

Abstract
Reactive oxygen species (ROS) have profound influences on cellular homeostasis. In excess, they can potentiate the oxidation of numerous molecules, including proteins, lipids, and nucleic acids, affecting function. Furthermore, ROS-mediated oxidation of proteins can directly or indirectly modulate gene expression via effects on redox-sensitive transcription factors or via effects on phospho-relay–mediated signal transduction. In doing so, ROS impact numerous fundamental cellular processes, and have thus been implicated as critical mediators of both homeostasis and disease pathogenesis. Vascular reduced nicotinamide adenine dinucleotide phosphate oxidase (NOX) is a major contributor of ROS within the lung. The generation of ROS in the pulmonary vasculature has a pivotal role in endothelial cell (EC) activation and function. Alterations in EC phenotype contribute to vascular tone, permeability, and inflammatory responses and, thus, have been implicated in numerous diseases of the lung, including pulmonary hypertension, ischemic–reperfusion injury, and adult respiratory distress syndrome. Thus, although a detailed understanding of NOX-derived ROS in pulmonary EC biology in the context of health and disease is nascent, there is mounting evidence implicating these enzymes as critical modifiers of diseases of the lung and pulmonary circulation. The purpose of this review is to focus specifically on known as well as putative roles for pulmonary EC NOX, with attention to studies on the intact lung.
Keywords: reduced nicotinamide adenine dinucleotide phosphate oxidase, pulmonary, endothelium
Clinical Relevance
NOX family members participate in a broad range of cell biological processes, including fibrosis, cytoskeletal rearrangements, cell migration, differentiation, growth, proliferation, and apoptosis. Their capacity to modify redox-sensitive signaling pathways and gene expression, as well as contribute to oxidative stress and injury, in part accounts for their diverse biological properties and their contribution to both health and disease.
The Reduced Nicotinamide Adenine Dinucleotide Phosphate Oxidase Family
A Source of Reactive Oxygen Species
Reactive oxygen species (ROS) are chemically reactive, oxygen-derived molecules that are generated during normal cellular metabolism via incomplete reduction of molecular oxygen. They consist of molecules with unpaired electrons, such as superoxide anions (O2−) and hydroxyl radical (OH), and molecules that have oxidizing propensity, but do not possess free electrons (e.g., hydrogen peroxide [H2O2] and hypochlorous acid [HOCl]). Superoxide anion is generally viewed as the primary ROS in cells, and is rapidly converted into other forms of ROS, including H2O2 and OH (1). In addition, O2− can react with other radicals, such as nitric oxide, (NO) to form the powerful oxidant, peroxynitrite (2). ROS are involved in the regulation of fundamental cellular activities, such as cell growth and differentiation (3). However, their overproduction and/or the presence of impaired antioxidant ability result(s) in “oxidative” stress, which can induce and propagate significant injury (4).
There are multiple enzymatic sources of ROS, including the enzymes of the respiratory chain (5, 6), xanthine oxidase (7, 8), cytochrome P450 (9), cyclo-oxygenase (10, 11), myeloperoxidase (12), uncoupled endothelial NO synthase (eNOS) (13–16), and reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) (17, 18). Of these, NOX family members are the only enzymes with the primary function of ROS generation: specifically, O2−. The effects of these enzymes are counterbalanced by endogenous antioxidant systems, which include the antioxidant enzymes catalase (19, 20), superoxide dismutases (21, 22), heme oxygenases (23, 24), thioredoxin (25, 26), and glutathione peroxidase (27). Under physiological conditions, ROS may have beneficial effects, playing a critical role in both signal transduction (28) and redox-sensitive gene expression (29, 30). The balance between the extent of ROS production and antioxidant processes determines the oxidative state of the cell. If there is disequilibrium shifted toward unopposed ROS, either from excess ROS production or insufficient antioxidant capacity, oxidative stress can occur. Oxidative stress can promote protein oxidation, lipid peroxidation, and DNA damage/mutations, leading to injury.
The NOX family of ROS-producing enzymes is increasingly recognized as a critical source of ROS in both physiologic and pathologic settings. The regulated production of ROS by NOX in phagocytic cells has been established as a critical determinant of efficient pathogen elimination (31–33). Inborn defects in this enzyme system are responsible for the development of chronic granulomatous disease (CGD), characterized by recurrent infections secondary to ineffective defense against bacterial and fungal pathogens (34, 35). The phagocytic oxidase, NOX2 (gp91phox), expressed predominantly in neutrophils and macrophages, is responsible for the respiratory burst necessary for microbicidal activity (36). The enzyme is inactive in resting leukocytes, but can be activated in response to microbes, such as bacterial and fungal pathogens. Activation is regulated in part by the assembly of the catalytic gp91phox subunit with the regulatory subunits (p22phox, p47phox, p40phox, and small GTPase Ras-related C3 botulinum toxin substrate [Rac]) (37–39). The enzyme catalyzes the transfer of electrons from NADPH to molecular oxygen, generating O2− and other forms of ROS secondarily (40) (Figure 1).
Figure 1.

Schematic of reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX)–mediated free radical production, domain structure, and putative molecular functions. Akt, protein kinase B; AP-1, activated protein-1; Et1, endothelin-1; HIF-1α, hypoxia-inducible factor 1-α; MAPK, mitogen-activated protein kinase; PTK, protein tyrosine kinase; Rac, Ras-related C3 botulinum toxin substrate; ROS, reactive oxygen species; RTK, receptor tyrosine kinase.
Signaling through ROS
ROS can modify signaling via the oxidation of reactive cysteines within certain target molecules, including phosphatases, kinases, and transcription factors, and thus NOX-derived ROS can influence both physiologic and pathophysiologic processes (41, 42). Post-translational modification of proteins by tyrosine phosphorylation is a major mechanism in the regulation of protein function, and thus is a critical determinant of many cell biological processes (43). Phosphorylation is dynamically regulated by the relative activities of specific protein tyrosine kinases and protein tyrosine phosphatases (44). The capacity of NOX-derived ROS to modify diverse redox-sensitive kinases and inhibit phosphatases accounts, in part, for the critical influence of NOX on a great number of cellular processes under homeostatic and pathologic conditions (45–47). In addition to these effects of NOX-derived ROS on signal transduction, NOX, via oxidation, can modify the activity of transcription factors containing cysteine residues, thus influencing redox-sensitive gene expression (48). Although contemporary cell and molecular biological techniques have rapidly expanded our understanding of the structure–function relationship, molecular regulations, and contribution of individual NOX isoforms in cell biologic processes, many critical questions on the roles of these enzymes in the intact lung remain to be determined.
Structure and Molecular Regulators
The NOX members (49–52) can be divided into three main groups based on domain organization (Figure 1). All NOX family members have transmembrane helices, which regulate electron transport across the membrane to generate O2− in the process of reducing oxygen. Within the six membrane-spanning domains of the molecule are two heme-binding sites (heme domain). NADPH and FAD binding sites are located within the cytoplasmic tail known as the flavin domain (53). This is the structural organization of NOX1, NOX3, NOX4, and the classic NOX2. In addition to this basic organization, NOX5 has a calcium-binding, calmodulin-like domain resulting in a calcium-regulated enzyme (54, 55). More recent studies point to phosphorylation of NOX5 as a key event in regulation of this enzyme, acting to sensitize it to lower levels of intracellular calcium (56). It is noteworthy that NOX5 is absent in the rodent genome, leading some investigators to speculate that it is a vestigial NOX isoform (57). An alternative interpretation is that this isoform has evolved for a highly specialized yet undefined function(s) (58). Because traditional loss-of-function studies in classic murine models of pulmonary disease are not possible for this isoform, the significance of NOX5 in the intact organism remains to be defined. The final group within the NOX family contains the dual oxygenase (DUOX) enzymes. These also share the NOX5-like calmodulin-like domain, in addition to peroxidase-homology domain, on the extracellular face of the membrane. This initially led to the suggestion that this domain may act as its own peroxidase (52). The exact function of the extracellular DUOX domain remains controversial (59), with additional studies suggesting that it may in fact function as a dismutase (60). For further details on the DUOX isoforms and their potential roles in airway biology, the reader is referred to the review by Fischer (61).
Multiple mechanisms regulate the generation of NOX-dependent ROS. The activity of NOX2 is regulated via its interactions with several regulatory subunits, including p22phox, p47phox, p67phox, p40phox, and the small GTPase Rac (62). Under resting conditions, the complex between NOX2 and p22phox at the membrane is inactive, and a second complex containing p47phox, p40phox, and p67phox is located within the cytosol. Upon stimulation, there is translocation of the cytosolic components to the membrane (63). The activation requires multiple molecular events, including protein phosphorylation, lipid metabolism, and guanine exchange by Rac. Protein kinase (PK) C isoforms are believed to be instrumental for p47phox phosphorylation; however, multiple other kinases have been implicated as upstream regulators of NOX activity in a context-specific manner, including mitogen-activated protein kinase (MAPKs; e.g., extracellular signal–regulated kinase 1/2 and p38 MAPK) (64). Thus, the enzyme can be activated both rapidly and with tight, multifaceted control.
