐quality evidence) (analysis performed by the trial authors) (SWiTCH 2012). The quality of the evidence was low due to high risk of bias and indirectness.
Switching to hydroxyurea and phlebotomy may have little or no effect on liver iron concentrations compared to continuing to receive RBC transfusions and chelation in children and adolescents with a history of stroke (median liver iron concentration: hydroxyurea: 17.3 mg Fe/g dry weight iron, IQR 10.0 to 30.6; transfusion: 17.3 mg Fe/g dry weight iron, IQR 8.8 to 30.7; one trial, 133 participants; low‐quality evidence) (analysis performed by the trial authors) (SWiTCH 2012). The quality of the evidence was low due to high risk of bias and indirectness.
SCD‐related AEs
Primary prevention
This outcome was not reported
Secondary preventions
We are very uncertain if switching to hydroxyurea and phlebotomy has any effect on the incidence of adverse events compared to continuing to receive RBC transfusions and chelation in children and adolescents with a history of stroke, RR 1.03 (95% CI 0.81 to 1.30) (one trial, 133 participants, very low‐quality evidence) (SWiTCH 2012) (Analysis 3.12). The quality of the evidence was very low due to high risk of bias, imprecision and indirectness.
Analysis 3.12.

Comparison 3 Hydroxyurea vs red blood cell transfusions, Outcome 12 Any SCD‐related adverse events ‐ secondary prevention.
Discussion
Silent cerebral infarcts (SCI) are the most common neurological injury in children and can occur in up to a third of adults with sickle cell disease (SCD). People with SCI have an increased risk for stroke and lower academic performance.
This Cochrane Review aimed to evaluate the literature on interventions to prevent SCI in people with SCD.
We identified five randomised controlled trials that met our inclusion criteria and included a total of 660 participants. The trials were published between 1998 and 2016. Two trials compared red blood cell (RBC) transfusions to standard care, one trial compared continuing transfusions to halting transfusions, and two trials compared hydroxyurea with phlebotomy to RBC transfusions with chelation.
The majority of participants had HbSS SCD, all trials were conducted in children and two trials additionally included adolescents. One trial focused on the prevention of infarcts, and three trials were for primary stroke prevention and one trial dealt with secondary stroke prevention. There were no randomised trials with haematopoietic stem cell transplantation (HSCT) therapy as a comparison.
Summary of main results
RBC transfusions versus standard care
Three randomised trials compared long‐term RBC transfusions to standard care. Two of these trials included children with no previous long‐term transfusions; one included children with SCI on magnetic resonance imaging (MRI) but normal transcranial doppler (TCD) velocities, and the other included children with abnormal TCD velocities. The third trial included children and adolescents on long‐term transfusion whose abnormal TCD velocities had normalised and compared continued transfusion to halted transfusion.
Two of the three trials were terminated early due to safety concerns.
The findings of the review led to the following main conclusions regarding RBC transfusions versus standard care.
Children with no previous long‐term transfusions and higher risk of stroke (abnormal TCD velocities or previous history of SCI)
Long‐term RBC transfusions may reduce the incidence of SCI in children with abnormal TCD velocities; but make little or no difference to the incidence of SCI in children with previous SCI on MRI and normal TCD velocities.
No deaths were reported in either trial.
Long‐term RBC transfusions may reduce the incidence of acute chest syndrome (ACS), and painful crisis.
Long‐term RBC transfusions probably reduce the incidence of clinical stroke.
Long‐term RBC transfusions may improve quality of life in children with previous SCI on MRI and normal TCD velocities, but make little or no difference to IQ scores.
Continued transfusions versus halted transfusions
Continuing long‐term RBC transfusions may reduce the incidence of SCI in children and adolescents whose TCD velocities have normalised.
We are very uncertain whether continuing RBC transfusions has any effect on all‐cause mortality, or clinical stroke in children and adolescents whose TCD velocities have normalised.
Several review outcomes were only reported in one of the trial arms (SCD‐related complications and alloimmunisation).
The trial did not report quality of life or cognitive function.
Hydroxyurea versus RBC transfusions
Primary prevention in children
There were no SCI, deaths or clinical strokes in either arm of the trial.
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on the risk of other SCD‐related complications (ACS or painful crisis).
The trial did not report quality of life or cognitive function.
Secondary prevention in children and adolescents
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on the risk of SCI, all‐cause mortality, or clinical stroke in children and adolescents with a history of stroke.
Switching to hydroxyurea and phlebotomy may increase the risk of SCD‐related serious adverse events.
The trial did not report quality of life or cognitive function.
Overall completeness and applicability of evidence
This review provides the most up‐to‐date assessment of the effectiveness and safety of interventions for the prevention SCI in children. We have identified one ongoing trial and we are awaiting results from another trial conducted in adults.
The results of this review can only be interpreted in consideration of the following factors:
The findings in this review can only be generalised to children with HbSS disease. Two trials also included adolescents (STOP 2 2005; SWiTCH 2012). Few children with HbSβº were included, however, they have symptoms that are clinically indistinguishable from HbSS, and are usually treated in the same way as children with HbSS.
Only one trial included new or enlarged SCI as a primary outcome (SIT 2014).
Only one of the trials assessed secondary prevention and was conducted in children who previously had an overt stroke (SWiTCH 2012).
All of the trials were conducted in high‐income countries (USA, Canada, France and the UK). The potential risks associated with transfusion therapy are increased in low‐income countries due to a lack of trained staff, modern equipment, sanitary conditions and clean, infection‐free blood products (Ansong 2013; Ohene‐Frempong 1999). Therefore, the risk‐benefit ratio will be different in low‐income countries to those in the included trials, and the results discussed in this review may not be generalisable to that setting.
Co‐inheritance of alpha thalassaemia may reduce the frequency of complications in individuals with HbSS (Rumaney 2014; Steinberg 2012). Only one trial reported co‐existence of alpha thalassaemia (at least one alpha globin gene missing) which occurred in 22% in the transfusion arm and 9% in the standard treatment arm (STOP 1998).
The prevention of SCI must be weighed carefully against the burden of regular blood transfusion. In the STOP trial, of the 192 children eligible to take part in the trial, 52 declined to undergo randomisation due to either reluctance of the child or their parents, or due to concern of the physician about compliance with the regimen (STOP 1998). In the SIT trial, of the 99 randomised to transfusion, 15 crossed over to observation, nine of which declined transfusion immediately after randomisation (SIT 2014). These figures may illustrate a relatively poor level of acceptance of this therapy.
The trials did not answer the question of how long RBC transfusion needs to be continued in this population, once commenced. The STOP 2 trial attempted to determine a safe age, or period, for discontinuation of chronic transfusion therapy for primary stroke prevention (STOP 2 2005). This trial was stopped two years early because a high proportion (39%) of participants reverted to an abnormal TCD (predicting a higher stroke risk) or had a stroke after transfusion was ceased. The trial authors suggested that transfusion therapy should be continued indefinitely (because the increased risk is presumed to be life‐long); however, eight participants (20%) of those who discontinued transfusion had no abnormal TCD over 25 months of observation. There is currently no way of predicting which individuals will require continuance of transfusion and which will not. Extended follow‐up analysis of the STOP trial also failed to identify predictors for lower‐risk groups (Lee 2006a).
Four of the five trials were stopped early. The STOP trial was terminated 16 months before the planned end date because of the high rate of stroke in the standard care arm (STOP 1998). The STOP 2 trial was terminated early over safety concerns because high risk TCD results developed in 14 participants and two participants had a stroke in the transfusion halted arm (STOP 2 2005). The SWiTCH trial was stopped early due to futility when hydroxyurea and phlebotomy showed no advantage in iron removal compared with long‐term transfusion and iron chelation (SWiTCH 2012). The TWiTCH trial was stopped early because after the first interim analysis, when 33% of participants had completed the trial, non‐inferiority was found, and non‐inferiority was confirmed again after 50% of participants had exited the trial (TWITCH 2016).
Children with severe vasculopathy were excluded from the TWiTCH trial, and only individuals with severe vasculopathy had a stroke in the SWiTCH trial (SWiTCH 2012; TWITCH 2016). So individuals with severe vasculopathy may not be candidates for hydroxyurea. Also, it is not known if the effectiveness of hydroxyurea with phlebotomy is sustained over the long term. In the SIT trial there was a 20% cross‐over rate to either transfusion or hydroxyurea therapy due to increasing disease severity in children receiving standard care (SIT 2014). Six per cent of children receiving standard care crossed over to regular monthly transfusions and 14% began receiving hydroxyurea treatment. We do not know how this may have influenced outcomes over the long term, but this increases uncertainty as to the efficacy of standard care compared to transfusions in these children.
Quality of the evidence
Overall the quality of the evidence was rated moderate to very low across different outcomes according to GRADE methodology (Table 1; Table 2; Table 3; Table 4; Table 5; Table 6). This was due to trials being at serious risk of bias; outcome estimates being imprecise (wide confidence intervals); and a serious risk of indirectness (no direct evidence for adults or children without HbSS) since the trials only included children and adolescents primarily with HbSS disease.
The HbSS and HbSβº thalassaemia genotypes account for approximately 70% of people with SCD. Individuals with these genotypes have a higher prevalence of SCI than individuals with HbSC and other milder forms of SCD. Nevertheless, the participant inclusion criteria for this review is broad and rates of SCI have been estimated to range from 3% to 38% in people with HbS/β‐thalassaemia and in 5% to 31% in people with HbSC (DeBaun 2012). A recent study in a cohort of 96 children with HbSC disease, estimated the prevalence of SCIs to be 13.5% (Guilliams 2015).
The outcome for clinical stroke in primary prevention in children with no previous long‐term transfusions and at higher risk of stroke (abnormal TCD velocities and presence of SCIs) was the only outcome rated as moderate‐quality evidence.
Children with no previous long‐term transfusions and higher risk of stroke (abnormal TCD velocities or previous history of SCI)
We considered all outcomes measured in the SIT trial (children with a previous history of SCI and normal TCD velocities) to be low‐quality evidence due to either serious risk of bias, indirectness or imprecision (SIT 2014).
In the STOP trial (children with abnormal TCD velocities), we considered three outcomes (SCI, ACS, clinical stroke) to be low‐quality evidence due to either serious risk of bias, indirectness or imprecision; and we considered two outcomes (all‐cause mortality, pain crisis) to be very low‐quality evidence due to serious risk of bias, indirectness and imprecision (STOP 1998). The trial did not report cognitive function or quality of life.
In children at higher risk of stroke (abnormal TCD velocities and SCIs), we considered one outcome (clinical stroke) as moderate‐quality evidence due to indirectness; and SCD‐related adverse events (ACS and painful crises) as low‐quality evidence due to serious risk of bias and indirectness.
Transfusions continued versus transfusions halted
We considered one outcome (SCI) as low‐quality evidence due to serious risk of bias and indirectness.
We considered two outcomes (all‐cause mortality, clinical stroke) as very low‐quality evidence due to serious risk of bias, indirectness and imprecision.
SCD‐related SAEs were partially reported in one of the trial arms and the trial did not report cognitive function or quality of life.
Hydroxyurea and phlebotomy versus RBC transfusions and iron chelation
Primary prevention
We considered four outcomes as very low‐quality evidence due to serious risk of bias, indirectness and imprecision. These were:
SCI
all‐cause mortality
clinical stroke
SCD‐related adverse events (ACS, painful crises)
The trial did not report cognitive function or quality of life.
Secondary prevention
We considered one outcome (pain crisis) to be low‐quality evidence due to serious risk of bias and indirectness
We considered four outcomes to be very low‐quality evidence due to serious risk of bias, indirectness, and imprecision. These were:
SCI;
all‐cause mortality;
ACS;
clinical stroke.
The trial did not report cognitive function or quality of life.
Potential biases in the review process
To our knowledge, our review process is free from bias. We conducted a comprehensive search; searching data sources (including multiple databases, and clinical trial registries) to ensure that all relevant trials would be captured. There were no restrictions for the language in which the paper was originally published. The relevance of each paper was carefully assessed and all screening and data extractions were performed in duplicate. We pre‐specified all outcomes and subgroups prior to analysis. There were insufficient numbers of included trials within the meta‐analyses for us to use a funnel plot to examine the risk of publication bias.
Agreements and disagreements with other studies or reviews
Significant challenges remain in advancing and managing the care of children and adults with SCD and neurological complications (DeBaun 2016a). A recent systematic review assessed the effect of SCI on intelligent quotient (IQ) (Kawadler 2016). Mean differences in scores confirmed that people with stroke had lower IQs than people with SCI, but people with SCI also had lower IQ scores than those with no SCI. The review also found that people with SCD generally had lower IQ scores than healthy controls. The review highlights the importance of current clinical practice to improve cognitive function in children with SCD including the use of TCD to identify people at high risk of stroke, long‐term transfusions, and hydroxyurea to reduce anaemia, painful crisis and disease‐specific adverse events that may also influence cognitive function (Kawadler 2016).
The investigators in the Cooperative Study in Sickle Cell Disease (CSSD) also established a link between MRI lesions and measurable global cognitive function (Armstrong 1996). Studies have reported that at least 27% of children with SCD have an SCI before six years of age (Kwiatkowski 2009). The Baby Hug trial also found neurocognitive decline increased with older age particularly between the ages of 12 and 24 months (Armstrong 2013a). Although MRI imaging was halted due to concern about sedation, in the subset of children who had an MRI, 13% had evidence of a silent infarct. In this report the authors also suggest that patterns seen in infants could extend well beyond into adulthood. As in the Kawadler review the authors suggest that as well as the role of infarction, additional studies should consider the effect that anaemia, and resulting haemolysis and nitric oxide availability, may have on cognitive function (Kawadler 2016). Since neurodevelopmental deficits appear early in life, interventions such as RBC transfusions, hydroxyurea and HST, should be evaluated shortly after diagnosis of SCD at an early age (Armstrong 2013a).
In this review it appears that long‐term transfusions may reduce the incidence of SCI in children with abnormal TCD velocities, probably reduce the incidence of stroke and may reduce SCD‐related adverse events of ACS and painful crisis. However, no association was found between long‐term RBC transfusions and measures of cognitive function in the one included trial that measured this outcome (SIT 2014). Most trials did not measure cognitive function or quality of life, and two trials have not yet reported quality of life (SWiTCH 2012; TWITCH 2016), which are important research gaps for consideration in future trials in order to establish whether evidence of therapies that prevent SCI or strokes, also improve cognitive function and quality of life.
Although in this review we found no trials that included interventions to prevent SCI in adults with SCD, a recent study found cumulative risk increases for SCI from 19% by eight years of age to 39% by 18 years of age with no apparent plateau being reached for risk of SCIs which are likely common in adults as well (Bernaudin 2015a; DeBaun 2016;DeBaun 2016a). Recent publications have identified a high prevalence of SCIs in adults and suggests that MRI screening should be part of standard care despite the lack of evidence‐based therapy, in order to help identify employment, vocational and disease management options in adults with SCI (DeBaun 2012; DeBaun 2016a). More research is needed to understand the clinical course and treatment options in adults (DeBaun 2012). This highlights another important research gap found in our review and the need for intervention trials to prevent SCI and identify management options in adults.
Authors' conclusions
Due to lack of evidence this review cannot comment on management for adults with HbSS disease or children and adults with HbSβº, HbSC or HbSβ+ disease.
Long‐term RBC transfusions may reduce the incidence of SCI in children with abnormal TCD velocities, but may have little or no effect in children with normal TCD velocities.
In children with no previous long‐term RBC transfusions and at high risk of stroke (previous SCI or abnormal TCD velocities), RBC transfusions probably reduce the risk of stroke and also may confer some additional advantage by reducing the rates of ACS and painful crisis. This must be balanced against the adverse effects and costs of a chronic transfusion regimen.
