

Abstract
Tyrosine kinase inhibitors are promising for the treatment of severe pulmonary hypertension. Their therapeutic effects are postulated to be due to inhibition of cell growth–related kinases and attenuation of vascular remodeling. Their potential vasodilatory activities have not been explored. Vasorelaxant effects of the tyrosine kinase inhibitors imatinib, sorafenib, and nilotinib were examined in isolated pulmonary arterial rings from normal and pulmonary hypertensive rats. Phosphorylation of myosin light chain phosphatase and myosin light chain was assessed by Western blots. Acute hemodynamic effects of imatinib were tested in the pulmonary hypertensive rats. In normal pulmonary arteries, imatinib reversed serotonin- and U46619-induced contractions in a concentration-dependent and endothelium-independent manner. Sorafenib and nilotinib relaxed U46619-induced contraction. Imatinib inhibited activation of myosin phosphatase induced by U46619 in normal pulmonary arteries. All three tyrosine kinase inhibitors concentration-dependently and completely reversed the spontaneous contraction of hypertensive pulmonary arterial rings unmasked by inhibition of nitric oxide synthase. Acute intravenous administration of imatinib reduced high right ventricular systolic pressure in pulmonary hypertensive rats, with little effect on left ventricular systolic pressure and cardiac output. We conclude that tyrosine kinase inhibitors have potent pulmonary vasodilatory activity, which could contribute to their long-term beneficial effect against pulmonary hypertension. Vascular smooth muscle relaxation mediated via activation of myosin light chain phosphatase (Ca2+ desensitization) appears to play a role in the imatinib-induced pulmonary vasodilation.
Keywords: tyrosine kinase inhibitors, pulmonary hypertension, vasodilation, SU5416, Ca2+ sensitization
Clinical Relevance
Tyrosine kinase inhibitors have been reported to be promising for the treatment of severe pulmonary hypertension as antivascular remodeling agents. Nothing, however, has been considered regarding their potential vasodilatory activities. Results of this study demonstrate that tyrosine kinase inhibitors have potent pulmonary vasodilatory activity, which could contribute to their long-term beneficial effect against pulmonary hypertension.
Pulmonary hypertension (PH) is characterized by progressive narrowing of small pulmonary arteries and arterioles, which results in increased pulmonary vascular resistance and right ventricular pressure overload. Despite recent advances in treatment severe PH remains debilitating and fatal (1). Major factors that contribute to the complex pathogenesis of pulmonary arterial narrowing are sustained vasoconstriction and fixed vascular remodeling.
Tyrosine kinase inhibitors (TKIs), such as imatinib, nilotinib, and sorafenib, are approved for the treatment of patients with malignant diseases (2, 3). Imatinib, an inhibitor of platelet-derived growth factor receptor (PDGFR), has also been reported as promising in the treatment of patients with severe PH (4). In addition, chronic treatment with imatinib or the tyrosine/serine/threonine kinase inhibitor sorafenib attenuates PH in rodent models (5–7). The therapeutic effects of these TKIs against PH are attributed to inhibition of cell growth–related factors and subsequent suppression of vascular remodeling (5, 7). It is possible, however, that TKIs also induce vascular smooth muscle cell relaxation and vasodilation because cell growth–related kinases are generally involved in cell contraction. For example, PDGFR and epidermal growth factor receptor (EGFR) are known to mediate smooth muscle cell contraction, and inhibition of these growth factor receptors induces vasodilation (8–11). In agreement with this idea, the onset of right ventricular systolic pressure lowering by chronic treatment with imatinib against established monocrotaline-induced PH was rather rapid (within 2 d) in a report by Schermuly and colleagues (7), suggesting its beneficial effects may not be explained solely by the regression of fixed vascular remodeling.
