

Abstract
The widely used herbicide, paraquat (PQ), is highly toxic and claims thousands of lives from both accidental and voluntary ingestion. The pathological mechanisms of PQ poisoning–induced acute lung injury (ALI) are not well understood, and the role of complement in PQ-induced ALI has not been elucidated. We developed and characterized a mouse model of PQ-induced ALI and studied the role of complement in the pathogenesis of PQ poisoning. Intraperitoneal administration of PQ caused dose- and time-dependent lung damage and mortality, with associated inflammatory response. Within 24 hours of PQ-induced ALI, there was significantly increased expression of the complement proteins, C1q and C3, in the lung. Expression of the anaphylatoxin receptors, C3aR and C5aR, was also increased. Compared with wild-type mice, C3-deficient mice survived significantly longer and displayed significantly reduced lung inflammation and pathology after PQ treatment. Similar reductions in PQ-induced inflammation, pathology, and mortality were recorded in mice treated with the C3 inhibitors, CR2-Crry, and alternative pathway specific CR2-fH. A similar therapeutic effect was also observed by treatment with either C3a receptor antagonist or a blocking C5a receptor monoclonal antibody. Together, these studies indicate that PQ-induced ALI is mediated through receptor signaling by the C3a and C5a complement activation products that are generated via the alternative complement pathway, and that complement inhibition may be an effective clinical intervention for postexposure treatment of PQ-induced ALI.
Keywords: acute lung injury, complement, inhibitor, mouse model, paraquat
Clinical Relevance
Paraquat (PQ) is widely used herbicide and can induce acute lung injury (ALI). Currently, the pathological mechanisms of PQ poisoning–induced ALI are not well understood, and the role of complement in PQ-induced ALI has not been elucidated. Our findings show, for the first time, that complement inhibition or treatment with anaphylatoxin receptor antagonist or monoclonal antibody reduced lung inflammation and pathology. This study opens new effective clinical intervention for postexposure treatment of PQ-induced ALI.
Paraquat (PQ) is the commercial name for N, N′-dimethyl-4, 4′-bipyridinium dichloride, and is one of the most widely used herbicides in the world, especially in the developing countries. PQ is highly toxic to humans and livestock, and since its introduction for agricultural use in 1962, PQ has claimed thousands of lives, with an overall mortality rate of 70% after poisoning (1). Such a high mortality rate results principally from the lack of specific detoxification agents.
After entering the body by ingestion, inhalation, or skin exposure, PQ rapidly distributes to most tissues. It reaches maximal plasma concentration within 90 minutes, and 50–70% is excreted by the kidney within 24 hours (2). Despite the high excretion rate, PQ is easily absorbed by pulmonary epithelial cells, and most deaths from acute PQ poisoning can be attributed to acute lung injury (ALI) or acute respiratory distress syndrome (ARDS), with features of neutrophil influx and increased vascular permeability with resultant pulmonary edema (3, 4).
Clinically, PQ intake toxicity can be divided into three categories (5): (1) Acute PQ intake, in which the dose exceeds 40 mg/kg bodyweight and results in rapid death (usually within 1–2 d) from pulmonary edema, cell infiltration and alveolar hemorrhage. Patients who do survive usually develop ARDS; (2) intermediate to severe PQ intake (20–40 mg/kg bodyweight), which is characterized by symptoms of pulmonary atelectasis resulting from the infiltration of profibroblasts that rapidly differentiate into fibroblasts. In some cases, these conditions can lead to fibrosis within 1–2 weeks. 3. PQ intake of less than 20 mg/kg bodyweight may result in no obvious infiltration and atelectasis, but symptoms can gradually develop into lung interstitial fibrosis and still result in slight damage to pulmonary functions.
Currently, the mechanisms underlying PQ poisoning are not well understood. For several decades, it was believed that PQ poisoning was related to the functions of several toxic products, including superoxide anions. For example, both reduced nicotinamide adenine dinucleotide phosphate oxidation and lipid peroxidation were thought to damage endothelial cells, with resultant ALI (6–8). However, mechanistic details of how PQ poisoning leads to ALI are lacking.
The complement system plays an important role in host defense and immune homeostatic mechanisms, but complement also contributes to the pathogenesis of many inflammatory and immunological disease conditions when excessively or inappropriately activated (9–13). The biological effector functions of complement are mediated primarily through the split products, C3a and C5a, which promote inflammation via direct and indirect mechanisms upon interaction with their receptors (14). In addition to being a potent chemoattractant, C5a can activate leukocytes, stimulate release of granzymes, and stimulate phagocytosis and respiratory burst of mononuclear cells (15). Furthermore, C5a induces mononuclear cells to express IL-1 and IL-8 in vitro, and enhances the release of LPS-induced IL-6 and TNF-α in vivo (16). The binding of C3a to its receptor (C3aR) has also been implicated in multiple proinflammatory processes, including secretion of lysozymes from leukocytes, release of histamine from mast cells, contraction of smooth muscle cells, and chemotaxis of eosinophils and mast cells (17).
Although complement activation is associated with many inflammatory disease processes, and is also a critical early step in ALI, little is known concerning the role of complement in PQ-induced ALI. In this study, we established a mouse model of acute PQ poisoning and investigated the correlation between complement activation and the early stages of PQ poisoning. Our results indicate that complement activation plays an important role in the onset of PQ-induced ALI, and that targeted complement inhibition represents a potential clinical intervention for PQ poisoning.
Materials and Methods
Mice ALI Model
For the dose-dependent PQ poisoning experiment, BALB/c mice (Laboratory Animal Center, Academy of Military Medical Science, Beijing, China) aged 6 to 8 weeks were administered PQ (Xianzhengda Limited Co., Shanghai, China) via intraperitoneal injection. PQ was dissolved in 500 μl of physiological saline at 10, 20, and 50 mg/kg bodyweight (PQ10, PQ20, and PQ50, respectively). The control group was treated with same volume of saline. For the PQ poisoning kinetics in relation to complement involvement studies, BALB/c mice were intraperitoneally injected with PQ20. All mice were killed at indicated times after PQ injection.
The Inhibition of Complement Activation
C3−/− mice and wild-type (WT) C57BL/6 mice (6 wk old) were poisoned with PQ20 by intraperitoneal administration, with saline as control. The mouse survival rate, concentration of proinflammatory cytokines, myeloperoxidase (MPO) activity, and histopathological analysis parameters were measured.