Like NOX2, NOX1 activity also requires regulatory subunits. NOX1 also complexes with p22phox (65), as well as NOXO1 and NOXA1, homologs of p47phox and p67phox, respectively (66). Unlike p47phox, NOXO1 binds to the membrane under unstimulated conditions, indicating that activation is not regulated by translocation (66). NOX3 requires p22phox (67) and appears to require NOXO1 binding (68), as demonstrated by the phenotypic similarities between NOXO1 inactivation and NOX3 deficiency (69). The role of cytosolic subunits in the activity of NOX3 remains unclear and data remain conflicting (67, 68). Importantly, unlike NOXO1-deficient mice, p47phox−/− mice do not share phenotypic similarities with NOX3-deficient animals, providing genetic evidence that p47phox is not obligatory for NOX3 function. The activity of the other NOX enzymes (NOX4, NOX5, and DUOX) does not appear to be modulated by regulatory subunits. NOX4 is constitutively active, and appears to be regulated both by expression and/or post-translational modifications (70, 71). The three NOX enzymes with calmodulin-like domains, NOX5, DUOX1, and DUOX2, are presumably regulated, in part, by calcium signaling (72, 73). Furthermore, phosphorylation has been demonstrated to sensitize NOX5 to the modulatory effects of calcium (56). Similarly, in vitro studies suggest that DUOX1 and DUOX2 activities are modified by the activity of kinases: specifically, PKA and PKC, respectively (74).
Recently, additional binding proteins that enhance the activity of NOX homologs have been identified. One such protein is polymerase (DNA-directed) delta interacting protein 2 (Poldip2), which associates with p22phox, NOX1, and NOX4, and has been shown to colocalize with p22phox at sites of NOX4 localization (75). Poldip2 increases NOX4 enzymatic activity (without effect on NOX1) in aortic vascular smooth muscle (VSM) cells, resulting in increased production of ROS. Furthermore, overexpression of Poldip2 results in Rho activation, enhancement of focal adhesions, and an increase in stress fiber formation. Poldip2 colocalizes with NOX4 and p22phox in focal adhesions and along stress fibers, and appears to be responsible for this specific arrangement, as NOX4 and p22phox localization and stress fiber formation are impaired when Poldip2 is depleted with small interfering RNA. These findings could have great implications on cell migration, growth, and vascular remodeling in large vessels, as well as in the lung, where Podip2 is also expressed (75). Further studies are required to understand the functional relevance of this regulator in the intact lung.
Nonphagocytic Nox
NOX isoforms represent a major source of ROS within the respiratory system. Because the lung functions to exchange molecular gases, specifically, oxygen and carbon dioxide, between the atmosphere and the circulation, it is exposed to various environmental insults, including pathogenic, gaseous, and particulate agents. Both the insults themselves and the host responses can contribute to tissue dysfunction and injury characteristic of pulmonary disease processes. The NOX/DUOX isoforms are expressed in multiple resident cells within the lung, and their expression and/or activity is dynamically regulated by diverse physiologic stimuli. Beyond the classic role of phagocytic NOX2 in the innate immune response, mounting evidence indicates that other NOX isoforms play a role in normal lung physiology as well as pathologic states. The existence of nonphagocytic NOX enzymatic activity and enzymes became recognized in the 1990s. Although it was well appreciated that neutrophils were the prototypical source of ROS, the production of ROS from other cell types, including vascular endothelial cells (ECs), became increasingly recognized. ROS play a role in the genesis of both EC vasodilatory dysfunction and EC activation. Work done in isolated pulmonary ECs initially provided evidence that ROS generated in the context of hypoxia–reoxygenation was derived from an NOX-like enzyme (76). Similar studies using pharmacologic inhibition of NOX-like enzyme activity provided evidence that an NOX enzyme(s) influenced EC activation and phenotype in vitro (77).
Distribution in Nonphagocytic Cells
All seven NOX family members have been identified to date (NOX1–NOX5 and DUOX1 and DUOX2) in select nonphagocytic cells (78). Our understanding of the cellular and tissue distribution of NOX family members under physiologic and pathologic states is in evolution. At baseline, messenger RNA (mRNA) for NOX2 (51), DUOX1 (79), and NOX4 (51) are detected in the intact lung. Although phagocytic cells, such as alveolar macrophages, likely contribute significantly to lung-associated NOX2, nonphagocytic cells, including pulmonary ECs, express NOX2 (80). NOX4 is expressed in pulmonary ECs (80, 81), smooth muscle cells (82), and pulmonary fibroblasts (83). In airway epithelial cells, DUOX1 and DUOX2 appear to dominate (84). NOX1 is expressed mainly within the colonic epithelium (85) but has been described in pulmonary ECs and type II airway epithelial cells (86). The tissue distribution of NOX3 under baseline conditions is restricted to the inner ear (87), but has been described in the lung in the context of a murine emphysema model. Specifically, loss of the Toll-like receptor (TLR) 4 in transgenic mice has been linked to increased expression of NOX3 in the intact lung and in pulmonary ECs in vitro (88). NOX5 mRNA expression is described in the testis and lymphoid tissues (54); however, this isoform is expressed in isolated ECs (89), and may be seen at lower levels in pulmonary ECs than NOX4 (81). It is clear that the expression of NOX isoforms within a cell or tissue can be modified by stimuli. Thus, in the context of disease, NOX isoform expression patterns may change. Similarly, relatively low mRNA expression of any given NOX isoform does not preclude a critical role in a particular cell type or organ system.
NOX AND ENDOTHELIAL FUNCTION
Due to its strategic location, the vascular endothelium plays a critical role in maintaining vascular homeostasis. ECs form a semipermeable barrier between the blood and interstitium, and dynamically regulate numerous biologic processes, both in the maintenance of homeostasis and during disease. Through the synthesis and secretion of paracrine factors, cytoskeleton dynamics, and alterations in expression of adhesion molecules, ECs regulate vascular tone, vascular permeability, blood fluidity, smooth muscle phenotype, inflammatory cytokine production, and chemotactic responses. As a consequence, perturbations of EC function contribute to the pathophysiology of vascular disease.
ROS-Dependent Signaling in ECs
Vascular ECs express the classic phagocytic NOX (NOX2), as well as NOX1, NOX4, and NOX5. NOX3 may be induced within pulmonary ECs (88). Mounting evidence points to a critical role for NOX-derived ROS in EC biology (90, 91) (Figure 2). The effects of NOX on EC function and phenotype appear to be both direct and indirect in nature. Many of the critical signaling molecules involved in modulation of EC activation and function are redox sensitive. These notably include transcription factors, such as NF-κB, activated protein-1 (AP-1), hypoxia-inducible factor (HIF)-1α, and p53 (92). Thus, NOX-derived ROS can influence gene expression in ECs via redox-sensitive transcriptional effects. Furthermore, kinases, such as the MAPK (p38 and c-Jun N-terminal kinase), protein kinase B (Akt), and Src, and protein phosphatases, including protein tyrosine phosphatase (PTP–PEST), are components of redox-sensitive signaling pathways (41, 93–98) and may be targets of NOX-derived ROS. Because stimuli contributing to EC dysfunction are also associated with increased NOX enzyme activity, and this class of enzymes is the major source of EC ROS, it follows that NOX-derived ROS are critical determinants of both physiologic and pathologic EC responses.
Figure 2.
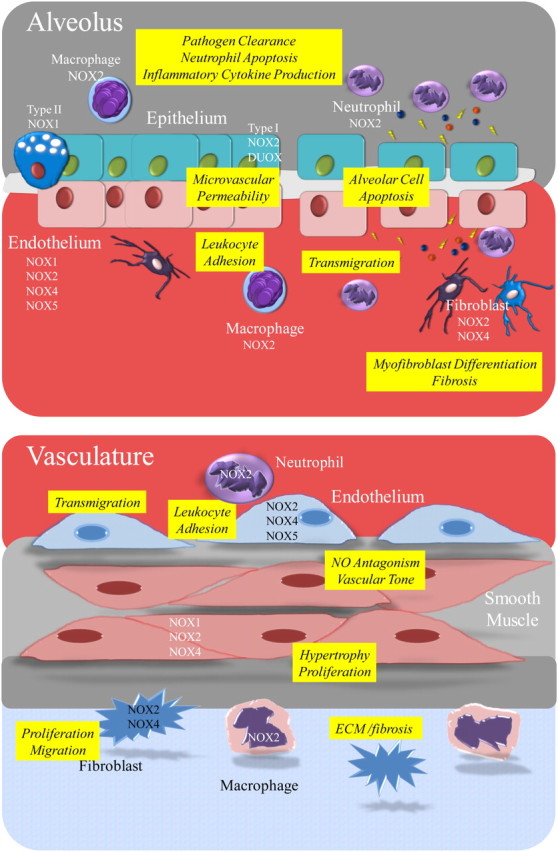
Schematic of NOX/dual oxygenase (DUOX) family member distribution within the alveolar and vascular compartments, and putative role of NOX in physiology and disease. ECM, extracellular matrix; NO, nitric oxide.
