

Abstract
Disease exacerbations and muscle wasting comprise negative prognostic factors of chronic obstructive pulmonary disease (COPD). Transient systemic inflammation and malnutrition have been implicated in skeletal muscle wasting after acute exacerbations of COPD. However, the interactions between systemic inflammation and malnutrition in their contributions to muscle atrophy, as well as the molecular basis underlying the transition of systemic inflammation to muscle atrophy, remain unresolved. Pulmonary inflammation was induced in mice by an intratracheal instillation of LPS to model acute disease exacerbation. Systemic inflammation, nutritional intake, and body and muscle weights were determined. Muscle inflammatory signaling and atrophy signaling were examined, and the effect of the muscle-specific inactivation of NF-κB on muscle atrophy was assessed in genetically modified mice. The intratracheal LPS instillation was followed by markedly elevated circulating cytokine concentrations and NF-κB activation in extrapulmonary tissues, including skeletal muscle. The administration of intratracheal LPS increased the expression of muscle E3 ubiquitin ligases, which govern muscle proteolysis, in particular MuRF1, and caused a rapid loss of muscle mass. Reduced food intake only partly accounted for the observed muscle atrophy, and did not activate NF-κB in muscle. Rather, plasma transfer experiments revealed the presence of NF-κB–signaling and atrophy-signaling properties in the circulation of intratracheal LPS–treated mice. The genetic inhibition of muscle NF-κB activity suppressed intratracheal LPS–induced MuRF1 expression and resulted in a significant sparing of muscle tissue. Systemic inflammation and malnutrition contribute to the muscle wasting induced by acute pulmonary inflammation via distinct mechanisms, and muscle NF-κB activation is required for the transition from inflammatory to muscle atrophy signaling.
Keywords: pulmonary inflammation, systemic inflammation, muscle atrophy, NF-κB, cytokines
Clinical Relevance
Disease exacerbations and muscle wasting comprise negative prognostic factors of chronic obstructive pulmonary disease (COPD). Transient systemic inflammation and malnutrition have been implicated in skeletal muscle wasting after acute exacerbations of COPD and in acute lung injury. The data described here reveal that systemic inflammation and malnutrition contribute to the muscle wasting induced by acute pulmonary inflammation via distinct mechanisms, and indicate that muscle NF-kB activation is required for the transition from inflammatory to muscle-atrophy signaling.
Acute exacerbations of chronic obstructive pulmonary disease (COPD) are accompanied by pulmonary and transient systemic inflammation and malnutrition, which have been implicated in the onset of stepwise weight loss, muscle wasting and increased mortality (1–6). The underlying mechanisms are unknown and difficult to unravel in patients during the acute phase of an exacerbation. In many instances, the acute loss of muscle mass is dependent on increased muscle protein breakdown mediated by the ubiquitin (Ub) 26S-proteasome system (UPS) (7). The rate-limiting enzymes of this process during acute muscle atrophy include the Ub-E3 ligation enzymes atrogin-1/MAFbx and MuRF1. The genetic deletion of these “atrogenes” attenuates muscle atrophy under various conditions (8, 9), and the increased expression of MuRF1 and atrogin-1 has been reported in quadriceps muscle during exacerbations of COPD (1). Inducible transcription factors, including NF-κB, have been implicated in the transcriptional regulation of atrogin-1 and MuRF1 (10). NF-κB is an essential regulator of cellular responses to inflammatory stimuli (11). TNF-α and IL-1β induce NF-κB activation (12, 13) and increase the expression of atrogin-1 and MuRF1 (8, 14) in the skeletal muscle of experimental models. Although the constitutive activation of NF-κB in skeletal muscle increases MuRF1 expression and causes muscle atrophy in mice (15), contradictory findings on NF-κB activity in muscle biopsies of patients with COPD have been reported (16, 17). These reports may reflect differences in phases of stable muscle mass versus active muscle loss, and active muscle loss likely occurs during COPD exacerbation, considering the decreases in muscle strength and activation of the UPS reported during acute exacerbation (1).
We hypothesized that muscle NF-κB activation plays a critical role in the muscle atrophy associated with an acute systemic inflammatory response during COPD exacerbations. Therefore, the first objective of this study was to model the pulmonary inflammation observed with an acute exacerbation in mice, and evaluate the degree of systemic inflammation. The second objective was to gauge the extent to which skeletal muscle responds to systemic inflammation by assessing muscle mass and atrophy-signaling responses, and to assess the relative contributions of circulating inflammatory factors and semistarvation to muscle NF-κB activation and atrophy signaling. The final and main objective was to determine the degree of NF-κB involvement in these processes, using mice with genetically repressed muscle NF-κB.
Materials and Methods
More detailed information is provided in the online supplement.
Animals, Treatment, and Tissue Collection
C57BL/6 mice were obtained from Charles River (Maastricht, The Netherlands). NF-κB–luciferase (18) and muscle IκBα-super repressor (MISR) (15) murine lines were bred and maintained in a C57BL/6 background. All studies were performed according to a protocol approved by the Institutional Animal Care Committee of Maastricht University. Acute lung inflammation was induced in anesthetized mice by the intratracheal instillation of 20 μg LPS (Escherichia coli, serotype O55:B5; Sigma, St. Louis, MO) dissolved in 50 μl sterile 0.9% NaCl, as described previously (19). In some experiments, animals were pair-fed to match the 24-hour food intake by intratracheal NaCl–treated or intratracheal LPS–treated mice. Mice were killed 4, 7, 24, 48, 72, 96, or 120 hours after LPS instillation, and a heart puncture was performed to collect blood for leukocyte isolation (as will be described) or to obtain plasma. Lungs were processed to assess local inflammation, as described previously (19). The soleus, gastrocnemius, tibialis anterior, and plantaris muscles were collected from both hind limbs, using standardized dissection methods.
Blood Cytokine and Chemokine Profile
Plasma cytokine and chemokine protein concentrations were quantified (n = 6 per group) using a Mouse Cytokine Multiplex Panel (Bio-Rad, Hercules, CA).