We therefore hypothesized that TKIs would have pulmonary vasodilatory activity and would effectively lower the high pulmonary vascular resistance in a rat model of severe PH. To test this hypothesis, we examined whether: (1) TKIs relaxed precontracted normotensive and hypertensive rat pulmonary arterial rings and (2) acute intravenous administration of imatinib reduced high pulmonary vascular resistance and pressure in pulmonary hypertensive rats. We used the vascular endothelial growth factor receptor blockade (by Sugen[SU]5416) plus a hypoxia/normoxia-exposed severe PH rat model, which more closely mimics human severe PH histologically and hemodynamically than the conventional chronically hypoxic and monocrotaline-injected models (12, 13). We also examined whether imatinib-induced vasodilation involved Ca2+ desensitization because recent studies suggest that Ca2+ sensitization (smooth muscle cell contraction mediated via inhibition of myosin light chain phosphatase [MLCP]) plays an essential role in the sustained phase of various agonist-induced vascular smooth muscle contractions (14, 15) and in the sustained vasoconstriction of PH (16, 17).
Methods
Animals
All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of South Alabama.
Two groups of male Sprague-Dawley rats (180–220 g) were used; one group was normal (untreated), and the other was injected subcutaneously with SU5416 (20 mg/kg) and exposed to hypoxia (10% O2) for 3 weeks. The rats were then returned to normoxia for an additional 2 weeks (SU5416/hypoxia/normoxia-exposed PH rats) (12, 13).
Experimental Protocols
Isometric tension study.
Twenty minutes after the addition of U46619 (thromboxane A2 analog; 100 nM) or serotonin (10 μM), concentration-response curves to imatinib (1–30 μM) were determined in endothelium-intact and endothelium-denuded pulmonary arterial rings from normal rats. Serotonin and the thromboxane analog were used because they are involved in the pathogenesis of various experimental and human forms of PH.
Concentration-response curves to imatinib, nilotinib, and sorafenib were constructed in endothelium-intact pulmonary arterial rings from normal and SU5416/hypoxia/normoxia-exposed rats. Because imatinib caused an endothelium-independent relaxation and because our preliminary results indicate that this is also the case for the other TKI inhibitors, the following experiments were performed under nitric oxide synthase-inhibited status. All rings received 200 μM of Nω-nitro-l-arginine (l-NNA) after 60 minutes of equilibration. l-NNA induced marked contractions in hypertensive but not normotensive pulmonary arteries (see Fig. E1 in the online supplement) (18). Concentration-response curves to TKIs (0.1–100 μM) were determined in normotensive pulmonary arterial rings precontracted by U46619 (100 nM) and in hypertensive pulmonary arterial rings precontracted by l-NNA.
Catheterized rats.
Acute hemodynamic effects of imatinib were tested in anesthetized, catheterized, SU5416/hypoxia/normoxia-exposed rats. After the baseline measurements, imatinib (20 or 50 mg/kg, total volume of 0.1 ml) was injected intravenously, and hemodynamic parameters were monitored for 10 minutes (the effect of imatinib reached a plateau within 5 min). In some cases, 5 mg/kg of imatinib, which was found to have no significant hemodynamic effect, was administered at least 15 minutes before the higher dose was examined. At the end of study, the rat was killed by an overdose of pentobarbital sodium, and the heart was collected for measurement of the right ventricle/left ventricle + septum (RV/LV+S) weight ratio as an index of RV hypertrophy.
Western blot analyses.
Pulmonary arterial rings from normal rats were isolated for Western blot analyses. Rings were suspended in muscle baths in the same manner as for the tension measurement experiments. After 60 minutes of equilibration, all rings were exposed to l-NNA (200 μM) for 10 minutes. They were then given three different treatments: (1) no treatment, (2) U46619 (100 nM) alone, and (3) U46619 followed by 30 μM imatinib. Thirty minutes after the various treatments, rings were snap frozen by immersion in a dry ice–acetone slurry containing 10% trichloroacetic acid and stored at −80°C for subsequent analyses (19).
Statistical Analysis
Values are means ± SEM. Comparisons between groups were made with Student's t test, repeated measure ANOVA, or ANOVA with Scheffe's post hoc test for multiple comparisons. Differences were considered significant at P < 0.05.
Results
Effects of Imatinib on Agonist-Induced Contractions in Pulmonary Arterial Rings
Imatinib concentration-dependently reversed serotonin-induced (Figure 1A) and U46619-induced (Figure 1B) contractions in pulmonary arterial rings isolated from normal rats. These relaxations were not reduced by endothelial removal (Figures 1A and 1B).
Figure 1.