Endothelial Cell Activation
ROS signaling shapes numerous critical aspects of EC phenotype that have been implicated in pulmonary disease states. Hallmarks of EC activation are the surface expression of adhesion molecules (e.g., intercellular adhesion molecule-1, vascular cell adhesion molecule-1, and E-selectin), and secretion of monocyte chemoattractants. EC activation is also characterized by alterations in EC permeability. ROS signaling promotes EC permeability changes, which are associated with reversible rounding of the cells and development of paracellular gaps. Regulation of EC permeability is mediated through dynamic changes in the actin cytoskeleton that generate centripetal tension, and alterations in adhesive junctional proteins, which tether the cells (110). In addition, EC activation is an essential component of the inflammatory response through critical recruitment of leukocytes to the affected tissue compartment, which, when present in excess, can contribute to tissue injury. Thus, EC activation and associated barrier disruption are characteristic events in injurious inflammatory states, such as ischemia–reperfusion (IR) injury, sepsis, atherosclerosis, hypertension, hyperoxia, and acute lung injury.
An expanding body of evidence supports the role of ROS in EC activation in response to a number of stimuli, including oscillatory shear stress, endotoxin, cytokines, such as TNF-α, cigarette smoke, and hypercholesterolemia (111–113). NOX enzyme activity is sensitive to regulation by numerous stimuli implicated in a wide variety of pathophysiologic conditions, including agonists associated with EC activation. NOX2, for example, is activated by several such agonists, including cytokines (e.g., TNF-α), circulating factors (e.g., vascular endothelial growth factor [VEGF] and thrombin), metabolic factors, such as glucose, and environmental and mechanical stressors, including hypoxia–reoxygenation and shear stress (114–117). In contrast, NOX4 appears to be constitutively active in ECs, although NOX4-dependent ROS production can increase in response to the bacterial endotoxin LPS (101). This isoform appears to directly interact with the LPS receptor, TLR4, upon ligand binding (100, 101).
Vasomotor Effects
NOX-mediated oxidative stress affects vascular function via the capacity of superoxide anion to rapidly react with NO to produce peroxynitrite, and thus decrease NO bioavailablity (103). In addition, NOX-mediated ROS can potentiate the ROS-generating capacity of other enzymes, including xanthine oxidoreductase and eNOS (104). The vasocontrictive agonists of G protein–coupled receptors, angiotensin II and endothelin I, both activate EC NOX (105, 106). Furthermore, NOX can potentiate eNOS uncoupling through its capacity to mediate oxidative degradation of the eNOS cofactor, tetrahydrobiopterin, resulting in an enzyme that generates O2− and not NO (107). In a similar vein, xanthine dehydrogenase can be converted to xanthine oxidase with the capacity to generate O2− via NOX-derived oxidation (108). Because eNOS-generated NO is a major mediator of endothelium-dependent vasorelaxation, NOX-derived O2− can negatively influence vasodilation through direct and indirect effects on both NO and eNOS, respectively.
IR
In vivo human data on the role of NOX in vascular function is limited. Recent studies support a critical role for NOX in EC responses to IR injury in humans. Specifically, Loukogeorgakis and colleagues (109) characterized EC-dependent, flow-mediated vasodilation after IR in vivo in healthy volunteers compared with patients with NOX2 or p47phox-dependent CGD. Patients with both forms of CGD fail to demonstrate decreases in flow-mediated dilation after IR, a process mediated by endothelial-derived NO. This provides the first evidence that NOX-derived ROS contribute to EC dysfunction in humans in response to IR. There are insufficient clinical data to determine if patients with CGD are protected from other forms of IR injury, such as myocardial infarction or cerebrovascular stroke.
Proliferation and Angiogenesis
NOX-dependent signaling has been implicated in other critical EC phenotypic responses. EC responses to proliferative agonists and angiogenic factors, such as angiotensin II, hypoxia, and VEGF, require NOX-derived ROS (115, 118–120). NOX2 and NOX4 both contribute to EC proliferation under basal conditions (70). NOX4 plays a critical role in angiogenic responses in human microvascular ECs in culture (121). In vivo, the capacity of VEGF to stimulate angiogenesis is impaired in NOX2-deficient mice (122). NOX-derived ROS may also play a role in the mobilization and differentiation of stem/progenitor cells during neovascularization through ROS-mediated signaling and angiogenic gene expression (123).
Compartmentalization in ECs
Many ROS are diffusible, frequently short-lived, and reactive, and thus the site of generation is critical for determining the specificity of signaling. The temporal and spatial coupling of ROS production and activation of redox-sensitive signaling pathways requires localization of NOX with redox-modulated protein targets. Thus, specific spatial compartmentalization of NOX may influence both its capacity to be activated and, moreover, target NOX-derived ROS to select effectors. Although evidence supports a critical role of NOX-derived ROS in response to receptor signaling, the mechanisms underlying receptor-mediated activation of NOX are incompletely defined. NOX localization is pivotal in EC migration, a cellular function necessary for wound repair, angiogenesis, and injury resolution. Rac1, p47phox, and NOX2 are critical for EC migration, and are localized to the focal complexes at the cytoplasmic projections and leading edge of migrating cells known as lamellipodia. During migration, p47phox is targeted to the scaffolding protein, tumor necrosis factor receptor–associated factor 4 (TRAF4), that binds the scaffold protein, hydrogen peroxide–inducible gene 5 (Hic-5), at the focal contact. Thus, localized ROS signaling at lamellipodia is mediated by a complex of p47phox-TRAF4-Hic5 (124). Such localization at focal complexes also appears to occur at membrane ruffles generated during migration. Here, p47phox interacts with the cytoskeletal protein, Wiskott-Aldrich syndrome protein family member 1, along with Rac1 and p21 protein (Cdc42/Rac)-activated kinase 1 (PAK1) (125). In response to VEGF, PAK1 phosphorylates p47phox, resulting in localized ROS at the site of membrane ruffling (125). Additional actin-binding proteins, such as Ras GTPase-activating-like protein, have also been shown to link the cytoskeleton to Rac1 and NOX2, and appear to be required for EC migration (126).
Caveolae and lipid rafts are microdomains within the cytoplasmic membranes enriched for signaling molecules, including G protein–coupled receptors and receptor tyrosine kinases, among others (127). Therefore, caveolae and lipid rafts function as signaling platforms by compartmentalizing components of the signaling apparatus. NOX family members have been identified within lipid rafts and caveolae, and thus these localized interactions can target NOX-dependent ROS to these signaling platforms (128). In addition to the contribution of NOX localization in EC migration, NOX recruitment to lipid rafts has been implicated in death receptor signaling in ECs. Engagement of multiple death receptors by ligands, including Fas ligand, TNF-α, and endostatin, results in receptor clustering within lipid rafts in ECs (129). For example, Fas ligand recruits Rac1, NOX2, and p47phox to these lipid microdomains in ECs, leading to increased NOX activity, increased ROS, and, ultimately, EC dysfunction. Thus, localization of NOX components and death receptors results in formation of a redox-signaling platform.
NOX-dependent ROS have been implicated in other subcellular compartments, including endosomes and the nucleus. In the case of IL-1β receptor, ligand binding triggers receptor endocytosis (130). Rac1 and NOX2 are recruited to the early endosome, where NOX-dependent ROS production leads to TRAF6 recruitment and, ultimately, NF-κB activation (131). In human ECs, NOX4 is predominantly nuclear, and appears to contribute to nuclear ROS production under basal and stimulated conditions (81). Compartmentalization of NOX within the nucleus could thus facilitate effects on redox-sensitive transcription factors, such as p53, AP-1, NF-κB, and HIF-1α. Thus, the specificity of NOX-dependent events and their capacity to contribute to redox-sensitive signaling platforms may result from compartmentalization of the enzymes and their targets into subcellular organelles, such as endosomes and the nucleus. Alternatively, interactions between NOX family members with scaffolding proteins or lipid microdomains, such as caveolae or lipid rafts, can account for temporal and spatial compartmentalization.
Lessons From The Intact Lung
Hypoxic Pulmonary Vasoconstriction
In addition to pulmonary EC–derived NOX2, VSM expresses NOX1, NOX2, and NOX4 (135, 136). NOX4 has been proposed by some to be the predominant VSM NOX isoform (82). These NOX isoforms are determinants of systemic VSM hypertrophic and phenotypic responses (137), and are unregulated in vascular disease, playing pivotal roles in systemic hypertension and atherosclerosis (91, 138). Early studies implicated pulmonary smooth muscle NOX activity in oxygen sensing, and a potential molecular determinant of hypoxic pulmonary vasoconstriction (HPV) (139, 140). Studies in NOX2 (gp91phox) knockouts revealed normal HPV (141), arguing against the role of NOX2 in oxygen sensing. More recent ex vivo studies of pulmonary vascular rings implicate NOX4 as a key regulator of basal pulmonary vascular tone and antagonist of HPV (142). The contribution of NOX as an oxygen sensor in the pulmonary circulation remains a topic of debate and investigation, and the reader is directed to Wolin and colleagues (143) for further review.