In children and adolescents at high risk of stroke who have had their TCD velocities normalised, continuing RBC transfusions may reduce the risk of SCI. No treatment duration threshold has been established for stopping transfusions.
We are very uncertain if switching to hydroxyurea with phlebotomy from long‐term RBC transfusions has any effect on this review's outcomes in children with previously abnormal TCD velocities who do not have a severe vasculopathy.
Switching to hydroxyurea with phlebotomy from long‐term RBC transfusions may increase the risk of SCD‐related adverse events in children and adolescents who have had a previous clinical stroke. However, we are very uncertain if switching to hydroxyurea with phlebotomy has any effect on other review outcomes (SCI, clinical stroke, all‐cause mortality).
Information from well‐designed RCTs of long‐term RBC blood transfusion regimens, hydroxyurea, or HSCT in people with SCD are desirable in order to make recommendations for the optimal use of interventions to prevent SCI. Recent improvements in methods of detecting high‐risk individuals are improving clinical outcome, but further research is needed to assess the relative risks and benefits of hydroxyurea in comparison with long‐term RBC transfusion therapy for prevention of SCI. Randomised trials comparing HSCT to hydroxyurea or RBC transfusions are also needed. Finally trials should include measurements of cognitive function and quality of life to determine if interventions are improving neurodevelopmental outcomes in people with SCD.
Acknowledgements
We thank the editorial base of the Cochrane Cystic Fibrosis and Genetic Disorders Review Group.
We thank the National Institute of Health Research (NIHR). This review is part of a series of reviews that have been funded by the NIHR Cochrane Programme Grant ‐ Safe and Appropriate Use of Blood Components. This research is also supported by the NIHR Oxford Biomedical Research Centre Programme. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. CENTRAL (the Cochrane Library) search strategy
#1 MeSH descriptor: [Anemia, Sickle Cell] explode all trees #2 MeSH descriptor: [Hemoglobin, Sickle] explode all trees #3 ("hemoglobin S" or "haemoglobin S" or "hemoglobin SC" or "haemoglobin SC" or "hemoglobin SE" or "haemoglobin SE" or "hemoglobin SS" or "haemoglobin SS" or "hemoglobin C disease" or "hemoglobin D disease" or "hemoglobin E disease" or "haemoglobin C disease" or "haemoglobin D disease" or "haemoglobin E disease" or "Hb SC" or HbSC or HbAS or HbSS or HbAC or "Hb SE" or "Hb SS" or "Hb C disease" or "Hb D disease" or "Hb E disease" or "SC disease" or "SC diseases") #4 (sickle or sicklemia or sicklaemia or sickled or sickling or meniscocyt* or drepanocyt*) #5 ((Hb S or HbS) near/3 (disease* or thalassemi* or thalassaemi*)) #6 #1 or #2 or #3 or #4 or #5 #7 MeSH descriptor: [Cerebral Infarction] explode all trees #8 MeSH descriptor: [Brain Infarction] this term only #9 MeSH descriptor: [Stroke] this term only #10 MeSH descriptor: [Stroke, Lacunar] this term only #11 ((ischemic or ischaemic or cerebrovascular) near/2 (event* or injur* or complication*)) #12 ((MRI or "magnetic resonance imaging" or neuroimaging or "white matter") near/3 abnormal*) #13 (cerebral vasculopath* or cerebrovascular accident* or cerebral vascular accident*) #14 ((cerebral or cerebellar or cerebrovascular or choroidal or hemispher* or cortical or subcortical or brain*) near/3 (infarct* or ischemi* or ischaemi* or stroke*)) #15 ((asymptomatic* or silent* or nonsymptomatic* or unsymptomatic* or non‐symptomatic* or quiet* or symptomfree or symptom‐free or symptomless or symptom‐less or occult or "free of symptom" or "free of symptoms" or subclinical* or covert* or incomplete*) near/5 (infarct* or ischemi* or ischaemi* or stroke*)) #16 #7 or #8 or #9 or #10 or #11 or #12 or #13 or #14 or #15 #17 #6 and #16
Appendix 2. MEDLINE (OvidSP) search strategy
1. exp Anemia, Sickle Cell/ 2. Hemoglobin, Sickle/ 3. (h?emoglobin S or h?emoglobin SC or h?emoglobin SE or h?emoglobin SS or h?emoglobin C disease or h?emoglobin D disease or h?emoglobin E disease or Hb SC or HbSC or HbAS or HbSS or HbAC or Hb SE or Hb SS or Hb C disease or Hb D disease or Hb E disease or SC disease*).tw,kf. 4. (sickle or sicklemia or sicklaemia or sickled or sickling or meniscocyt* or drepanocyt*).tw,kf. 5. ((Hb S or HbS) adj3 (disease* or thalass?emi*)).tw,kf. 6. or/1‐5 7. exp Cerebral Infarction/ 8. Brain Infarction/ 9. Stroke/ 10. Stroke, Lacunar/ 11. ((ischemic or ischaemic or cerebrovascular) adj2 (event* or injur* or complication*)).tw,kf. 12. ((MRI or magnetic resonance imaging or neuroimaging or white matter) adj3 abnormal*).tw,kf. 13. (cerebral vasculopath* or cerebrovascular accident* or cerebral vascular accident*).tw,kf. 14. ((cerebral or cerebellar or cerebrovascular or choroidal or hemispher* or cortical or subcortical or brain*) adj3 (infarct* or ischemi* or ischaemi* or stroke*)).tw,kf. 15. ((asymptomatic* or silent* or nonsymptomatic* or unsymptomatic* or non‐symptomatic* or quiet* or symptomfree or symptom‐free or symptomless or symptom‐less or occult or "free of symptom" or "free of symptoms" or subclinical* or covert* or incomplete*) adj5 (infarct* or ischemi* or ischaemi* or stroke*)).tw,kf. 16. or/7‐15 17. 6 and 16 18. randomized controlled trial.pt. 19. controlled clinical trial.pt. 20. randomi*.tw. 21. placebo.ab. 22. clinical trials as topic.sh. 23. randomly.ab. 24. groups.ab. 25. trial.tw. 26. or/18‐25 27. exp animals/ not humans/ 28. 26 not 27 29. 17 and 28
Appendix 3. PubMed search strategy
#1 ("hemoglobin S" OR "haemoglobin S" OR "hemoglobin SC" OR "haemoglobin SC" OR "hemoglobin SE" OR "haemoglobin SE" OR "hemoglobin SS" OR "haemoglobin SS" OR "hemoglobin C disease" OR "hemoglobin D disease" OR "hemoglobin E disease" OR "haemoglobin C disease" OR "haemoglobin D disease" OR "haemoglobin E disease" OR "Hb SC" OR HbSC OR HbAS OR HbSS OR HbAC OR "Hb SE" OR "Hb SS" OR "Hb C disease" OR "Hb D disease" OR "Hb E disease" OR "SC disease" OR "SC diseases" OR sickle* OR sickled OR sickling OR meniscocyt* OR drepanocyt*) #2 (("Hb S" OR HbS OR sickl*) AND (disease* OR thalassemi* OR thalassaemi*)) #3 #1 OR #2 #4 ((ischemic or ischaemic or cerebrovascular) AND (event* or injur* or complication*)) #5 ((MRI or "magnetic resonance imaging" or neuroimaging or "white matter") AND abnormal*) #6 (cerebral vasculopath* or cerebrovascular accident* or cerebral vascular accident*) #7 ((cerebral or cerebellar or cerebrovascular or choroidal or hemispher* or cortical or subcortical or brain*) AND (infarct* or ischemi* or ischaemi* or stroke*)) #8 ((asymptomatic* or silent* or nonsymptomatic* or unsymptomatic* or non‐symptomatic* or quiet* or symptomfree or symptom‐free or symptomless or symptom‐less or occult or "free of symptom" or "free of symptoms" or subclinical* or covert* or incomplete*) AND (infarct* or ischemi* or ischaemi* or stroke*)) #9 #4 OR #5 OR #6 OR #7 OR #8 #10 ((random* OR blind* OR "control group" OR placebo* OR "controlled study" OR groups OR trial* OR "systematic review" OR "meta‐analysis" OR metaanalysis OR "literature search" OR medline OR pubmed OR cochrane OR embase) AND (publisher[sb] OR inprocess[sb] OR pubmednotmedline[sb])) #11 #3 AND #9 AND #10
Appendix 4. Embase (OvidSP) search strategy
1. exp Sickle Cell Anemia/ 2. Hemoglobin S/ 3. (h?emoglobin S or h?emoglobin SC or h?emoglobin SE or h?emoglobin SS or h?emoglobin C disease or h?emoglobin D disease or h?emoglobin E disease or Hb SC or HbSC or HbAS or HbSS or HbAC or Hb SE or Hb SS or Hb C disease or Hb D disease or Hb E disease or SC disease*).tw. 4. (sickle or sicklemia or sickled or sickling or meniscocyt* or drepanocyt*).tw. 5. ((Hb S or HbS or sickl*) adj3 (disease* or thalass?emi*)).tw. 6. or/1‐5 7. exp Cerebral Infarction/ 8. exp Cerebrovascular Accident/ 9. ((ischemic or ischaemic or cerebrovascular) adj2 (event* or injur* or complication*)).tw. 10. ((MRI or magnetic resonance imaging or neuroimaging or white matter) adj3 abnormal*).tw,kf. 11. (cerebral vasculopath* or cerebrovascular accident* or cerebral vascular accident*).tw. 12. ((cerebral or cerebellar or cerebrovascular or choroidal or hemispher* or cortical or subcortical or brain*) adj3 (infarct* or ischemi* or ischaemi* or stroke*)).tw. 13. ((asymptomatic* or silent* or nonsymptomatic* or unsymptomatic* or non‐symptomatic* or quiet* or symptomfree or symptom‐free or symptomless or symptom‐less or occult or "free of symptom" or "free of symptoms" or subclinical* or covert* or incomplete*) adj5 (infarct* or ischemi* or ischaemi* or stroke*)).tw. 14. or/7‐13 15. 6 and 14 16. crossover‐procedure/ or double‐blind procedure/ or randomized controlled trial/ or single‐blind procedure/ 17. (random* or factorial* or crossover* or cross over* or cross‐over* or placebo* or doubl* blind* or singl* blind* or assign* or allocat* or volunteer*).mp. 18. 16 or 17 19. 15 and 18 20. limit 19 to embase
Appendix 5. CINAHL (EBSCOHost) search strategy
S1 (MH "Anemia, Sickle Cell+") S2 TX ("hemoglobin S" or "haemoglobin S" or "hemoglobin SC" or "haemoglobin SC" or "hemoglobin SE" or "haemoglobin SE" or "hemoglobin SS" or "haemoglobin SS" or "hemoglobin C disease" or "hemoglobin D disease" or "hemoglobin E disease" or "haemoglobin C disease" or "haemoglobin D disease" or "haemoglobin E disease" or "Hb SC" or HbSC or HbAS or HbSS or HbAC or "Hb SE" or "Hb SS" or "Hb C disease" or "Hb D disease" or "Hb E disease" or "SC disease" or "SC diseases" or sickle or sicklemia or sicklaemia or sickled or sickling or meniscocyt* or drepanocyt*) S3 TX ((Hb S or HbS or sickl*) N3 (disease* or thalass?emi*)) S4 S1 OR S2 OR S3 S5 (MH "Infarction") S6 (MH "Cerebral Ischemia+") S7 (MH "Stroke+") S8 ((ischemic or ischaemic or cerebrovascular) N2 (event* or injur* or complication*)) S9 ((MRI or "magnetic resonance imaging" or neuroimaging or "white matter") N3 abnormal*) S10 (cerebral vasculopath* or cerebrovascular accident* or cerebral vascular accident*) S11 ((cerebral or cerebellar or cerebrovascular or choroidal or hemispher* or cortical or subcortical or brain*) N3 (infarct* or ischemi* or ischaemi* or stroke*)) S12 ((asymptomatic* or silent* or nonsymptomatic* or unsymptomatic* or non‐symptomatic* or quiet* or symptomfree or symptom‐free or symptomless or symptom‐less or occult or "free of symptom" or "free of symptoms" or subclinical* or covert* or incomplete*) N5 (infarct* or ischemi* or ischaemi* or stroke*)) S13 S5 OR S6 OR S7 OR S8 OR S9 OR S10 OR S11 OR S12 S14 S4 AND S13 S15 (MH "Clinical Trials+") S16 PT Clinical trial S17 TX (clinic* N1 trial* OR controlled N1 trial*) S18 TX ( (singl* N1 blind*) or (singl* N1 mask*) ) or TX ( (doubl* N1 blind*) or (doubl* N1 mask*) ) or TX ( (tripl* N1 blind*) or (tripl* N1 mask*) ) or TX ( (trebl* N1 blind*) or (trebl* N1 mask*) ) S19 TX (randomi* OR placebo*) S20 (MH "Random Assignment") S21 TX (random* N2 (allocat* OR assign*)) S22 (MH "Quantitative Studies") S23 S15 or S16 or S17 or S18 or S19 or S20 or S21 or S22 S24 S14 and S23
Appendix 6. Transfusion Evidence Library search strategy
sickle AND (cerebral OR ischemic OR ischaemic OR ischemia OR ischaemia OR cerebrovascular OR stroke OR infarct OR infarction OR MRI OR magnetic OR neuroimaging)
Appendix 7. LILACS search strategy
tw:(sickle) AND (instance:"regional") AND ( db:("LILACS") AND type_of_study:("clinical_trials"))
Appendix 8. Web of Science CPCI‐S search strategy
TOPIC: (sickle OR sickled OR sickling) AND TOPIC: (ischemic OR ischaemic OR ischemia OR ischaemia OR stroke OR strokes OR infarct OR infarcts OR infarction OR hemispher* OR cerebral OR cerebrovascular OR subcortical OR cortical OR choroidal OR MRI OR magnetic OR neuroimaging) AND TOPIC: (random OR randomly OR randomised OR randomized OR blind OR blinded OR control group OR placebo OR controlled study OR groups OR trial OR trials OR systematic review OR meta‐analysis OR metaanalysis OR literature search OR medline OR pubmed OR cochrane OR embase)
Appendix 9. ClinicalTrials.gov search strategy
Search Terms: ischemic OR ischaemic OR ischemia OR ischaemia OR stroke OR strokes OR infarct OR infarcts OR infarction OR hemispheric OR cerebral OR cerebrovascular OR subcortical OR cortical OR choroidal OR MRI OR magnetic resonance imaging OR neuroimaging Study Type: Interventional Studies Condition: sickle
Appendix 10. WHO ICTRP search strategy
[Title: ischemic OR ischaemic OR ischemia OR ischaemia OR stroke OR strokes OR infarct OR infarcts OR infarction OR hemispheric OR cerebral OR cerebrovascular OR subcortical OR cortical OR choroidal OR MRI OR magnetic resonance imaging OR neuroimaging Condition: sickle OR SCD Recruitment Status: ALL] OR [Title: sickle OR SCD Condition: ischemic OR ischaemic OR ischemia OR ischaemia OR stroke OR strokes OR infarct OR infarcts OR infarction OR hemispheric OR cerebral OR cerebrovascular OR subcortical OR cortical OR choroidal Recruitment Status: ALL] OR [Title OR Condition: sickle OR SCD Intervention: MRI OR magnetic resonance imaging OR neuroimaging Recruitment Status: ALL]
Data and analyses
Comparison 1.