RNA Isolation and Assessment of Gene Expression
RNA was isolated from homogenized lung and muscle tissue for cDNA preparation and subsequent quantitative PCR analysis (primer sequences of transcripts of interest are provided in Table E1 in the online supplement). The relative DNA starting quantities of the samples were derived from the standard curve based on the threshold cycle values, using MyiQ analysis software (Bio-Rad). The expression of the genes of interest was normalized to the geometric average of at least three reference genes (β-actin, glyceraldehyde 3–phosphate dehydrogenase, and cyclophilin A) by geNorm software (20).
Leukocyte Isolation and Tissue Preparation for the Detection of NF-κB Transcriptional Activity
Leukocytes were isolated from whole blood using Polymorphprep (Axis-Shield, Oslo, Norway), and were lysed in luciferase assay buffer. Tissues were homogenized (Polytron, Kinematica, Switzerland) in luciferase buffer. Luciferase activity was measured in supernatants of the lysates, according to the manufacturer’s protocol (Promega, Madison, WI), using a tube luminometer (Lumat LB 9507; Berthold Technologies, Bad Wildbad, Germany).
Plasma Transfer Experiments
C2C12 murine myoblasts were cultured and differentiated into myotubes, as previously described (21). Differentiation medium (i.e., Dulbecco’s Modified Eagle’s Medium containing 0.5% FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin) was supplemented with plasma (10% final; vol/vol) of mice after intratracheal LPS or intratracheal NaCl (control) treatment in the presence of 50 U/ml heparin (Leo, Breda, The Netherlands). Myotubes (when differentiated for 5 days) were incubated in presence of plasma for 24 hours, cells were lysed for RNA extraction, and quantitative PCR analyses were performed as already described.
Statistical Analysis
Raw data were entered into SPSS (version 11.0; SPSS, Inc., Chicago, IL) for statistical analysis. Values as expressed in the graphs were subjected to Mann-Whitney U tests to detect statistically significant differences.
Results
Pulmonary Inflammation after Intratracheal Instillation of LPS
The intratracheal instillation of LPS induced extensive neutrophil infiltration of the lungs, which was maximal after 48 hours (Figure 1A). The expression levels of cytokines and chemokines, including TNF-α, IL-6, CXCL1, ICAM-1, and CXCL2, increased as early as 4 hours after LPS instillation (Figure 1B, parts 1–5), which coincided with elevated IκBα mRNA transcript levels (Figure 1B, part 6). Finally, a rapid and sustained increase of NF-κB transcriptional activity (Figure 1C) was observed after intratracheal LPS instillation in the lung homogenates of NF-κB–luciferase reporter mice.
Figure 1.
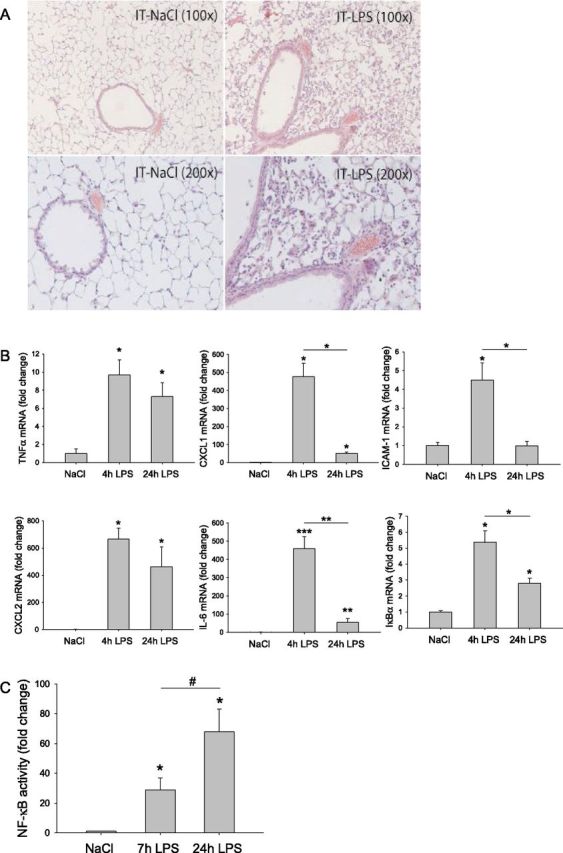
Pulmonary inflammation after intratracheal instillation of LPS. Mice were subjected to the intratracheal instillation of NaCl or LPS, as described in Materials and Methods. (A) Inflammatory cell infiltration was assessed in the lungs 48 hours after the intratracheal instillation, using hematoxylin and eosin staining, followed by light microscopy. (B) Inflammatory and NF-κB–sensitive gene products were assessed in lung mRNA at the indicated time points (n = 3) by quantitative PCR, normalized to geNorm, and expressed as fold change of control. (C) Alternatively, using lung homogenates of transgenic NF-κB reporter mice prepared at the indicated time points after intratracheal LPS (n = 6), NF-κB transcriptional activity was assessed by the measurement of luciferase enzyme activity (normalized to total protein concentration), expressed as fold change compared with intratracheal NaCl. #P < 0.05 as indicated or *P < 0.05, **P < 0.01, or ***P < 0.001, compared with control (intratracheal NaCl), unless otherwise indicated. CXCL1, chemokine (C-X-C motif) ligand 1; ICAM-1, intercellular adhesion molecule 1; RLU, relative light units.
Increased Concentrations of Circulating Inflammatory Mediators after Pulmonary Inflammation
To investigate whether acute pulmonary inflammation was accompanied by a systemic inflammatory response, concentrations of circulating proinflammatory cytokines were determined (Figure 2A). A general pattern of increased plasma concentrations was apparent within 4 hours after intratracheal LPS (e.g., TNFα, IL-1α, CXCL1, and IL-6), although IL-1β, regulated upon activation, normal T-cell expressed, and secreted (RANTES), and granulocyte colony–stimulating factor (G-CSF) concentrations lagged and were not apparent at the earliest time point. The increases in circulating cytokines were maximal at 24–48 hours after the intratracheal LPS challenge, involved as much as a 75-fold induction over NaCl control values, and returned to control concentrations by 72 hours after intratracheal LPS. In addition, compared with intratracheal NaCl control activity, NF-κB transcriptional activity was increased 2-fold in isolated circulating leukocytes of intratracheal LPS–treated NF-κB–luciferase reporter mice (Figure 2B). Lung and leukocyte NF-κB transcriptional activities were correlated after intratracheal LPS challenge (Figure 2C).
