

Summary
Capillary Electrophoresis is a powerful methodology for quantification and structural characterization of highly anionic polysaccharides. Separation of saccharides under conditions of electrophoretic flow, typically achieved under low pH1,2, are charge-based. Resolution of components is often superior to flow-based techniques, such as liquid chromatography. During the heparin contamination crisis, capillary electrophoresis was one of the key methodologies used to identify whether or not heparin lots were contaminated3. Here we describe a method for isolation of sulfated heparin/heparan sulfate saccharides from urine, their digestion by deployment of heparinase enzymes4, resolution of species through use of orthogonal digestions, and analysis of the resulting disaccharides by capillary electrophoresis.
Keywords: heparin, heparan sulfate, pentosan polysulfate, capillary electrophoresis, heparinase, Alcian Blue
1. Introduction
Sulfated polysaccharides have been shown to be potentially important therapies for a number of diseases, including thrombosis5, cancer6, and interstitial cystitis7. However, while pharmacodynamics markers are available for some indications (such as anti-Xa activity), a systematic understanding of the pharmacology of complex polysaccharides, particularly highly sulfated sugars, such as heparin, pentosan polysulfate, and heparin derivatives are necessary for understanding tissue distribution, pharmacokinetics, and elimination pathways. In addition, a convergence of –omics-based analysis of biological samples (a bottom-up approach) with a structure-based analysis of protein-glycan interactions (a top-down approach)8 is enabling the more specific description of these drugs and enabling the creation of optimized therapeutic modalities for a range of diseases, including anti-virals9, anti-inflammatories10, and anti-thrombotics11.
Most recently, techniques such as capillary electrophoresis12, nuclear magnetic reasonance13, and mass spectrometry2,14 have been applied to the structural analysis of anionic complex polysaccharides, including to define structure-activity relationships, for comparison of products, and for the detection of contaminants, such as oversulfated chondroitin sulfate15 (Figure 1). Implementation of these methods is critical for understanding the biological roles of complex polysaccharides and elucidating new uses for them in a range of diseases. Additionally, these methods enable development of true assessments of pharmacokinetics since the composition of complex polysaccharides, such as heparin, is known to be altered after in vivo administration because of differential filtration in the kidney16.
Figure 1. Disaccharide units of representative complex polysaccharides.
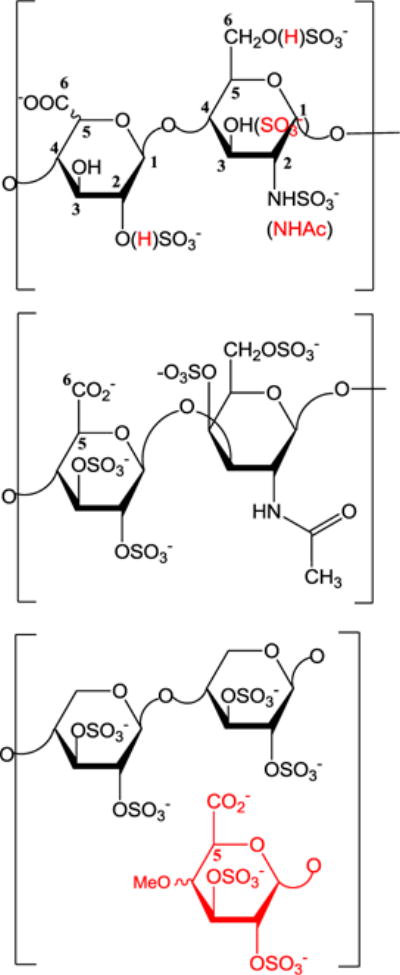
(Top) Disaccharide unit of heparin/heparan sulfate. Differential O-sulfonation and the presence of either N-acetylation or sulfation yields significant structural diversity. (Middle) Oversulfated chondroitin sulfate (OSCS). OSCS, a contaminant in heparin contains primarily a tetrasulfated disaccharide unit. (Bottom) Pentosan polysulfate is a 1→4 linked β-D-xylopyranose with laterally substituted 4-methylglucopyranosyluronic acid units glycosidically linked to the 2 position of the main chain at every 10th xylopyranose unit on average.