Concentration-relaxation curves to imatinib in endothelium-intact (closed circles) and -denuded (open circles) pulmonary arterial rings from normal rats precontracted with serotonin (A) and U46619 (B). Values are means ± SE (n = 4 in each group).
Effects of Imatinib on MLCP Activity and MLC Phosphorylation
MLCP activity and MLC phosphorylation were measured as indices for Ca2+ sensitivity and smooth muscle contractility, respectively. In agreement with previous studies (19), U46619 increased MLCP activity (as reflected by an increase in phosphorylation of its regulatory subunit MYPT1 at Thr850) (Figure 2A) as well as the phosphorylation of MLC (Figure 2B) in pulmonary arterial rings from normal rats. Imatinib (30 μM) significantly reduced the U46619-induced increased phosphorylation of MYPT1Thr850 and MLC.
Figure 2.

Effects of imatinib (30 μM) on U46619-induced increase in phosphorylation of the regulatory subunit of (A) myosin light chain phosphatase (MYPT1) and (B) myosin light chain (MLC) in pulmonary arteries from normal rats. Top panels show representative immunoblots for phosphorylated (p) and total MYPT1 and MLC. Bottom panels show calculated densitometric ratios of pMYPT1/MYPT1 and pMLC/MLC. Values are means ± SE (n = 5–6 in each group) normalized to the ratio for controls (without U46619 and imatinib) as a value of 1. *P < 0.05.
Effects of Various TKIs on Contractions in Pulmonary Arterial Rings
Three different TKIs—imatinib, nilotinib, and sorafenib—reversed U46619-induced contractions in endothelium-intact pulmonary arterial rings from normal rats pretreated with l-NNA (Figure 3A). All SU5416/hypoxia/normoxia-exposed rats developed severe PH (RV/LV+S ratio, 0.64 ± 0.03 [n = 9] versus 0.20 ± 0.02 [n = 4] for control) with occlusive neointimal lesions in small pulmonary arteries and arterioles (12, 13). In contrast to no contraction by l-NNA in normotensive pulmonary arterial rings, the NOS inhibitor caused marked contraction in hypertensive rings from SU5416/hypoxia/normoxia-exposed PH rats (Figure E1), which is similar to what is observed in pulmonary arterial rings isolated from chronically hypoxic rats (18). This phenomenon can be interpreted as evidence of high spontaneous tone that is opposed by endogenously produced nitric oxide in hypertensive pulmonary arterial rings under resting tensions in physiological salt solution. In other words, l-NNA unmasks the spontaneous contraction of hypertensive pulmonary arteries. All TKIs concentration-dependently and completely reversed the spontaneous contraction in pulmonary arterial rings isolated from SU5416/hypoxia/normoxia-exposed PH rats (Figure 3B). Nilotinib and sorafenib were more potent than imatinib in reversing the U-46619–induced and the spontaneous contractions in normotensive and hypertensive pulmonary arteries, respectively.
Figure 3.

Concentration-relaxation curves to tyrosine kinase inhibitors, imatinib (open circles; n = 8), nilotinib (closed circles; n = 4), and sorafenib (open triangles; n = 4). All inhibitors showed concentration-dependent relaxations of pulmonary arterial rings from normal rats precontracted with U46619 (A) and from SU5416/hypoxia/normoxia-exposed PH rats precontracted with nitro-l-arginine (B). Values are means ± SE. *P < 0.001 versus sorafenib and nilotinib.
Acute Hemodynamic Effects of Imatinib in Catheterized PH Rats
Compared with normotensive rats, SU5416/hypoxia/normoxia-exposed rats had very high RVSP (20 ± 3 versus 100 ± 5 mm Hg, respectively; n = 4 and 11, respectively; P < 0.001) with markedly reduced cardiac index (CI) (119 ± 12 versus 59 ± 8 ml/min/kg, respectively; n = 4 each; P < 0.001), normal LVSP (138 ± 9 versus 140 ± 6 mm Hg, respectively; n = 4 and 11, respectively; not significant [n.s.]), and HR (372 ± 23 versus 347 ± 19 mm Hg, respectively; n = 4 and 11; n.s.). All PH rats showed severe RV hypertrophy as reflected by high RV/LV+S ratio compared with normal rats (0.20 ± 0.02 versus 0.55 ± 0.02, respectively; n = 4 and 11, respectively; P < 0.001).