Figure 2.
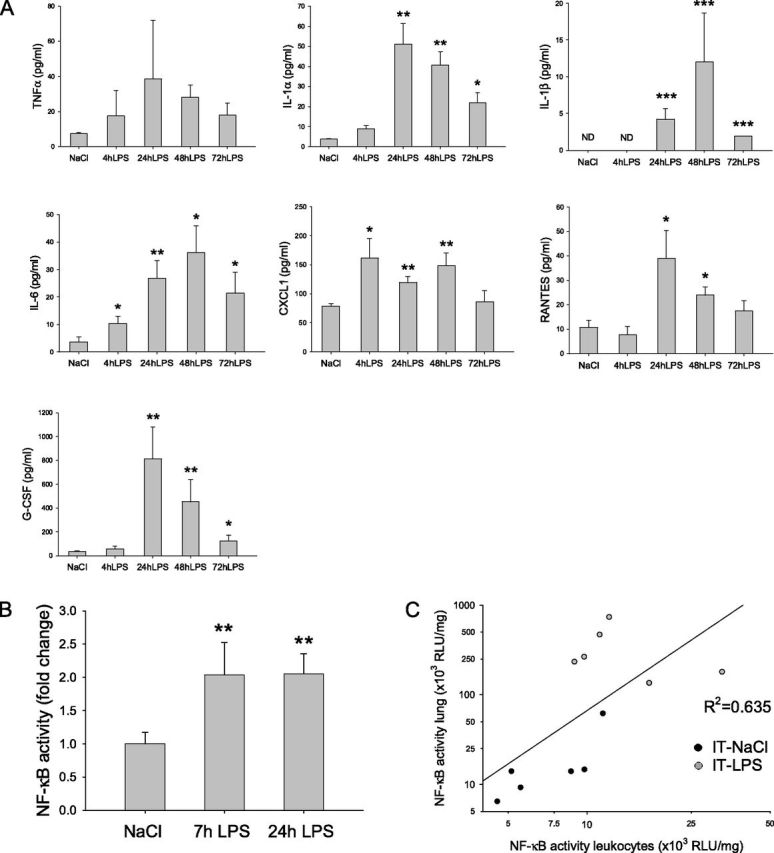
Increased concentrations of circulating inflammatory mediators after pulmonary inflammation. (A) Mice were subjected to the intratracheal instillation of NaCl or LPS, and blood was collected after terminal anesthesia at the indicated time points (n = 6). Circulating cytokines and chemokines were determined in plasma. (B) Circulating leukocytes were isolated from the blood of NF-κB reporter mice 7 hours or 24 hours after intratracheal instillation (n = 6), and luciferase activity was assessed, normalized to total protein, and expressed as fold change of intratracheal NaCl. (C) The correlation between luciferase activity in lung homogenates and circulating leukocytes of NF-κB reporter mice was assessed after intratracheal instillation (C).*P < 0.05, **P < 0.01, and ***P < 0.001, compared with control (intratracheal NaCl). Correlation at 7 hours, R2 = 0.635, P < 0.05. G-CSF, granulocyte colony–stimulating factor; CXCL1, chemokine (C-X-C motif) ligand 1; RANTES, regulated upon activation, normal T cell expressed, and secreted; RLU, relative light units.
Muscle Atrophy in Response to Systemic Inflammation Occurs Mainly Independent of Semistarvation
The acute systemic inflammatory response was also accompanied by a rapid loss of body weight (bw) (Figure 3A), which was maximal after 48 hours. The recovery of bw began by 72 to 96 hours after treatment, and was not fully completed after 120 hours. Food intake was slightly reduced by the experimental procedure itself (intratracheal NaCl) during the first 24 hours, but rapidly recovered to habitual levels of food intake (Figure 3B). In contrast, intratracheal LPS caused a more pronounced and sustained reduction in food intake. Pair-feeding experiments revealed that decreased food intake accounted for approximately 60% (24 hours) and approximately 20% (72 hours) of the reduction in bw observed after intratracheal LPS (Figure 3C).
Figure 3.
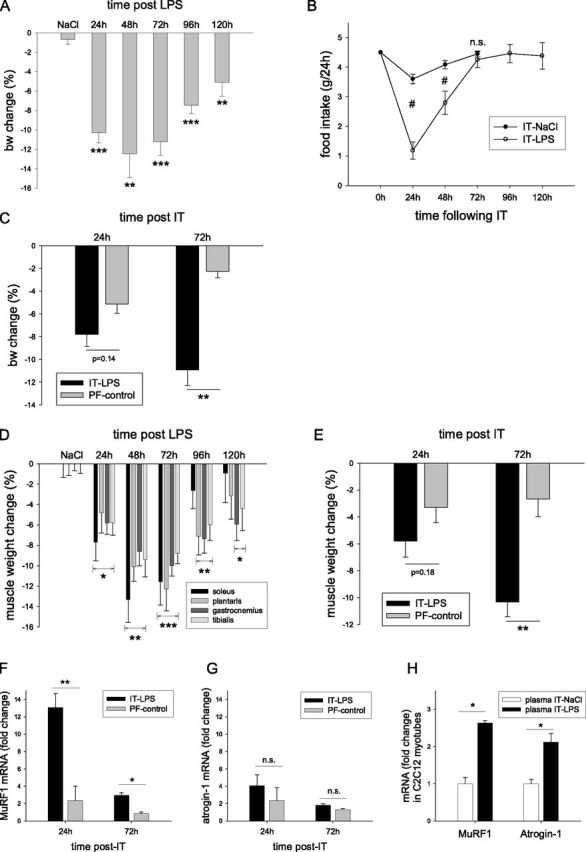
Muscle atrophy in response to systemic inflammation occurs mainly independent of semistarvation. Mice were subjected to the intratracheal instillation of NaCl or LPS, and body weights (bw) (A) and food weights (B) were measured every 24 hours for 5 days (n = 6–8). (C) At the indicated time points, mice were terminally anesthetized, and muscle weights were determined. Alternatively, mice were pair-fed (PF) by providing the amounts of food consumed during 24 hours by intratracheal NaCl–treated or intratracheal LPS–treated animals (n = 4), and the change (%) in bw (D) or muscle mass of the soleus–plantaris–gastrocnemius complex (E) between intratracheal NaCl and intratracheal LPS (intratracheal LPS) and the PF groups for intratracheal NaCl and intratracheal LPS (PF-control) was calculated. Muscle ring finger 1 (MuRF1) (F) and atrogin-1 (G) mRNA transcript levels were determined and normalized to geNorm in plantaris muscles of intratracheal NaCl, intratracheal LPS, and the respective PF-treated groups at 24 hours and 72 hours, and the intratracheal LPS and intratracheal LPS PF (PF-control) expression levels were expressed as fold change compared with intratracheal NaCl or intratracheal NaCl PF, respectively (n = 4–6). (H) Alternatively, plasma (10% final; vol/vol) derived from control (intratracheal NaCl) or intratracheal LPS mice was added to in vitro cultured, 5-day differentiated C2C12 myotubes (n = 6), and MuRF1 or atrogin-1 mRNA concentrations were determined. *P < 0.05, **P < 0.01, and ***P < 0.001, compared with control values (intratracheal NaCl), unless otherwise indicated. #P < 0.01, between intratracheal NaCl and intratracheal LPS.