Pulmonary Hypertension
Growing evidence implicates NOX-derived ROS in the pathogenesis of pulmonary hypertension (PH). Hypoxia, a known inducer of PH, increases O2− production in ECs in culture via a NOX-dependent pathway (76). The capacity of intact vessels to respond to the vasoconstrictor, endothelin-I (Et-I), is dependent on NOX2 expression and lost in vessels derived from NOX2−/− mice (132). In intact animals, NOX2−/− mice are protected from hypoxia-induced endothelial dysfunction, indicating that NOX-derived ROS is upstream of impaired NO-dependent relaxation in the pulmonary circulation (133). Furthermore, deletion of NOX2 in mice is protective from the effects of chronic hypoxia, indicating that NOX2-derived ROS is a critical determinant in the pathogenesis of vascular dysfunction in response to chronic hypoxia (134). Importantly, NOX4 expression has been demonstrated to be significantly elevated in the pulmonary vascular media in patients with idiopathic pulmonary arterial hypertension (144). These same studies provide evidence that NOX4 is increased in isolated pulmonary artery smooth muscle cells in response to hypoxia and is a critical determinant of both pulmonary artery smooth muscle cell ROS and proliferation in vitro. Interestingly, NOX4 is a transcriptional target of HIF-1α, and hypoxia-induced proliferation of VSM cells is NOX4 dependent (145). Thus, evidence continues to mount supporting the role of NOX isoforms in pathologic vascular remodeling in PH and as potential modulators in HPV.
Pulmonary Inflammation and Lung Injury
The recognition that human CGD was attributable to dysfunctional NOX (gp91phox/NOX2 in the X-linked form and p47phox in the recessive form) clearly highlights the critical role of NOX family members in host responses to pathogens (146). Pulmonary infectious complications are hallmarks of this disease and a major cause of morbidity and mortality. Although it is clear that NOX2 contributes to immune responses in humans, the characterization of gp91phox- and p47phox-deficient animals has provided evidence that NOX2-derived ROS may also suppress neutrophilic inflammation, thus playing an apparently paradoxical anti-inflammatory role. Mice deficient in NOX2 are more susceptible to Staphylococcus and Aspergillus infections, and develop elevated and persistent infection, with inflammatory responses to these organisms consistent with the role of NOX2 in organism clearance (147). Intriguingly, these mice (gp91phox−/−) demonstrate improved outcomes when challenged with other bacterial pathogens, such as Streptococcus pneumoniae (148), suggesting that NOX-derived ROS are not necessarily required for antimicrobial defense with these specific pathogens. Furthermore, despite the fact that pneumococcal pneumonia in gp91phox−/− animals is associated with increased inflammatory cytokine production, increased neutrophilic infiltrates, and lower rates of neutrophilic apoptosis, decreased NOX-derived ROS is associated with improved disease outcomes. Taken together, these results suggest that, in addition to bacterial clearance, NOX-generated ROS play a role in suppression of neutrophilic infiltrates within the lung, and thus have a potential anti-inflammatory role.
Further evidence supporting the capacity of NOX2-dependent ROS to influence the magnitude of inflammatory responses within the lung derives from studies of influenza virus in gp91phox−/− mice (149). These animals demonstrate a heightened inflammatory response, with increased macrophage and neutrophil numbers and reduced evidence of apoptosis. Again, in the face of a more robust inflammatory response, gp91phox deficiency results in improved resolution of the virus as well as lung function (149). Both pulmonary and systemic inflammatory responses to systemic infectious insults have been characterized in gp91phox−/− and p47phox−/− mice with the gram-negative bacteria, Escherichia coli, or its bacterial toxin, LPS (150, 151). Again, loss of NOX-derived ROS results in increased neutrophilic infiltration into the lung and alveolar space with associated increases in the chemoattractant chemokines and decreased bacterial clearance. Importantly, gp91phox−/− and p47phox−/− mice were protected from sepsis-induced microvascular permeability changes, indicating that NOX-derived ROS is necessary for microvascular injury in vivo (150).
NF-κB is a critical component of the endothelial inflammatory response and an essential component of the innate immune response (99). EC expression of adhesion molecules and inflammatory cytokines elaborated during lung injury are, in part, NF-κB dependent. ROS is a known trigger for the activation of NF-κB transcription factor complex (92). Recent studies have identified an obligatory role of NOX family members in efficient activation of NF-κB in response to engagement of pathogen recognition receptors: specifically, TLR4 (100). In particular, Park and colleagues (101) have demonstrated that LPS signaling via TLR4 requires NOX4 for both ROS production and NF-κB activation. Moreover, they provide evidence of a physical association between NOX4 and TLR4. Loss of NOX4 via RNA interference in human ECs blocks both LPS-induced adhesion molecule expression (specifically, intercellular adhesion molecule-1) and generation of proinflammatory cytokines, including IL-8 and monocyte chemotactic protein-1 (101). In addition to its role in pathogen recognition, TLR4 also participates in recognition of noninfectious ligands, such as the free fatty acids (102). Recently, NOX4 has been implicated in the capacity of free fatty acids to activate NF-κB and generate inflammatory responses in human ECs via a TLR4-dependent pathway (102). Thus, NF-κB–dependent vascular inflammation triggered by both infectious and noninfectious insults requires NOX4-dependent ROS.
Noninfectious models of lung injury and inflammation have also incriminated NOX family members as critical in vivo determinants of both lung inflammation and tissue remodeling. NOX2 has been implicated as a determinant of acid aspiration–induced lung injury (152). Here, p47phox−/− animals demonstrate enhanced neutrophilic infiltrates and proinflammatory cytokines/chemokines after acid installation into the lung, as well as enhanced bronchoalveolar albumin, suggestive of alveolar–capillary leak. In a more chronic injury model of cigarette exposure, NOX-derived ROS again play a role in regulating the magnitude of inflammatory response (153). Surprisingly, loss of gp91phox or p47phox results in enhanced sensitivity to cigarette smoke–induced alveolar damage, and correlates with increased signaling via the TLR4–NFκ-B pathway. It is noteworthy that the deletion of gp91phox and p47phox is associated with enhanced expression of NOX4 after cigarette smoke exposure in these animals, suggesting the potential for a compensatory pathway and identifying a potential confounder. p47phox deficiency is also protective in the face of IR injury, with decreased neutrophilic infiltration and permeability changes (154). Analysis of NOX1-deficient transgenic mice indicate that NOX1, and not NOX2, is critical for hyperoxia-induced lung injury in vivo (86). NOX1 is predominantly expressed within ECs and type II epithelial cells within the lung, and its deficiency is associated with decreased hyperoxia-induced ROS, both in vivo and in isolated epithelial and endothelial cells in vitro. Furthermore, NOX1 deficiency results in blunting of stress kinase activation/phosphorylation (e.g., extracellular signal–regulated kinase 1/2 and Jun N-terminal kinase) and alveolar epithelial and endothelial cell apoptosis, implicating NOX1-derived ROS in signaling in this model of lung injury. Intriguingly, NOX2 deficiency is not found to be protective in this noninfectious model (86).
Pulmonary Fibrosis
In addition to the role of NOX family members in these early stages of lung injury (e.g., inflammatory cell infiltration, microvascular permeability, and alveolar cell apoptosis), recent evidence supports the role of NOX4 in the later fibrogenic response to lung injury. Hecker and colleagues (155) recently demonstrated that NOX4 is up-regulated in noninfectious models of lung injury and in humans with the fibrotic lung disease, idiopathic pulmonary fibrosis. NOX4 mRNA is expressed at increased levels in fibroblasts isolated from patients with idiopathic pulmonary fibrosis (156). Loss-of-function analysis in vitro suggests that NOX4 plays an obligatory role in fibroblast proliferation, migration, and production of both collagen and profibrotic cytokines (156). Furthermore, using genetic and pharmacologic antagonism of NOX4 in vivo, this group provides evidence that NOX4 is required for pulmonary fibrosis contributing to myofibroblast differentiation and extracellular matrix production.
Thus, evidence from cell culture data implicating the contribution of NOX-dependent ROS in the activation of ECs in response to various agonists arises from a combination of approaches. These have included the use of antioxidants, pharmacologic inhibitors of the enzymes, dominant negative Rac1, RNA interference, and cells derived from transgenic animals. In vivo studies with genetic deletion of gp91phox or p47phox also implicate NOX-derived ROS in endothelial activation in vivo, but interpretation of such studies is currently limited since cell/tissue–restricted knockout animals are not yet available. In vivo data indicate that NOX family members play pivotal roles in both early and late events in infectious and noninfectious modes of lung injury, vascular tone/remodeling, and pulmonary fibrosis. Development and characterization of cell type–restricted and conditional transgenics will be critical to define the relevant NOX isoforms and relevant cell types responsible for these phenotypes in vivo and ultimately define the role of these family members in distinct stages of injury, remodeling, and resolution within the lung.