Red blood cell transfusions vs standard care
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Proportion of participants developing new or progressive SCI lesions | 2 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 1.1 Normal TCD velocities | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Abnormal TCD velocities | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 SCD ‐ related serious adverse events ‐ acute chest syndrome | 2 | 326 | Risk Ratio (IV, Random, 95% CI) | 0.24 [0.12, 0.49] |
| 2.1 Normal TCD velocities | 1 | 196 | Risk Ratio (IV, Random, 95% CI) | 0.20 [0.08, 0.51] |
| 2.2 Abnormal TCD velocities | 1 | 130 | Risk Ratio (IV, Random, 95% CI) | 0.30 [0.11, 0.87] |
| 3 SCD‐related serious adverse events ‐ pain crisis | 2 | 326 | Risk Ratio (IV, Random, 95% CI) | 0.63 [0.42, 0.95] |
| 3.1 Normal TCD velocities | 1 | 196 | Risk Ratio (IV, Random, 95% CI) | 0.56 [0.40, 0.78] |
| 3.2 Abnormal TCD velocities | 1 | 130 | Risk Ratio (IV, Random, 95% CI) | 0.90 [0.44, 1.86] |
| 4 Clinical stroke | 2 | 326 | Risk Ratio (IV, Random, 95% CI) | 0.12 [0.03, 0.49] |
| 4.1 Normal TCD velocities | 1 | 196 | Risk Ratio (IV, Random, 95% CI) | 0.14 [0.02, 1.12] |
| 4.2 Abnormal TCD velocities | 1 | 130 | Risk Ratio (IV, Random, 95% CI) | 0.10 [0.01, 0.73] |
| 5 Any transfusion‐related adverse events ‐ antibody development | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 5.1 Normal TCD velocities | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Any transfusion‐related adverse events ‐ transfusion reactions | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 6.1 Normal TCD velocities | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 2.
Transfusions continued vs transfusions halted
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Proportion of participants developing new or progressive SCI lesions | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 2 All‐cause mortality | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 3 Clinical stroke | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected |
Comparison 3.
Hydroxyurea vs red blood cell transfusions
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Proportion of participants developing new or progressive SCI lesions ‐ secondary prevention | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 2 All‐cause mortality ‐ secondary prevention | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 3 SCD‐related SAEs ‐ Acute chest syndrome ‐ primary prevention | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 4 SCD‐related SAEs ‐ Acute chest syndrome ‐ secondary prevention | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 SCD‐related SAEs ‐ Pain crisis ‐ primary prevention | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 6 SCD‐related SAEs ‐ Pain crisis ‐ secondary prevention | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Total SCD‐related SAEs ‐ primary prevention | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 8 Total SCD‐related SAEs ‐ secondary prevention | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Clinical stroke ‐ secondary prevention | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Transfusion‐related complications ‐ serum ferritin ‐ primary prevention | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 11 Transfusion‐related complications ‐ liver iron concentration ‐ primary prevention | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 12 Any SCD‐related adverse events ‐ secondary prevention | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods |
Trial design: RCT Trial grouping: parallel Open label: yes Cluster RCT: no |
|
| Participants | Participant flow: 1210 registered for screening; 1074 had screening MRI evaluated by neuroradiology committee; 675 had normal, 20 indeterminate MRI; 379 had infarct‐like lesions on screening MRI; 291 had infarct‐like lesions adjudicated by neurology committee; 220 had pre‐randomisation MRIs adjudicated by neuroradiology committee; 196 underwent randomisation Baseline characteristics RBC transfusions
Standard care
Included criteria: children aged 5 ‐ 15 years, confirmed diagnosis of haemoglobin SS or haemoglobin Sβº thalassaemia, and at least 1 infarct‐like lesion on the screening MRI scan defined as an MRI signal abnormality that was at least 3 mm in one dimension and that was visible in two planes on fluid‐attenuated inversion recovery (FLAIR) T2‐weighted images, as determined by agreement of two of the three trial neuroradiologists Excluded criteria: history of focal neurologic deficit associated with an infarct on brain MRI, a seizure disorder, treatment with hydroxyurea in the previous 3 months, a history of regular transfusion therapy, or imaging or non‐imaging transcranial doppler measurement that was above the trial‐defined thresholds |
|
| Interventions |
Intervention characteristics RBC transfusions
Standard care
|
|
| Outcomes |
Primary outcome: the recurrence of infarct or haemorrhage as determined by neuroimaging, clinical evidence of permanent neurologic injury, or both. A new infarct had to meet the criteria for a SCI; an enlarged SCI was defined as a previously identified SCI that increased by at least 3 mm along any linear dimension in any plane on MRI; TIA, included in secondary analyses of neurologic outcomes, defined as an event that resulted in focal neurologic deficits that lasted less than 24 hours, did not result in abnormalities on T2‐weighted or FLAIR images that were indicative of an acute infarct, and had no other reasonable medical explanation Secondary outcomes: changes in cognition, assessed by measurement of IQ scores with the Wechsler Abbreviated Scale of Intelligence12 or the Wechsler Preschool and Primary Scale of Intelligence III; also assessed scores on the Behavior Rating Inventory of Executive Function (BRIEF) |
|
| Identification |
Sponsorship source: supported by grants from the National Institute of Neuro‐logical Disorders and Stroke (5U01NS042804, 3U01NS042804 (American Recovery Reinvestment ACT supplementary grant) to Dr DeBaun); the Institute of Clinical and Translational Scienc‐es, National Center for Research Resources, and the National Center for Advancing Translational Sciences, Clinical and Trans‐lational Research; NIH Roadmap for Medical Research (UL1TR000448, to Washington University; UL1TR001079, to Johns Hopkins University; and UL1TR000003, to the Children’s Hospital of Philadelphia); and Research and Development in the National Health Service, UK Country: USA, Canada, France and the UK Setting: paediatric outpatients with SCD Comments: 29 centres; recruitment: December 2004 to May 2010.The last participant enrolled completed the exit visit on July 29, 2013.Trial registration: NCT00072761 & ISRCTN52713285 Mean length of follow‐up: Children were followed for a median of 3 years. Power Calculation: a sample size of 204 participants (102 in each group) would give the trial 85% power to detect a decrease of at least 86% in the prevalence of the primary endpoint, assuming a 10% dropout rate and a cross‐over rate of 16% from transfusion to observation and 3% from observation to transfusion, at a 2‐tailed nominal alpha level of 0.05 Authors name: Dr M.R. DeBaun Institution: Department of Pediat‐rics, Division of Hematology–Oncology, Vanderbilt–Meharry Center of Excellence in Sickle Cell Disease, Monroe Carell Jr. Children’s Hospital Email: m.debaun@vanderbilt.edu Address: Department of Pediatrics, Division of Hematology–Oncology, Vanderbilt–Meharry Center of Excellence in Sickle Cell Disease, Monroe Carell Jr. Children’s Hospital at Vanderbilt, 2200 Children’s Way, Rm. 11206 DOT, Nashville, TN 37232 |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization assignments were provided by the statistical data co‐ordinating center with the use of a permuted block design, with stratification according to site, age, and sex. Participants were assigned in a 1:1 ratio to the observation group or the transfusion group" |
| Allocation concealment (selection bias) | Low risk | Judgement Comment: assignments were provided by the statistical data co‐ordinating centre |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Judgement comment: by the nature of the treatments used (blood transfusions vs observation), it is impractical to make SIT trial blinded (masked) |
| Blinding of outcome assessment (detection bias) SCI, stroke, and all‐cause mortality | Low risk | Members of neuroradiology and neurology committees, who were unaware of the trial‐group assignments, adjudicated neurologic and MRI findings All‐cause mortality is an objective outcome unaffected by blinding of the outcome assessor |
| Blinding of outcome assessment (detection bias) All outcomes except SCI, stroke and all‐cause mortality | High risk | Judgement comment: unblinded trial |
| Incomplete outcome data (attrition bias) Stroke and SCI | Low risk | All outcomes reported. All participants accounted for, conducted an ITT and per protocol analysis for the primary outcomes The primary endpoint was ascertained for 185 of the 196 participants (94%). Of the 99 participants randomly assigned to the transfusion group, 90 started receiving transfusions within 4 weeks after assignment. The crossover rate from transfusion to observation was 15% (15 of 99 participants); 9 participants declined blood transfusion, and 6 crossed over to observation at a median time of 34 days |
| Incomplete outcome data (attrition bias) All outcomes except stroke and SCI | Low risk | All outcomes reported. All participants accounted for, conducted an ITT and per protocol analysis for the primary outcomes The primary endpoint was ascertained for 185 of the 196 participants (94%). Of the 99 participants randomly assigned to the transfusion group, 90 started receiving transfusions within 4 weeks after assignment. The crossover rate from transfusion to observation was 15% (15 of 99 participants); 9 participants declined blood transfusion, and 6 crossed over to observation at a median time of 34 days |
| Selective reporting (reporting bias) | Low risk | Protocol available and all planned outcomes reported |
| Other bias | Unclear risk | Judgement comment: among participants in the observation group, 32% received transfusions (a median of three transfusions each), including 6 participants who crossed over to regular monthly transfusions at a median of 1.7 years. During the course of the trial, hydroxyurea was started in 14 of 97 participants (14%) in the observation group and in 3 of 99 (3%) in the transfusion group because of disease severity. Exclusion criteria included treatment with hydroxyurea. Not clear how long or when treatment began ‐ possible contamination and unknown effect on outcomes. 6 also crossed over to regular transfusion giving 20% cross‐over rate to either hydroxyurea or transfusion |
| Methods |
Trial design: RCT Trial grouping: parallel group Open label: yes Cluster RCT: no Mean (SD) length of follow‐up: transfusion arm: 21.0 (5.7) months; standard care: 18.3 (7.0) months Analysis: 4 interim analyses and one final analysis were planned. The date of the first analysis changed from 20 months to 14 months after recruitment began |
|
| Participants | Participant flow: screened: N = 1934; eligible: N = 206 randomised: N = 130 Baseline characteristics RBC transfusions
Standard care
Included criteria: children 2 ‐ 16 years of age and who had been given a diagnosis of SCA or sickle βº thalassaemia at high risk of stroke with a blood flow velocity of at least 200 cm per second on 2 TCD trials. Only children with an MRI at trial entry were included in the SCI analysis Excluded criteria: history of stroke, had an indication for or contraindication to long‐term transfusion, were receiving other treatments that affected the risk of stroke, were infected with HIV, had been treated for seizures, were pregnant, or had a serum ferritin concentration above 500 ng per mL Pretreatment: baseline Hb was higher in the standard care group. Alpha thalassaemia was more common RBC transfusion group |
|
| Interventions |
Intervention characteristics RBC transfusions
Standard care
|
|
| Outcomes |
Primary outcome: cerebral infarction and intracranial haemorrhage Secondary outcomes: death, transfusion‐related adverse events |
|
| Identification |
Sponsorship source: supported by Cooperative Agreements (U10 HL 52193 and U10 HL 52016) with the National Heart, Lung, and Blood Institute. Country: USA & Canada Setting: Paediatric outpatients treating children with sickle cell disease aged 2 ‐ 16 years of age with HbSS or HbSβº thalassaemia Comments: declarations of interest: None; published trial registration: No registration found; Authors name: Dr Robert J. Adams Institution: Department of Neurology, Medical College of Georgia Email: rjadams@mcg.edu Address: Department of Neurology, Medical College of Georgia, 1467 Harper St., HB‐2060, Augusta, GA 30912‐3200 |
|
| Notes | Screening began in January 1995 and ended in November 1996. The trial was to run to December 1998 but was stopped in September 1997 "Estimates of stroke risk for patients randomized to standard care were obtained by fitting an exponential model to the follow‐up of TCD (1) patients follow‐up, it was estimated that 47% of patients in this group should develop stroke on study. Assuming transfusion prevents 70% of these strokes, 14% of the patients randomized to transfusion should have strokes on study. Taken together, these values imply that a sample size of 46 per treatment arm should provide the desired statistical power of 90% to detect a 70% reduction in stroke incidence at a type I error rate of 0.05 for a two‐sided test" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The DCC developed permuted blocks within which treatment allocations were randomly and evenly assigned. The blocks themselves were randomly assigned to each of the 12 Centers." |
| Allocation concealment (selection bias) | Low risk | "After telephone verification that the patient was eligible and acquisition of written parental consent, the DCC ran a short randomization program and provided the Investigator with the trial group assignment." " Permuted blocks are used to blind Investigators to the potential treatment assignment of each patient while preserving approximate balance within and across Centers. The DCC provided the Clinical Center Investigator and patient with the treatment assignment." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Judgement Comment: clinicians and participants were unblinded |
| Blinding of outcome assessment (detection bias) SCI, stroke, and all‐cause mortality | Low risk | A panel of physicians with no knowledge of the children’s treatment assignments who were not affiliated with the trial centres determined whether an event was a stroke. The primary endpoints were cerebral infarction and intracranial haemorrhage. "The protocol was intended to identify all neurologic events. A panel of physicians with no knowledge of the children’s treatment assignments who were not affiliated with the trial centres determined whether an event was a stroke." |
| Blinding of outcome assessment (detection bias) All outcomes except SCI, stroke and all‐cause mortality | High risk | Judgement comment: unblinded trial |
| Incomplete outcome data (attrition bias) Stroke and SCI | Unclear risk | Quote: "Only those subjects who had an MRI of the brain at the time of randomization were included in this analysis. Since the question being addressed was secondary to the trial, ITT analysis was not used; treatment classification was based on actual study experience. Five subjects randomized to the transfusion therapy arm were managed with standard care. The parents of 3 children refused transfusion therapy, another child was not compliant with monthly appointments, and 1 child who developed a delayed transfusion reaction could not be provided phenotypically matched blood. For this analysis, all 5 of these patients are considered to be in the standard care treatment arm." Judgement comment: this is a subgroup analysis that included only those with an MRI and done by treatment and not ITT |
| Incomplete outcome data (attrition bias) All outcomes except stroke and SCI | Low risk | Judgement comment: an ITT analysis was used, despite 12 participants crossing over between groups (2) or withdrawing from the trial (10). Reasons were provided. 10 participants from the transfusion group withdrew from the trial because of problems with compliance (n = 4), multiple alloantibodies (n = 1), ineligibility (n = 1) or other unspecified reasons (n = 4). Two participants from the standard care group crossed over to the transfusion group, one on the second day due to diagnosis of subacute intracerebral hematoma and the other after 12 months for treatment of leg ulcers |
| Selective reporting (reporting bias) | Unclear risk | Judgement comment: no protocol available and no prospective trial registration |
| Other bias | Unclear risk | Quote: "Since equipment or imaging programs were upgraded during the course of the study, the quality of later MRI studies was frequently better than those done earlier. As a result, some lesions seen on later studies could, in retrospect, be seen on those previously reported as showing no abnormality. In that case, the earlier result was changed to reflect the latest reading." Judgement comment: "Four interim analyses and one final analysis were planned, with the Lan–DeMets approximation of the O’Brien–Fleming stopping boundary. The date of the first analysis was changed from 20 months to 14 months after recruitment began." "Because of the high rate of stroke in the standard‐care group and the significant effect of transfusion found at the second interim analysis, the data safety and monitoring board recommended that the trial be stopped 16 months before the planned date of December 1998 so that transfusion could be offered to children in the standard‐care group." It is unclear if there could be an effect on the estimate with a change in readings; or early termination of the trial, even though recommended by the Safety committee |
| Methods |