Postural and locomotive hind limb muscles (musculus soleus, musculus plantaris, musculus gastrocnemius, and musculus tibialis) also displayed a rapid decline in weight, which was maximal at 48–72 hours after intratracheal LPS (Figure 3D). The muscle weights of pair-fed animals revealed that reduced food intake accounted for approximately 55% of decreased muscle weight at 24 hours, and for approximately 25% of the maximal muscle atrophy observed at 72 hours (Figure 3E).
The expression of MuRF1 and atrogin-1, which are rate-limiting enzymes in UPS-mediated muscle proteolysis (Figures 3F and 3G), was slightly induced by reduced food intake (pair-fed control mice), but only during the first 24 hours. Conversely, MuRF1 mRNA expression was robustly up-regulated at both 24 and 72 hours after the intratracheal LPS challenge (Figure 3F), whereas a more modest increase in atrogin-1 mRNA was observed (Figure 3G). Although the induction of atrogin-1 was on the same order of magnitude as observed in muscles of pair-fed control mice, MuRF1 expression after intratracheal LPS was largely independent of semistarvation.
To assess whether atrophy-inducing factors are present in the circulation of intratracheal LPS–treated animals, plasma transfer experiments were performed (Figure 3H). Compared with control mice, plasma obtained from intratracheal LPS–challenged mice caused a greater than 2-fold induction in MuRF1 and atrogin-1 expression in cultured C2C12 myotubes, revealing that atrophy-signaling properties are contained within the plasma of intratracheal LPS–treated animals.
Systemic Inflammation Is Accompanied by Increased Inflammatory Gene Expression and NF-κB Activation in Skeletal Muscle
To evaluate systemic inflammation further, various peripheral organs and tissues were collected from NF-κB reporter mice. A 2- to 3-fold increase was observed in response to intratracheal LPS in the heart, liver, ileum, and pancreas, which was less apparent in the kidney and jejunum, and absent in epididymal fat (Figure 4A). Importantly, an increased transcriptional activity of NF-κB was also observed in skeletal muscle of NF-κB reporter mice compared with intratracheal NaCl control mice. Muscle mRNA concentrations of CXCL1 and IκBα were significantly increased 24 hours after the induction of acute pulmonary inflammation (Figure 4B), and IκBα mRNA concentrations revealed a rapid and transient increase after intratracheal LPS instillation (Figure 4C), confirming NF-κB activation in skeletal muscle. Importantly, increased IκBα transcript levels were not observed in the muscle of pair-fed animals, suggesting that muscle NF-κB activation was not the result of semistarvation (Figure 4D). Plasma transfer experiments revealed increased IκBα mRNA concentrations in myotubes incubated with plasma derived from intratracheal LPS–challenged animals compared with intratracheal NaCl–challenged animals (Figure 4E). These data demonstrate that muscle NF-κB is activated in response to pulmonary inflammation, which accompanies a muscle-wasting program associated with systemic inflammation as opposed to nutritional deprivation.
Figure 4.
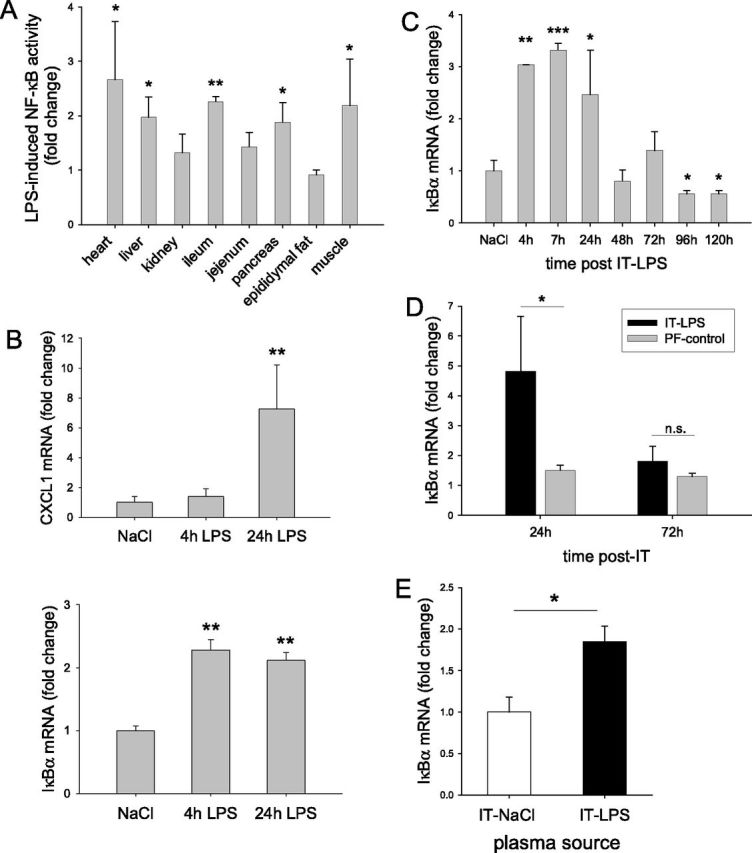
Systemic inflammation is accompanied by increased inflammatory gene expression and NF-κB activation in skeletal muscle. (A) Peripheral tissues were collected from NF-κB reporter mice 24 hours after intratracheal NaCl or intratracheal LPS (n = 6), and luciferase activity was determined, normalized to protein, and expressed as fold change compared with intratracheal NaCl concentrations for the respective tissues. (B and C) In separate experiments, skeletal muscle was dissected from wild-type (WT) mice after terminal anesthesia at the indicated time points (B, n = 6; C, n = 3–6), and mRNA transcript levels of inflammatory (CXCL1) and NF-κB sensitive (IκBα) genes was determined, normalized to geNorm, and expressed as fold change compared with intratracheal NaCl. (D) Similarly, IκBα mRNA concentrations were determined in animals subjected to PF and full treatment (n = 4–6), and are expressed as in Figure 3F. (E) Alternatively, IκBα mRNA concentrations were determined in myotubes (n = 6) cultured in the presence of plasma obtained from control (intratracheal NaCl) or intratracheal LPS–challenged mice. *P < 0.05, **P < 0.01, and ***P < 0.001, compared with control values (intratracheal NaCl), unless otherwise indicated.