The current method employs isolation of heparin/heparin sulfate from urine, quantification of the amount of material by Alcian Blue, digestion of the material into its components by the use of heparinases and analysis by capillary zone electrophoresis in an uncoated fused silica capillary. Under conditions of low pH, separation is dictated by analyte electrophoretic mobility almost exclusively1,2. Due to the fact that all saccharide components have a net negative charge, due to the presence of carboxylate and sulfate moieties, separation is conducted under reverse polarity. In addition, supplementation of the low pH (pH2.5) buffer with dextran sulfate and tris prevents non-specific adsorption of analytes, enabling symmetrical peaks shapes and accurate quantification.
The net result is that separation under these conditions in capillary electrophoresis is orthogonal to more traditional separation techniques, such as HPLC. In capillary electrophoresis, the most highly sulfated species migrate through the capillary the fastest and are detected first. While many reports have indicated that this technology enables the resolution of the basic saccharide structures in heparin and related materials, there are a number of considerations:
Due to the potential dynamic range of the components in the sample (i.e. 50% of the sample for the principal component compared to 0.1% for some of the minor components), one needs to take care with regards to the amount of sample that is placed on the capillary to ensure quantitative results;
There is little/no resolution of α and β anomers in CE;
To accurately separate and quantify constituent saccharides requires the implementation of distinct digests, increasing the potential for the introduction of error. The introduction of an internal standard is essential.
Due to the presence of multiple isomeric disaccharide units within acidic polysaccharides, the resolving power of CE can be amplified through the use of polysaccharide lyases, such as the heparinases. Digestion of heparin-based materials, followed by CE analysis, enables exact quantification, compositional analysis, and determination of the presence of contaminants17, even if highly related to heparin (Figure 1). Taken together, capillary electrophoresis is an important approach to the identification and analysis of complex, anionic polysaccharides.
2. Materials
Note 1: Prepare all solutions using ultrapure water and analytical grade reagents. Prepare and store all reagents at room temperature, unless otherwise indicated.
2.1 Isolation of HS From Urine
10× Benzonase buffer (500mM Tris-Cl [8.0], 200mM NaCl, 20mM MgCl2)
Benzonase (25u/μl)
Bovine kidney heparan sulfate [Note 2: Standard selected should be as close as possible in composition to that of samples]
Proteinase K
0.1 M CaCl2 (10× dilution in ultrapure water of 1M solution)
Vivapure Q mini H columns [Note 3: Column is from Santarus; equivalent materials are available and should be strong anion exchange]
50mM Ammonium Acetate: Dissolve 385.4mg dissolved in 90ml of ultrapure water. Q.S. to 100 mL
2M NaCl:11.66g dissolved in 75ml of ultrapure water. Adjust volume to 100ml.
Vivaspin 5000 (MWCO 3000)
Speedvac
2.2 Alcian Blue Dot Blot to Quantify Yield
Nitrocellulose membrane (0.45 μM pore size; 8×11.5 cm)
Easy Titer ELIFA System
CTAB solution (1% (w/v) hexadecyltrimethylammonium bromide): 30% (v/v) 1-propanol. Add 30mL 1-propanol to 70 mL of ultrapure water and mix well. To 50 mL of 30% 1-propanol add 500 mg of CTAB, mix well with a stir bar.
Wash buffer (50mM Tris-Acetate/ 150 mM NaCl, pH 7.4)- 10ml of 0.5M Tris-acetate stock (6.06g Tris-Base dissolved in 80ml of water, pH adjusted to 7.4 with acetic acid and volume adjusted to 100ml with ultrapure water) + 7.5ml of 2M NaCl + 82.5ml of ultrapure water) for a total of 100ml.