The acute effects of intravenous imatinib (20 and 50 mg/kg, bolus) on RVSP, LVSP, and CI were tested in anesthetized SU5416/hypoxia/normoxia-exposed severe pulmonary hypertensive rats. Imatinib (50 mg/kg) significantly lowered high RVSP (from 105 ± 7 to 87 ± 5 mm Hg; P < 0.05), with only minor effects on LVSP (from 140 ± 9 to 134 ± 10 mm Hg; n.s.), HR (from 367 ± 24 to 347 ± 19 mm Hg; n.s.), and CI (from 59 ± 8 to 59 ± 10 mm Hg; n.s.) (Figure 4A). Lower doses of imatinib (5 and 20 mg/kg) had no and a smaller but significant RVSP-lowering effect, respectively (Figure 4B).
Figure 4.
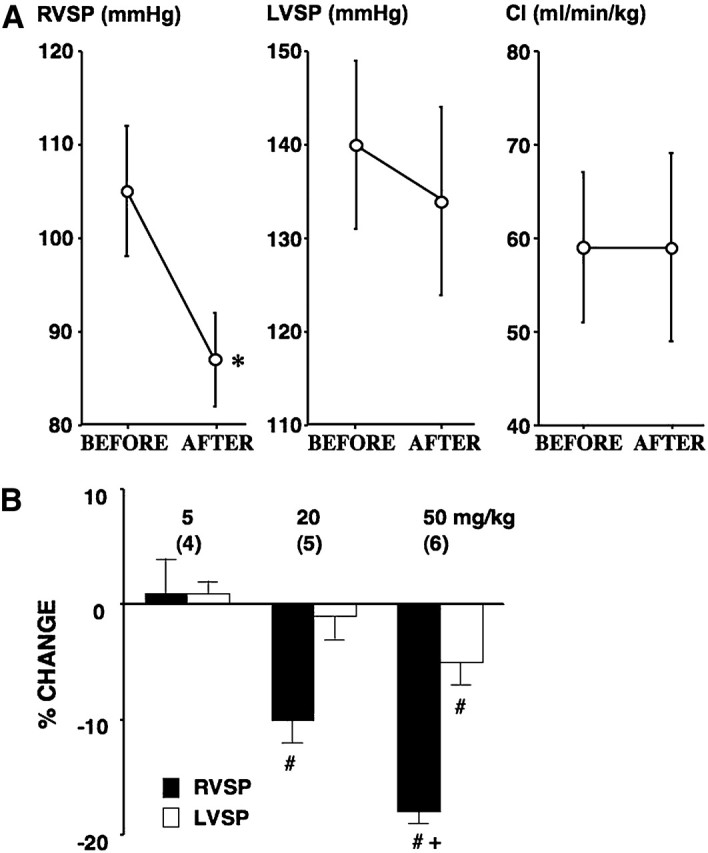
(A) Effects of imatinib (50 mg/kg, intravenous bolus) on right ventricular systolic pressure (RVSP) (left panel; n = 6), left ventricular systolic pressure (LVSP) (middle panel; n = 6), and cardiac index (CI) (right panel; n = 4) in SU5416/hypoxia/normoxia-exposed PH rats. Values are means ± SE. *P < 0.05 versus BEFORE value. (B) Effects of imatinib (5, 20, and 50 mg/kg, intravenous bolus) on RVSP (closed bar) and LVSP (open bar) in SU5416/hypoxia/normoxia-exposed PH rats. Percentage change is expressed as a difference in values between baseline and after imatinib divided by each baseline value. Values are means ± SE of n = 6. #P < 0.05 versus baseline value. +P < 0.05 versus 20 mg/kg. Number of rats examined is indicated in parentheses.
Discussion
This study demonstrated that the TKIs imatinib, nilotinib, and sorafenib concentration-dependently and completely relaxed precontracted normotensive and hypertensive pulmonary arterial rings from normal and SU5416/hypoxia/normoxia-exposed PH rats, respectively. Endothelium-independent vascular smooth muscle relaxation mediated via activation of MLCP (Ca2+ desensitization) appeared to play a role in the imatinib-induced pulmonary vasorelaxation. In addition, acute intravenous administration of imatinib effectively reduced the high RVSP without major effects on systemic arterial pressure and cardiac output in SU5416/hypoxia/normoxia-exposed rats with severe PH.