In CR2-Crry– and CR2-factorH (fH)–targeting inhibition study, there were four groups of BALB/c mice. The mice were intravenously administered PBS (PQ + PBS group), CR2-Crry (0.25 mg/mouse) (PQ + Crry group), or CR2-fH (0.8 mg/mouse) (PQ + CR2-fH group) immediately after intraperitoneal injection of 20 mg/kg PQ; the control group was treated at the same injection schedule with the same volume of saline. The survival rate and histological examination were studied in both targeting inhibition studies. In addition, the distribution of CR2-Crry was evaluated in the CR2-Crry–targeting inhibition study.
For the blockade experiment of anaphylatoxin receptor signaling, four groups of mice were designated (PQ + C3aR antagonist, PQ + C5aR monoclonal antibody [mAb], PQ + PBS, saline group). Mice received 600 μg/kg C5aR mAb (C5aR mAb; Hycult Biotechnology, Uden, The Netherlands) 20/70 or 1 mg/kg C3aR antagonist (SB290157; Calbiochem, Darmstadt, Germany) as intravenous injection 50 minutes before and 2 hours after intraperitoneal injection of 20 mg/kg PQ or PBS (18). The survival rate and histological injury were studied in each group, and proinflammatory cytokines in PQ + C3aR antagonist and PQ + PBS groups were conducted using the BD CBA mouse inflammation kit system (San Jose, CA).
Measurement Assays
Relative lung weight was measured and calculated as previously described (19). Cytokines in serum were measured using the BD CBA mouse inflammation kit system (San Jose, CA). Total protein quantification of bronchoalveolar lavage fluid (BALF), MPO activity, and C3c in serum were measured as previously described (20). Lung tissue damage was observed by light microscopy and electron microscopy, according to conventional procedures (19). Immunohistochemistry for complement component distribution and indirect immunofluorescence staining for CR2-Crry were performed as previously described, with minor modification (21). For details, see the online supplement.
125I–CR2-Crry Biodistribution
CR2-Crry was labeled with 125I by the IODO-GEN method (Amersham Biosciences, Pittsburgh, PA), as previously described (21), and was injected into mice for 125I-CR2-Crry biodistribution analysis. For details, see the online supplement.
Detection of C3aR mRNA and C5aR mRNA Expression
Total RNA was isolated from lung tissue and relative quantitative real-time PCR was performed. The relative C3aR and C5aR expression data were analyzed using the 2−∆∆CT method (22). For details, see the online supplement.
Statistical Analysis
All analyses were performed with GraphPad Prism software (GraphPad Inc., San Diego, CA). The difference between different dose groups and different time groups was analyzed by one-way ANOVA. The significance between survival curves was analyzed by Kaplan-Meier survival analysis with log-rank test.
Results
Intraperitoneal Administration of PQ causes ALI in Mice
We first established a mouse model of acute PQ poisoning by intraperitoneal injection of saline-diluted PQ at PQ10, PQ20, and PQ50. Control mice received saline only. Animals were killed 48 hours after injection, at which time we determined weight loss, relative lung weight, protein content of BALF, MPO activity of lung samples, and histopathological changes in lung sections. There was a significant loss of weight in all PQ treated groups at 48 hours after injection (Figure 1A). Compared with saline controls, at 48 hours after PQ injection, the PQ20 and PQ50 groups also had significantly higher lung index values (Figure 1B), increased BALF total protein content (Figure 1C), and higher MPO activity (Figure 1D). Lung index was assessed as an indication of congestion and edema in lung tissue. Increased BALF protein concentration indicates enhancement of vascular permeability, whereas lung MPO activity is a measure of neutrophil infiltration (23). Thus, PQ poisoning resulted in a dose-dependent increase in mouse lung congestion and edema, vascular permeability, and neutrophil infiltration.
Figure 1.

Dose-dependent effect of paraquat (PQ) on acute lung injury (ALI). Mice were injected intraperitoneally with normal saline (N) or saline-diluted PQ solution at a dose of 10 mg/kg (PQ10), 20 mg/kg (PQ20), or 50 mg/kg (PQ50), and mice were killed for analysis 48 hours later. Weight loss (A), relative lung weight (B), bronchoalveolar lavage fluid (BALF) total protein (C), and myeloperoxidase (MPO) activity (D) were analyzed. *P < 0.05, **P < 0.01 between 0 and 48 hours after PQ injection (A), or between the PQ injection groups and the normal control group (B–D). Data presented are means (±SD); n = 8 per group.
PQ-induced lung injury was further characterized by histopathology (see Figure E1 in the online supplement). At 48 hours after PQ poisoning, injured lung tissue presented as typical diffuse alveolar damage and ALI. Mice from the saline group had normal pulmonary structure and intact alveoli epithelium (Figures E1A and E1E). In the PQ10 group (Figures E1B and E1F), there was no significant damage other than slightly widened lung septa, sparse denaturation of alveoli epithelial cells, and infiltration of inflammatory cells. In the PQ20 group (Figures E1C and E1G), there was more severe lung damage than in the PQ10 group. Lungs from the PQ20 group were mainly characterized by denaturation and diffuse alveoli collapse, with significantly widened septa, aggravated interstitial edema, and infiltration of inflammatory cells. In the PQ50 group (Figures E1D and E1H), the lungs were even more severely damaged, with infiltration of inflammatory cells accompanied by large quantities of exudates and severe edema, especially around vessels. Furthermore, large quantities of erythrocytes in bronchiole tubes and vessels, hemorrhage in lung interstitial tissue, and increased or fused alveoli walls were observed in lungs from the PQ50 group. Notably, none of the three PQ groups presented with hyaline membrane formation or fibrous tissue proliferation. ALI after PQ poisoning was further characterized through ultrastructural studies. At 48 hours after PQ20 poisoning, mouse lungs presented with obvious injury; pulmonary epithelial cells had vacuolar degeneration, with nuclei moving to the edges, mitochondrial swelling, and denatured endoplasmic reticulum (Figures E1I–E1J). Interstitial edema was characterized by the existence of intercellular vacuolization and exudates, including damage to the blood–gas barrier, with cell debris falling off to alveolar spaces (Figure E1K). Neutrophil infiltration was also identified in the interstitial edema in this group (Figure E1L).
The kinetics of ALI after PQ poisoning were studied in a separate set of experiments using a 20 mg/kg PQ dose. The histopathological presentation of ALI progressed with time (Figure E2). At 4 hours after PQ poisoning, a small number of inflammatory cells and slight vascular congestion were seen in lung tissue (Figure E2A). At 12 hours after PQ injection, degeneration was seen in some pneumocytes, lung septa were widened, and infiltration of inflammatory cells increased (Figure E2B). At 24 hours (Figure E2C) and 48 hours (Figure E2D) after PQ poisoning, lung injury was aggravated and diffused, with bronchiole epithelial cell collapse, large amounts of exudate, serious edema around vessels, and large quantities of inflammatory cells, mainly infiltrating neutrophils.