Future Directions
Our current knowledge of the functional role of NOX isoforms in vivo generally, and in the lung specifically, has been shaped predominantly by our understanding of the localization of the isoforms in situ and functional analysis of global knockout mice. The limitations of both types of analysis should be recognized. Prudent interpretation of isoform localization studies is necessary in light of the potential for nonspecific antibody cross-reactivity. In addition, transgenic animals with global loss of function cannot address the role of isoforms within defined cells or compartments. Furthermore, in vitro studies suggest that loss of one isoform may modify expression of alternative isoforms, confounding interpretation of such results. Thus, although evidence continues to mount regarding the importance of NOX family members in redox signaling and oxidative stress, both in health and disease, critical questions remain unanswered. At the molecular and cellular levels, a greater knowledge of function of individual family members and tissue distribution is needed. Furthermore, better understanding of the physiologic stimuli and regulators of their expression and activity is critical, as is the need to characterize and define the role of subcellular compartmentalization in regulation of effects. Currently, one major obstacle in NOX research is the lack of available specific inhibitors. A greater understanding of both the function and regulation of the different NOX family members will be valuable to developing selective and specific inhibitors. Tissue-restricted gene deletion and selective pharmacologic and/or molecular inhibitors will be necessary to more clearly define the contribution of individual NOX family members to pulmonary injury and repair processes. Ultimately, selective pharmacologic or molecular inhibitors will be required to move the field into an in-depth understanding of the role of these enzymes in human physiology and disease states.
Conclusions
NOX family members participate in a broad range of cell biological processes, including fibrosis, cytoskeletal rearrangements, cell migration, differentiation, growth, proliferation, and apoptosis. Their capacity to modify redox-sensitive signaling pathways and gene expression, as well as contribute to oxidative stress and injury, in part accounts for their diverse biological properties and their contribution to both health and disease. During the last decade and a half, our understanding of the role and regulation of these enzymes has grown exponentially. Recent in vivo animal studies and limited human studies have generated greater insight into the contribution of NOX family members in development and resolution of diverse forms of vascular and lung pathology. The development of selective and specific inhibitors will allow for a greater understanding of the contribution of these ROS-generating enzymes, both in human health and disease states.
Footnotes
This work was supported by National Heart, Lung, and Blood Institute grants K08HL088320 (R.D.) and R0HL049441 (P.M.H.).
Originally Published in Press as DOI: 10.1165/rcmb.2010-0331RT on April 12, 2012
References
- 1.Bors W, Saran M, Lengfelder E, Spottl R, Michel C. The relevance of the superoxide anion radical in biological systems. Curr Top Radiat Res Q 1974;9:247–309. [PubMed] [Google Scholar]
- 2.Pryor WA, Squadrito GL. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am J Physiol 1995;268:L699–L722. [DOI] [PubMed] [Google Scholar]
- 3.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 2007;39:44–84. [DOI] [PubMed] [Google Scholar]
- 4.Gerschman R, Gilbert D, Nye SW, Dwyer P, Fenn WO. Oxygen poisoning and X-irradiation: a mechanism in common. 1954. Nutrition 2001;17:162. [PubMed] [Google Scholar]
- 5.Littauer A, de Groot H. Release of reactive oxygen by hepatocytes on reoxygenation: three phases and role of mitochondria. Am J Physiol 1992;262:G1015–G1020. [DOI] [PubMed] [Google Scholar]
- 6.Nohl H, Breuninger V, Hegner D. Influence of mitochondrial radical formation on energy-linked respiration. Eur J Biochem 1978;90:385–390. [DOI] [PubMed] [Google Scholar]
- 7.Winterbourn CC, Sutton HC. Iron and xanthine oxidase catalyze formation of an oxidant species distinguishable from OH·: comparison with the Haber-Weiss reaction. Arch Biochem Biophys 1986;244:27–34. [DOI] [PubMed] [Google Scholar]
- 8.Pederson TC, Aust SD. The role of superoxide and singlet oxygen in lipid peroxidation promoted by xanthine oxidase. Biochem Biophys Res Commun 1973;52:1071–1078. [DOI] [PubMed] [Google Scholar]
- 9.Bondy SC, Naderi S. Contribution of hepatic cytochrome P450 systems to the generation of reactive oxygen species. Biochem Pharmacol 1994;48:155–159. [DOI] [PubMed] [Google Scholar]
- 10.Cheung K, Archibald AC, Robinson MF. The origin of chemiluminescence produced by neutrophils stimulated by opsonized zymosan. J Immunol 1983;130:2324–2329. [PubMed] [Google Scholar]
- 11.Niwa K, Haensel C, Ross ME, Iadecola C. Cyclooxygenase-1 participates in selected vasodilator responses of the cerebral circulation. Circ Res 2001;88:600–608. [DOI] [PubMed] [Google Scholar]
- 12.Paul BB, Strauss RR, Jacobs AA, Sbarra AJ. Function of H(2)O(2), myeloperoxidase, and hexose monophosphate shunt enzymes in phagocytizing cells from different species. Infect Immun 1970;1:338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Munzel T, Li H, Mollnau H, Hink U, Matheis E, Hartmann M, Oelze M, Skatchkov M, Warnholtz A, Duncker L, et al. Effects of long-term nitroglycerin treatment on endothelial nitric oxide synthase (NOS III) gene expression, NOS III–mediated superoxide production, and vascular NO bioavailability. Circ Res 2000;86:E7–E12. [DOI] [PubMed] [Google Scholar]
- 14.Vergnani L, Hatrik S, Ricci F, Passaro A, Manzoli N, Zuliani G, Brovkovych V, Fellin R, Malinski T. Effect of native and oxidized low-density lipoprotein on endothelial nitric oxide and superoxide production: key role of L-arginine availability. Circulation 2000;101:1261–1266. [DOI] [PubMed] [Google Scholar]
- 15.Pritchard KA, Groszek L, Smalley DM, Sessa WC, Wu M, Villalon P, Wolin MS, Stemerman MB. Native low-density lipoprotein increases endothelial cell nitric oxide synthase generation of superoxide anion. Circ Res 1995;77:510–518. [DOI] [PubMed] [Google Scholar]
- 16.Vasquez-Vivar J, Martasek P, Whitsett J, Joseph J, Kalyanaraman B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: an EPR spin trapping study. Biochem J 2002;362:733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baehner RL, Nathan DG, Karnovsky ML. The metabolic and bactericidal defect in chronic granulomatous disease. Vox Sang 1969;17:35–36. [PubMed] [Google Scholar]
- 18.Baehner RL, Gilman N, Karnovsky ML. Respiration and glucose oxidation in human and guinea pig leukocytes: comparative studies. J Clin Invest 1970;49:692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loew O. A new enzyme of general occurrence in organisms. Science 1900;11:701–702. [DOI] [PubMed] [Google Scholar]
- 20.Foulkes EC, Lemberg R. The formation of choleglobin and the role of catalase in the erythrocyte. Proc R Soc Lond B Biol Sci 1949;136:435–448. [DOI] [PubMed] [Google Scholar]
- 21.McCord JM, Fridovich I. Superoxide dismutase: an enzymic function for erythrocuprein (hemocuprein). J Biol Chem 1969;244:6049–6055. [PubMed] [Google Scholar]
- 22.McCord JM, Fridovich I. The utility of superoxide dismutase in studying free radical reactions. I. Radicals generated by the interaction of sulfite, dimethyl sulfoxide, and oxygen. J Biol Chem 1969;244:6056–6063. [PubMed] [Google Scholar]
- 23.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol 1997;37:517–554. [DOI] [PubMed] [Google Scholar]
- 24.Yoshida T, Kikuchi G. Sequence of the reaction of heme catabolism catalyzed by the microsomal heme oxygenase system. FEBS Lett 1974;48:256–261. [DOI] [PubMed] [Google Scholar]
- 25.Laurent TC, Moore EC, Reichard P. Enzymatic synthesis of deoxyribonucleotides. IV. Isolation and characterization of thioredoxin, the hydrogen donor from Escherichia coli B. J Biol Chem 1964;239:3436–3444. [PubMed] [Google Scholar]
- 26.Casagrande S, Bonetto V, Fratelli M, Gianazza E, Eberini I, Massignan T, Salmona M, Chang G, Holmgren A, Ghezzi P. Glutathionylation of human thioredoxin: a possible crosstalk between the glutathione and thioredoxin systems. Proc Natl Acad Sci USA 2002;99:9745–9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mills GC. Hemoglobin catabolism. I. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. J Biol Chem 1957;229:189–197. [PubMed] [Google Scholar]