NF-κB–Dependent Muscle Atrophy after Pulmonary Inflammation
To investigate further the potential role of NF-κB in muscle wasting associated with acute pulmonary inflammation, transgenic mice expressing a nondegradable mutant form of IκBα specifically in muscle (MISR mice) were subjected to intratracheal LPS challenge. The loss of bw was attenuated in MISR mice after acute pulmonary inflammation, compared with wild-type (WT) control mice, despite similar decreases in food intake (Figures E1A and E1B). The increase in CXCL1 mRNA concentrations observed in WT muscle in response to intratracheal LPS was abolished in muscles of MISR mice, reflecting the efficient inhibition of NF-κB–mediated gene expression in muscle (Figure 5A). Conversely, circulating CXCL1 concentrations in MISR mice were indistinguishable from those in WT mice after intratracheal LPS (Figure 5B), which rules out important contributions of skeletal muscle to circulating CXCL1 concentrations.
Figure 5.
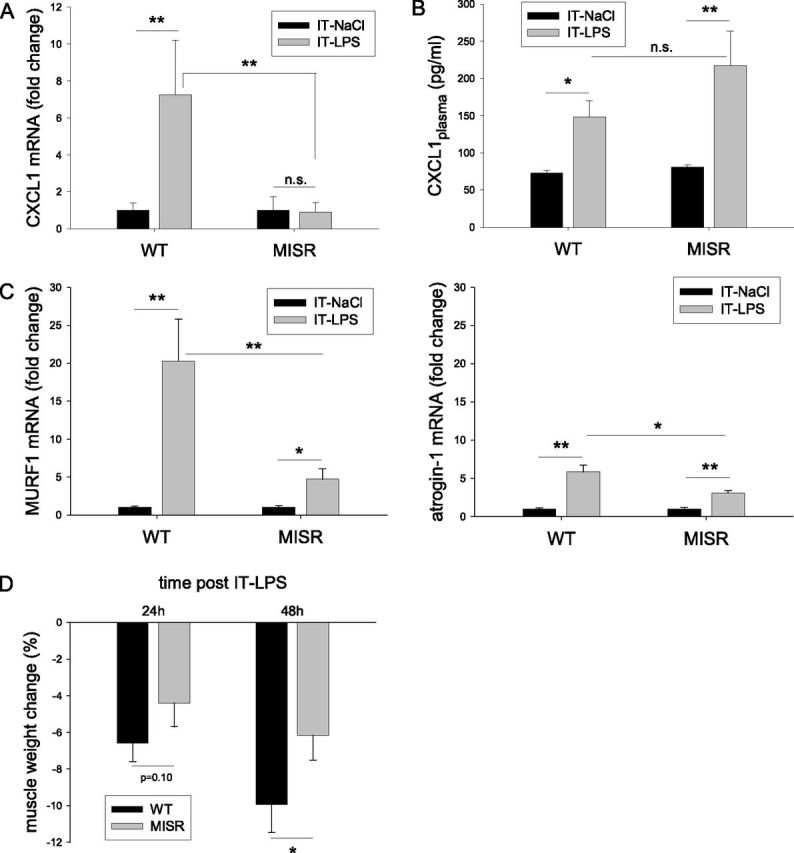
NF-κB–dependent muscle atrophy after pulmonary inflammation. Muscle IκBα-super repressor (MISR) or transgene negative littermate control (WT, wild type) mice were subjected to intratracheal NaCl or intratracheal LPS for 24 hours. mRNA concentrations of CXCL1 (A) or of MuRF1 and atrogin-1 (C) were determined and normalized to geNorm in excised skeletal muscle (n = 8). (B) Circulating CXCL1 protein concentrations (n = 8) were assessed in plasma. (D) Muscle weights were determined at 24 hours and 48 hours after intratracheal (n = 8), and expressed as percent change compared with intratracheal NaCl. *P < 0.05 and **P < 0.01, compared with control values (intratracheal NaCl), unless otherwise indicated.