Trial design: RCT Trial grouping: parallel Open label: yes Cluster RCT: no Mean length of follow‐up: the median time from randomisation to an end‐point event was 3.2 months (range, 2.1 to 10.1), and the mean (SD) was 4.5 (2.6) months |
|
| Participants |
Baseline characteristics Transfusions continued
Transfusions halted
Thalassaemia presence reported but not by treatment arm Included criteria: children whose Doppler studies normalized after 30 or more months of transfusion were eligible for the present trial. In addition, children who had not participated in the previous STOP trial whose condition met the criteria for eligibility and treatment were also eligible for the present trial. Adequate participation in a transfusion program (≥ 24 transfusions in 30 months and Hb S < 30% in at least 20 of the 30 months); 2 Normal TCD examinations at least 2 weeks apart while receiving transfusions within 4 months of randomisation; age, 5 ‐ 20 years; consent to participate in trial Excluded criteria: prior stroke; Indication for chronic transfusion; contraindication for chronic transfusion; moderate‐to‐severe intracranial arterial disease on MRA. The STOP II trial excluded people with severe artery stenosis detected on MRA thus eliminating a known risk factor for SCI |
|
| Interventions |
Intervention characteristics Transfusions continued
Transfusions halted
|
|
| Outcomes | Primary outcome: composite endpoint was a stroke (cerebral infarction or intracranial haemorrhage) or reversion to abnormal velocity on TCD ultrasonography, defined as 2 consecutive studies with abnormal velocities, 3 consecutive studies with an average velocity of 200 cm per second or more, or 3 consecutive inadequate studies plus evidence of severe stenosis on MRA Secondary outcomes: also reports deaths, acute chest syndrome and transfusion adverse events | |
| Identification |
Sponsorship source: supported by grants (U01 HL 052193 and U01 HL 052016) from the National Heart, Lung, and Blood Institute Country: USA and Canada Setting: Multicentre extension study of STOP trial conducted in 23 centres (including the 12 centres in STOP) Authors name: Dr Robert J Adams Institution: Medical College of Georgia Email: rjadams@mcg.edu Address: Department of Neurology, Medical College of Georgia, 1429 Harper St., HF 1154, Augusta, GA 30912, Declarations of interest: No potential conflict of interest relevant to this article was reported |
|
| Notes | Children were monitored by transcranial Doppler examinations after transfusions were halted and by resuming transfusions if the examination indicated a high risk of stroke This trial was an extension of the previous STOP trial, in which children with abnormal velocities on TCD ultrasonographic examination were administered transfusions to prevent a first stroke.The trial was meant to last 54 months and involve 50 participants in each group, with 60 of the participants enrolled during the first 12 months and 40 during the next 24 months; after recruitment ended, there were 18 months of follow‐up. 4 interim analyses and 1 final analysis were planned for the composite endpoint. The trial was stopped on the advice of the data safety and monitoring committee because of concern about safety at the fourth interim analysis with 79 participants enrolled Trial registration: no registration found. Power calculation: for a 54‐month trial involving 50 participants in each group, with 60 of the participants enrolled during the first 12 months and 40 during the next 24 months; after recruitment ended, there were 18 months of follow‐up. Analysis: the trial was stopped by the National Heart, Lung, and Blood Institute on the advice of the data safety and monitoring committee because of concern about safety at the fourth interim analysis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Participants were stratified at randomisation according to the presence or absence of ischaemic lesions on MRI; random, permuted blocks of four or six participants were used within each group as defined by MRI. Institutional balancing with a tolerance of two participants per site was imposed to maintain an approximate balance in treatment assignments at each site. Eligible participants underwent randomization with equal probability of continuing or halting transfusion." |
| Allocation concealment (selection bias) | Unclear risk | Judgement comment: method of allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Judgement comment: participants and personnel were unblinded |
| Blinding of outcome assessment (detection bias) SCI, stroke, and all‐cause mortality | Low risk | "All images were reviewed for the presence, size, and location of ischemic lesions by 2 experts who were unaware of the treatment assignment. Review was done independently, and in case of disagreement, the 2 experts reviewed the images jointly until a consensus was reached. When annual studies or those obtained at the time of a clinical event were read, they were compared with previously obtained images. Stroke was defined as persistent abnormalities or transient neurologic symptoms accompanied by a new cerebral lesion appropriate to the patient’s clinical presentation. Silent infarcts were defined by evidence of cerebral infarction on MRI in patients without a compatible history of a cerebrovascular event." Judgement comment: all‐cause mortality unaffected by blinding ‐ objective outcome |
| Blinding of outcome assessment (detection bias) All outcomes except SCI, stroke and all‐cause mortality | High risk | Judgement comment: unblinded for outcomes that were not SCI, stroke, or mortality |
| Incomplete outcome data (attrition bias) Stroke and SCI | Low risk | One participant in each group had no follow‐up brain MRI |
| Incomplete outcome data (attrition bias) All outcomes except stroke and SCI | Unclear risk | An ITT analysis was performed. There was no statistically significant difference in the mean follow‐up time of participants in the continued transfusion (464 days) and transfusion‐halted (523 days) groups. Data on nine participants assigned to no continued transfusion who did not have a primary end‐point event were censored: five of these participants resumed chronic transfusion and four started treatment with hydroxyurea. Of 38 participants assigned to continued transfusion, 5 discontinued participation in the trial. N = 14 (17%) of participants discontinued or censored |
| Selective reporting (reporting bias) | Unclear risk | Judgement comment: primary outcomes reported. Did not state secondary outcomes. Not clear if all adverse events reported and also censored data not contributing to outcome reporting |
| Other bias | Unclear risk | Judgement comment: the trial was stopped by the National Heart, Lung, and Blood Institute on the advice of the data safety and monitoring committee because of concern about safety at the fourth interim analysis |
| Methods |
Trial design: RCT Trial grouping: parallel group Open Label: yes Cluster RCT: no Total duration of trial treatment was 30 months after randomisation, with a final trial visit scheduled 6 months after discontinuation of trial treatments Non‐inferiority trial |
|
| Participants | Screened: N = 202; enrolled: N = 161; randomised: N = 134 Baseline characteristics RBC transfusions
Hydroxyurea
Included criteria: paediatric participants with severe forms of SCA (HbSS, HbS/βº‐thalassaemia, HbS/OArab;); age range of 5.0 ‐ 18.9 years, inclusive, at the time of enrolment; initial (primary) completed overt clinical stroke after the age 12 months with documented infarction on brain CT or MRI; at least 18 months of chronic monthly erythrocyte transfusions since primary stroke; transfusional iron overload, defined as a previously documented liver iron concentration ≥ 5.0 mg Fe per g of dry weight liver or serum ferritin ≥ 500 ng/mL on 2 independent measurements; adequate monthly erythrocyte transfusions with average HbS ≤ 45% (the upper limit of the established academic community standard) for the past 6 months before enrolment; parent or guardian willing and able to provide informed consent with verbal or written assent from the child (< 18 years of age), and subject willing and able to provide informed consent (≥ 18 years of age); ability to comply with trial‐related treatments, evaluations, and follow‐up Excluded criteria: inability to receive or tolerate chronic RBC transfusion therapy; inability to take or tolerate daily oral hydroxyurea; clinical and laboratory evidence of hypersplenism (temporary); abnormal laboratory values at initial evaluation (temporary); current participation in other therapeutic clinical trials; current use of other therapeutic agents for SCD (e.g. arginine, decitabine, magnesium); any condition or chronic illness, such as a positive tuberculin (PPD) test, which in the opinion of the investigator makes participation ill‐advised; inability or unwillingness to complete required screening studies, including blood tests, brain MRI/MRA, and liver biopsy; a sibling enrolled in SWiTCH |
|
| Interventions |
RBC transfusions
Hydroxyurea
|
|
| Outcomes |
Primary outcome: composite primary endpoint of secondary stroke recurrence rate and quantitative liver iron concentration. Secondary outcomes: non‐stroke neurological events, non‐neurological sickle cell clinical events, quality of life evaluation, and measures of organ function |
|
| Identification |
Sponsorship source: National Heart, Lung, and Blood Institute grants U01‐HL078787 (R.E.W.) and U01‐HL078987 (R.W.H.) Country: USA Setting: 26 paediatric outpatients treating children with SCD Comments: declarations of interest: The authors declare no competing financial interests. Trial registration: ClinicalTrials.gov NCT00122980 Authors name: Russell E. Ware Institution: Center for Global Health, Baylor College of Medicine and Texas Children’s Hospital, Email: reware@bcm.edu Address: Russell E. Ware, MD, PhD, Director, Centerfor Global Health, Baylor College of Medicine and Texas Children’s Hospital, 1102 Bates St, Ste FC‐1145, Houston, TX 77030 |
|
| Notes | "Study exit examinations were performed in 112 children (75%) who did not experience an adjudicated stroke and the examinations revealed few changes. Only 1 subject (alternative treatment) developed a new subcortical lacuna, consistent with silent infarction occurring during the study treatment period. Similarly, only 1 subject had progressive vasculopathy (evolved from grade 0 to grade 4); this subject was also in the alternative treatment arm." Analysis: because reduction in LIC was not superior on hydroxyurea/phlebotomy, the DSMB concluded that the composite primary endpoint would not be met and recommended trial closure. NHLBI closed SWiTCH. N = 40 did not complete treatment phase in transfusion/iron chelation arm and N = 43 did not complete treatment phase in hydroxyurea/phlebotomy arm Power calculation: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Judgement comment: method of sequence generation not reported |
| Allocation concealment (selection bias) | Unclear risk | Judgement comment: method of allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Judgement comment: by the nature of the trial treatments used to prevent recurrent stroke (blood transfusions vs hydroxyurea), it is impractical to make SWiTCH a blinded (masked) trial |
| Blinding of outcome assessment (detection bias) SCI, stroke, and all‐cause mortality | Low risk | Judgement comment: the inclusive independent stroke adjudication process for all suspected new neurological events is a novel feature of the trial. Stroke recurrence is a primary endpoint but also is a critical safety endpoint for the SWiTCH trial. Accordingly, it was necessary to develop an inclusive process by which all potential stroke events were recognized and systematically adjudicated using a standardised protocol and masked consultants. Participants who develop any acute neurological change are promptly evaluated for possible stroke. In addition, site personnel are provided with a written script to use at each interval clinic visit, to ensure that subjects and families are asked each month about any signs and symptoms of stroke. After a new neurological event is suspected, the stroke adjudication process begins. The clinical history and neurological exam are reviewed by 3 independent neurologists without knowledge of the imaging findings. Simultaneously, the radiological evaluation is reviewed by 3 independent masked neuroradiologists without knowledge of the clinical history or neurological examination. Only after their independent consensus opinions are formed are these two opinions reconciled into a final stroke adjudication decision; a diagnosis of stroke requires new neurological findings with corresponding radiological changes |
| Blinding of outcome assessment (detection bias) All outcomes except SCI, stroke and all‐cause mortality | High risk | Judgement comment: the SWiTCH principal investigator was masked to all treatment‐specific results, including laboratory tests and clinical events. In addition, all investigators at the peripheral clinical sites are masked to trial treatment results outside of their own clinical centre |
| Incomplete outcome data (attrition bias) Stroke and SCI | Low risk | The primary statistical analyses of efficacy and safety will be performed on the ITT population, which consists of all subjects who were randomised to a trial treatment and for whom outcome data are available |
| Incomplete outcome data (attrition bias) All outcomes except stroke and SCI | Low risk | The primary statistical analyses of efficacy and safety will be performed on the ITT population, which consists of all subjects who were randomised to a trial treatment and for whom outcome data are available |
| Selective reporting (reporting bias) | High risk | Judgement comment: several secondary outcomes not reported (i.e. quality of life, growth and development, organ damage, transfusion‐related, chelation‐related and phlebotomy related complications) |
| Other bias | Unclear risk | Judgement comment: the DSMB concluded that the composite primary trial endpoint would not be met and recommended trial closure. NHLBI closed SWiTCH ‐ N = 40 did not complete treatment phase in transfusion/iron chelation arm and N = 43 did not complete treatment phase in hydroxyurea/phlebotomy arm. More participants had moya‐moya in the hydroxyurea arm (11 participants) than the transfusion arm (5 participants), it was not known if there was a difference between treatment arms in the number of participants with other types of severe vasculopathy |
| Methods |
Trial design: RCT Trial grouping: parallel group Open Label: yes Cluster RCT :no Duration: 24 months after randomisation with a 6‐month visit after completing exit studies |
|
| Participants | Screened: N = 159; excluded: N = 38; randomised: N = 121 Baseline characteristics RBC transfusions
Hydroxyurea
Included criteria: inclusion criteria: children aged 4 ‐ 16 years with severe forms of SCA (HbSS, HbSβº thalassaemia, HbSOArab),documented index (pre‐treatment) abnormally high TCD velocity by TCD ultrasonography. An abnormally high index TCD is defined as TCD V greater than or equal to 200 cm/s, or abnormally high TCDi V greater than or equal to 185cm/sec, or TCD maximum V greater than or equal to 250 cm/s. At least 12 months of chronic monthly RBC transfusions since the index abnormal TCD examination. Adequate monthly erythrocyte transfusions with average HbS level less than or equal to 45% (the upper limit of the established academic community standard) for the past 6 months before enrolment. Parent or guardian willing and able to provide informed consent with verbal or written assent from the child. Ability to comply with trial‐related treatments, evaluations, and follow‐up Excluded criteria: completed overt clinical stroke or TIA. Inability to obtain TCD velocities due to anatomical abnormalities such as: a) inadequate bone windows; b) previous revascularisation procedures. Known severe vasculopathy or moya‐moya disease on brain MRA. Inability to receive or tolerate chronic RBC transfusion therapy, due to any of the following: Multiple RBC alloantibodies making cross‐matching difficult or impossible; RBC autoantibodies making cross‐matching difficult or impossible; religious objection to transfusions that preclude their chronic use; non‐compliance with transfusions over the past 6 months before enrolment (temporary exclusion); inability to take or tolerate daily oral hydroxyurea, including: known allergy to hydroxyurea therapy; positive serology to HIV infection; malignancy; current lactation; previous stem cell transplant or other myelosuppressive therapy; clinical and laboratory evidence of hypersplenism (temporary exclusions): a) palpable splenomegaly greater than 5 cm below the left costal margin; transfusion requirement > 250 mL/kg over the previous 12 months; abnormal laboratory values at initial evaluation (temporary exclusions): pre‐transfusion haemoglobin concentration < 80 g/L WBC count less than 3.0 x 109/L; absolute neutrophil count < 1.5 x 109/L; platelet count < 100 x 109/L; serum creatinine more than twice the upper limit for age OR greater than or equal to 1.0 mg/d; current participation in other therapeutic clinical trials; current use of other therapeutic agents for SCD (e.g. arginine, decitabine, magnesium) Subjects must have been off hydroxyurea for at least 3 months prior to enrolment. Any condition or chronic illness, such as a positive tuberculin (PPD) test, which in the opinion of the CI makes participation ill‐advised. Inability or unwillingness to complete required screening and exit studies, including TCD ultrasonography, brain MRI/MRA, liver MRI and blood tests. A sibling enrolled in TWiTCH; pregnancy or unwillingness to use a medically acceptable form of contraception if sexually active (male OR female). Pretreatment: alpha thalassaemia not reported. Mild to moderate vasculopathy: 6 (10%) in RBC arm and 4 (7%) in hydroxyurea arm. Most baseline demographic, clinical, and laboratory characteristics were similar between treatment groups, except for higher white blood cell and absolute neutrophil counts and higher bilirubin concentrations in the alternative group |
|