8M Guanidine Chloride. To make an 8M solution in water, heat the solution to 35°C for approximately 30 minutes. Q.S. to 100 mL with ultrapure water.
18 mM Sulfuric acid/ 0.25% Triton X100- Dissolve 2.5ml of 10% Triton X-100 in 100ml of ultrapure water. Add 0.1ml of Concentrated sulphuric acid.
Staining solution: 5 ml Alcian Blue stock + 5 ml 8M Guanidine Chloride + 90 ml 18 mM Sulfuric acid/ 0.25% Triton X100. This solution should be prepared fresh for every use.
2.3 Digestion of Heparin/Heparan Sulfate by Heparinase Cocktail
1. Heparinase I (4.2.2.7), Heparinase II(4.2.2.7), and Heparinase III(4.2.2.8) from Pedobacter heparinus are purified as recombinant enzymes from E. coli18–20. [Note 4: The three heparinases have overlapping substrate specificities. Employing all three enzymes results in cleavage of 90- >99% of the glycosidic bonds of heparin/heparan sulfate. The enzymes are lyases, meaning cleavage results in the formation of a Δ4,5 double bond that is UV-absorbing [λmax= 232 nm].
2. 2-O sulfatase and Δ4,5 glycuronidase from Pedobacter heparinus are purified as recombinant enzymes from E. coli21,22. [Note 5: Δ4,5 glycuronidase cleaves only Δ4,5 double bonds that do not contain a 2-O sulfate.]
3. Enzyme Cocktail Buffer 10× stock (250 mM NaOAc, 10 mM CaOAc2). 2g of anhydrous sodium acetate and 158.16mg Calcium acetate (anhydrous) are dissolved in 80ml water. The pH is adjusted to 7.0 with 1N acetic acid. Bring up volume to 100ml with water.
2.4 Analysis by Capillary Electrophoresis
Agilent 3D Capillary Electrophoresis or equivalent.
Extended light path capillary, 75 μm I.D., 72 cm
Water, CE grade
1N Sodium hydroxide, CE grade
Phosphoric acid, CE grade
Tris Base
Dextran sulfate
Heparin disaccharide standards. [Note 6: These are commercially available or can be isolated from heparin or chemically synthesized]
4. Methods
4.1 HS isolation from urine
[Note 6: An optional pre-step is to heat inactivate the sample through incubation at 52°C for 1.5 hrs. This inactivates many viruses, if present. HS isolation should occur in a BSL-2 hood, regardless].
Nuclease treatment: To 360 μl of urine sample add 40μl of 10× Benzonase buffer (see materials)
Incubate Overnight at 37°C. In parallel set up a control reaction with 50ng of bovine kidney HS in PBS in place of sample as positive control.
Protease treatment: To the above samples add 20 μl proteinase K (20mg/ml) and 20 μl CaCl2 (0.1M).
Incubate at 55°C for 2hrs. [Note 7: Precipitation may be observed in the sample. If this is the case, spinning of the sample to remove precipitate is warranted].
After digestion, enrichment of HS is completed using ion exchange spin columns. Equilibrate Vivapure Q mini H columns with 200 μl of 50mM Ammonium Acetate. Spin at 2000× g for 2 min. Load samples (approximately 450 μl) onto the column and spin at 2000 × g for 2min. Wash columns with 200 μl 50mM Ammonium Acetate, three times, each time spinning at 2000 × g for 2 min. and discarding the flow-through. Elute bound HS with 200 μl of 2M NaCl. [Note 8: An optional step is to do a second 2M elution to ensure all HS material is collected. In our experience, this is not required. Also, addition of urea at after step 4 can help ensure that HS bound to proteins is released].
Desalting: Vivaspin 5000 (MWCO 3000) filters are rinsed with 500 μl water 3× (each time accompanied by a spin at 15000×g 20min, discarding the flow-through). The crude HS prep is loaded and spun at 13,500×g. Add 500 μl of 50mM Ammonium Acetate and spin at 15000×g for approximately 35 min. repeat the step 5 times.