The physical and pathological indices also changed over time after PQ20 treatment. Lung index gradually increased, and reached significance at 24 hours (P < 0.01) and 48 hours (P < 0.01) after PQ poisoning (Figure 2A). For BALF total protein, the increase was moderate up to 24 hours, although there was no apparent difference (P > 0.05); however, total protein content increased dramatically by 48 hours after PQ administration (P < 0.01), and was almost 10-fold higher than that at 0 hours (Figure 2B). The MPO activity was also increased significantly by 12 hours (P < 0.01), and continued to increase at 24 and 48 hours (P < 0.01) (Figure 2C).
Figure 2.
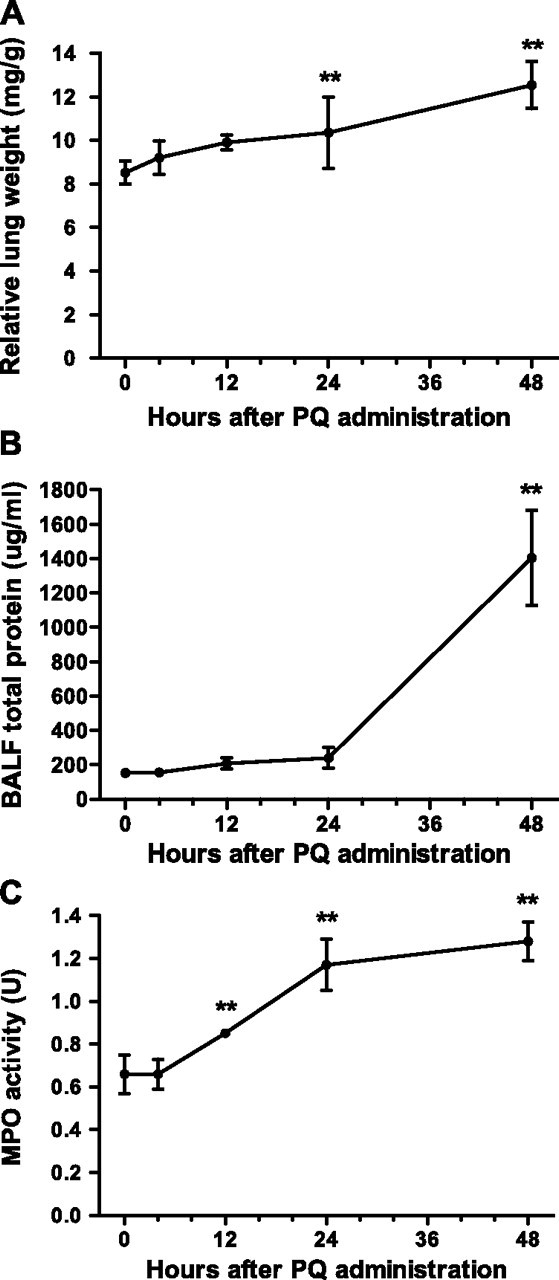
Time-dependent progression of ALI after PQ poisoning. ALI was assessed at different times after PQ20 treatment by lung index (A), BALF total protein (B), and MPO activity (C). *P < 0.05 or **P < 0.01, relative to the corresponding 0-hour group. Data presented are means (±SD); n = 8 per group.
Involvement of Complement in PQ-Induced ALI in Mice
In initial studies to investigate the potential role of complement in our mouse model of PQ-induced ALI, we measured complement deposition, complement receptor expression in lung tissue, and complement activation in serum. Deposition of C1q and C3, and expression of C5aR in lungs of PQ20- and saline-treated control mice was determined by immunohistochemistry (Figures 3A–3I). At time 0 hours, low levels of C1q and C3 were detected on smooth muscle cells, with low levels of C1q also seen on bronchial epithelial cells, and low level expression of C5aR was detected in interstitial tissue. However, there was increased deposition of all proteins at 4 and 24 hours after PQ20 treatment, with more intense staining at 24 hours compared with 4 hours. C3 staining was detected on the endothelium, with some inflammatory cells visible in interstitial tissue at 4 hours after PQ poisoning, and with staining detectable in injured bronchiole epithelium and lung interstitial tissue at 24 hours. The strongest C1q-specific staining was found in lung interstitial tissue, bronchiole epithelial base membrane, and lung endothelium. In addition, deposition of mannan-binding lectin (MBL)–A was detected, but no apparent MBL-A staining was observed after PQ poisoning (Figures 3J–3L). The staining profile for C5aR was similar to that for C1q and C3, with increased endothelial and interstitial staining at 4 hours, which was further increased at 48 hours after PQ treatment. In addition, we measured serum levels of the complement activation product, C3c, at different time points after PQ20 poisoning. During the complement activation process, C3 converts to C3a and C3b, and C3b is then lysed to become iC3b and soluble C3f. The iC3b is further lysed into cell membrane–attached C3 dg and soluble C3c. Elevated levels of C3c have been found in many clinical inflammatory diseases (24), and are an indicator of complement activation (25). In this study, C3c levels began to increase at 12 hours (P < 0.05), reached a peak at 24 hours, and were sustained for 48 hours after PQ injection (P < 0.01) (Figure 3M).
Figure 3.
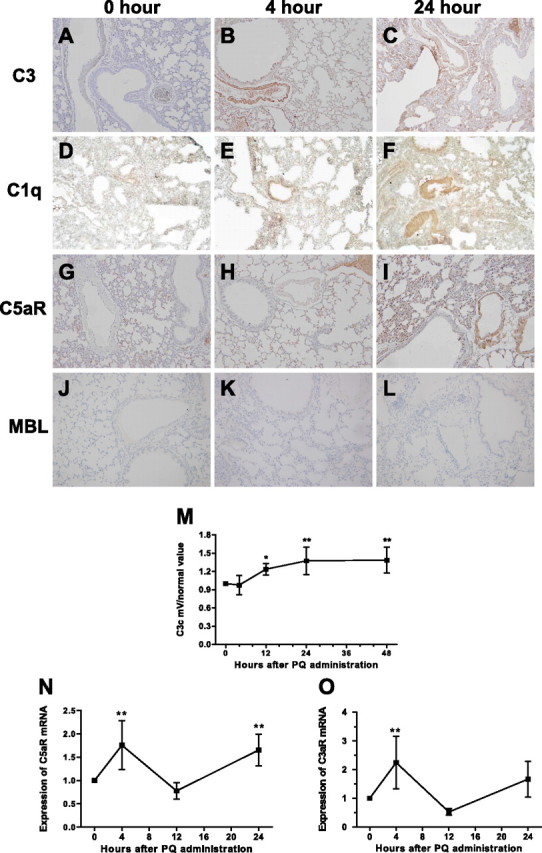
Complement activation at early timepoint after PQ injection. (A–L) Immunohistochemical staining for C3, C1q, C5aR and mannan-binding lectin (MBL) in lung and the C3c level in serum after PQ20 poisoning. C3 deposition was strongly elevated at 4 and 24 hours after PQ injection (A–C). C1q depoistion (D–F) was also elevated on interstitial, epithelial base membrane, and lung endothelium at 4 and 24 hours after PQ20 treatment. Expression of C5aR (G–I) was elevated 4 hours after PQ20 treatment, with a further increase in staining seen at 24 hours after PQ20 treatment. No MBL deposition was detected in any group of mice lung (J–L). (M) Changes of C3c levels in serum at indicated times. (N–O) C5aR and C3aR mRNA expression in the lung. Relative C5aR mRNA and C3aR mRNA expression were determined in lung tissue at indicated time points after PQ administration; mRNA expression was assessed by relative real-time quantitative PCR analysis. *P < 0.05 or **P < 0.01, relative to the corresponding 0-hour group. Data presented are means (±SD); n = 7–9 per group. Magnification, ×200.