| Interventions |
Intervention characteristics RBC transfusions
Hydroxyurea
|
|
| Outcomes |
Primary outcome: TCD time‐averaged mean velocity on the index side defined as the cerebral hemisphere with the higher mean arterial velocity at baseline assessment Secondary outcomes: TCD velocity on the non‐index side, new stroke or non‐stroke neurological events, new brain MRI/MRA lesions, hepatic iron overload, sickle‐related events, neuropsychological status, quality of life, growth, and treatment‐related complications |
|
| Identification |
Sponsorship source: National Heart, Lung, and Blood Institute through grants R01 HL‐095647 (REW) and R01 HL‐095511 (BRD) Country: USA & Canada Setting: 26 paediatric hospital and health centres in children with abnormal TCD velocities greater or equal to 200 cm/s but no vasculopathy Comments: declarations of interest: All authors declared trial registration: ClinicalTrials.gov, number NCT01425307; . Authors name: Russell E Ware Institution: Cincinnati Children’s Hospital Medical Center Email: russell.ware@cchmc.org Address: Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA |
|
| Notes |
Power calculation: 100 participants (50 per treatment arm) who complete the 24‐month post‐randomisation follow‐up period will provide at least 90% power to test the non‐inferiority hypothesis under reasonable scenarios Analysis: 2 planned interim analyses after 33% and 67% of participants had completed exit studies At the first scheduled interim analysis, non‐inferiority was shown and the sponsor terminated the trial after 50% had exited and repeat analysis confirmed the first interim analysis. We did analyses in the ITT population, except for a planned per‐protocol analysis of TCD velocities, which excluded participants who exited the trial early In hydroxyurea group 2 participants did not reach MTD because of medication non‐adherence leading to trial withdrawal and 1 participant had an early adjudicated TIA during the overlap period while on both hydroxyurea and transfusions. 6 events in 6 children were deemed TIA (3 in each group) Classified in Ware appendix as SAEs ‐ include infections/fever, neurological, hepatobiliary disease, splenomegaly/splenectomy In the standard group, average serum ferritin remained stable at 2713 ng/mL at baseline and 2674 ng/mL at trial exit, and average liver iron concentration increased slightly from 8·5 mg Fe per g dry weight liver to 11·3 mg Fe per g dry weight liver (P = 0·052). 19 adverse events were attributed to deferasirox chelation treatment, which occurred in nine (15%) participants assigned to the standard group, 18 adverse events were attributed to phlebotomy procedures, which occurred in 14 participants (23%) assigned to receive hydroxyrea.In the alternative group, the average serum ferritin decreased from 3080 ng/mL at baseline to 1276 ng/mL at trial exit (P < 0·0001), and average hepatic iron decreased from 11·3 mg Fe per g dry weight liver to 9·5 mg Fe per g dry weight liver P = 0·001). Compared with the standard group, iron overload improved more in the alternative group, with a difference in ferritin change of –1047 ng/mL (95% CI –1524 to –570; P < 0·001) and a difference in liver iron change of –4·3 mg Fe per g dry weight liver(–6·1 to –2·5; P = 0·001; table 2) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was done centrally, stratified by site with a block size of four, and we used an adaptive randomisation scheme to balance the covariates of baseline age and TCD velocity (appendix)." Judgement comment: participants will be randomised to treatment arm by means of an adaptive randomisation algorithm to maintain balance between treatment groups with respect to site, age, and the mean of 2 screening maximum TCD velocities. Participants are randomised to either the standard or alternative treatment arm at an approximately 1 to 1 ratio. Randomisation is stratified within site to achieve approximate balance with respect to 2 baseline factors: (1) the mean of the 2 screening TCD velocity values; and (2) participant age at enrolment. Block sizes will be fixed at 4 to maintain equal sample sizes in the 2 arms within a given site every fourth randomisation. Random block size was not employed due to the small number of participants excepted at each clinical site. Assignment within a given block of 4 will be randomly ordered and will vary from block to block. The first 8 participants to be randomised will be assigned to a treatment arm based on the site‐specific unconstrained random allocation scheme |
| Allocation concealment (selection bias) | Low risk | Judgement comment: from appendix: this process assures that the differences in mean baseline age and mean baseline TCD velocity between the 2 arms, after the participant’s randomisation, do not exceed a critical value determined by a statistical probability distribution. Adaptive randomisation and trial‐wide imbalance constraints and other constraints appear to ensure that allocation is statistically determined. Randomisation was done centrally, stratified by site with a block size of four, and we used an adaptive randomisation scheme to balance the co‐variates of baseline age and TCD velocity |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Judgement comment: participants and personnel are unblinded |
| Blinding of outcome assessment (detection bias) SCI, stroke, and all‐cause mortality | Low risk | Judgement comment: TCD examinations were done just before transfusions or phlebotomy, and all were read centrally by observers masked to treatment assignment and previous TCD results. All new potential stroke events were assessed with careful neurological evaluation and brain MRI/MRA examinations, then adjudicated centrally by a panel of expert reviewers. Independent and then consensus opinions were obtained from neurologists and neuroradiologists masked to trial treatment. Brain MRI/MRA examinations at trial exit allowed us to confirm that no strokes had been missed by the adjudication process Judgement comment: al‐cause mortality is an objective outcome unaffected by blinding of the outcome assessor |
| Blinding of outcome assessment (detection bias) All outcomes except SCI, stroke and all‐cause mortality | High risk | Judgement comment: unblinded trial |
| Incomplete outcome data (attrition bias) Stroke and SCI | Low risk | All randomised participants included in safety and efficacy analysis Transfusion arm: 0 lost to follow‐up; 8 discontinued intervention; 3 adjudicated TIA; 1 TCD velocity >240 cm/s; 2 non‐adherence; 1 difficulty finding matched blood; 1 chose to withdraw Hydroxyurea arm: 6 discontinued intervention; 3 adjudicated TIA; 3 non‐adherence |
| Incomplete outcome data (attrition bias) All outcomes except stroke and SCI | Low risk | All randomised participants included in safety and efficacy analysis. Transfusion arm: 0 lost to follow‐up; 8 discontinued intervention; 3 adjudicated TIA; TCD velocity >240 cm/s; 2 non‐adherence; 1 difficulty finding matched blood; 1 chose to withdraw Hydroxyurea arm: 6 discontinued intervention; 3 adjudicated TIA; 3 non‐adherence |
| Selective reporting (reporting bias) | Unclear risk | Judgement comment: some outcomes will be reported in future papers |
| Other bias | Unclear risk | Judgement comment: children with severe vasculopathy were excluded from TWiTCH trial during screening, so these children might not be suitable candidates for hydroxyurea. Mean age of the trial participants was slightly older than the peak age for primary stroke (about 5 ‐ 6 years), yet still within the published range.The duration of hydroxyurea therapy without transfusions was relatively short; longer follow‐up is clearly needed to establish whether these findings are maintained over time. Trial stopped early based on TCD velocities, an accepted surrogate for primary stroke risk in children with SCA, still be some uncertainty with regards to effectiveness for stroke prevention in certain populations and over timeAfter full enrolment and when 37% of the participants had exited the trial, the first scheduled interim analysis showed that the stopping boundary had been passed and non‐inferiority was shown. After 50% of participants had exited, repeat analyses supported these findings and the trial was terminated by NHLBI. Remaining participants then completed all exit studies before discontinuing protocol‐directed trial treatment. In total, the standard group included 42 participants who completed trial treatment, 11 who had truncated treatment, and 8 who exited early; the alternative group included 41 participants who completed treatment, 13 who had truncated treatment, and 6 who exited early |
IQR: interquartile range ITT: intention‐to‐treat MRA: magnetic resonance angiography MRI: magnetic resonance imaging MTA: maximum tolerated dose RBC: red blood cell RCT: randomised controlled trial SCA: sickle cell anaemia SCD: sickle cell disease SCI: silent cerebral infarcts TCD: transcranial doppler TIA: transient ischaemic attack WBC: white blood cell
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Adams 1999 | Not a RCT |
| BABY HUG 2011 | Not designed to assess SCI‐related outcomes |
| Bernaudin 2015 | Not a RCT |
| Bernaudin 2016 | Not a RCT |
| Kawadler 2016 | systematic review |
| NCT00004485 | Not a RCT |
| NCT00402480 | Not a RCT |
| NCT01340404 | Not an RCT |
| NCT01801423 | Not a RCT |
| NCT02560935 | Not designed to assess SCI‐related outcomes |
| NCT02675790 | Not designed to assess SCI‐related outcomes |
| Steinberg 2010 | Not a RCT |
RCT: randomised controlled trial
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | Randomised controlled trial |
| Participants |
Inclusion criteria: completion of all components of the Phase 1 trial (NCT00528801); Wechsler Adult Intelligence Scale (WAIS) III‐Performance IQ (PIQ) score ≤ 90, hemoglobin ≤ to 9.0 g/dL. People who did not complete Phase I of the trial are eligible for enrolment in this trial if they meet all of the following criteria: capable of giving informed consent for the trial; willing to undergo transfusion therapy for 6 months; African descent; proficient/fluent in English; haemoglobin electrophoresis confirming haemoglobin SS or SB0 (≤ 15%); WAIS III‐PIQ score ≤ 90; hemoglobin ≤ 9.0 g/dL; MMSE score of ≥ 20; POMS score on the Depression‐Dejection Subscale ≤ 40 Exclusion criteria: history of life threatening or serious transfusion complications; lack of venous access; current enrolment in the Arginine study (NCT00513617); pregnant; refusal of transfusion; history of unexplained severe haemolytic transfusion reaction; history of serious allergic, pulmonary transfusion reaction requiring hospitalisation; positive auto‐immune haemolytic anaemia (direct Coombs test with IgG and complement); multiple (3 or more) clinically significant allo‐antibodies due to common antigens (e.g. EC, Kel), uncommon, clinically significant antibody that results in difficulty in finding matched units (e.g. anti‐JKB); currently taking hydroxyurea and not on a stable dose in the 6 months before trial entry; creatinine level ˃ 1.7 mg/dL; ferritin level ˃ 1500 ng/mL or quantitative liver iron level ˃ 7 mg/g dry weight and not currently on iron chelation therapy. This is a pilot transfusion in which only 6 months of transfusion will be utilized. The likelihood of iron overload induced toxicity from the transfusions over the 6 months is very small. Furthermore, ferritin is disproportionately elevated in SCD and overestimates the iron burden. Therefore, a quantitative liver iron or ferritin level (or both) has been included as criteria for exclusion.) Major infarct identified on Phase I MRI. Currently on Procrit or related drug that stimulates RBC production In addition to the exclusion criteria listed above, people who did not complete Phase I (or who completed Phase I more than 1 year prior to enrolment into this trial) are disqualified for enrolment in this trial if they meet any of the following criteria: overt stroke; previous evidence of an abnormal MRI or computed axial tomography (CT) scan other than small periventricular or watershed lesions; history of head injury that resulted in neurological symptoms or medical visit; abnormal neurological exam with focal findings; alcohol consumption exceeding 14 drinks/week if female or 21 drinks/week if male; drug abuse, as defined as using non‐prescribed medication; history of claustrophobia and/or presence of metallic implants such as pacemakers, surgical aneurysm clips, or known metal fragments embedded in the body; baseline blood pressure greater than 140/90 mm Hg on 2 repeated measurements (a second measurement is needed only if the first is greater than 140/90 mm Hg); history of uncontrolled hypertension; any long‐term disorder that may result in neurocognitive or brain dysfunction that is not secondary to SCD (including any of the following: inflammatory arterial disorders (e.g. lupus, polyarteritis); history of cancer requiring chemotherapy and/or radiation; untreated hyperlipidaemia; diabetes; ongoing active infection such as HIV, tuberculosis, or sarcoidosis; history of long‐term blood transfusion; long‐term kidney failure/dialysis; long‐term lung disease characterized by a need for oxygen; morbid obesity (i.e. weight greater than 115 kg); heart disease (including a history of congestive heart failure, history of severe coronary artery disease characterized by angioplasty or surgery, or history of angina); active hepatitis or liver failure; acquired or congenital immune deficiency; history of psychoses (e.g. delusions, hallucinations) and/or schizophrenia; neurodegenerative disorder; genetic disorder associated with neurocognitive dysfunction such as Down Syndrome; other long‐term illness or disorder other than SCD that will adversely affect the person's performance in the trial) |
| Interventions | RBC transfusions, standard care |
| Outcomes | Safety and benefits of transfusion therapy on neurocognitive function Frequency and severity of acute sickle cell events |
| Notes | The primary author has been contacted for the full trial report and results |
MMSE: Mini‐Mental Status Examination POMS: Profile of Mood States RBC: red blood cell RCT: randomised controlled trial SCD: sickle cell disease
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | Hydroxyurea to Prevent Brain Injury in Sickle Cell Disease (HUPrevent) |
| Methods | RCT |
| Participants |
Inclusion criteria: participant must have sickle cell anaemia (haemoglobin SS) or sickle Beta‐zero (null) thalassaemia (haemoglobin S‐B0) as confirmed at the local institution by haemoglobin analysis after 6 months of age. Participant must be 9 ‐ 48 months of age. All screening procedures except MRI can be completed between 9 and 12 months of age, with the exception of the MRI, for which the child must have reached the age of 12 months. Informed consent must be signed by the participant's legally authorized guardian acknowledging written consent to join the trial. Inclusion criteria for randomisation participant must be 12 ‐ 54 months of age. Participant must have successfully completed screening procedures (TCD, MRI of the brain, neurology exam, and cognitive evaluation) Exclusion criteria: history of a focal neurologic event lasting more than 24 hours with medical documentation or a history of prior overt stroke. Other neurological problems, such as neurofibromatosis, lead poisoning, non‐febrile seizure disorder, or tuberous sclerosis. Known HIV infection. Treatment with anti‐sickling drugs or hydroxyurea within 3 months or anticipated treatment during the course of the trial. Chronic blood transfusion therapy, ongoing or planned. Poor adherence likely per his/her hematologist and trial co‐ordinator based on previous compliance in clinic appointments and following advice. Presence or planned permanent (or semi‐permanent) metallic structures attached to their body. (e.g. braces on teeth), which their physicians believe will interfere with the MRI of the brain. History of two or more TCD studies with a velocity ≥ 200 cm/s by the non‐imaging technique, or ≥ 185 cm/s for the imaging technique or a indeterminate TCD. Significant cytopenias (ANC < 1500/µl, platelets < 150,000/µl, reticulocytes < 80,000/µl, unless the haemoglobin is > 9 g/dL). Cytopenias will be considered transient exclusions. Other significant organ system dysfunction, known allergy or intolerance of hydroxyurea, significant prematurity (gestational age of < 32 weeks). Exclusion criteria for randomisation participants whose MRI show a silent or overt cerebral infarct. Participants who have a non‐imaging TCD trial with a velocity ≥ 185 cm/s or a TCD that is indeterminate. Participants with abnormal kidney function (creatinine > 0.8 mg/dL) Significant cytopenias (ANC) < 1500/µl, platelets < 150,000/µl, reticulocytes < 80,000/µl, unless the haemoglobin is > 9 g/dL). Cytopenias will be considered transient exclusions |
| Interventions | Hydroxyurea, placebo |
| Outcomes | Primary outcome measures: CNS complication (time frame: 3 years). A composite of abnormally elevated cerebral blood flow velocity as measured by TCD ultrasound, SCI, or stroke Secondary outcome measures: proportion of participants with SAEs attributed to trial procedures (time frame: 3 years). Also evaluating the safety of trial procedures including the sedation required to obtain MRI of the brain in young children and administration of hydroxyurea Proportion of participants undergoing randomisation (time frame: 6 months). Evaluating the proportion of screened participants that undergo randomisation to hydroxyurea or placebo |
| Starting date | October 2011 |
| Contact information | James F. CasellaRainey Professor of Pediatric Hematology, Johns Hopkins University, jcasella@jhmi.eduBaltimore, Maryland, USA, 21287 |
| Notes | Estimated completion date: October 2017 |
ANC: absolute neutrophil count CNS: central nervous system MRI: magnetic resonance imaging RCT: randomised controlled trial SAEs: severe adverse events SCI: silent cerebral infarcts TCD: transcranial doppler
Contributions of authors
Lise Estcourt: searching; selection of trials; eligibility assessment; content expert, and review content development.