Collect the retentate after each spin (× 5) in a separate eppendorf tube, do not pool the ammonium acetate elutions.
Speedvac dry each collection. [Note 8: Visible material should be white to off white].
Resuspend pellet, if present, in 100 μl of 50mM Ammonium Acetate.
Sample and Standard formulation: Prepare purified HS standards (45, 22.5, 11.25, 5.62, 2.81, 1.4, 0.7, 0 μg/ml) in 50mM Ammonium Acetate. After isolation of HS material from urine, prepare duplicates of three dilutions for each sample: neat, 1:3, and 1:10. Dilution should be made using ultrapure water. Total volume for each should be 250–300 μL (enough to comfortably transfer 100 μL for sample/dot analysis). Load samples in duplicates. Prepare standards of heparin via serial dilution of stock (4.5 mg/mL): 45, 22.5, 11.25, 5.62, 2.81, 1.4, 0.7, and 0 μg/ml. Dilutions should be made using ultrapure water. Total volume for each should be 250–300 μL (enough to comfortably transfer 100 μL for sample/dot analysis). Load standards in duplicates.
4.2 DOT BLOT Assay to Quantify HS
Activate Nitrocellulose membrane (0.4μ) filter via addition of CTAB buffer for 5 min. Add enough solution to cover the membrane.
Wash 3× with wash buffer (50mM Tris-acetate/150mM NaCl, pH 7.4). For each wash, allow the membrane to soak for 15min.
Mount activated membrane in the Easy Titer ELIFA system. Transfer 100μl sample or std to blot. Apply vacuum until membrane sample completely passes through. Remove membrane from ELIFA system and stain with alcian blue solution (see materials). Allow the membrane to stand for 15–60min on a shaker. De-stain membrane in wash buffer (3× 5–10min per wash/shaker). Dry membrane, scan and determine intensity of staining (Figure 2). [Note 9: Quantification is by number of sulfate groups. Thus, to ensure as accurate of quantification as possible requires the use of a standard with a similar sulfate-to-carboxylate ratio].
Figure 2.
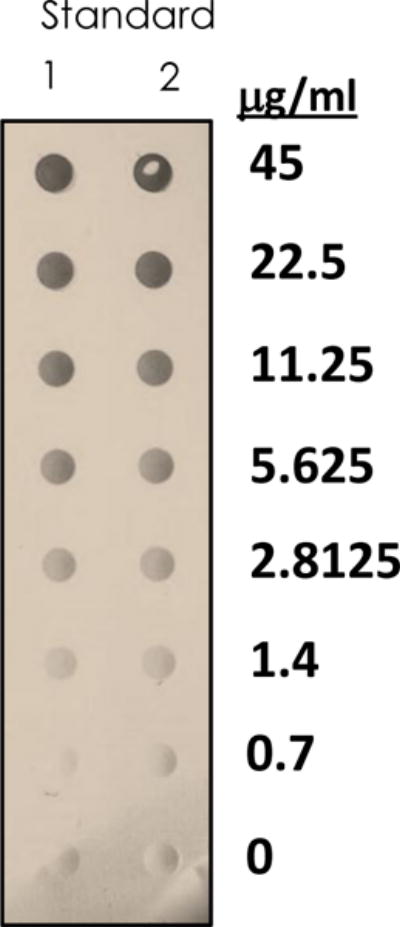
Representative dot blot for the quantification of acidic polysaccharides isolated from urine. Standards run simultaneously enable the creation of a standard curve (via densitometry), allowing for a (semi-)quantitative estimate of the amount of material isolated.
4.3 Heparinase digestion
Note 11: Heparinase enzymes used here are His-Tagged for easy removal. Enzymes can be left in the reaction mixture but may affect baseline on CE analysis.]