As observed previously, a robust 20-fold induction of muscle MuRF1 mRNA occurred in response to the intratracheal LPS challenge. This was reduced to a 5-fold induction in MISR mice (Figure 5C). Atrogin-1 mRNA was induced 6-fold by the intratracheal LPS challenge, and NF-κB inhibition in MISR mice reduced this to a 3-fold induction (Figure 5C). Importantly, the reduced expression of MuRF1 and atrogin-1 was accompanied by an attenuation of skeletal muscle atrophy in MISR mice compared with WT littermates (Figure 5D). In addition, the increased expression of microtubule-associated protein 1 light chain 3 beta (LC3B, 30%), BCL2/adenovirus E1B 19-kD interacting protein 3 (Bnip3, 65%), and GABAA receptor-associated protein like 1 (GabarapL1, 570%), which operate in autophagy–lysosomal proteolysis, was differentially affected by NF-κB inhibition (Figure E2A). Finally, whereas a loss of muscle weight progressed between 24 hours and 48 hours after intratracheal LPS in WT animals (−6.6% ± 1.0% versus −9.9% ± 1.5%, respectively; P < 0.05), muscle weight did not change significantly during this phase in MISR mice (−4.4% ± 1.3% versus −6.2% ± 1.3%, respectively; no significance). Moreover, the decrease in muscle fiber size in response to the intratracheal LPS challenge, which was observed in all fiber types, was strongly attenuated in MISR mice (Figure E2B). These data demonstrate that the inhibition of muscle NF-κB activation prevents inflammatory and atrophy signaling in skeletal muscle, and results in the significant sparing of muscle tissue after acute pulmonary inflammation.
Discussion
The results of this study demonstrate that acute pulmonary inflammation is accompanied by malnutrition, a rapid and potent systemic inflammatory response, and muscle atrophy, which consists of both semistarvation-dependent and semistarvation-independent mechanisms. The latter are likely initiated by circulating inflammatory mediators as part of a systemic inflammatory response, as was revealed by plasma transfer experiments. Importantly, muscle NF-κB activation occurs independently of semistarvation, and muscle atrophy (signaling) caused by systemic inflammation, but not semistarvation, is fully prevented by the inhibition of NF-κB activation in skeletal muscle.
Pulmonary and systemic inflammation has been implicated in acute COPD exacerbations (2, 5, 6). The intratracheal instillation of LPS in mice or rats is an established model of acute pulmonary inflammation (19, 22), and is known to involve the NF-κB activation of resident as well as infiltrating immune cells (23, 24). In line with these observations, elevated NF-κB transcriptional activity and increased concentrations of transcripts encoding inflammatory and NF-κB–responsive genes were detected in lung homogenates of intratracheal LPS–treated animals. Similar findings have also been reported in lungs of patients with COPD, in particular during exacerbations (5, 25), and are often accompanied by elevated circulating concentrations of humoral inflammatory mediators (2, 6). Indeed, increased plasma concentrations of cytokines and chemokines such as TNF-α, IL-1α/β, IL-6, CXCL1, RANTES, and G-CSF were detectable after the local inflammatory response in the airways of intratracheal LPS–treated mice. The systemic inflammatory response was further characterized by increased NF-κB activity in circulating leukocytes, which reflects their activated state. In line with these observations, increased proinflammatory cytokine production by leukocytes isolated from the blood of patients with pulmonary inflammation has been reported (26, 27). Interestingly, the NF-κB transcriptional activity in lung tissue and circulating leukocytes was correlated, suggesting an initially coordinated inflammatory response of the pulmonary and circulatory compartments. The elevated systemic inflammatory status was also reflected in skeletal muscle, because CXCL1 and IκBα mRNA concentrations as well as NF-κB transcriptional activity were increased.
Several lines of evidence suggest an important contribution of systemic inflammation to muscle atrophy after acute pulmonary inflammation. In our model, the appearance of inflammatory cytokines including TNF-α, IL-1α, IL-1β, and IL-6 in the circulation preceded and accompanied muscle atrophy, suggesting their potential involvement in the loss of muscle tissue. In support of this idea, studies in which these cytokines were captured in the circulation of animals suffering from endotoxemia, or conversely, in which these cytokines were administered to healthy animals (28, 29), revealed the attenuation or induction of atrophy and muscle proteolysis, respectively. Although NF-κB activation was documented in the skeletal muscle of septic mice (30), the intracellular molecules mediating the conversion of inflammatory stimuli to atrophy signaling remain unresolved in vivo. In our work, elevated concentrations of circulating TNF-α, IL-1, and IL-6 coincided with evidence of NF-κB signaling in skeletal muscle up to 24 hours after intratracheal LPS. NF-κB signaling was also observed in in vitro cultured C2C12 myotubes when incubated with plasma obtained from intratracheal LPS–challenged mice, suggesting that NF-κB signaling in skeletal muscle after acute pulmonary inflammation results from inflammatory mediators contained within the circulatory compartment. Although we did not perform blocking experiments to address their identity, in vitro studies by others suggest these may concern TNF-α or IL-1, whereas little evidence exists for NF-κB activation by IL-6 (12, 13). Importantly, in the present study, the inhibition of muscle NF-κB activation resulted in a significant sparing of muscle mass in MISR mice after intratracheal LPS. Of interest, atrophy in response to pulmonary inflammation was observed in muscles of different fiber compositions, and the NF-κB–dependent reversal of muscle atrophy occurred regardless of muscle fiber type. Because previous work demonstrated the NF-κB dependency of the TNF-α–induced loss of myosin heavy chain in cultured skeletal muscle (12, 31), we are tempted to speculate that circulating TNF-α or IL-1, but not IL-6, is responsible for the NF-κB–dependent atrophy in our model. Our data are in line with an earlier report in which an important role for NF-κB in muscle maintenance was established, including the induction of muscle atrophy after the constitutive activation of muscle NF-κB (15). Therefore, our data demonstrate for the first time in vivo, to the best of our knowledge, that the inhibition of muscle NF-κB activation significantly attenuates skeletal muscle atrophy during conditions of systemic inflammation, and reveals that muscle NF-κB activation is required for the transition of systemic inflammatory cues to muscle atrophy.