Patricia Fortin: searching; selection of trials; eligibility assessment; and review content development.
Sally Hopewell: methodological expert and review development.
Marialena Trivella: statistical and methodological expert and review development
Carolyn Doree: protocol development, search methods and strategies
Miguel Abboud: protocol development, review development and content expert.
Sources of support
Internal sources
-
NHS Blood and Transplant, Research and Development, UK.
To fund the work of the Systematic Review Initiative (SRI)
External sources
-
National Institute for Health Research, UK.
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
-
National Institute for Health Research (NIHR) Cochrane Programme Grant, UK.
To provide funding for systematic review authors and methodological support from the Centre for Statistics in Medicine, Oxford
Declarations of interest
Lise Estcourt: partly funded by the NIHR Cochrane Programme Grant ‐ Safe and Appropriate Use of Blood Components.
Patricia Fortin: funded by the NIHR Cochrane Programme Grant ‐ Safe and Appropriate Use of Blood Components.
Sally Hopewell: partly funded by the NIHR Cochrane Programme Grant ‐ Safe and Appropriate Use of Blood Components.
Marialena Trivella: partly funded by the NIHR Cochrane Programme Grant ‐ Safe and Appropriate Use of Blood Components.
Miguel Abboud: has received research funding from Mast Therapeutics, Eli Lilly, Shire, Dilaforette, he also serves on advisory boards and steering committees for Eli Lilly, Pfizer, Astra Zeneca and Novo. These activities had no relationship to the management of neurologic complications in sickle cell disease.
New
References
References to studies included in this review
- Beverung LM, Strouse JJ, Hulbert ML, Neville K, Liem RI, Inusa B, et al. Health‐related quality of life in children with sickle cell anemia: impact of blood transfusion therapy. American Journal of Hematology 2015;90(2):139‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bhatnagar P, Casella EB, Arking DE, Casella JF. Genome‐wide association for silent cerebral infarction (SCI) in sickle cell disease: the silent infarct transfusion trial (SIT) cohort. Blood 2009;114(22):2563. 19762504 [Google Scholar]; Bhatnagar P, Purvis S, Barron‐Casella E, DeBaun MR, Casella JF, Arking DE, et al. Genome‐wide association study identifies genetic variants influencing F‐cell levels in sickle‐cell patients. Journal of Human Genetics 2011;56(4):316‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bhatnagar P, Purvis S, Barron‐Casella E, DeBaun MR, Casella JF, Arking DE, et al. Genome‐wide association study identifies genetic variants influencing F‐cell levels in sickle‐cell patients. Journal of Human Genetics 2011;56(4):316‐23. Supplementary information. http://www.nature.com/jhg/journal/v56/n4/suppinfo/jhg201112s1.html. [DOI] [PMC free article] [PubMed] [Google Scholar]; Casella JF, King AA, Barton B, White DA, Noetzel MJ, Ichord RN, et al. Design of the silent cerebral infarct transfusion (SIT) trial. Pediatric Hematology & Oncology 2010;27(2):69‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]; DeBaun MR, Gordon M, McKinstry RC, Noetzel MJ, White DA, Sarnaik SA, et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. New England Journal of Medicine 2014;371(8):699‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]; DeBaun MR, Sarnaik SA, Rodeghier MJ, Minniti CP, Howard TH, Iyer RV, et al. Associated risk factors for silent cerebral infarcts in sickle cell anemia: low baseline hemoglobin, sex, and relative high systolic blood pressure. Blood 2012;119(16):3684‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]; Eigbire‐Molen O, Darbari DS, Ponisio MR, Milchenko MV, Rodeghier MJ, Casella JF, et al. Progressive loss of brain volume in children with sickle cell anemia: a report from the silent cerebral infarct transfusion trial cohort. Blood 2015;126(23):546. [DOI] [PubMed] [Google Scholar]; Glassberg JA, Wang J, Cohen R, Richardson LD, DeBaun MR. Risk factors for increased ED utilization in a multinational cohort of children with sickle cell disease. Academic Emergency Medicine 2012;19(6):664‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]; ISRCTN52713285. Silent Cerebral Infarct Multi‐Center Clinical Trial. www.isrctn.com/ISRCTN52713285 24 August 2004. [Protocol/ serial number: U01NS42804]; Kawadler JM, Clark CA, McKinstry RC, Kirkham FJ. Brain atrophy in paediatric sickle cell anaemia: findings from the silent infarct transfusion (SIT) trial. British Journal of Haematology2016; Vol. Apr 7 [Epub ahead of print]. [DOI: 10.1111/bjh.14039] [DOI] [PubMed]; King AA, Rodeghier MJ, Panepinto JA, Strouse JJ, Casella JF, Quinn CT, et al. Silent cerebral infarction, income, and grade retention among students with sickle cell anemia. American Journal of Hematology 2014;89(10):188‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]; King, AA, Strouse JJ, Rodeghier MJ, Compas BE, Casella JF, McKinstry RC, et al. Parent education and biologic factors influence on cognition in sickle cell anemia. American Journal of Hematology 2014;89(2):162‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kirkham FJ, Darekar E, Kija R, Bhadresha R, McSwiggan J, Cox SE, et al. Brain T2‐weighted signal intensity ratio in children with sickle cell disease with and without stroke. European Journal of Paediatric Neurology 2011;15 Suppl 1:S60. [Abstract no: P07.3] [Google Scholar]; Land V, Heijboer H, Fijnvandraat K, DeBaun MR, Casella JF. Transfusions for silent cerebral infarcts in sickle cell anemia. New England Journal of Medicine 2014;371(19):1841‐2. [DOI] [PubMed] [Google Scholar]; Liem RI, Liu J, Gordon MO, Vendt BA, McKinstry RC 3rd, Kraut MA, et al. Reproducibility of detecting silent cerebral infarcts in pediatric sickle cell anemia. Journal of Child Neurology 2014;29(12):1685‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]; NCT00072761. Silent Cerebral Infarct Transfusion Multi‐Center Clinical Trial (SIT). clinicaltrials.gov/show/NCT00072761 Date first received: 10 November 2003. ; Ogunsile FJ, DeBaun MR, Currie K, Rodeghier MJ. Parvovirus B19 infection associated with silent cerebral infarcts: a secondary analysis of the silent cerebral infarct trial cohort. Blood 2015;126(23):3410. [DOI] [PubMed] [Google Scholar]; Quinn CT, McKinstry RC, Dowling MM, Ball WS, Kraut MA, Casella JF, et al. Acute silent cerebral ischemia occurs more frequently than silent cerebral infarction in children with sickle cell anemia. Blood 2010;116(21):268. [Google Scholar]; Roberts DO, Covert B, Rodeghier M, Parmar N, DeBaun MR, Thompson AA, et al. Randomization is not associated with socio‐economic and demographic factors in a multi‐center clinical trial of children with sickle cell anemia. Pediatric Blood & Cancer 2014;61(9):1529‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]; Thangarajh M, Yang G, Fuchs D, Ponisio MR, McKinstry RC, Jaju A, et al. Magnetic resonance angiography‐defined intracranial vasculopathy is associated with silent cerebral infarcts and glucose‐6‐phosphate dehydrogenase mutation in children with sickle cell anaemia. British Journal of Haematology 2012;159(3):352‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]; Vendt BA, McKinstry RC, Ball WS, Kraut MA, Prior FW, Barton B, et al. Silent cerebral infarct transfusion (SIT) trial imaging core: application of novel imaging information technology for rapid and central review of MRI of the brain. Journal of Digital Imaging 2009;22(3):326‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wolf RB, Saville BR, Roberts DO, Fissell RB, Kassim AA, Airewele G, et al. Factors associated with growth and blood pressure patterns in children with sickle cell anemia: Silent Cerebral Infarct Multi‐Center Clinical Trial cohort. American Journal of Hematology 2015;90(1):2‐7. [DOI] [PubMed] [Google Scholar]; Land V, Heijboer H, Fijnvandraat K, DeBaun MR, Casella JF. Transfusions for silent cerebral infarcts in sickle cell anemia. New England journal of medicine 2014;371(19):1841‐2. [DOI] [PubMed] [Google Scholar]
- Abboud M, Cure J, Gallagher D, Berman B, Hsu L, Wang W, et al. Magnetic resonance angiography (MRA) in children with abnormal transcranial doppler (TCD) velocities in the 'STOP' study. Blood 1999;94(10 Suppl 1):645a. [DOI] [PubMed] [Google Scholar]; Abboud MR, Cure J, Gallagher D, Berman B, Hsu L, Wang W, et al. Magnetic resonance angiography in children with abnormal TCD in the STOP study. National Sickle Cell Disease Program Annual Meeting Conference Proceedings; 1999 Mar;. 1999:49. [Google Scholar]; Adamkiewicz T, Abboud M, Barredo J, Cavalier ME, Peterson J, Rackoff B, et al. Serum ferritin in children with SCD on chronic transfusion: correlation with serum alanine aminotransferase and liver iron (STOP/STOP II liver iron ancillary study). 35th Anniversary Convention of the National Sickle Cell Disease Program and the Sickle Cell Disease Association of America; 2007 Sep 17‐22; Washington DC. 2007:316. [Google Scholar]; Adamkiewicz T, Abboud MR, Barredo JC, Kirby‐Allen M, Alvarez OA, Casella JF, et al. Serum ferritin in children with sickle cell disease on chronic transfusion: measure of iron overload or end organ injury? STOP/ STOP II liver iron ancillary study. Blood2006; Vol. 108, issue 11. [Abstract no: 791]; Adams R, Brambilla D, McKie V, Files B, Vichinsky E, Abboud M, et al. Stroke prevention in sickle cell disease (STOP): final results. Blood 2000;96(11 Pt 1):10a. [Google Scholar]; Adams R, McKie V, Files B, Vichinsky E, Abboud M, Hsu L, et al. Stroke prevention in sickle cell disease (STOP): Results of transcranial doppler ultrasound screening stroke risk. Stroke; a Journal of Cerebral Circulation 1998;29(1):308. [Google Scholar]; Adams RJ. Lessons from the stroke prevention trial in sickle cell anemia (STOP) study. Journal of Child Neurology 2000;15(5):344‐9. [DOI] [PubMed] [Google Scholar]; Adams RJ, Brambilla D. Stroke prevention in sickle cell anaemia: the 'STOP' trial. 22nd International Joint Conference on Stroke and Cerebral cCrculation; 1997 Feb 6‐8; Anaheim, California. 1997. [Google Scholar]; Adams RJ, Brambilla D. Stroke prevention trial in sickle cell anemia: STOP. National Sickle Cell Disease Program Annual Meeting Conference Proceedings; 1996 Mar. 1996:19. [Google Scholar]; Adams RJ, Brambilla D, McKie V, Files B, Vichinsky E, Abboud M, et al. Risk of stroke in children with sickle cell disease and abnormal transcranial doppler ultrasound (TCD). Blood 1999;94(10 Suppl 1):419a. [Google Scholar]; Adams RJ, Brambilla D, McKie V, Files B, Vichinsky E, Abboud M, et al. Stroke prevention in sickle cell disease (STOP): follow up after closure of the trial. Blood 1998;92(10 Suppl 1):527a. [Google Scholar]; Adams RJ, Brambilla D, McKie VC, Files B, Vichinsky E, Abboud M, et al. Stroke prevention in sickle cell disease (STOP): baseline characteristics of trial patients. Blood 1997;90(10 Suppl 1):123a. [Google Scholar]; Adams RJ, Brambilla D, Miller ST. Optimizing primary stroke prevention in children with sickle cell. 28th Annual Meeting of the National Sickle Cell Disease Program; 2005 April 9‐13; Cincinnati, Ohio.. 2005:3. [Google Scholar]; Adams RJ, Brambilla D, Vichinsky E, Abboud M, Pegelow C, Carl EM, et al. Risk of stroke and conversion to abnormal TCD in children screened with transcranial doppler (TCD) during the STOP study. National Sickle Cell Disease Program Annual Meeting Conference Proceedings; 2002 Sept. 2002:3. [Google Scholar]; Adams RJ, Brambilla DJ, McKie V, File B, Vichinsky E, Abboud M. Stroke prevention in sickle cell disease (STOP): Baseline characteristics of trial patients. National Sickle Cell Disease Program Annual Meeting Conference Proceedings; 1997 Sept. 1997:41. [Google Scholar]; Adams RJ, Brambilla DJ, STOP II Investigators. The study design of optimizing primary stroke prevention in children with sickle cell anemia. 27th Annual Meeting of the National Sickle Cell Disease Program; 2004 Apr 18‐21; Los Angeles, California. 2004:174. [Google Scholar]; Adams RJ, Brambilla DJ, Vichinsky E, Abboud M, Pegelow C, Carl EM, et al. Stroke prevention trial in sickle cell anemia (STOP Study): risk of stroke in 1933 children screened with transcranial doppler (TCD). National Sickle Cell Disease Program Annual Meeting Conference Proceedings; 1999 Mar. 1999:54. [Google Scholar]; Adams RJ, Virgil CM, Brambilla D, Carl E, Gallagher D, Nichols FT, et al. Stroke prevention trial in sickle cell anemia. Controlled Clinical Trials 1998;19(1):110‐29. [DOI] [PubMed] [Google Scholar]; Files B, Brambilla D, Kutlar A, Miller S, Pegelow C, Vichinsky E, et al. A randomized trial of chronic transfusion to prevent stroke in sickle cell anemia: changes in ferritin with chronic transfusion. Blood 1998;92(10 Suppl 1):528a. [Google Scholar]; Gates A, Rogers MA, Puczynski M. Stroke prevention trial in sickle cell anemia: comments on effects of chronic transfusion on pain. Journal of Pediatrics 2002;141(5):742‐3. [DOI] [PubMed] [Google Scholar]; Hyacinth HI, Gee BE, Voeks JH, Adams RJ, Hibbert J. High frequency of RBC transfusions in the STOP study was associated with reduction in serum biomarkers of neurodegeneration, vascular remodeling and inflammation. Blood2012; Vol. 120, issue 21. [Abstract no: 244]; Kutlar A, Harbi J, Jackson B, Holley L, Gallagher D, Clair B, et al. Laboratory parameters in patients randomized in the STOP study and their modification by transfusion. Blood 2000;96(11 Pt 2):18b‐9b. [Google Scholar]; Kutlar A, Harbin J, Jackson BB, Holley LGD, Clair B, Brambilla D, et al. Baseline laboratory parameters in patients randomized to the STOP study and their modification by transfusion. National Sickle Cell Disease Program Annual Meeting Conference Proceedings; 2000 Apr. 2000:82a. [Google Scholar]; Kwiatkowski JL, Granger S, Brambilla D, Brown RC, Miller S, Adams RJ, et al. Elevated blood flow velocity in the anterior cerebral artery and stroke risk in sickle cell disease: extended analysis from the STOP trial. British Journal of Haematology 2006;134(3):333‐9. [DOI] [PubMed] [Google Scholar]; Kwiatkowski JL, Morales K, Brambilla DJ, Files B, Adamkiewicz T, Adams RJ. Long‐term follow‐up of transcranial doppler ultrasonography in children with sickle cell disease: Results of the STOP and STOP II patient cohorts. Blood 2002;100(11 Pt 1):663a. [Google Scholar]; Lee MT, Piomelli S, Granger S, Miller ST, Harkness S, Brambilla DJ, et al. Stroke prevention trial in sickle cell anemia (STOP): extended follow‐up and final results. Blood 2006;108(3):847‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lee SB, Ramsingh D, Kutlar A, Holley L, McKie VC, Adams RJ. C‐reactive protein in children with sickle cell disease at risk for stroke in the STOP study. Stroke; a Journal of Cerebral Circulation 2002;33(1):373. [Google Scholar]; Lezcano NE, Odo N, Kutlar A, Brambilla D, Adams RJ. Transfusion lowers plasma‐free hemoglobin in children with sickle cell disease at risk for stroke. Stroke; a Journal of Cerebral Circulation 2006;37(2):695‐6. [DOI] [PubMed] [Google