Based on these data, we next analyzed C3aR and C5aR gene expression after PQ poisoning. We compared C3aR mRNA and C5aR mRNA levels in lung tissue by relative real-time quantitative PCR in samples isolated at 0, 4, 12, and 24 hours after PQ20 treatment. The mRNA expression of both C5aR (Figure 3N) and C3aR (Figure 3O) was significantly enhanced in lung tissue 4 hours after PQ treatment (P < 0.01), but levels returned to normal by 12 hours after PQ treatment. C5aR mRNA levels, however, were increased again at 24 hours after PQ treatment (P < 0.01). Importantly, C5aR mRNA expression correlated with protein expression data for C5aR. Thus, complement activation and deposition, as well as anaphylatoxin receptor expression, correlate with lung injury.
C3 Deficiency Decreases Lung Inflammation and Attenuates PQ-Induced Lung Injury
All complement pathways converge at the C3 cleavage step, and to further investigate the role of complement in the pathogenesis of PQ-induced ALI, we determined the effect of C3 deficiency on the outcome of PQ poisoning. Compared with WT mice, C3−/− mice presented with significantly less lung inflammation and injury as a result of PQ20 poisoning. Gross examination of lungs and hematoxylin and eosin staining of mouse lung sections revealed much less focal hemorrhage (dark red coloration), vascular congestion, and inflammatory cell infiltration in C3−/− mice compared with WT mice after PQ poisoning (Figures 4A–4H). A significant decrease in neutrophil infiltration in lungs from C3−/− mice compared with WT mice was demonstrated by measurement of MPO activity at 30 hours after PQ treatment, and there was no difference in MPO levels between saline-treated control mice and PQ-treated C3−/− mice (P < 0.01) (Figure 4I). The serum concentration of the proinflammatory cytokines, IL-6, TNF-α, IL-10, and monocyte chemoattractant protein-1, were also significantly reduced in C3−/− mice compared with WT mice 4 hours after PQ poisoning (Figure 4J). Finally, C3 deficiency significantly increased survival times, with 25% mortality in the C3−/− group (P < 0.01) compared with 100% mortality in the WT group at 60 hours after PQ treatment (Figure 4K).
Figure 4.

Effect of C3 deficiency on lung injury, inflammation, and survival after PQ poisoning. Wild-type (WT) C57 BL/6 mice and C3−/− mice were treated intraperitoneally with PQ20 or saline. (A–D) Gross appearance of mouse lung sections 30 hours after PQ or saline treatment: (A) WT mice + PQ; (B) WT mice + saline; (C) C3−/− mice + PQ; (D) C3−/− mice + saline. (E–H) Hematoxylin and eosin staining of lung sections: (E) WT mice + PQ; (F) WT mice + saline; (G) C3−/− mice + PQ; (H) C3−/− mice + saline. Representative images shown, n = 3. (I) MPO activity in lung samples 30 hours after PQ or saline administration in WT or C3 −/− mice (n = 3 in each group). (J) Serum concentrations of proinflammatory cytokines 4 hours after PQ poisoning (n = 6 in each group). (K) Survival analysis after PQ injection. (n = 8 per group). *P < 0.05 and **P < 0.01 relative to the corresponding the C3−/− + PQ group with WT mice + PQ group. Data presented are means (±SD). Magnification, ×200 (E–H).
Targeted Complement Inhibition Alleviates Lung Injury and Prolongs Survival of Mice with PQ-Induced ALI
To investigate the role of complement in the pathogenesis of PQ-induced ALI in a more clinically relevant paradigm, we treated PQ-poisoned mice with CR2-Crry, a complement inhibitory fusion protein that targets to sites of complement activation and inhibits all three complement pathways at the C3 activation step (20). In these experiments, 0.25 mg CR2-Crry was administered intravenously immediately after PQ injection. Examination of lungs from PBS-treated mice 30 hours after PQ injection revealed dark red coloration with evident hemorrhagic foci on the surface (Figure 5B), whereas lungs from CR2-Crry–treated mice (Figure 5C) appeared normal (Figure 5A). Hematoxylin and eosin staining revealed severe tissue damage, with focal hemorrhage and infiltration of neutrophils in lungs from the PQ + PBS group (Figure 5F), whereas lungs from the PQ + CR2-Crry group appeared almost normal, with only slight parenchymal expansion and minimal pulmonary neutrophil congestion (Figure 5G). Furthermore, whereas a significant level of C3 deposition was seen in lungs from PQ + PBS–treated mice (Figure 5J), only minimal C3 deposition was observed on smooth muscle cells in lungs from PQ + CR2-Crry–treated mice (Figure 5K), similar to that seen in control mice (Figure 5I). In addition, PQ-poisoned mice treated with CR2-Crry survived significantly longer than mice treated with PBS, with 3-day survival rates of 80 and 0%, respectively (Figure 5M) (P < 0.01). We also performed a study in which PQ-poisoned mice were treated with multiple doses of CR2-Crry (at 0, 8, and 24 hours after PQ injection), but there was no significant difference in any measurement compared with mice treated with a single injection (data not shown).
Figure 5.