- Enzyme Cocktail preparation: Mix Hep I (approximately 800 mU), Hep II (approximately 800 mU), and Hep III (approximately 600 mU)with 10× cocktail buffer (250 mM NaOAc, 10 mM CaOAc2) to a total volume of 50 μL. [Note11: An example calculation is as follows –
- 4.4 μL Hep I (1.2 mg/mL, specific activity 154.6 μmoles/min*mg)
- 9.2 μL Hep II (5.6/mL, specific activity 7.8 μmoles/min*mg)
- 1.9 μL Hep III (3.5 mg/mL, specific activity 88 μmoles/min*mg)
- 25 μL of EC buffer
- 9.5 μL of ultrapure water (Vt= 50μL)]
Digestion: The digestion reaction is setup in a total volume of 60 μl containing the following - 3 μL of enzyme cocktail (Table 1), 6 μL of 10× cocktail buffer, 6 μL of 10 mg/ml heparin/heparan sulfate sample, and 45 μL of ultrapure water mixed in a 1.5 mL centrifuge tube. The centrifuge tube should be closed tightly and incubated at 30°C for16 hours.
An aliquot of the digested material is set aside, while the rest of the digest undergo subsequent enzymatic treatment with either 2-O-S, or Δ4,5 glycuronidase (3μL, Table 1). The setup for this enzymatic modification is as follows- 17 μL of the digest from step 2 is added to each of three reactions:
Incubate each reaction for 6 hours at 30°C.
Nickle spin column cleanup: Digested material is prepared for CE analysis by passing through a nickel spin column (Qiagen) equilibrated with water. In brief, add 600μL of water to nickel spin column. Spin at 2000 rpm for 2 min. Replace flow through tube (make sure that no residual water is present). Add sample to the column. Spin at 2000 rpm for 2min to elute. His-tagged enzyme should bind to the column and be removed.
Table 1.
Orthogonal Digestions
| Digest | 2-O Sulfatase | Δ4,5 Glycuronidase | Water |
|---|---|---|---|
| 1 | – | – | 3 |
| 2 | 3 | – | – |
| 3 | – | 3 | – |
3.3 Capillary Electrophoresis
-
1
All analyses are performed on an Agilent 3D CE system or equivalent equipped with a photodiode array detector.
-
2
Electrophoretic separations are completed using an uncoated fused-silica capillary with a length of 72 cm (75 μm diameter), at 25°C.
-
3
Prior to daily analysis, wash capillary with ultrapure water for 15 minutes.
-
4
Ensure removal of residual material on the capillary (to ensure smooth electrophoretic flow) by washing capillary with 0.1 N NaOH. Remove NaOH via washing with water.
-
5
Wash capillary with running buffer (50 mM Tris, 10 μM dextran sulfate (10,000 MW) at pH 2.5) for 15 minutes.
[Note 12: If a large (>10) number of samples are being run, steps 3–5 should be repeated to ensure removal of residual material adsorbed to the capillary surface. Failure to do so will interfere with the electrophoretic separation].
-
6
The sample for analysis is injected at a pressure of 20 mbar for 30 seconds. [Note 13: Pressure injection ensures that all digested material, regardless of charge, enters the capillary].
-
7
Separations are carried out at 30 kV in negative polarity by applying the sample at the cathode. Detection is conducted by monitoring absorbance at 232 nm. Digestion with the heparinases results in the formation of a UV-detectable, Δ4,5 double bond (digest 1, Figure 3). Treatment with the 2-O sulfatase results in a shift to longer migration times of those species that contain a 2-O sulfate21 (digest 2, Figure 3). Treatment with the a Δ4,5 glycuronidase results in the disappearance of species that do not contain a 2-O sulfate22 (digest 3).
-
8
Detection and quantification of the individual species is completed by injection of standards and through integration of the area under the curve. The general migration time for disaccharide species is trisulfated<disulfated<monosulfated<non-sulfated. In addition, sulfate positioning affects migration with 2-O-sulfate<N-sulfate<6-O-sulfate. Finally tetrasaccharide species tend to migrate between the trisulfated disaccharide and disulfated disaccharides.