- 28.Nathan C. Specificity of a third kind: reactive oxygen and nitrogen intermediates in cell signaling. J Clin Invest 2003;111:769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med 2008;45:549–561. [DOI] [PubMed] [Google Scholar]
- 30.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol 2008;4:278–286. [DOI] [PubMed] [Google Scholar]
- 31.Babior BM, Peters WA. The O2–producing enzyme of human neutrophils: further properties. J Biol Chem 1981;256:2321–2323. [PubMed] [Google Scholar]
- 32.Cheson BD, Curnette JT, Babior BM. The oxidative killing mechanisms of the neutrophil. Prog Clin Immunol 1977;3:1–65. [PubMed] [Google Scholar]
- 33.Babior BM. The respiratory burst of phagocytes. J Clin Invest 1984;73:599–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curnutte JT, Kipnes RS, Babior BM. Defect in pyridine nucleotide dependent superoxide production by a particulate fraction from the cranulocytes of patients with chronic granulomatous disease. N Engl J Med 1975;293:628–632. [DOI] [PubMed] [Google Scholar]
- 35.Curnutte JT, Babior BM. Chronic granulomatous disease. Adv Hum Genet 1987;16:229–297. [DOI] [PubMed] [Google Scholar]
- 36.Rotrosen D, Yeung CL, Leto TL, Malech HL, Kwong CH. Cytochrome b558: the flavin-binding component of the phagocyte NADPH oxidase. Science 1992;256:1459–1462. [DOI] [PubMed] [Google Scholar]
- 37.Park JW, Babior BM. The translocation of respiratory burst oxidase components from cytosol to plasma membrane is regulated by guanine nucleotides and diacylglycerol. J Biol Chem 1992;267:19901–19906. [PubMed] [Google Scholar]
- 38.Okamura N, Babior BM, Mayo LA, Peveri P, Smith RM, Curnutte JT. The p67-phox cytosolic peptide of the respiratory burst oxidase from human neutrophils: functional aspects. J Clin Invest 1990;85:1583–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.el Benna J, Ruedi JM, Babior BM. Cytosolic guanine nucleotide–binding protein Rac2 operates in vivo as a component of the neutrophil respiratory burst oxidase: transfer of Rac2 and the cytosolic oxidase components p47phox and p67phox to the submembranous actin cytoskeleton during oxidase activation. J Biol Chem 1994;269:6729–6734. [PubMed] [Google Scholar]
- 40.McPhail LC, DeChatelet LR, Shirley PS. Further characterization of NADPH oxidase activity of human polymorphonuclear leukocytes. J Clin Invest 1976;58:774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fialkow L, Chan CK, Rotin D, Grinstein S, Downey GP. Activation of the mitogen-activated protein kinase signaling pathway in neutrophils: role of oxidants. J Biol Chem 1994;269:31234–31242. [PubMed] [Google Scholar]
- 42.Haque SJ, Flati V, Deb A, Williams BR. Roles of protein-tyrosine phosphatases in Stat1 alpha-mediated cell signaling. J Biol Chem 1995;270:25709–25714. [DOI] [PubMed] [Google Scholar]
- 43.Zor U, Ferber E, Gergely P, Szucs K, Dombradi V, Goldman R. Reactive oxygen species mediate phorbol ester–regulated tyrosine phosphorylation and phospholipase A2 activation: potentiation by vanadate. Biochem J 1993;295:879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiarugi P. PTPs versus PTKs: the redox side of the coin. Free Radic Res 2005;39:353–364. [DOI] [PubMed] [Google Scholar]
- 45.Nasmith PE, Mills GB, Grinstein S. Guanine nucleotides induce tyrosine phosphorylation and activation of the respiratory burst in neutrophils. Biochem J 1989;257:893–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grinstein S, Furuya W. Tyrosine phosphorylation and oxygen consumption induced by G proteins in neutrophils. Am J Physiol 1991;260:C1019–C1027. [DOI] [PubMed] [Google Scholar]
- 47.Fialkow L, Chan CK, Grinstein S, Downey GP. Regulation of tyrosine phosphorylation in neutrophils by the NADPH oxidase: role of reactive oxygen intermediates. J Biol Chem 1993;268:17131–17137. [PubMed] [Google Scholar]
- 48.Sun Y, Oberley LW. Redox regulation of transcriptional activators. Free Radic Biol Med 1996;21:335–348. [DOI] [PubMed] [Google Scholar]
- 49.Hohn DC, Lehrer RI. NADPH oxidase deficiency in X-linked chronic granulomatous disease. J Clin Invest 1975;55:707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Banfi B, Maturana A, Jaconi S, Arnaudeau S, Laforge T, Sinha B, Ligeti E, Demaurex N, Krause KH. A mammalian H+ channel generated through alternative splicing of the NADPH oxidase homolog NOH-1. Science 2000;287:138–142. [DOI] [PubMed] [Google Scholar]
- 51.Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of NOX3, NOX4, and NOX5. Gene 2001;269:131–140. [DOI] [PubMed] [Google Scholar]
- 52.Edens WA, Sharling L, Cheng G, Shapira R, Kinkade JM, Lee T, Edens HA, Tang X, Sullards C, Flaherty DB, et al. Tyrosine cross-linking of extracellular matrix is catalyzed by DUOX, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J Cell Biol 2001;154:879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu H, Qiu H, Yoon HW, Huang S, Bunn HF. Identification of a cytochrome b-type NAD(P)H oxidoreductase ubiquitously expressed in human cells. Proc Natl Acad Sci USA 1999;96:14742–14747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banfi B, Molnar G, Maturana A, Steger K, Hegedus B, Demaurex N, Krause KH. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. J Biol Chem 2001;276:37594–37601. [DOI] [PubMed] [Google Scholar]
- 55.Banfi B, Tirone F, Durussel I, Knisz J, Moskwa P, Molnar GZ, Krause KH, Cox JA. Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5). J Biol Chem 2004;279:18583–18591. [DOI] [PubMed] [Google Scholar]
- 56.Jagnandan D, Church JE, Banfi B, Stuehr DJ, Marrero MB, Fulton DJ. Novel mechanism of activation of NADPH oxidase 5: calcium sensitization via phosphorylation. J Biol Chem 2007;282:6494–6507. [DOI] [PubMed] [Google Scholar]
- 57.Kawahara T, Quinn MT, Lambeth JD. Molecular evolution of the reactive oxygen–generating NADPH oxidase (NOX/DUOX) family of enzymes. BMC Evol Biol 2007;7:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fulton DJ. NOX5 and the regulation of cellular function. Antioxid Redox Signal 2009;11:2443–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Donko A, Peterfi Z, Sum A, Leto T, Geiszt M. Dual oxidases. Philos Trans R Soc Lond B Biol Sci 2005;360:2301–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ameziane-El-Hassani R, Morand S, Boucher JL, Frapart YM, Apostolou D, Agnandji D, Gnidehou S, Ohayon R, Noel-Hudson MS, Francon J, et al. Dual oxidase-2 has an intrinsic Ca2+-dependent H2O2-generating activity. J Biol Chem 2005;280:30046–30054. [DOI] [PubMed] [Google Scholar]
- 61.Fischer H. Mechanisms and function of DUOX in epithelia of the lung. Antioxid Redox Signal 2009;11:2453–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Diekmann D, Abo A, Johnston C, Segal AW, Hall A. Interaction of Rac with p67phox and regulation of phagocytic NADPH oxidase activity. Science 1994;265:531–533. [DOI] [PubMed] [Google Scholar]
- 63.Zhao X, Bey EA, Wientjes FB, Cathcart MK. Cytosolic phospholipase A2 (cPLA2) regulation of human monocyte NADPH oxidase activity: cPLA2 affects translocation but not phosphorylation of p67(phox) and p47(phox). J Biol Chem 2002;277:25385–25392. [DOI] [PubMed] [Google Scholar]
- 64.Yamamori T, Inanami O, Nagahata H, Cui Y, Kuwabara M. Roles of p38 MAPK, PKC and PI3-K in the signaling pathways of NADPH oxidase activation and phagocytosis in bovine polymorphonuclear leukocytes. FEBS Lett 2000;467:253–258. [DOI] [PubMed] [Google Scholar]
- 65.Ambasta RK, Kumar P, Griendling KK, Schmidt HH, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem 2004;279:45935–45941. [DOI] [PubMed] [Google Scholar]
- 66.Cheng G, Lambeth JD. NOXO1, regulation of lipid binding, localization, and activation of Nox1 by the Phox homology (PX) domain. J Biol Chem 2004;279:4737–4742. [DOI] [PubMed] [Google Scholar]
- 67.Ueno N, Takeya R, Miyano K, Kikuchi H, Sumimoto H. The NADPH oxidase Nox3 constitutively produces superoxide in a p22phox-dependent manner: its regulation by oxidase organizers and activators. J Biol Chem 2005;280:23328–23339. [DOI] [PubMed] [Google Scholar]
- 68.Cheng G, Ritsick D, Lambeth JD. Nox3 regulation by NOXO1, p47phox, and p67phox. J Biol Chem 2004;279:34250–34255. [DOI] [PubMed] [Google Scholar]
- 69.Kiss PJ, Knisz J, Zhang Y, Baltrusaitis J, Sigmund CD, Thalmann R, Smith RJ, Verpy E, Banfi B. Inactivation of NADPH oxidase organizer 1 results in severe imbalance. Curr Biol 2006;16:208–213. [DOI] [PubMed] [Google Scholar]
- 70.Petry A, Djordjevic T, Weitnauer M, Kietzmann T, Hess J, Gorlach A. NOX2 and NOX4 mediate proliferative response in endothelial cells. Antioxid Redox Signal 2006;8:1473–1484. [DOI] [PubMed] [Google Scholar]