Scholar]; Miller S, Wright E, Abbou M, Berman B, Files B, Scher C, et al. Impact of chronic transfusion on non‐neurological events during the stroke prevention trial in sickle cell anemia (STOP). Pediatric Research 2000;47(4 Pt 2):251A. [Google Scholar]; Miller S, Wright E, Abboud M, Berman B, Files B, Scher C, et al. Impact of chronic transfusion on non‐neurological events during the stroke prevention trial in sickle cell anemia (STOP). National Sickle Cell Disease Program Annual Meeting Conference Proceedings; 2000 Apr. 2000:61a. [Google Scholar]; NCT00000592. Stroke Prevention in Sickle Cell Anemia (STOP 1). clinicaltrials.gov/ct2/show/NCT00000592 Date first received: 27 October 1999. ; National Heart, Lung, and Blood Institute (NHLBI). Clinical alert: periodic transfusions lower stroke risk in children with sickle cell anemia. https://www.nlm.nih.gov/databases/alerts/sickle97.html (accessed prior to 09 February 2017):www.nlm.nih.gov/databases/alerts/sickle97.html. ; Olivieri N, Brambilla D, McKie V, Piomelli S, Kutlar A, Files B, et al. Changes in cerebral blood flow velocities during chronic transfusion therapy to prevent stroke in sickle cell disease. Blood 2000;96(11 Pt 1):486a. [Google Scholar]; Pegelow C, Adams R, Hsu L, McKie V, Wang W, Zimmerman R, et al. Children with silent infarct and elevated transcranial doppler ultrasonography velocity are at increased risk of subsequent infarctive events. National Sickle Cell Disease Program Annual Meeting Conference Proceedings; 1999 Mar. 1999:137. [Google Scholar]; Pegelow CH, Wang W, Granger S, Hsu LL, Vichinsky E, Moser FG, et al. Silent infarcts in children with sickle cell anemia and abnormal cerebral artery velocity. Archives of Neurology 2001;58(12):2017‐21. [DOI] [PubMed] [Google Scholar]; Sarnaik SA. Prevention of stroke by transfusions in children with sickle cell anemia. New England Journal of Medicine 1998;339:1477‐8. [PubMed] [Google Scholar]; Vichinsky E, Luban E, Wright E, Olivieri N, Driscoll C, Pegelow, et al. Prospective red cell phenotype matching in STOP ‐ a multi‐centre transfusion trial. National Sickle Cell Disease Program Annual Meeting Conference Proceedings; 1999 Mar. 1999:163. [Google Scholar]; Vichinsky E, Luban N, Wright E, Olivieri N, Driscoll C, Pegelow C, et al. Prospective red cell phenotype matching in STOP ‐ a multi‐center transfusion trial. Blood 1998;92(10 Suppl 1):528a. [Google Scholar]; Vichinsky E, Luban NL, Wright E, Olivieri N, Driscoll C, Pegelow CH, et al. Prospective RBC phenotype matching in a stroke‐prevention trial in sickle cell anemia: a multicenter transfusion trial. Transfusion 2001;41(9):1086‐92. [DOI] [PubMed] [Google Scholar]; Wang W, Morales K, Olivier N, Styles L, Scher C, Adams R, et al. Effect of chronic transfusion on growth in children with sickle cell anemia: results of the STOP trial. National Sickle Cell Disease Program Annual Meeting Conference Proceedings; 2002 Sept. 2002:100. [Google Scholar]
- Abboud MR, Yim E, Adams RJ. The progression and development of silent infarcts in children with sickle cell anemia is prevented by chronic transfusions and is unrelated to level of hemolysis. Blood2008; Vol. 12. [Abstract no: 712]; Abboud MR, Yim E, Musallam KM, Adams RJ. Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from STOP II. Blood 2011;118(4):894‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]; Adamkiewicz T. Transcranial doppler measures in patients with sickle cell disease at high risk for stroke and receiving hydroxyurea: the HyRetro ancillary study. 52nd Ash Annual Meeting and Exposition; 2010 Dec 4‐7; Orlando, Florida. 2010. [Abstract no: 1620] [Google Scholar]; Adams R, Fullerton H, Kwiatkowski, Voeks J. Development of high risk TCD in sickle cell disease. Stroke; a Journal of Cerebral Circulation2015; Vol. 46, issue Suppl 1. [Abstract no: 17]; Adams RJ, Brambilla D. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. New England Journal of Medicine 2005;353(26):2769‐78. [DOI] [PubMed] [Google Scholar]; Adams RJ, Lackland DT, Brown L, Brown D, Voeks J, Fullerton H, et al. Transcranial doppler re‐screening of subjects who participated in STOP and STOP II. American Journal of Hematology 2016;91(12):1191‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]; Brown C, Miller S, Kwiatkowski J, Brambilla D, Adams R. Optimizing primary stroke prevention in sickle cell anemia (STOP 2): an argument for prolonged transfusion?. 29th Annual Meeting of the National Sickle Cell Disease Program; 2006 Apr 8‐12; Memphis, USA.. 2006:78. [Google Scholar]; Kanter J, Kwiatkowski J, Fullerton HJ, Voeks J, Debenham E, Brown LJ, Adams RJ. Impact of TCD screening protocol on the incidence of hemorrhagic stroke in children and young adults with sickle cell disease. Blood 2015;126(23):3402. [Google Scholar]; Kwiatkowski JL, Kanter J, Fullerton HJ, Voeks J, Debenham E, Brown DG, et al. Ischemic stroke in children and young adults with sickle cell disease (SCD) in the post‐STOP era. Blood 2015;126(23):68. [DOI] [PubMed] [Google Scholar]; NCT00006182. Stroke prevention in sickle cell anemia (STOP 2). clinicaltrials.gov/show/NCT00006182 Date first received: 21 August 2000. ; Sayer G, Bowman L, Clair B, Cail A, Blanchard B, Natrajan K, et al. Long term outcome of patients enrolled into STOP and STOP II trials: a single center experience. Blood2012; Vol. 120, issue 21. [Abstract no: 3219]
- Aygun B, Mortier NA, Kesler K, Lockhart A, Schultz WH, Cohen A, et al. Therapeutic phlebotomy is safe in children with sickle cell anaemia and can be effective treatment for transfusional iron overload. British Journal of Haematology 2015;169(2):262‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]; Aygun B, Mortier NA, Kesler K, Schultz WH, Alvarez OA, Rogers ZR, et al. Therapeutic phlebotomy in children with sickle cell anemia, stroke, and iron overload: the SWiTCH experience. 53rd Ash Annual Meeting and Exposition; 2011 Dec 10‐13; San Diego, California.. 2011. [Abstract no: 1044] [Google Scholar]; Helton KJ, Adams RJ, Kesler KL, Lockhart A, Aygun B, Driscoll C, et al. Magnetic resonance imaging/angiography and transcranial Doppler velocities in sickle cell anemia: results from the SWiTCH trial. Blood 2014;124(6):891‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]; NCT00122980. Stroke with transfusions changing to hydroxyurea (SWiTCH). clinicaltrials.gov/ct2/show/NCT00122980 Date first received: 20 July 2005. ; National Institutes of Health. Stroke prevention study in children with sickle cell anemia, iron overload stopped early. www.nih.gov/news/health/jun2010/nhlbi‐03.htm (accessed prior to 09 February 2017). ; Ware RE, Helms RW. Stroke with transfusions changing to hydroxyurea (SWiTCH): a phase 3 randomised clinical trial for treatment of children with sickle cell anemia. American Journal of Hematology2011; Vol. 86, issue 11. [Abstract no: 844] [DOI] [PMC free article] [PubMed]; Ware RE, Helms RW. Stroke with transfusions changing to hydroxyurea (SWiTCH): a phase 3 randomized clinical trial for treatment of children with sickle cell anemia, previous stroke, and iron overload. Blood 2010;116(21):367. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ware RE, Helms RW, SWiTCH Investigators. Stroke with transfusions changing to hydroxyurea (SWiTCH). Blood 2012;119(17):3925‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ware RE, McMurray MA, Schultz WH, Alvarez OA, Aygun B, Cavalier ME, et al. Academic community standards for chronic transfusion therapy in children with sickle cell anemia and stroke. Blood2006; Vol. 108, issue 11. [Abstract no: 1213]; Ware RE, Schultz W, Yovetich N, Mortier NA, Alvarez O, Hilliard L, et al. Stroke with transfusions changing to hydroxyurea (SWiTCH): a phase III randomized clinical trial for treatment of children with sickle cell anemia, stroke, and iron overload. Pediatric Blood & Cancer 2011;57(6):1011‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aygun B, Wruck LM, Schultz WH, Mueller BU, Brown C, Luchtman‐Jones L, et al. Chronic transfusion practices for prevention of primary stroke in children with sickle cell anemia and abnormal TCD velocities. American Journal of Hematology 2012;87(4):428‐30. [DOI] [PubMed] [Google Scholar]; Helton K, Roberts D, Schultz WH, Davis BR, Kalfa TA, Pressel SL, et al. Effects of Chronic Transfusion Therapy on MRI and MRA in Children with Sickle Cell Anemia at Risk for Primary Stroke: Baseline Imaging from theTwitch Trial. Blood 2014;124(21):4052. [Google Scholar]; Imran H, Aygun B, Davis BR, Pressel SL, Schultz WH, Jackson S, et al. Effects of chronic transfusion therapy on transcranial doppler ultrasonography velocities in children with sickle cell anemia at risk for primary stroke: Baseline findings from the Twitch trial. Blood 2014;124(21):87. [Google Scholar]; NCT01425307. Transcranial doppler (TCD) with transfusions changing to hydroxyurea [TCD with transfusions changing to hydroxyurea (TWiTCH): a phase III randomized trial to compare standard therapy (erythrocyte transfusions) with alternative therapy (hydroxyurea) for the maintenance of lowered TCD velocities in pediatric subjects with sickle cell anemia and abnormal pre‐treatment TCD velocities]. clinicaltrials.gov/ct2/show/NCT01425307 Date first received: 19 August 2011. ; Ware RE, Davis BR, Schultz WH, Brown C, Aygun B, Sarnaik SA, et al. TCD with transfusions changing to hydroxyurea (TWiTCH): hydroxyurea therapy as an alternative to transfusions for primary stroke prevention in children with sickle cell anemia. Blood 2015;126(23):3. [Google Scholar]; Ware RE, Davis BR, Schultz WH, Brown RC, Aygun B, Sarnaik S, et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia ‐ TCD with Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open‐label, phase 3, non‐inferiority trial. Lancet 2016;387(10019):661‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wood JC, Cohen AR, Pressel SL, Aygun B, Imran H, Luchtman‐Jones L, et al. Organ iron accumulation in chronically transfused children with sickle cell anaemia: baseline results from the TWiTCH trial.. British Journal of Haematology 2016;172(1):122‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wood JC, Pressel S, Rogers ZR, Odame I, Kwiatkowski JL, Lee MT, et al. Liver iron concentration measurements by MRI in chronically transfused children with sickle cell anemia: baseline results from the TWiTCH trial.. American Journal of Hematology 2015;90(9):806‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
- Adams RJ, Carl EM, McKie VC, Odo NA, Kutlar A, Phillips M, Brambilla D. A pilot trial of hydroxyurea to prevent strokes in children with sickle cell anemia. National Sickle Cell Disease Program Annual Meeting Conference Proceedings; 1999 Mar. 1999:53. [Google Scholar]
- Adams RJ, Luden J, Miller S, Wang W, Rees R, Li D, et al. TCD in infants: a report from the Baby Hug study. 28th Annual Meeting of the National Sickle Cell Disease Program; 2005 Apr 9‐13; Cincinnati, Ohio.. 2005:105. [Google Scholar]; Alvarez O, Miller ST, Wang WC, Luo Z, McCarville MB, Schwartz GJ, et al. Effect of hydroxyurea treatment on renal function parameters: results from the multi‐center placebo‐controlled BABY HUG clinical trial for infants with sickle cell anemia. Pediatric Blood & Cancer 2012;59(4):668‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]; Armstrong FD, Elkin TD, Brown RC, Glass P, Rana S, Casella JF, et al. Developmental function in toddlers with sickle cell anemia. Pediatrics 2013;131(2):e406‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kalpatthi R, Thompson B, Lu M, Wang WC, Patel N, Kutlar A, et al. Comparison of hematologic measurements between local and central laboratories: data from the BABY HUG trial. Clinical Biochemistry 2013;46(3):278‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lebensburger JD, Miller ST, Howard TH, Casella JF, Brown RC, Lu M, et al. Influence of severity of anemia on clinical findings in infants with sickle cell anemia: analyses from the BABY HUG study. Pediatric Blood & Cancer 2012;59(4):675‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lederman HM, Connolly MA, Ware RE, Luchtman‐Jones L, Goldsmith JC. Effects of hydroxyurea (HU) on lymphocyte subsets and the immune response to pneumococcal, measles, mumps and rubella vaccination in the pediatric hydroxyurea phase III clinical trial ‐ BABY HUG. Blood2012; Vol. 120, issue 21. [Abstract no: 243]; McGann PT, Flanagan JM, Howard TA, Dertinger SD, He J, Kulharya AS, et al. Genotoxicity associated with hydroxyurea exposure in infants with sickle cell anemia: results from the BABY‐HUG Phase III Clinical Trial. Pediatric Blood & Cancer 2012;59(2):254‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sheehan VA, Luo Z, Flanagan JM, Howard TA, Thompson BW, Wang WC, et al. Genetic modifiers of sickle cell anemia in the BABY HUG cohort: influence on laboratory and clinical phenotypes. American Journal of Hematology 2013;88(7):571‐6. [DOI] [PubMed] [Google Scholar]; Thornburg CD, Files BA, Luo Z, Miller ST, Kalpatthi R, Iyer R, et al. Impact of hydroxyurea on clinical events in the BABY HUG trial. Blood 2012;120(22):4304‐10; quiz 4448. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang WC, Oyeku S, Luo Z, Boulet SL, Miller ST, Casella J, et al. Hydroxyurea is associated with lower costs of care of young children with sickle cell anemia. Pediatrics 2013;132(4):67783. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang WC, Pavlakis SG, Helton KJ, McKinstry R, Casella J, Adams RJ, et al. MRI abnormalities of the brain in one‐year‐old children with sickle cell anemia. Pediatric Blood & Cancer 2008;51(5):643‐6. [DOI] [PubMed] [Google Scholar]; Wang WC, Ware RE, Miller ST, Iyer RV, Casella JF, Minniti CP, et al. Hydroxycarbamide in very young children with sickle cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011;377(9778):1663‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernaudin F, Verlhac S, Ducros‐Miralles E, Delatour RP, Dalle JH, Petra E, et al. French national Drepagreffe trial: cognitive performances and neuroimaging at enrollment and after 12 months on transfusion program or transplantation (AP‐HP: NCT 01340404). Blood2015; Vol. 126, issue 23:544.
- Bernaudin F, Verlhac S, Ducros ME, Peffault DR, Dalle JH, Paillard C, et al. Trial comparing HSCT vs transfusions in sickle cell anemia (SCA) patients with abnormal cerebral velocities: cerebral vasculopathy outcome at 1 year. Bone Marrow Transplantation 2016;51:S49‐50. [Google Scholar]
- Kawadler JM, Clayden JD, Clark CA, Kirkham FJ. Intelligence quotient in paediatric sickle cell disease: a systematic review and meta‐analysis. Developmental Medicine & Child Neurology 2016;58(7):672‐9. [DOI] [PubMed] [Google Scholar]
- NCT00004485. Bone marrow transplantation in treating children with sickle cell disease [Phase I/II study of induction of stable mixed chimerism after bone marrow transplantation from HLA‐identical donors in children with sickle cell disease]. clinicaltrials.gov/ct2/show/NCT00004485 Date first received: 18 October 1999.