Figure 3.
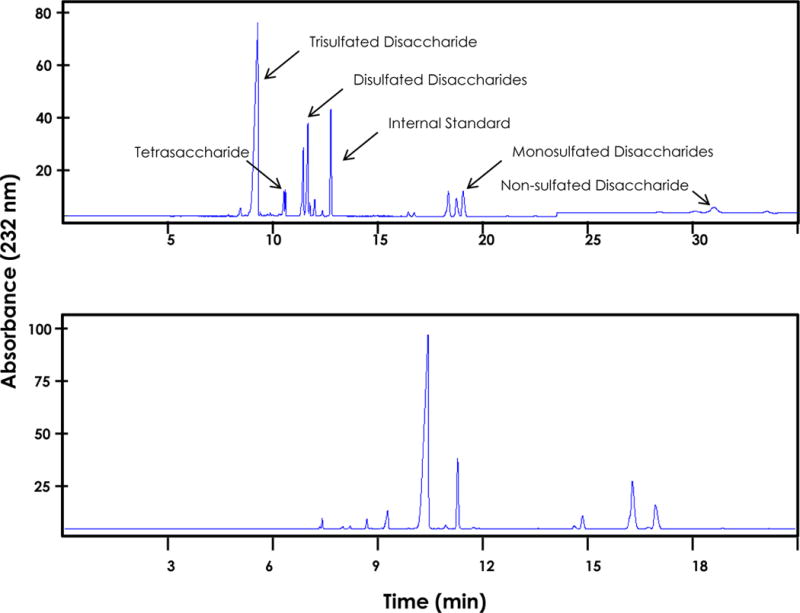
Representative electropherograms of disaccharide analysis of heparin by capillary electrophoresis. (Top) Digestion with the three heparinases yields primarily trisulfated disaccharide with decreasing amounts of other disaccharide products. A tetrasaccharide, containing a 3-O sulfate, is observed between the major disaccharide products. (Bottom) Digestion with the 2-O sulfatase moves the major disaccharide peak to a longer migration time, enabling the resolution of minor products.
[Note 14: Tetrasaccharide species typically migrate between the trisulfated disaccharide and the disulfated disaccharides].
References
- 1.Ampofo SA, Wang HM, Linhardt RJ. Disaccharide compositional analysis of heparin and heparan sulfate using capillary zone electrophoresis. Analytical biochemistry. 1991;199:249–255. doi: 10.1016/0003-2697(91)90098-e. [DOI] [PubMed] [Google Scholar]
- 2.Rhomberg AJ, Ernst S, Sasisekharan R, Biemann K. Mass spectrometric and capillary electrophoretic investigation of the enzymatic degradation of heparin-like glycosaminoglycans. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:4176–4181. doi: 10.1073/pnas.95.8.4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guerrini M, et al. Oversulfated chondroitin sulfate is a contaminant in heparin associated with adverse clinical events. Nature biotechnology. 2008;26:669–675. doi: 10.1038/nbt1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ernst S, Langer R, Cooney CL, Sasisekharan R. Enzymatic degradation of glycosaminoglycans. Critical reviews in biochemistry and molecular biology. 1995;30:387–444. doi: 10.3109/10409239509083490. [DOI] [PubMed] [Google Scholar]
- 5.Petitou M, et al. Synthesis of thrombin-inhibiting heparin mimetics without side effects. Nature. 1999;398:417–422. doi: 10.1038/18877. [DOI] [PubMed] [Google Scholar]
- 6.Zacharski LR, Ornstein DL. Heparin and cancer. Thrombosis and haemostasis. 1998;80:10–23. [PubMed] [Google Scholar]
- 7.Anderson VR, Perry CM. Pentosan polysulfate: a review of its use in the relief of bladder pain or discomfort in interstitial cystitis. Drugs. 2006;66:821–835. doi: 10.2165/00003495-200666060-00006. [DOI] [PubMed] [Google Scholar]
- 8.Robinson LN, et al. Harnessing glycomics technologies: integrating structure with function for glycan characterization. Electrophoresis. 2012;33:797–814. doi: 10.1002/elps.201100231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shukla D, et al. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999;99:13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 10.Nelson RM, et al. Heparin oligosaccharides bind L- and P-selectin and inhibit acute inflammation. Blood. 1993;82:3253–3258. [PubMed] [Google Scholar]