- 71.Wingler K, Wunsch S, Kreutz R, Rothermund L, Paul M, Schmidt HH. Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radic Biol Med 2001;31:1456–1464. [DOI] [PubMed] [Google Scholar]
- 72.Si J, Fu X, Behar J, Wands J, Beer DG, Souza RF, Spechler SJ, Lambeth D, Cao W. NADPH oxidase NOX5-S mediates acid-induced cyclooxygenase-2 expression via activation of NF-kappaB in Barrett's esophageal adenocarcinoma cells. J Biol Chem 2007;282:16244–16255. [DOI] [PubMed] [Google Scholar]
- 73.Ha EM, Lee KA, Park SH, Kim SH, Nam HJ, Lee HY, Kang D, Lee WJ. Regulation of DUOX by the Galphaq-phospholipase Cbeta-Ca2+ pathway in Drosophila gut immunity. Dev Cell 2009;16:386–397. [DOI] [PubMed] [Google Scholar]
- 74.Rigutto S, Hoste C, Grasberger H, Milenkovic M, Communi D, Dumont JE, Corvilain B, Miot F, De Deken X. Activation of dual oxidases Duox1 and Duox2: differential regulation mediated by camp-dependent protein kinase and protein kinase C–dependent phosphorylation. J Biol Chem 2009;284:6725–6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassegue B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res 2009;105:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zulueta JJ, Yu FS, Hertig IA, Thannickal VJ, Hassoun PM. Release of hydrogen peroxide in response to hypoxia-reoxygenation: role of an NAD(P)H oxidase–like enzyme in endothelial cell plasma membrane. Am J Respir Cell Mol Biol 1995;12:41–49. [DOI] [PubMed] [Google Scholar]
- 77.Jones SA, O'Donnell VB, Wood JD, Broughton JP, Hughes EJ, Jones OT. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am J Physiol 1996;271:H1626–H1634. [DOI] [PubMed] [Google Scholar]
- 78.Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med 2007;43:319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J 2003;17:1502–1504. [DOI] [PubMed] [Google Scholar]
- 80.Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, Ahmad M, Edwards J, Wolin MS. Cytosolic NADPH may regulate differences in basal Nox oxidase–derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol 2005;288:H13–H21. [DOI] [PubMed] [Google Scholar]
- 81.Pendyala S, Gorshkova IA, Usatyuk PV, He D, Pennathur A, Lambeth JD, Thannickal VJ, Natarajan V. Role of Nox4 and Nox2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid Redox Signal 2009;11:747–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hoidal JR, Brar SS, Sturrock AB, Sanders KA, Dinger B, Fidone S, Kennedy TP. The role of endogenous NADPH oxidases in airway and pulmonary vascular smooth muscle function. Antioxid Redox Signal 2003;5:751–758. [DOI] [PubMed] [Google Scholar]
- 83.Dhaunsi GS, Paintlia MK, Kaur J, Turner RB. NADPH oxidase in human lung fibroblasts. J Biomed Sci 2004;11:617–622. [DOI] [PubMed] [Google Scholar]
- 84.Harper RW, Xu C, Eiserich JP, Chen Y, Kao CY, Thai P, Setiadi H, Wu R. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett 2005;579:4911–4917. [DOI] [PubMed] [Google Scholar]
- 85.Banfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem 2003;278:3510–3513. [DOI] [PubMed] [Google Scholar]
- 86.Carnesecchi S, Deffert C, Pagano A, Garrido-Urbani S, Metrailler-Ruchonnet I, Schappi M, Donati Y, Matthay MA, Krause KH, Barazzone Argiroffo C. NADPH oxidase-1 plays a crucial role in hyperoxia-induced acute lung injury in mice. Am J Respir Crit Care Med 2009;180:972–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Paffenholz R, Bergstrom RA, Pasutto F, Wabnitz P, Munroe RJ, Jagla W, Heinzmann U, Marquardt A, Bareiss A, Laufs J, et al. Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev 2004;18:486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang X, Shan P, Jiang G, Cohn L, Lee PJ. Toll-like receptor 4 deficiency causes pulmonary emphysema. J Clin Invest 2006;116:3050–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.BelAiba RS, Djordjevic T, Petry A, Diemer K, Bonello S, Banfi B, Hess J, Pogrebniak A, Bickel C, Görlach A. NOX5 variants are functionally active in endothelial cells. Free Radic Biol Med 2007;42:446–459. [DOI] [PubMed] [Google Scholar]
- 90.Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci 2003;24:471–478. [DOI] [PubMed] [Google Scholar]
- 91.Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol 2003;285:R277–R297. [DOI] [PubMed] [Google Scholar]
- 92.Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. FASEB J 1996;10:709–720. [DOI] [PubMed] [Google Scholar]
- 93.El Benna J, Han J, Park JW, Schmid E, Ulevitch RJ, Babior BM. Activation of p38 in stimulated human neutrophils: phosphorylation of the oxidase component p47phox by p38 and ERK but not by JNK. Arch Biochem Biophys 1996;334:395–400. [DOI] [PubMed] [Google Scholar]
- 94.Gozal E, Forman HJ, Torres M. ADP stimulates the respiratory burst without activation of ERK and AKT in rat alveolar macrophages. Free Radic Biol Med 2001;31:679–687. [DOI] [PubMed] [Google Scholar]
- 95.Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Respir Crit Care Med 2002;166:S4–S8. [DOI] [PubMed] [Google Scholar]
- 96.Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K, Griendling KK. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J Biol Chem 1999;274:22699–22704. [DOI] [PubMed] [Google Scholar]
- 97.Catarzi S, Biagioni C, Giannoni E, Favilli F, Marcucci T, Iantomasi T, Vincenzini MT. Redox regulation of platelet-derived-growth-factor-receptor: role of NADPH-oxidase and c-Src tyrosine kinase. Biochim Biophys Acta 2005;1745:166–175. [DOI] [PubMed] [Google Scholar]
- 98.Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. Biofactors 2003;17:287–296. [DOI] [PubMed] [Google Scholar]
- 99.Liu SF, Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol 2006;290:L622–L645. [DOI] [PubMed] [Google Scholar]
- 100.Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol 2004;173:3589–3593. [DOI] [PubMed] [Google Scholar]
- 101.Park HS, Chun JN, Jung HY, Choi C, Bae YS. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc Res 2006;72:447–455. [DOI] [PubMed] [Google Scholar]
- 102.Maloney E, Sweet IR, Hockenbery DM, Pham M, Rizzo NO, Tateya S, Handa P, Schwartz MW, Kim F. Activation of NF-kappaB by palmitate in endothelial cells: a key role for NADPH oxidase–derived superoxide in response to TLR4 activation. Arterioscler Thromb Vasc Biol 2009;29:1370–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA 1990;87:1620–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Munzel T, Daiber A, Ullrich V, Mulsch A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler Thromb Vasc Biol 2005;25:1551–1557. [DOI] [PubMed] [Google Scholar]
- 105.Rueckschloss U, Quinn MT, Holtz J, Morawietz H. Dose-dependent regulation of NAD(P)H oxidase expression by angiotensin II in human endothelial cells: protective effect of angiotensin II type 1 receptor blockade in patients with coronary artery disease. Arterioscler Thromb Vasc Biol 2002;22:1845–1851. [DOI] [PubMed] [Google Scholar]
- 106.Duerrschmidt N, Wippich N, Goettsch W, Broemme HJ, Morawietz H. Endothelin-1 induces NAD(P)H oxidase in human endothelial cells. Biochem Biophys Res Commun 2000;269:713–717. [DOI] [PubMed] [Google Scholar]
- 107.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 2003;111:1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McNally JS, Saxena A, Cai H, Dikalov S, Harrison DG. Regulation of xanthine oxidoreductase protein expression by hydrogen peroxide and calcium. Arterioscler Thromb Vasc Biol 2005;25:1623–1628. [DOI] [PubMed] [Google Scholar]
- 109.Loukogeorgakis SP, van den Berg MJ, Sofat R, Nitsch D, Charakida M, Haiyee B, de Groot E, MacAllister RJ, Kuijpers TW, Deanfield JE. Role of NADPH oxidase in endothelial ischemia/reperfusion injury in humans. Circulation 2010;121:2310–2316. [DOI] [PubMed] [Google Scholar]
- 110.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 2001;91:1487–1500. [DOI] [PubMed] [Google Scholar]
- 111.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 2000;87:840–844. [DOI] [PubMed] [Google Scholar]
- 112.Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol 2003;91:7A–11A. [DOI] [PubMed] [Google Scholar]
- 113.Zhang H, Park Y, Wu J, Chen X, Lee S, Yang J, Dellsperger KC, Zhang C. Role of TNF-alpha in vascular dysfunction. Clin Sci (Lond) 2009;116:219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Van Buul JD, Fernandez-Borja M, Anthony EC, Hordijk PL. Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxid Redox Signal 2005;7:308–317. [DOI] [PubMed] [Google Scholar]
- 115.Ushio-Fukai M. Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovasc Res 2006;71:226–235. [DOI] [PubMed] [Google Scholar]
- 116.Diebold I, Djordjevic T, Petry A, Hatzelmann A, Tenor H, Hess J, Gorlach A. Phosphodiesterase 2 mediates redox-sensitive endothelial cell proliferation and angiogenesis by thrombin via Rac1 and NADPH oxidase 2. Circ Res 2009;104:1169–1177. [DOI] [PubMed] [Google Scholar]