- NCT00402480. Hydroxyurea to prevent stroke in children with sickle cell anemia and elevated TCD flow velocity [Effects of hydroxyurea on the prevention of primary stroke in children with sickle cell anemia and elevated transcranial doppler (TCD) flow velocity]. clinicaltrials.gov/ct2/show/NCT00402480 Date first received: 21 November 2006.
- NCT01340404. Allogeneic genoidentical stem cell transplantation in children with sickle‐cell anemia and cerebral vasculopathy [A multicenter study comparing the results of allogeneic stem cell genoidentical in children with sickle cell anemia and cerebral vascular disease detected by transcranial doppler]. clinicaltrials.gov/ct2/show/NCT01340404 Date first received: 21 April 2011.
- NCT01801423. Sickle cell disease ‐ stroke prevention in Nigeria trial (SPIN) [Primary prevention of strokes in Nigerian children with sickle cell disease affiliated titles: sickle cell disease ‐ stroke prevention in Nigeria (SPIN) trial]. clinicaltrials.gov/ct2/show/NCT01801423 Date first received: 26 February 2013.
- NCT02560935. Primary prevention of stroke in children with SCD in Sub‐Saharan Africa II (SPRING). clinicaltrials.gov/ct2/show/NCT02560935 Date first received: 2 September 2015.
- NCT02675790. Low dose hydroxyurea for secondary stroke prevention in children with sickle cell disease in Sub‐Saharan Africa (SPRINT). clinicaltrials.gov/ct2/show/NCT02675790 Date first received: 8 January 2016.
- Steinberg MH, McCarthy WF, Castro O, Ballas SK, Armstrong FD, Smith W, et al. The risks and benefits of long‐term use of hydroxyurea in sickle cell anemia: A 17.5 year follow‐up. American Journal of Hematology 2010;85(6):403‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies awaiting assessment
- NCT00850018. Examining cognitive function and brain abnormalities in adults with sickle cell disease ‐ pilot intervention study [Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell disease ‐ a pilot intervention study]. clinicaltrials.gov/ct2/show/NCT00850018 Date first received: 20 February 2009. ; Vichinsky E, Neumay L, Gold JI, Weiner MW, Kasten J, Truran D, et al. A Randomized trial of the safety and benefit of transfusion vs. standard care in the prevention of sickle cell‐related complications in adults: a preliminary report from the phase I NHLBI comprehensive sickle cell centers (cscc) study of neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adult patients with sickle cell disease. Blood 2010;116(21):1321. 20410507 [Google Scholar]; Vichinsky E, Neumayr L, Gold JI, Weiner MW, Kasten J, Truran D. A randomised trial of the safety and benefit of transfusion vs. standard care in the prevention of sickle cell‐related complications in adults: a preliminary report from the phase II NHLBI comprehensive sickle cell centers (CSCC) study of neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adult patients with sickle cell disease. 52nd Ash Annual Meeting and Exposition; 2010 Dec 4‐7; Orlando, Florida.. 2010. [Abstract no: 3221] [Google Scholar]
References to ongoing studies
- NCT01389024. Hydroxyurea to prevent brain injury in sickle cell disease (HUPrevent) [Hydroxyurea to prevent central nervous system (CNS) complications of sickle cell disease in children]. clinicaltrials.gov/ct2/show/NCT01389024 Date first received: 30 June 2011.
Additional references
- Abboud MR, Yim E, Mussallam KM, Adams RJ. Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from STOP II. Blood 2011;118(4):894‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam S, Jonassaint J, Kruger H, Kail M, Orringer EP, Eckman JR, et al. Surgical and obstetric outcomes in adults with sickle cell disease. American Journal of Medicine 2008;121(10):916‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. New England Journal of Medicine 1998;339(1):5‐11. [DOI] [PubMed] [Google Scholar]
- Adekile AD, Yacoub F, Gupta R, Sinan T, Haider MZ, Habeeb Y, et al. Silent brain infarcts are rare in Kuwaiti children with sickle cell disease and high Hb F. Americal Journal of Hematology 2002;70(3):228‐31. [DOI] [PubMed] [Google Scholar]
- Akinsheye I, Alsultan A, Solovieff N, Ngo D, Baldwin CT, Sebastiani P, et al. Fetal hemoglobin in sickle cell anemia. Blood 2011;118(1):19‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansong D, Akoto AO, Ocloo D, Ohene‐Frempong K. Sickle cell disease: management options and challenges in developing countries. Mediterranean Journal of Hematology and Infectious Diseases 2013;5(1):e2013062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong FD Jr, Thompson RJ, Wang W, Zimmerman R, Pegelow CH, Miller S, et al. Cognitive functioning and brain magnetic resonance imaging in children with sickle Cell disease. Neuropsychology Committee of the Cooperative Study of Sickle Cell Disease. Pediatrics 1996;97(6 Pt 1):864‐70. [PubMed] [Google Scholar]
- Armstrong FD, Elkin TD, Brown RC, Glass P, Rana S, Casella JF, et al. Developmental function in toddlers with sickle cell anemia. Pediatrics 2013;131(2):e406‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernaudin F, Socie G, Kuentz M, Chevret S, Duval M, Bertrand Y, et al. Long‐term results of related myeloablative stem‐cell transplantation to cure sickle cell disease. Blood 2007;110(7):2749‐56. [DOI] [PubMed] [Google Scholar]
- Bernaudin F, Verlhac S, Arnaud C, Kamdem A, Chevret S, Hau I, et al. Impact of early transcranial Doppler screening and intensive therapy on cerebral vasculopathy outcome in a newborn sickle cell anemia cohort. Blood 2011;117:1130‐40; quiz 1436. [DOI] [PubMed] [Google Scholar]
- Bernaudin F, Verlhac S, Arnaud C, Kamdem A, Vasile M, Kasbi F, et al. Chronic and acute anemia and extracranial internal carotid stenosis are risk factors for silent cerebral infarcts in sickle cell anemia. Blood 2015;125(10):1653‐61. [DOI] [PubMed] [Google Scholar]
- Casella JF, King AA, Barton B, White DA, Noetzel MJ, Ichord RN, et al. Design of the silent cerebral infarct transfusion (SIT) trial. Pediatric Hematology & Oncology 2010;27(2):69‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravorty S, Williams TN. Sickle cell disease: a neglected chronic disease of increasing global health importance. Archives of Disease in Childhood 2015;100(1):48‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. New England Journal of Medicine 1995;332(20):1317‐22. [DOI] [PubMed] [Google Scholar]
- Chou ST. Transfusion therapy for sickle cell disease: a balancing act. Hematology 2013;2013:439‐46. [DOI] [PubMed] [Google Scholar]
- Chou ST, Jackson T, Vege S, Smith‐Whitley K, Friedman DF, Whesthoff CM. High prevalence of red blood cell alloimmunization in sickle cell disease despite transfusion from Rh‐matched minority donors. Blood 2013;122(6):1062‐71. [DOI] [PubMed] [Google Scholar]
- Veritas Health Innovation. Covidence. Version accessed prior to 28 November 2016. Melbourne, Australia: Veritas Health Innovation, 2015.
- DeBaun MR, Derdeyn CP, McKinstry RC 3rd. Etiology of strokes in children with sickle cell anemia. Mental Retardation & Developmental Disabilities Research Reviews 2006;12(3):192‐9. [DOI] [PubMed] [Google Scholar]
- DeBaun MR, Armstrong FD, McKinstry RC, Ware RE, Vichinsky E, Kirkham FJ. Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood 2012;119(20):4587‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBaun MR, Gordon M, McKinstry RC, Noetzel MJ, White DA, Sarnaik SA, et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. New England Journal of Medicine 2014;371(8):699‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBaun MR, Kirkham FJ. Central nervous system complications and management in sickle cell disease. Blood 2016;127(7):829‐38. [DOI] [PubMed] [Google Scholar]
- DeBaun MR, King AA. Prevention of central nervous system sequelae in sickle cell disease without evidence from randomized controlled trials:the case for a team‐based learning collaborative. Hematology American Society of Hematology Education Program 2016;2016(1):632‐639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks JJ, Higgins JPT, Altman DG on behalf of the Cochrane Statistical Methods Group. Chapter 9: Analysing data and undertaking meta‐analysis. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
- Dowling MM, Quinn CT, Plumb P, Rogers ZR, Rollins NK, Koral K, et al. Acute silent cerebral ischemia and infarction during acute anemia in children with and without sickle cell disease. Blood 2012;120(19):3891‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field JJ, Nathan DG. Advances in sickle cell therapies in the hydroxyurea era. Molecular Medicine 2014;20 Suppl 1:S37‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenette PS, Atweh GF. Sickle cell disease: old discoveries, new concepts, and future promise. Journal of Clinical Investigation 2007;117(4):850‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravitz L, Pincock S. Sickle‐cell disease. Nature 2014;515(7526):S1. [DOI] [PubMed] [Google Scholar]
- Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa. A neglected cause of early childhood mortality. American Journal of Preventive Medicine 2011;41(6 Suppl 4):S398‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams KP, Fields ME, Hulbert ML. Higher‐than‐expected prevalence of silent cerebral infarcts in children with hemoglobin SC disease. Blood 2015;125(2):416‐7. [DOI] [PubMed] [Google Scholar]
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration 2011. Available from handbook.cochrane.org.
- Higgins JPT, Deeks JJ, editor(s). Chapter 7: Selecting studies and collecting data. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
- Higgins JPT, Altman DG, Sterne JAC, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
- Higgins JPT, Deeks JJ, Altman DG, editor(s). Chapter 16: Special topics in statistics. In: Higgins JPT, Green S, editor(s). Cochrane Handbook of Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
- Howard J. Transfusion therapy in sickle cell disease. 25th Regional Congress of the International Society of Blood Transfusion in Conjunction with the 33rd Annual Conference of the British Blood Transfusion Society; 2015 Jun 27 ‐ Jul 01; London, United Kingdom. 2015. [Google Scholar]
- Kato GJ, McGowan V, Machado RF, Little JA, Taylor J, Morris CR, et al. Lactate dehydrogenase as a biomarker of hemolysis‐associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood 2006;107(6):2279‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato GJ, Hsieh M, Machado R, Taylor J 6th, Little J, Butman JA, et al. Cerebrovascular disease associated with sickle cell pulmonary hypertension. American Journal of Hematology 2006;81(7):503‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski JL, Zimmerman RA, Pollock AN, Seto W, Smith‐Whitley K, Shults J, et al. Silent infarcts in young children with sickle cell disease. British Journal of Haematology 2009;146(3):300‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MT, Piomelli S, Granger S, Miller ST, Harkness S, Brambilla DJ, et al. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow‐up and final results. Blood 2006;108(3):847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org. The Cochrane Collaboration..
- Leikin SL, Gallagher D, Kinney TR, Sloane D, Klug P, Rida W. Mortality in children and adolescents with sickle cell disease. Cooperative Study of Sickle Cell Disease. Pediatrics 1989;84(3):500‐8. [PubMed] [Google Scholar]
- Marouf R, Gupta R, Haider MZ, Adekile AD. Silent brain infarcts in adult Kuwaiti sickle cell disease patients. American Journal of Hematology 2003;73(4):240‐3. [DOI] [PubMed] [Google Scholar]
- Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta‐Analyses: the PRISMA Statement. Annals of Internal Medicine 2009;151(4):264‐9. [DOI] [PubMed] [Google Scholar]
- National Institute for Health and Care Excellence (NICE). Sickle cell disease. http://cks.nice.org.uk/sickle‐cell‐disease. UK: NICE, 2010; Vol. (accessed 10 April 2016).
- Ohene‐Frempong K. Sickle cell disease in the United States of America and Africa. Hematology (American Society of Hematology Education Program). American Society of Hematology, 1999:64‐72. [Google Scholar]
- Oringanje C, Nemecek E, Oniyangi O. Hematopoietic stem cell transplantation for people with sickle cell disease. Cochrane Database of Systematic Reviews 2016, Issue 5. [DOI: 10.1002/14651858.CD007001.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815‐34. [DOI] [PubMed] [Google Scholar]
- Pegelow CH, Wang W, Granger S, Hsu LL, Vichinsky E, Moser FG, et al. Silent infarcts in children with sickle cell anemia and abnormal cerebral artery velocity. Archives of Neurology 2001;58(12):2017‐21. [DOI] [PubMed] [Google Scholar]
- Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model‐based map and population estimates. Lancet 2012;381(9861):142‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasants S. Epidemiology: a moving target. Nature 2014;515(7526):S2‐3. [DOI] [PubMed] [Google Scholar]
- Porter J, Garbowski M. Consequences and management of iron overload in sickle cell disease. ASH Education Program Book 2013;1:447‐56. [DOI] [PubMed] [Google Scholar]
- Pule GD, Mowla S, Novitzky N, Wiysonge CS, Wonkam A. A systematic review of known mechanisms of hydroxyurea‐induced fetal hemoglobin for treatment of sickle cell disease. Expert Review of Hematology 2015;8(5):669‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DC, Williams TN, Gladwin MT. Sickle‐cell disease. Lancet 2010;376(9757):2018‐31. [DOI] [PubMed] [Google Scholar]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.1. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
- Rumaney MB, Ngo Bitoungui VJ, Vorster AA, Ramesar R, Kengne AP, Ngogang J, et al. The co‐inheritance of alpha‐thalassemia and sickle cell anemia Is associated with better hematological indices and lower consultations rate in Cameroonian patients and could improve their survival. PLoS ONE 2014;9:e100516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheunemann LP, Ataga KI. Delayed hemolytic transfusion reaction in sickle cell disease. American Journal of the Medical Sciences 2010;339(3):266‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and ‘Summary of findings’ tables. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
- Sparkenbaugh E, Pawlinski R. Interplay between coagulation and vascular inflammation in sickle cell disease. British Journal of Haematology 2013;162(1):3‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. American Journal of Hematology 2012;87(8):795‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterne JAC, Egger M, Moher D, editor(s). Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time‐to‐event data into meta‐analysis. Trials2007; Vol. 8:16. [DOI] [PMC free article] [PubMed]
- Ubesie A, Emodi I, Ikefuna A, Ilechukwu G, Ilechukwu G. Prevalence of human immunodeficiency virus transmission among transfused children with sickle cell anemia in Enugu Nigeria. Annals of Medical and Health Sciences Research 2012;2(2):109‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land V, Mutsaerts HJ, Engelen M, Heijboer H, Roest M, Hollestelle MJ, et al. Risk factor analysis of cerebral white matter hyperintensities in children with sickle cell disease. British Journal of Haematology 2016;172(2):274‐84. [DOI] [PubMed] [Google Scholar]
- Venketasubramanian N, Prohovnik I, Hurlet A, Mohr JP, Piomelli S. Middle cerebral artery velocity changes during transfusion in sickle cell anemia. Stroke 1994;25(11):2153‐8. [DOI] [PubMed] [Google Scholar]
- Walters MC, Hardy K, Edwards S, Adamkiewicz T, Barkovich J, Bernaudin F, et al. Pulmonary, gonadal, and central nervous system status after bone marrow transplantation for sickle cell disease. Biology of Blood and Marrow Transplantation 2010;16(2):263‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WC, Ware RE, Miller ST, Iyer RV, Casella JF, Minniti CP, et al. Hydroxycarbamide in very young children with sickle‐cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011;377(9778):1663‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]