- 11.Sundaram M, et al. Rational design of low-molecular weight heparins with improved in vivo activity. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:651–656. doi: 10.1073/pnas.252643299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schirm B, Benend H, Watzig H. Improvements in pentosan polysulfate sodium quality assurance using fingerprint electropherograms. Electrophoresis. 2001;22:1150–1162. doi: 10.1002/1522-2683()22:6<1150::AID-ELPS1150>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 13.Yates EA, et al. 1H and 13C NMR spectral assignments of the major sequences of twelve systematically modified heparin derivatives. Carbohydrate research. 1996;294:15–27. doi: 10.1016/s0008-6215(96)90611-4. [DOI] [PubMed] [Google Scholar]
- 14.Kuberan B, et al. Analysis of heparan sulfate oligosaccharides with ion pair-reverse phase capillary high performance liquid chromatography-microelectrospray ionization time-of-flight mass spectrometry. Journal of the American Chemical Society. 2002;124:8707–8718. doi: 10.1021/ja0178867. [DOI] [PubMed] [Google Scholar]
- 15.Nemes P, Hoover WJ, Keire DA. High-throughput differentiation of heparin from other glycosaminoglycans by pyrolysis mass spectrometry. Analytical chemistry. 2013;85:7405–7412. doi: 10.1021/ac401318q. [DOI] [PubMed] [Google Scholar]
- 16.Kandrotas RJ. Heparin pharmacokinetics and pharmacodynamics. Clinical pharmacokinetics. 1992;22:359–374. doi: 10.2165/00003088-199222050-00003. [DOI] [PubMed] [Google Scholar]
- 17.Aich U, Shriver Z, Tharakaraman K, Raman R, Sasisekharan R. Competitive inhibition of heparinase by persulfonated glycosaminoglycans: a tool to detect heparin contamination. Analytical chemistry. 2011;83:7815–7822. doi: 10.1021/ac201498a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Godavarti R, et al. Heparinase III from Flavobacterium heparinum: cloning and recombinant expression in Escherichia coli. Biochemical and biophysical research communications. 1996;225:751–758. doi: 10.1006/bbrc.1996.1246. [DOI] [PubMed] [Google Scholar]
- 19.Ernst S, et al. Expression in Escherichia coli, purification and characterization of heparinase I from Flavobacterium heparinum. The Biochemical journal. 1996;315(Pt 2):589–597. doi: 10.1042/bj3150589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sasisekharan R, Bulmer M, Moremen KW, Cooney CL, Langer R. Cloning and expression of heparinase I gene from Flavobacterium heparinum. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:3660–3664. doi: 10.1073/pnas.90.8.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myette JR, et al. The heparin/heparan sulfate 2-O-sulfatase from Flavobacterium heparinum. Molecular cloning, recombinant expression, and biochemical characterization. The Journal of biological chemistry. 2003;278:12157–12166. doi: 10.1074/jbc.M211420200. [DOI] [PubMed] [Google Scholar]
- 22.Myette JR, et al. Molecular cloning of the heparin/heparan sulfate delta 4,5 unsaturated glycuronidase from Flavobacterium heparinum, its recombinant expression in Escherichia coli, and biochemical determination of its unique substrate specificity. Biochemistry. 2002;41:7424–7434. doi: 10.1021/bi012147o. [DOI] [PubMed] [Google Scholar]