CR2-Crry and CR2-factorH (fH) alleviate lung injury with less C3 deposition and prolong survival of mice with PQ-induced ALI, respectively. (A–D) Gross appearance of mouse lung 30 hours after PQ injection(n = 3 in each group): (A) normal mouse group; (B) PQ + PBS group; (C) PQ + CR2-Crry group; (D) PQ + CR2-fH group. (E–H) Histological examinations of lung tissues of mice 30 hours after PQ injection: (E) normal mouse group; (F) PQ + PBS group; (G) PQ + CR2-Crry group, (H) PQ + CR2-fH group. (I–L) C3 deposition at lung tissues of mice 30 hours after PQ injection (n = 3 in each group): (I) normal mouse group; (J) PQ + PBS group; (K) PQ + CR2-Crry group; (L) PQ + CR2-fH group. (M) Survival analysis in ALI model with CR2-Crry or CR2-fH intravenous administration 0 hours after PQ poisoning. **P < 0.01 and *** P < 0.001 relative to the corresponding PQ + CR2-Crry group or PQ + CR2-fH group with PQ + PBS group. Data presented are means (±SD); n = 5 per group. Magnification, ×200 (E–L).
The alternative pathway of complement activation appears to play a key role in causing tissue injury in a number of inflammatory conditions. We investigated the role of the alternative pathway in PQ poisoning by treating mice with CR2-fH, a targeted complement inhibitor that is specific for the alternative pathway. PQ-poisoned mice were injected intravenously with 0.8 mg CR2-fH immediately after PQ administration. We used an increased dose compared with CR2-Crry based on previously published efficacy data (26). At 30 hours after PQ injection, lungs from CR2-fH–treated mice appeared similar to lungs from CR2-Crry–treated mice, with minimal evidence of any focal hemorrhage, and only slight parenchymal expansion with minimal pulmonary neutrophil congestion (Figures 5D and 5H). C3 staining intensity and distribution was also similar between CR2-fH– and CR2-Crry–treated mice (Figure 5L). CR2-fH treatment also significantly improved survival (P < 0.001) of PQ-poisoned mice, and to a similar extent as CR2-Crry (Figure 5M).
The complement inhibitors used in the above studies have been shown to target to sites of complement activation via the CR2 moiety that binds the complement activation products, iC3b and C3 d (27). We therefore performed a CR2-Crry biodistribution study to investigate lung targeting and to determine whether there was any evidence of complement activation and C3 deposition at other anatomical sites after PQ poisoning. Radiolabeled CR2-Crry was administered intravenously immediately after PQ injection. At 4 hours after administration, 125I-labeled CR2-Crry was present at relatively high concentrations in the blood, and was also detected in most other tissues (Figure 6A). However, at 24 hours after administration, when circulatory levels had decreased, 125I-CR2-Crry was present at significantly higher levels (P < 0.01) in the lung compared with all other tissues. These data show preferential targeting (and retention) of CR2-Crry to the lung, and further indicate that there are no other sites of major complement activation after PQ poisoning in our model. CR2-Crry targeting to the lung was also confirmed by indirect immunofluorescence microscopy. At 4 hours after PQ and CR2-Crry administration, CR2-Crry was detected in lung interstitial tissue and on lung endothelium (Figure 6B). There was no evidence of CR2-Crry binding in lungs from control mice treated with PBS + CR2-Crry or PBS only (Figures 6C and 6D).
Figure 6.

Localization of CR2-Crry in tissues of PQ-poisoned mice treated with CR2-Crry immediately after PQ administration. (A) Biodistribution of CR2-Crry in main tissues of mice by radiolabeled 125I-CR2-Crry (2 μg) intravenous injection into control mice and groups of mice with 20 mg/kg PQ administration at 0 hours (n = 4–6 per group). Groups of mice were killed at 4 and 24 hours after 125I-CR2-Crry injection, and serum and other main tissues were collected for analysis. (B–D) Localization of CR2-Crry in lung tissues of mice 4 hours after PQ injection by indirect immunofluorescent staining with 7G6 antibody in PQ + CR2-Crry group (B), PQ + PBS group (C), and normal mouse group (D). CR2-Crry deposition is denoted by green fluorescence on interstitial tissue and vascular endothelium; n = 4–6 in each group. Data presented are means (±SD) (A). Magnification, ×200 (B–D).
Blockade of Anaphylatoxin Receptor Signaling Suppresses Lung Inflammation and Attenuates PQ-Induced ALI
Data presented previously here show that both C3aR mRNA and C5aR mRNA expression is up-regulated after PQ administration, suggesting a role for the complement activation products, C3a and C5a, in PQ-induced ALI. We therefore investigated the effect of C3aR and C5aR blockade on PQ poisoning. Gross examination and microscopic examination of lungs 30 hours after PQ injection revealed that treatment with either C3aR antagonist or blocking anti-C5aR antibody reduced focal hemorrhage, pulmonary neutrophil congestion, and parenchymal expansion (Figures 7A–7H). Furthermore, 4 hours after PQ20 poisoning, mice treated with C3aR antagonist had significantly reduced serum levels of IL-6, IL-10, monocyte chemoattractant protein-1, and TNF-α compared with control mice treated with saline (Figure 7I) (P < 0.01). Finally, both C3aR blockade and C5aR blockade significantly improved the survival of PQ-poisoned mice, with almost 50% long-term survival in the PQ + C3aR antagonist group (P < 0.05) and 25% survival in the PQ + C5aR mAb group (P < 0.01), compared with 0% survival in the PQ + PBS group (Figure 7J). To identify if there was additive effect in the combination of C5aR mAb and C3aR antagonist, we also tested a combination of C5aR mAb and C3aR antagonist treatment. The results show similar survival rates when compared with each treatment alone (data not shown). There was thus no additive effect in combining the treatment.
Figure 7.

Blockade of the binding of anaphylatoxin with their receptors alleviates lung injury and prolongs survival of mice with PQ-induced ALI. Four groups of mice were designed, with 11–13 mice in each group (saline, PQ + PBS, PQ + C3aR antagonist, and PQ + C5aR monoclonal antibody [mAb] groups). Mice received C5aR mAb 20/70 (600 μg/kg) or C3aR antagonist (1 mg/kg) by intravenous injection 50 minutes before and 2 hours after intraperitoneal injection of 20 mg/kg PQ. (A–D) Gross appearance of mouse lung 30 hours after PQ injection (n = 3). (A) PBS group, (B) PQ + PBS group, (C) PQ + C3aR antagonist group, and (D) PQ + C5aR antibody group. (E–H) Histological examinations of lung tissues of mice 30 hours after PQ injection in each group (n = 3). (E) PBS group, (F) PQ + PBS group, (G) PQ + C3aR antagonist group, and (H) PQ + C5aR antibody group. (I) Comparison of proinflammatory cytokine concentration in serum of PQ + PBS and PQ + C3aR antagonist groups 4 hours after PQ poisoning (n = 6). (J) Survival analysis of mice in each group after PQ administration (n = 8–10). *P < 0.05 and **P < 0.01 relative to the corresponding PQ + C3aR antagonist group or PQ + C5aR antibody group with PQ + PBS group. Data presented are means (±SD). Magnification, ×200 (E–H).