Imatinib is approved for the treatment of patients with chronic myeloid leukemia and gastrointestinal stromal tumors (2, 3). It inhibits PDGFR, c-Abelson tyrosine kinase (c-Abl), and c-kit at low concentrations (∼1 μM). In addition, it may inhibit other kinases, such as EGFR, Src family kinases, and protein kinase C (PKC), at higher concentrations (20, 21). Recently, chronic administration of imatinib has been reported to be promising for the clinical treatment of severe PH (4). Chronically administered high doses of imatinib (50 and 100 mg/kg) reverse the established PH induced by monocrotaline in rats and by chronic hypoxia in mice (5, 7). There are also positive clinical case reports of chronic treatment with imatinib in patients with severe PH (4, 21).
It is assumed that the beneficial effect of imatinib against PH is due to inhibition of PDGFR and the prevention of cell proliferation (5, 7). There are, however, several possible mechanisms by which imatinib could also elicit pulmonary vasodilation. These include: (1) inhibition of PDGFR-mediated elevation of intracellular Ca2+ levels (22); (2) inhibition of other off-target protein kinases, such as EGFR, Src, and PKC, which may regulate intracellular Ca2+ levels and Ca2+ sensitivity (8, 23, 24); and (3) inhibition of c-Abl–mediated actin polymerization (25). We found in this study that imatinib completely reversed agonist-induced contractions in isolated normal rat pulmonary arteries in an endothelium-independent manner. Consistent with previous studies in systemic arterial rings (26, 27), U46619-induced pulmonary arterial ring contraction was accompanied by increases in phosphorylation of MLC and MYPT1Thr850, the regulatory subunit of MLC phosphatase (MLCP). Imatinib (30 μM) reversed not only the contraction and the increased phosphorylation of MLC but also the increased phosphorylation of MYPT1Thr850. These results suggest that imatinib is an effective endothelium-independent pulmonary vasorelaxant and that Ca2+ desensitization (mediated through dephosphorylation of MYPT1Thr850 and activation of MLCP) may contribute to its action. However, mechanisms of imatinib-induced vasodilation may be different among different arteries and mechanisms of contraction. It is also suggested that inhibition of other off-targeted kinases (e.g., EGFR, Src family kinases, and PKC), rather than simply its major targets (i.e., PDGFR, c-kit, and c-Abl), may account for the vasorelaxant activity of imatinib because the lower concentrations (1 and 3 μM) that should be sufficient to inhibit the major targeted kinases (23) had little effect against the agonist-induced contractions.
In addition to our in vitro finding that a high concentration of imatinib (30 μM) completely reversed the spontaneous contraction of hypertensive pulmonary arterial rings, we found that high doses (20 and 50 mg/kg) but not a low dose (5 mg/kg) of imatinib effectively reduced the high RVSP in SU5416/hypoxia/normoxia-exposed PH rats. The low dose (5 mg/kg) is close to the usual clinical dose for the treatment of patients with leukemia (400 mg/d) and the dose currently being used in clinical trials in patients with PH. The high dose (50 mg/kg) has been reported to be effective in the chronic treatment of pulmonary hypertensive rodent models (5, 7). In these preclinical studies, lower doses (1 and 10 mg/kg) had little or no effect in reversing the established PH. Taken together, these observations suggest that high concentrations and doses of imatinib (much higher than those effectively inhibiting the major targeted kinases) are required to exert its vasodilatory action, which may have played an important role in its chronic beneficial effect against preclinical models of PH. If this is the case for human PH, one can predict that a low dose (400 mg/d or ∼5 mg/kg) of imatinib against PH in clinical trials may not be as effective as expected. In fact, a very recent report demonstrates that low doses of imatinib (200 or 400 mg/d) in patients with PH failed to show significant improvement in the primary endpoint (6-min walking distance) with no significant reduction in pulmonary arterial pressure (28).