- 117.Duerrschmidt N, Stielow C, Muller G, Pagano PJ, Morawietz H. NO-mediated regulation of NAD(P)H oxidase by laminar shear stress in human endothelial cells. J Physiol 2006;576:557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dimmeler S, Zeiher AM. Reactive oxygen species and vascular cell apoptosis in response to angiotensin II and pro-atherosclerotic factors. Regul Pept 2000;90:19–25. [DOI] [PubMed] [Google Scholar]
- 119.Schafer M, Schafer C, Ewald N, Piper HM, Noll T. Role of redox signaling in the autonomous proliferative response of endothelial cells to hypoxia. Circ Res 2003;92:1010–1015. [DOI] [PubMed] [Google Scholar]
- 120.Colavitti R, Pani G, Bedogni B, Anzevino R, Borrello S, Waltenberger J, Galeotti T. Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. J Biol Chem 2002;277:3101–3108. [DOI] [PubMed] [Google Scholar]
- 121.Datla SR, Peshavariya H, Dusting GJ, Mahadev K, Goldstein BJ, Jiang F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler Thromb Vasc Biol 2007;27:2319–2324. [DOI] [PubMed] [Google Scholar]
- 122.Tojo T, Ushio-Fukai M, Yamaoka-Tojo M, Ikeda S, Patrushev N, Alexander RW. Role of gp91phox (Nox2)-containing NAD(P)H oxidase in angiogenesis in response to hindlimb ischemia. Circulation 2005;111:2347–2355. [DOI] [PubMed] [Google Scholar]
- 123.Ushio-Fukai M, Urao N. Novel role of NADPH oxidase in angiogenesis and stem/progenitor cell function. Antioxid Redox Signal 2009;11:2517–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wu RF, Xu YC, Ma Z, Nwariaku FE, Sarosi GA, Terada LS. Subcellular targeting of oxidants during endothelial cell migration. J Cell Biol 2005;171:893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wu RF, Gu Y, Xu YC, Nwariaku FE, Terada LS. Vascular endothelial growth factor causes translocation of p47phox to membrane ruffles through WAVE1. J Biol Chem 2003;278:36830–36840. [DOI] [PubMed] [Google Scholar]
- 126.Ikeda S, Yamaoka-Tojo M, Hilenski L, Patrushev NA, Anwar GM, Quinn MT, Ushio-Fukai M. IQGAP1 regulates reactive oxygen species–dependent endothelial cell migration through interacting with Nox2. Arterioscler Thromb Vasc Biol 2005;25:2295–2300. [DOI] [PubMed] [Google Scholar]
- 127.Shaul PW, Anderson RG. Role of plasmalemmal caveolae in signal transduction. Am J Physiol 1998;275:L843–L851. [DOI] [PubMed] [Google Scholar]
- 128.Milovanova T, Chatterjee S, Hawkins BJ, Hong N, Sorokina EM, Debolt K, Moore JS, Madesh M, Fisher AB. Caveolae are an essential component of the pathway for endothelial cell signaling associated with abrupt reduction of shear stress. Biochim Biophys Acta 2008;1783:1866–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang AY, Yi F, Zhang G, Gulbins E, Li PL. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension 2006;47:74–80. [DOI] [PubMed] [Google Scholar]
- 130.Oakley FD, Smith RL, Engelhardt JF. Lipid rafts and caveolin-1 coordinate interleukin-1beta (IL-1beta)–dependent activation of NFkappaB by controlling endocytosis of Nox2 and IL-1beta receptor 1 from the plasma membrane. J Biol Chem 2009;284:33255–33264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li Q, Spencer NY, Oakley FD, Buettner GR, Engelhardt JF. Endosomal Nox2 facilitates redox-dependent induction of NF-kappaB by TNF-alpha. Antioxid Redox Signal 2009;11:1249–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu JQ, Erbynn EM, Folz RJ. Chronic hypoxia-enhanced murine pulmonary vasoconstriction: role of superoxide and gp91phox. Chest 2005;128(Suppl 6):594S–596S. [DOI] [PubMed] [Google Scholar]
- 133.Fresquet F, Pourageaud F, Leblais V, Brandes RP, Savineau JP, Marthan R, Muller B. Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br J Pharmacol 2006;148:714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox). Am J Physiol Lung Cell Mol Physiol 2006;290:L2–L10. [DOI] [PubMed] [Google Scholar]
- 135.Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II–induced superoxide formation and redox-sensitive signaling pathways. Circ Res 2001;88:888–894. [DOI] [PubMed] [Google Scholar]
- 136.Sorescu D, Somers MJ, Lassegue B, Grant S, Harrison DG, Griendling KK. Electron spin resonance characterization of the NAD(P)H oxidase in vascular smooth muscle cells. Free Radic Biol Med 2001;30:603–612. [DOI] [PubMed] [Google Scholar]
- 137.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 1994;74:1141–1148. [DOI] [PubMed] [Google Scholar]
- 138.Guzik TJ, West NE, Black E, McDonald D, Ratnatunga C, Pillai R, Channon KM. Vascular superoxide production by NAD(P)H oxidase: association with endothelial dysfunction and clinical risk factors. Circ Res 2000;86:E85–E90. [DOI] [PubMed] [Google Scholar]
- 139.Mohazzab KM, Wolin MS. Properties of a superoxide anion-generating microsomal NADH oxidoreductase, a potential pulmonary artery PO2 sensor. Am J Physiol 1994;267:L823–L831. [DOI] [PubMed] [Google Scholar]
- 140.Marshall C, Mamary AJ, Verhoeven AJ, Marshall BE. Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol 1996;15:633–644. [DOI] [PubMed] [Google Scholar]
- 141.Archer SL, Reeve HL, Michelakis E, Puttagunta L, Waite R, Nelson DP, Dinauer MC, Weir EK. O2 sensing is preserved in mice lacking the gp91 phox subunit of NADPH oxidase. Proc Natl Acad Sci USA 1999;96:7944–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ahmad M, Kelly MR, Zhao X, Kandhi S, Wolin MS. Roles for Nox4 in the contractile response of bovine pulmonary arteries to hypoxia. Am J Physiol Heart Circ Physiol 2010;298:H1879–H1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wolin MS, Ahmad M, Gupte SA. Oxidant and redox signaling in vascular oxygen sensing mechanisms: basic concepts, current controversies, and potential importance of cytosolic NADPH. Am J Physiol Lung Cell Mol Physiol 2005;289:L159–L173. [DOI] [PubMed] [Google Scholar]
- 144.Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, Selbitz AC, Schermuly RT, Ghofrani HA, Kwapiszewska G, et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res 2007;101:258–267. [DOI] [PubMed] [Google Scholar]
- 145.Diebold I, Petry A, Hess J, Gorlach A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol Biol Cell 2010;21:2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Babior BM, Curnutte JT. Chronic granulomatous disease—pieces of a cellular and molecular puzzle. Blood Rev 1987;1:215–218. [DOI] [PubMed] [Google Scholar]
- 147.Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet 1995;9:202–209. [DOI] [PubMed] [Google Scholar]
- 148.Marriott HM, Hellewell PG, Whyte MK, Dockrell DH. Contrasting roles for reactive oxygen species and nitric oxide in the innate response to pulmonary infection with Streptococcus pneumoniae. Vaccine 2007;25:2485–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Snelgrove RJ, Edwards L, Rae AJ, Hussell T. An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur J Immunol 2006;36:1364–1373. [DOI] [PubMed] [Google Scholar]
- 150.Gao XP, Standiford TJ, Rahman A, Newstead M, Holland SM, Dinauer MC, Liu QH, Malik AB. Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by gram-negative sepsis: studies in p47phox−/− and gp91phox−/− mice. J Immunol 2002;168:3974–3982. [DOI] [PubMed] [Google Scholar]
- 151.Zhang WJ, Wei H, Frei B. Genetic deficiency of NADPH oxidase does not diminish, but rather enhances, LPS-induced acute inflammatory responses in vivo. Free Radic Biol Med 2009;46:791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Segal BH, Davidson BA, Hutson AD, Russo TA, Holm BA, Mullan B, Habitzruther M, Holland SM, Knight PR. Acid aspiration–induced lung inflammation and injury are exacerbated in NADPH oxidase–deficient mice. Am J Physiol Lung Cell Mol Physiol 2007;292:L760–L768. [DOI] [PubMed] [Google Scholar]
- 153.Yao H, Edirisinghe I, Yang SR, Rajendrasozhan S, Kode A, Caito S, Adenuga D, Rahman I. Genetic ablation of NADPH oxidase enhances susceptibility to cigarette smoke–induced lung inflammation and emphysema in mice. Am J Pathol 2008;172:1222–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yang Z, Sharma AK, Marshall M, Kron IL, Laubach VE. NADPH oxidase in bone marrow-derived cells mediates pulmonary ischemia–reperfusion injury. Am J Respir Cell Mol Biol 2009;40:375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, Thannickal VJ. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 2009;15:1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Amara N, Goven D, Prost F, Muloway R, Crestani B, Boczkowski J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 2010;65:733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]