Discussion
PQ is a widely used herbicide which is highly toxic to humans and livestock. PQ poisoning presents as ALI, and claims many lives every year due principally to lack of an efficient clinical treatment and unknown pathogenic mechanisms. In recent years, ALI has been linked with complement activation (28, 29), but ALI can be dependent on different mechanisms, depending on how it is induced. For example, LPS-induced ALI (the most commonly used model of ALI) is neutrophil dependent, but complement independent (30), whereas immune complex–induced ALI and ALI secondary to ischemic injury has been shown to have a complement dependence (20, 31). The immunopathogenesis of PQ-induced ALI is very poorly understood, and the aim of the current study was to investigate the role of complement in PQ-induced ALI and the potential of complement inhibition for clinical intervention. In this study, we initially established a dose-dependent PQ-induced mouse model of ALI, and analyzed the kinetics of PQ toxicity. The results show that injuries induced by PQ are similar to those seen in clinical patients with ALI, such as lung congestion and edema, increased vascular permeability, and extensive neutrophil infiltration. Using this model, we demonstrated an important role for the complement system in the pathogenesis of PQ-induced ALI.
Complement can be activated via three distinct pathways: the classical, alternative, and lectin pathways. All pathways converge at the cleavage and activation of C3, with the subsequent generation of complement activation products with biological effector functions, namely, C3 opsonins, soluble C3a and C5a anaphylatoxins, and the cytolytic membrane attack complex. In the current study, we detected the complement activation and used specific complement inhibitors to demonstrate a key role for the alternative pathway of complement activation in PQ-induced ALI, as well as important roles for the complement activation products, C3a and C5a. Thus, the alternative pathway and the complement anaphylatoxins represent potential therapeutic targets for the treatment of PQ poisoning.
Although LPS-induced ALI has been shown to be complement independent in a mouse model, C1q levels are elevated 4 hours after LPS induction of ALI (29). C1q is the recognition molecule for the classical pathway, and is also recognized by receptors expressed on phagocytes, granulocytes, endothelial cells, and vascular smooth muscle cells. After tissue damage, C1q can reach levels of more than 35 μg/ml, and stimulates neutrophils, eosinophils, and other cells to produce reactive oxygen species, which could aggravate tissue injury (32, 33). C1q also directly interacts with endothelial cells, and specific blockade of C1q can protect against permeability edema and pulmonary injury (34). In our model, we observed intense C1q staining in lung endothelium and interstitial tissue by 24 hours after PQ poisoning. MBL belongs to the collectin family, and activates complement system after binding the carbohydrates on the surface of microorganisms (35). To identify if the MBL activation pathway participated the complement activation, MBL-A, which was found elevated in acute phase after LPS induction (36), was detected. The results showed no significant MBL-A staining after PQ poisoning, which indicates that MBL activation pathway did not participate in the complement activation induced after PQ poisoning (data not shown). Although these data, together with previous data, may indicate a role for the classical pathway in PQ-induced injury, the fact remains that CR2-Crry (inhibitor of all pathways) and CR2-fH (alternative pathway inhibitor) were similarly protective. Thus, although we cannot rule out a role for the classical pathway in initiating complement activation, the alternative pathway is key for driving injury in this model, possibly through amplification of the classical pathway.
Both C3a and C5a have well documented and multifunctional roles in lung inflammation, and, among other things, can increase vascular permeability, recruit and activate leukocytes, activate endothelial cells, up-regulate expression of adhesion molecules and cytokines, and induce goblet cells to secrete mucus (37). In this context, PQ-induced ALI was associated with high MPO activity, extensive neutrophil infiltration, and increased serum levels of proinflammatory cytokines. We therefore investigated the effect of C3aR and C5aR blockade on PQ-induced ALI, and our data demonstrating that blockade of either receptor was protective indicate an important role for both anaphylatoxins in PQ-induced ALI. Furthermore, PQ poisoning resulted in increased expression of both C3aR and C5aR. Both receptors are expressed on immune cells, as well as nonmyeloid cells, such as endothelial and epithelial cells (15, 38), and up-regulation of C5aR has been documented in pathological conditions, such as sepsis (39) and in response to lung toxins, such as cigarette smoke (40). Increased receptor expression is thought to predispose to elevated production of proinflammatory cytokines (40), and the rapid up-regulation of both C3aR and C5aR could therefore contribute to the increased proinflammatory milieu within the lungs. Although C3a is generally a less potent mediator than C5a, the serum concentration of C3 is 10 times higher than that of C5 (41), and the expression of C3aR is higher than that of C5aR at 4 hours after PQ administration. The data demonstrate that C3a and C5a, generated via the alternative activation pathway, both play key roles in PQ-induced ALI, and that blocking the effect of either peptide is similarly protective. Although C5a is generally regarded as a more potent proinflammatory mediator than C3a, these findings are in keeping with previous studies in a rat model of ARDS (42). In these previous studies, the authors demonstrated that C3a had profound systemic hemodynamic and systemic cytokine effects, whereas C5a exerted direct intralung modulation of neutrophil infiltration and cytokine production (42). In addition, importantly, we show that blockade of both C3a and C5a production by inhibition of C3 activation offers similar levels of protection, whether all complement activation pathways (CR2-Crry) or only the alternative pathway (CR2-fH) is inhibited.
Overall agreement from animal studies on the mechanisms of lung injury and death after acute PQ poisoning is that oxidative stress and inflammation play major roles (43–45). Accordingly, therapeutic interventions employing antioxidants/free radical scavengers and lung surfactants have been shown to prolong survival and/or increase eventual survival rate in a dose-dependent fashion. Nevertheless, Cho and colleagues (46) found that inhaled nitric oxide improves the survival of PQ-injured rats without altering inflammation parameters. There are, however, apparent contradictions in the effects of anti-inflammatory agents. Dinis-Oliveira and colleagues (47) found that dexemethosone improved survival of PQ-poisoned rats due to its anti-inflammatory activity, and another specific anti-inflammatory agent, Montelukast, increased the 7-day survival of PQ-poisoned rats from 30 to 80% by blocking the action of leukotriene D4 in the lungs and bronchial tubes (48). However, anti-inflammatory agent, prostaglandin E1 analog, accelerated death in PQ poisoning (49). Importantly, however, it was recently reported that immunosuppressive therapy saved approximately 25% of human patients from PQ poisoning (43). Data from these studies indicate that the inflammatory response after PQ poisoning is complex, and that some aspects of the response can be protective, whereas others can be injurious. The data reported here suggest that complement activation upon PQ poisoning is a part of excessive inflammatory response that is mainly harmful, and therefore inhibition of complement activation could alleviate early phase lung injury and improve survival. Thus, although our data show that complement inhibition does not provide complete protection, it may create an important time window for application of other effective treatments and interventions.