An unexpected and potentially important observation is that the blood pressure–lowering effect of imatinib in SU5416/hypoxia/normoxia-exposed severe PH rats was relatively pulmonary selective. Although it is unclear how imatinib preferentially reduced the high RVSP, one possible explanation is that it may inhibit the activity of a kinase/signaling pathway that regulates Ca2+ sensitivity (e.g., Src/Rho kinase pathway [24]) that is up-regulated in the hypertensive pulmonary arteries but not in the normotensive systemic arteries. Considering that clinical application of the higher doses of imatinib are unacceptable due to its potential adverse side effects (29), it may be important in future studies to identify these kinase(s) and signaling pathway(s) to develop more specifically targeted and effective therapies.
We also examined pulmonary vasorelaxant effects of two other TKIs: nilotinib, an inhibitor of PDGFR and c-Abl (30), and sorafenib, an inhibitor of PDGF and vascular endothelial growth factor receptors as well as the serine/threonine kinases Raf-1 and b-Raf (31). We found that these TKIs relaxed precontracted normotensive and hypertensive pulmonary arteries more potently than imatinib did. The relaxation by all three TKIs was apparently at least largely independent of nitric oxide synthesis. Further studies are required to elucidate more detailed mechanisms of the relaxation. These results support our hypothesis that TKIs have a potent pulmonary vasodilatory action through inhibition of smooth muscle cell growth factor–related kinases and suggest that nilotinib and sorafenib might be more effective than imatinib in treating patients with PH.
CONCLUSION
This study demonstrated that TKIs, which are promising candidates for the treatment of severe PH as antigrowth factor and antiproliferating agents, are potent pulmonary vasodilators. This new information may be important in interpreting the chronic beneficial effects of TKIs in the clinical treatment of PH.
Footnotes
This work was supported by a grant from the American Heart Association (0765477Z) and by funds from the Department of Pharmacology and Center for Lung Biology, University of South Alabama.
This article has an online supplement, which is accessible from this issue's table of contents online at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2010-0371OC on March 4, 2011
References
- 1.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004;351:1425–1436. [DOI] [PubMed] [Google Scholar]
- 2.Cohen MH, Williams G, Johnson JR, Duan J, Gobburu J, Rahman A, Benson K, Leighton J, Kim SK, Wood R, et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin Cancer Res 2002;8:935–942. [PubMed] [Google Scholar]
- 3.Dagher R, Cohen M, Williams G, Rothmann M, Gobburu J, Robbie G, Rahman A, Chen G, Staten A, Griebel D, et al. Approval summary: imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res 2002;8:3034–3038. [PubMed] [Google Scholar]
- 4.Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med 2005;353:1412–1413. [DOI] [PubMed] [Google Scholar]
- 5.Klein M, Schermuly RT, Ellinghaus P, Milting H, Riedl B, Nikolova S, Pullamsetti SS, Weissmann N, Dony E, Savai R, et al. Combined tyrosine and serine/threonine kinase inhibition by sorafenib prevents progression of experimental pulmonary hypertension and myocardial remodeling. Circulation 2008;118:2081–2090. [DOI] [PubMed] [Google Scholar]
- 6.Moreno-Vinasco L, Gomberg-Maitland M, Maitland ML, Desai AA, Singleton PA, Sammani S, Sam L, Liu Y, Husain AN, Lang RM, et al. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol Genomics 2008;33:278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest 2005;115:2811–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fernandez-Patron C. Therapeutic potential of the epidermal growth factor receptor transactivation in hypertension: a convergent signaling pathway of vascular tone, oxidative stress, and hypertrophic growth downstream of vasoactive G-protein-coupled receptors? Can J Physiol Pharmacol 2007;85:97–104. [DOI] [PubMed] [Google Scholar]
- 9.Florian JA, Watts SW. Epidermal growth factor: a potent vasoconstrictor in experimental hypertension. Am J Physiol 1999;276:H976–H983. [DOI] [PubMed] [Google Scholar]