Acute loss of muscle mass typically relies on increased muscle proteolysis by the Ub 26S-proteasome pathway, which depends to a large extent on the rate-limiting E3 Ub ligases atrogin-1 and MuRF1 (10). Atrogin-1 and MuRF1 expression were induced 4-fold and 13-fold, respectively, in skeletal muscle of animals subjected to intratracheal LPS instillation in our study. Similar increases in atrogin-1 and MuRF1 expression have been reported in acute COPD exacerbations (1), which also involve a systemic inflammatory response. Importantly, circulating mediators were likely responsible for triggering proteolysis, because incubation with plasma from intratracheal LPS–challenged mice induced the increased expression of MuRF1 and atrogin-1 in cultured myotubes. TNF-α and IL-1 have been reported to induce atrogin-1 (14, 32) and MuRF1 (8) in skeletal muscle. The induction of atrogin-1 appears to involve transcriptional regulators other than NF-κB (33). In line with those reports, atrogin-1 expression levels in muscle after intratracheal LPS only slightly differed between MISR and WT mice. Conversely, muscle atrophy induced by the constitutive activation of muscle NF-κB relies on the increased expression of MuRF1 (15). Accordingly, the present data reveal a strong suppression of MuRF1 mRNA accumulation in response to intratracheal LPS treatment in muscles of MISR compared with WT mice, despite a similar degree of circulating inflammatory cytokines. This result strongly suggests that the blockade of muscle NF-κB activity attenuates muscle atrophy in response to pulmonary inflammation by preventing the full transcriptional activation of the MuRF1 promoter.
In addition to Ub 26S-proteasome–mediated proteolysis, a complementary role was recently attributed to the autophagy–lysosomal pathway in skeletal muscle atrophy (34), including inflammation-induced muscle catabolism (35). In line with Doyle and colleagues (35), the expression levels of the autophagy-related genes LC-3B, Bnip3, and GabarapL1 were increased in skeletal muscle during pulmonary inflammation, but interestingly, displayed a differential dependence on muscle NF-κB. The NF-κB–independent elevation of Bnip3 transcripts may be the consequence of Forkhead box O3 (FOXO3), a transcriptional activation in response to semistarvation (36). Conversely, this accumulation of LC-3B and GabarapL1 mRNA was inhibited or attenuated, respectively, in muscles of MISR mice. In contrast to Bnip3, the increased expression of these Ub-like proteins is not sufficient to initiate autophagosome formation, but is considered necessary for the progression of autophagy. As such, suppressed concentrations of LC-3B and GabarapL1 may reflect the coordination of autophagy with attenuated Ub 26S-proteasome–mediated proteolysis in MISR muscle, and suggest an indirect dependence on NF-κB activation. Indeed, little evidence exists for the transcriptional regulation of LC-3B and GabarapL1 by NF-κB (37).
The semistarvation observed in response to pulmonary inflammation may have resulted from a loss of appetite as a consequence of fever associated with the response to systemic inflammation. Although starvation clearly affects muscle mass (38), our pair-feeding studies demonstrated that semistarvation could account at most for 55% and 25% of the muscle atrophy observed 24 hours and 72 hours after the induction of pulmonary inflammation, respectively. In skeletal muscle of pair-fed animals, the expression of atrogin-1 and MuRF1 was only marginally (∼ 2-fold) increased, in contrast to the much stronger induction, particularly in MuRF1 expression (13-fold), observed after intratracheal LPS. In addition, the experimental conditions of plasma transfer experiments were conducted in the presence of excess nutrients provided by the culture medium, indicating that atrophy signaling was likely induced by factors other than low nutrient concentrations in the circulation. Together, these data suggest that the induction of atrogin-1, and in particular MuRF1 expression after intratracheal LPS challenge, is largely independent of semistarvation. IκBα and CXCL1 expression levels were not affected in the skeletal muscle of pair-fed treated animals, whereas these transcripts were clearly induced by acute pulmonary inflammation. This indicates that muscle NF-κB activation is specific for inflammation-associated, but not starvation-associated, muscle atrophy, and suggests that the loss of muscle mass resulting from semistarvation does not rely on NF-κB activation. These findings explain why muscle atrophy in MISR mice was not completely prevented, because the residual loss of muscle may have resulted from semistarvation, and this loss was comparable based on the food intake of MISR and WT mice after intratracheal LPS. Indeed, the remaining muscle atrophy in MISR mice after the intratracheal LPS challenge matched the extent of atrophy induced by semistarvation alone. Altogether, these results support the notion that the semistarvation-independent component of muscle atrophy relies on NF-κB activation and subsequent MuRF1 expression, and is the consequence of systemic inflammation.
These findings reveal that therapeutic strategies using integrated nutritional and anti-inflammatory approaches may prevent the loss of skeletal muscle during acute inflammatory conditions such as COPD exacerbations by targeting independent signaling pathways in skeletal muscle.
Additional material
Supplementary data supplied by authors.
Acknowledgments
The authors thank M.C.J.M. Kelders, F. Snepvangers, and A. van Essen for technical assistance.