In summary, we report on the development of a model of PQ-induced ALI and demonstrate, for the first time, a role for complement in the pathogenesis of PQ poisoning. The study also identifies a complement activation pathway and complement activation products that represent potential therapeutic targets for the treatment of acute PQ poisoning, and possibly for postexposure prevention of PQ-induced ALI.
Acknowledgments
The authors thank Dr. Zaihai Xu for valuable comments and Yuchuan Li for technical assistance.
Footnotes
This work was supported by National Natural Science Foundation of China grants 30972616 and 81071538), 863 Project (2007AA02Z144).
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2010-0444OC on March 18, 2011
References
- 1.Onyeama HP, Oehme FW. A literature review of paraquat toxicity. Vet Hum Toxicol 1984;26:494–502. [PubMed] [Google Scholar]
- 2.Chan BS, Lazzaro VA, Seale JP, Duggin GG. The renal excretory mechanisms and the role of organic cations in modulating the renal handling of paraquat. Pharmacol Ther 1998;79:193–203. [DOI] [PubMed] [Google Scholar]
- 3.Dinis-Oliveira RJ, Remiao F, Carmo H, Duarte JA, Navarro AS, Bastos ML, Carvalho F. Paraquat exposure as an etiological factor of Parkinson's disease. Neurotoxicology 2006;27:1110–1122. [DOI] [PubMed] [Google Scholar]
- 4.Dinis-Oliveira RJ, De Jesus Valle MJ, Bastos ML, Carvalho F, Sanchez Navarro A. Kinetics of paraquat in the isolated rat lung: influence of sodium depletion. Xenobiotica 2006;36:724–737. [DOI] [PubMed] [Google Scholar]
- 5.Sittipunt C. Paraquat poisoning. Respir Care 2005;50:383–385. [PubMed] [Google Scholar]
- 6.Suntres ZE. Role of antioxidants in paraquat toxicity. Toxicology 2002;180:65–77. [DOI] [PubMed] [Google Scholar]
- 7.Bus JS, Aust SD, Gibson JE. Paraquat toxicity: proposed mechanism of action involving lipid peroxidation. Environ Health Perspect 1976;16:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonneh-Barkay D, Reaney SH, Langston WJ, Di Monte DA. Redox cycling of the herbicide paraquat in microglial cultures. Brain Res Mol Brain Res 2005;134:52–56. [DOI] [PubMed] [Google Scholar]
- 9.Ward PA. The dark side of C5a in sepsis. Nat Rev Immunol 2004;4:133–142. [DOI] [PubMed] [Google Scholar]
- 10.Robbins RA, Russ WD, Rasmussen JK, Clayton MM. Activation of the complement system in the adult respiratory distress syndrome. Am Rev Respir Dis 1987;135:651–658. [DOI] [PubMed] [Google Scholar]
- 11.Linton SM, Morgan BP. Complement activation and inhibition in experimental models of arthritis. Mol Immunol 1999;36:905–914. [DOI] [PubMed] [Google Scholar]
- 12.Welch TR. Complement in glomerulonephritis. Nat Genet 2002;31:333–334. [DOI] [PubMed] [Google Scholar]
- 13.Hawlisch H, Wills-Karp M, Karp CL, Kohl J. The anaphylatoxins bridge innate and adaptive immune responses in allergic asthma. Mol Immunol 2004;41:123–131. [DOI] [PubMed] [Google Scholar]
- 14.Hawlisch H, Belkaid Y, Baelder R, Hildeman D, Gerard C, Kohl J. C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity 2005;22:415–426. [DOI] [PubMed] [Google Scholar]
- 15.Guo RF, Riedemann NC, Ward PA. Role of C5a-C5aR interaction in sepsis. Shock 2004;21:1–7. [DOI] [PubMed] [Google Scholar]
- 16.Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol 2005;23:821–852. [DOI] [PubMed] [Google Scholar]
- 17.Kildsgaard J, Hollmann TJ, Matthews KW, Bian K, Murad F, Wetsel RA. Cutting edge: targeted disruption of the C3a receptor gene demonstrates a novel protective anti-inflammatory role for C3a in endotoxin-shock. J Immunol 2000;165:5406–5409. [DOI] [PubMed] [Google Scholar]
- 18.Baelder R, Fuchs B, Bautsch W, Zwirner J, Kohl J, Hoymann HG, Glaab T, Erpenbeck V, Krug N, Braun A. Pharmacological targeting of anaphylatoxin receptors during the effector phase of allergic asthma suppresses airway hyperresponsiveness and airway inflammation. J Immunol 2005;174:783–789. [DOI] [PubMed] [Google Scholar]
- 19.Dinis-Oliveira RJ, Remiao F, Duarte JA, Ferreira R, Navarro AS, Bastos ML, Carvalho F. P-glycoprotein induction: an antidotal pathway for paraquat-induced lung toxicity. Free Radic Biol Med 2006;41:1213–1224. [DOI] [PubMed] [Google Scholar]
- 20.De Steenwinkel JE, De Knegt GJ, Ten Kate MT, Van Belkum A, Verbrugh HA, Hernandez-Pando R, Van Soolingen D, Bakker-Woudenberg IA. Immunological parameters to define infection progression and therapy response in a well-defined tuberculosis model in mice. Int J Immunopathol Pharmacol 2009;22:723–734. [DOI] [PubMed] [Google Scholar]
- 21.Atkinson C, Song H, Lu B, Qiao F, Burns TA, Holers VM, Tsokos GC, Tomlinson S. Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection. J Clin Invest 2005;115:2444–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(t)) method. Methods 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 23.Goldblum SE, Wu KM, Jay M. Lung myeloperoxidase as a measure of pulmonary leukostasis in rabbits. J Appl Physiol 1985;59:1978–1985. [DOI] [PubMed] [Google Scholar]
- 24.Padilla-Docal B, Dorta-Contreras AJ, Fundora-Hernandez H, Noris-Garcia E, Bu-Coifiu-Fanego R, Gonzalez-Hernandez M, Rodriguez-Rey A. C3c intrathecal synthesis evaluation in patients with multiple sclerosis. Arq Neuropsiquiatr 2007;65:800–802. [DOI] [PubMed] [Google Scholar]
- 25.Markiewski MM, Mastellos D, Tudoran R, DeAngelis RA, Strey CW, Franchini S, Wetsel RA, Erdei A, Lambris JD. C3a and C3b activation products of the third component of complement (C3) are critical for normal liver recovery after toxic injury. J Immunol 2004;173:747–754. [DOI] [PubMed] [Google Scholar]