- 10.Sauro MD, Thomas B. Tyrphostin attenuates platelet-derived growth factor-induced contraction in aortic smooth muscle through inhibition of protein tyrosine kinase(s). J Pharmacol Exp Ther 1993;267:1119–1125. [PubMed] [Google Scholar]
- 11.Berk BC, Alexander RW. Vasoactive effects of growth factors. Biochem Pharmacol 1989;38:219–225. [DOI] [PubMed] [Google Scholar]
- 12.Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, Voelkel NF, McMurtry IF. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 2007;100:923–929. [DOI] [PubMed] [Google Scholar]
- 13.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 2001;15:427–438. [DOI] [PubMed] [Google Scholar]
- 14.Puetz S, Lubomirov LT, Pfitzer G. Regulation of smooth muscle contraction by small GTPases. Physiology (Bethesda) 2009;24:342–356. [DOI] [PubMed] [Google Scholar]
- 15.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin ii: modulated by g proteins, kinases, and myosin phosphatase. Physiol Rev 2003;83:1325–1358. [DOI] [PubMed] [Google Scholar]
- 16.McMurtry IF, Abe K, Ota H, Fagan KA, Oka M. Rho kinase-mediated vasoconstriction in pulmonary hypertension. Adv Exp Med Biol 2010;661:299–308. [DOI] [PubMed] [Google Scholar]
- 17.Oka M, Fagan KA, Jones PL, McMurtry IF. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. Br J Pharmacol 2008;155:444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oka M, Hasunuma K, Webb SA, Stelzner TJ, Rodman DM, McMurtry IF. EDRF suppresses an unidentified vasoconstrictor mechanism in hypertensive rat lungs. Am J Physiol 1993;264:L587–L597. [DOI] [PubMed] [Google Scholar]
- 19.Tsai MH, Jiang MJ. Rho-kinase-mediated regulation of receptor-agonist-stimulated smooth muscle contraction. Pflugers Arch 2006;453:223–232. [DOI] [PubMed] [Google Scholar]
- 20.Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 2005;105:2640–2653. [DOI] [PubMed] [Google Scholar]
- 21.Patterson KC, Weissmann A, Ahmadi T, Farber HW. Imatinib mesylate in the treatment of refractory idiopathic pulmonary arterial hypertension. Ann Intern Med 2006;145:152–153. [DOI] [PubMed] [Google Scholar]
- 22.Hughes AD. Increase in tone and intracellular Ca2+ in rabbit isolated ear artery by platelet-derived growth factor. Br J Pharmacol 1995;114:138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito A, Shimokawa H, Nakaike R, Fukai T, Sakata M, Takayanagi T, Egashira K, Takeshita A. Role of protein kinase C-mediated pathway in the pathogenesis of coronary artery spasm in a swine model. Circulation 1994;90:2425–2431. [DOI] [PubMed] [Google Scholar]
- 24.Knock GA, Shaifta Y, Snetkov VA, Vowles B, Drndarski S, Ward JP, Aaronson PI. Interaction between src family kinases and Rho-kinase in agonist-induced Ca2+-sensitization of rat pulmonary artery. Cardiovasc Res 2008;77:570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang DD, Anfinogenova Y. Physiologic properties and regulation of the actin cytoskeleton in vascular smooth muscle. J Cardiovasc Pharmacol Ther 2008;13:130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neppl RL, Lubomirov LT, Momotani K, Pfitzer G, Eto M. Somlyo AV. Thromboxane A2-induced bi-directional regulation of cerebral arterial tone. J Biol Chem 2009;284:6348–6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson DP, Susnjar M, Kiss E, Sutherland C. Walsh MP. Thromboxane A2-induced contraction of rat caudal arterial smooth muscle involves activation of Ca2+ entry and Ca2+ sensitization: Rho-associated kinase-mediated phosphorylation of mypt1 at thr-855, but not thr-697. Biochem J 2005;389:763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghofrani HA, Morrell NW, Hoeper MM, Olschewski H, Peacock AJ, Barst RJ, Shapiro S, Golpon H, Toshner M, Grimminger F, et al. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am J Respir Crit Care Med 2010;182:1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kerkela R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, Walters B, Shevtsov S, Pesant S, Clubb FJ, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med 2006;12:908–916. [DOI] [PubMed] [Google Scholar]
- 30.Deremer DL, Ustun C, Natarajan K. Nilotinib: a second-generation tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia. Clin Ther 2008;30:1956–1975. [DOI] [PubMed] [Google Scholar]
- 31.Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 2006;5:835–844. [DOI] [PubMed] [Google Scholar]