Footnotes
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2011-0119OC on April 26, 2012
References
- 1.Crul T, Testelmans D, Spruit MA, Troosters T, Gosselink R, Geeraerts I, Decramer M, Gayan-Ramirez G. Gene expression profiling in vastus lateralis muscle during an acute exacerbation of COPD. Cell Physiol Biochem 2010;25:491–500. [DOI] [PubMed] [Google Scholar]
- 2.Oudijk EJ, Lammers JW, Koenderman L. Systemic inflammation in chronic obstructive pulmonary disease. Eur Respir J Suppl 2003;46:5s–13s. [DOI] [PubMed] [Google Scholar]
- 3.Soler-Cataluna JJ, Martinez-Garcia MA, Roman Sanchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax 2005;60:925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vermeeren MA, Wouters EF, Geraerts-Keeris AJ, Schols AM. Nutritional support in patients with chronic obstructive pulmonary disease during hospitalization for an acute exacerbation; a randomized controlled feasibility trial. Clin Nutr 2004;23:1184–1192. [DOI] [PubMed] [Google Scholar]
- 5.Wedzicha JA, Seemungal TA. COPD exacerbations: defining their cause and prevention. Lancet 2007;370:786–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wouters EF, Groenewegen KH, Dentener MA, Vernooy JH. Systemic inflammation in chronic obstructive pulmonary disease: the role of exacerbations. Proc Am Thorac Soc 2007;4:626–634. [DOI] [PubMed] [Google Scholar]
- 7.Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 2004;18:39–51. [DOI] [PubMed] [Google Scholar]
- 8.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001;294:1704–1708. [DOI] [PubMed] [Google Scholar]
- 9.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA 2001;98:14440–14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol 2005;37:1974–1984. [DOI] [PubMed] [Google Scholar]
- 11.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 2006;25:6680–6684. [DOI] [PubMed] [Google Scholar]
- 12.Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS. NF-kappaB–induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 2000;289:2363–2366. [DOI] [PubMed] [Google Scholar]
- 13.Langen RC, Schols AM, Kelders MC, Wouters EF, Janssen-Heininger YM. Inflammatory cytokines inhibit myogenic differentiation through activation of nuclear factor–kappaB. FASEB J 2001;15:1169–1180. [DOI] [PubMed] [Google Scholar]
- 14.Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, Reid MB. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J 2005;19:362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai D, Frantz JD, Tawa NE, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 2004;119:285–298. [DOI] [PubMed] [Google Scholar]
- 16.Agusti A, Morla M, Sauleda J, Saus C, Busquets X. NF-kappaB activation and iNOS upregulation in skeletal muscle of patients with COPD and low body weight. Thorax 2004;59:483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plant PJ, Brooks D, Faughnan M, Bayley T, Bain J, Singer L, Correa J, Pearce D, Binnie M, Batt J. Cellular markers of muscle atrophy in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2010;42:461–471. [DOI] [PubMed] [Google Scholar]
- 18.Carlsen H, Moskaug JO, Fromm SH, Blomhoff R. In vivo imaging of NF-kappa B activity. J Immunol 2002;168:1441–1446. [DOI] [PubMed] [Google Scholar]
- 19.Haegens A, Heeringa P, van Suylen RJ, Steele C, Aratani Y, O’Donoghue RJ, Mutsaers SE, Mossman BT, Wouters EF, Vernooy JH. Myeloperoxidase deficiency attenuates lipopolysaccharide-induced acute lung inflammation and subsequent cytokine and chemokine production. J Immunol 2009;182:7990–7996. [DOI] [PubMed] [Google Scholar]
- 20.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002;3:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langen RC, Van Der Velden JL, Schols AM, Kelders MC, Wouters EF, Janssen-Heininger YM. Tumor necrosis factor–alpha inhibits myogenic differentiation through MyoD protein destabilization. FASEB J 2004;18:227–237. [DOI] [PubMed] [Google Scholar]
- 22.Starcher B, Williams I. A method for intratracheal instillation of endotoxin into the lungs of mice. Lab Anim 1989;23:234–240. [DOI] [PubMed] [Google Scholar]
- 23.Blackwell TS, Lancaster LH, Blackwell TR, Venkatakrishnan A, Christman JW. Differential NF-kappaB activation after intratracheal endotoxin. Am J Physiol 1999;277:L823–L830. [DOI] [PubMed] [Google Scholar]
- 24.Poynter ME, Irvin CG, Janssen-Heininger YM. A prominent role for airway epithelial NF-kappa B activation in lipopolysaccharide-induced airway inflammation. J Immunol 2003;170:6257–6265. [DOI] [PubMed] [Google Scholar]
- 25.Caramori G, Romagnoli M, Casolari P, Bellettato C, Casoni G, Boschetto P, Chung KF, Barnes PJ, Adcock IM, Ciaccia A, et al. Nuclear localisation of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax 2003;58:348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oudijk EJ, Nijhuis EH, Zwank MD, van de Graaf EA, Mager HJ, Coffer PJ, Lammers JW, Koenderman L. Systemic inflammation in COPD visualised by gene profiling in peripheral blood neutrophils. Thorax 2005;60:538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Godoy I, Donahoe M, Calhoun WJ, Mancino J, Rogers RM. Elevated TNF-alpha production by peripheral blood monocytes of weight-losing COPD patients. Am J Respir Crit Care Med 1996;153:633–637. [DOI] [PubMed] [Google Scholar]
- 28.Fong Y, Moldawer LL, Marano M, Wei H, Barber A, Manogue K, Tracey KJ, Kuo G, Fischman DA, Cerami A, et al. Cachectin/TNF or IL-1 alpha induces cachexia with redistribution of body proteins. Am J Physiol 1989;256:R659–R665. [DOI] [PubMed] [Google Scholar]
- 29.Tsujinaka T, Fujita J, Ebisui C, Yano M, Kominami E, Suzuki K, Tanaka K, Katsume A, Ohsugi Y, Shiozaki H, et al. Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice. J Clin Invest 1996;97:244–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Penner CG, Gang G, Wray C, Fischer JE, Hasselgren PO. The transcription factors NF-kappaB and AP-1 are differentially regulated in skeletal muscle during sepsis. Biochem Biophys Res Commun 2001;281:1331–1336. [DOI] [PubMed] [Google Scholar]
- 31.Li YP, Reid MB. NF-kappaB mediates the protein loss induced by TNF-alpha in differentiated skeletal muscle myotubes. Am J Physiol Regul Integr Comp Physiol 2000;279:R1165–R1170. [DOI] [PubMed] [Google Scholar]
- 32.Li W, Moylan JS, Chambers MA, Smith J, Reid MB. Interleukin-1 stimulates catabolism in C2C12 myotubes. Am J Physiol Cell Physiol 2009;297:C706–C714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. FOXO transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004;117:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sandri M. Autophagy in health and disease: 3. Involvement of autophagy in muscle atrophy. Am J Physiol Cell Physiol 2010;298:C1291–C1297. [DOI] [PubMed] [Google Scholar]
- 35.Doyle A, Zhang G, Abdel Fattah EA, Eissa NT, Li YP. Toll-like receptor 4 mediates lipopolysaccharide-induced muscle catabolism via coordinate activation of ubiquitin–proteasome and autophagy–lysosome pathways. FASEB J 2011;25:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. FOXO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007;6:458–471. [DOI] [PubMed] [Google Scholar]
- 37.Comb WC, Cogswell P, Sitcheran R, Baldwin AS. IKK-dependent, NF-kappaB–independent control of autophagic gene expression. Oncogene 2011;30:1727–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jagoe RT, Lecker SH, Gomes M, Goldberg AL. Patterns of gene expression in atrophying skeletal muscles: response to food deprivation. FASEB J 2002;16:1697–1712. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.