- 26.Huang Y, Qiao F, Atkinson C, Holers VM, Tomlinson S. A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury. J Immunol 2008;181:8068–8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiao F, Atkinson C, Song H, Pannu R, Singh I, Tomlinson S. Complement plays an important role in spinal cord injury and represents a therapeutic target for improving recovery following trauma. Am J Pathol 2006;169:1039–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun L, Guo RF, Gao H, Sarma JV, Zetoune FS, Ward PA. Attenuation of IgG immune complex–induced acute lung injury by silencing C5aR in lung epithelial cells. FASEB J 2009;23:3808–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bolger MS, Ross DS, Jiang H, Frank MM, Ghio AJ, Schwartz DA, Wright JR. Complement levels and activity in the normal and LPS-injured lung. Am J Physiol Lung Cell Mol Physiol 2007;292:L748–L759. [DOI] [PubMed] [Google Scholar]
- 30.Rittirsch D, Flierl MA, Day DE, Nadeau BA, McGuire SR, Hoesel LM, Ipaktchi K, Zetoune FS, Sarma JV, Leng L, et al. . Acute lung injury induced by lipopolysaccharide is independent of complement activation. J Immunol 2008;180:7664–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fleming SD, Mastellos D, Karpel-Massler G, Shea-Donohue T, Lambris JD, Tsokos GC. C5a causes limited, polymorphonuclear cell-independent, mesenteric ischemia/reperfusion–induced injury. Clin Immunol 2003;108:263–273. [DOI] [PubMed] [Google Scholar]
- 32.Lindgren S, Laurell AB, Eriksson S. Complement components and activation in primary biliary cirrhosis. Hepatology 1984;4:9–14. [DOI] [PubMed] [Google Scholar]
- 33.Otabor I, Tyagi S, Beurskens FJ, Ghiran I, Schwab P, Nicholson-Weller A, Klickstein LB. A role for lipid rafts in C1q-triggered O2− generation by human neutrophils. Mol Immunol 2004;41:185–190. [DOI] [PubMed] [Google Scholar]
- 34.Oroszlan M, Daha MR, Cervenak L, Prohaszka Z, Fust G, Roos A. MBL and C1q compete for interaction with human endothelial cells. Mol Immunol 2007;44:1150–1158. [DOI] [PubMed] [Google Scholar]
- 35.Ikeda K, Sannoh T, Kawasaki N, Kawasaki T, Yamashina I. Serum lectin with known structure activates complement through the classical pathway. J Biol Chem 1987;262:7451–7454. [PubMed] [Google Scholar]
- 36.Liu H, Jensen L, Hansen S, Petersen SV, Takahashi K, Ezekowitz AB, Hansen FD, Jensenius JC, Thiel S. Characterization and quantification of mouse mannan-binding lectins (MBL-A and MBL-C) and study of acute phase responses. Scand J Immunol 2001;53:489–497. [DOI] [PubMed] [Google Scholar]
- 37.Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol 2007;171:715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monsinjon T, Gasque P, Chan P, Ischenko A, Brady JJ, Fontaine MC. Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J 2003;17:1003–1014. [DOI] [PubMed] [Google Scholar]
- 39.Guo RF, Riedemann NC, Bernacki KD, Sarma VJ, Laudes IJ, Reuben JS, Younkin EM, Neff TA, Paulauskis JD, Zetoune FS, et al. . Neutrophil C5a receptor and the outcome in a rat model of sepsis. FASEB J 2003;17:1889–1891. [DOI] [PubMed] [Google Scholar]
- 40.Allen-Gipson DS, Floreani AA, Heires AJ, Sanderson SD, MacDonald RG, Wyatt TA. Cigarette smoke extract increases C5a receptor expression in human bronchial epithelial cells. J Pharmacol Exp Ther 2005;314:476–482. [DOI] [PubMed] [Google Scholar]
- 41.Abe M. Complement activation and inflammation. Rinsho Byori 2006;54:744–756. [PubMed] [Google Scholar]
- 42.Proctor LM, Strachan AJ, Woodruff TM, Mahadevan IB, Williams HM, Shiels IA, Taylor SM. Complement inhibitors selectively attenuate injury following administration of cobra venom factor to rats. Int Immunopharmacol 2006;6:1224–1232. [DOI] [PubMed] [Google Scholar]
- 43.Agarwal R, Srinivas R, Aggarwal AN, Gupta D. Immunosuppresive therapy in lung injury due to paraquat poisoning: a meta-analysis. Singapore Med J 2007;48:1000–1005. [PubMed] [Google Scholar]
- 44.Dinis-Oliveira RJ, Pontes H, Bastos ML, RemiĐo F, Duarte JA, Carvalho F. An effective antidote for paraquat poisonings: the treatment with lysine acetylsalicylate. Toxicology 2009;255:187–193. [DOI] [PubMed] [Google Scholar]
- 45.Saibara T, Toda K, Wakatsuki A, Ogawa Y, Ono M, Onishi S. Protective effect of 3-methyl-1-phenyl-2-pyrazolin-5-one, a free radical scavenger, on acute toxicity of paraquat in mice. Toxicol Lett 2003;143:51–54. [DOI] [PubMed] [Google Scholar]
- 46.Cho JH, Yang DK, Kim L, Ryu JS, Lee HL, Lim CM, Koh YS. Inhaled nitric oxide improves the survival of the paraquat-injured rats. Vascul Pharmacol 2005;42:171–178. [DOI] [PubMed] [Google Scholar]
- 47.Dinis-Oliveira RJ, Duarte JA, RemiĐo F, SÃnchez-Navarro A, Bastos ML, Carvalho F. Single high dose dexamethasone treatment decreases the pathological score and increases the survival rate of paraquat-intoxicated rats. Toxicology 2006;227:73–85. [DOI] [PubMed] [Google Scholar]
- 48.Ahmed AA. Protective effect of montelukast on paraquat-induced lung toxicity in rats. Biosci Trends 2009;3:63–72. [PubMed] [Google Scholar]
- 49.Williams JH, Fairshter RD, Ulich TR, Crosby S, Chen M, Rosario L, Vaziri ND. Adverse effects of (15S)-15-methyl-prostaglandin E1 in normal and paraquat-exposed rats. Toxicol Appl Pharmacol 1988;92:330–334. [DOI] [PubMed] [Google Scholar]