

Abstract
DLC1 is a RhoGAP-containing tumor suppressor that inhibits angiogenesis by repressing VEGF production in epithelial cells. Here we report the roles of DLC1 in endothelial cells. Silencing of DLC1 (siDLC1) enhances cell migration but reduces tube formation activities of human umbilical vein endothelial cells (HUVECs). Biochemically, RhoA activity and paxillin protein level are markedly increased in siDLC1 HUVECs. Although further silencing of RhoA restores the cell migration phenotype, the tube formation defect and up-regulated paxillin level remain unchanged. On the other hand, paxillin knockdown rescues tube formation and migration phenotypes but not the up-regulated RhoA activity. These results indicate that DLC1 regulates endothelial cell migration through RhoA and paxillin independently and controls tube formation mainly via paxillin. To further determine endothelial DLC1's function, we have generated endothelial specific knockout mice (DLC1-Tek). DLC1-Tek mice appear to be normal and healthy but their angiogenesis processes are compromised as shown in gel plug and aortic ring sprouting assays. Analysis of endothelial cells isolated from DLC1-Tek mice has further affirmed the cellular and biochemical phenotypes established in siDLC1 HUVECs. Our studies have demonstrated a positive regulatory role of endothelial DLC1 in angiogenesis.
Keywords: DLC1, RhoA, paxillin, focal adhesion, tube formation, angiogenesis
1. Introduction
Endothelial cells play a wide range of critical roles in regulating vascular development and function. They are essential for developing new blood vessels during embryogenesis known as vasculogenesis and growing new vessels from existing ones known as angiogenesis. Angiogenesis is regulated by complex signaling networks within endothelial cells initiated by cues from other cell types and from the extracellular environment. These cues detected by receptors on endothelial cells initiate intracellular signaling cascades that often integrate with signaling pathways generated from cell adhesion interactions. Together they activate various processes in endothelial cells and promote cell proliferation and directional migration, which lead to tube formation, followed by loop formation, and eventually vascular stabilization. Angiogenesis is an important natural process in growth and development of tissues, as well as in wound healing processes. It also occurs in diseases, such as cancer, diabetic blindness, and rheumatoid arthritis.
Deleted in liver cancer 1 (DLC1, also known as ARHGAP7 and STARD12 in humans and p122RhoGAP in rats) is a RhoGAP-containing tumor suppressor [1, 2, 5, 9, 10] that is often down-regulated in a variety of cancers, such as those of the liver, lung, breast, stomach, brain, colon and prostate due to either genomic deletion or aberrant DNA methylation [5, 10]. In addition, missense and nonsense mutations were detected in colon and prostate cancer patients [11]. DLC1 negatively regulates RhoA GTPase activity through its RhoGAP domain, which is essential for DLC1's tumor suppression function [12, 14]. Its RhoGAP activity is tightly regulated through phosphorylation by CDK5 [20] or ERK [15] as well as binding partners such as tensins [3, 18], which also recruit DLC1 to focal adhesions. On the other hand, not all suppression roles rely on controlling RhoA function. DLC1 negatively regulates angiogenesis by suppressing VEGF expression in epithelial cells [17]. This is accomplished by inhibiting the EGFR-MEK-HIF pathway and is independent from the RhoA pathway [17]. The studies also reveal a role of DLC1 in angiogenesis. Total knockout of DLC1 in mice results in embryonic lethality with defects in the brain, heart, and placental blood vessels [4, 16]. In contrast, another family member DLC2 is not required for embryogenesis and tissue development but plays a critical role in angiogenesis [13].
Here, we have used the human umbilical vein endothelial cell (HUVEC) system to understand the roles of DLC1 in endothelial cells and have further demonstrated its functions in endothelial-specific DLC1 knockout mice. The in vitro cell culture studies reveal the critical roles of DLC1 in endothelial cell migration and tube formation. While the mutant mice in our in vivo studies appear to be normal and healthy, their angiogenesis processes are compromised. Together, our studies identify the molecular mechanisms involving DLC1, RhoA, and paxillin in regulating endothelial cell migration and angiogenesis.
2. Materials and methods
2.1 Mice
To produce endothelial-specific DLC1 knockout mouse, mice carrying the DLC1loxp/loxp allele established in our laboratory were crossbred with B6.Cg-Tg(Tek-cre1Ywa/J) from Jackson laboratory to generate DLC1-Tek mice. These mice were bred and maintained under specific pathogen-free conditions at the animal facility of University of California, Davis All the mice related manipulations were performed with protocols approved by the animal ethics committee at the University of California, Davis.
2.2 Cell culture and reagents
HUVECs and primary mouse endothelial cells were cultured in endothelial cell growth medium (ScienCell). GenMuteTM siRNA&DNA transfection reagent (SignaGen) was used for transfections. DLC1, RhoA and paxillin siRNAs were purchased from Sigma-Aldrich. The RhoA Pull-down Activation Assay Biochem Kit and RhoA G-LISA Activation Assay Kit were purchased from Cytoskeleton.
2.3 Immunoblot analysis
Total cell lysates were resolved by SDS-PAGE and transferred to nitrocellulose membranes (Amersham Pharmacia Biotech) by electroblotting. The membranes were blocked with 5% non-fat milk and probed with corresponding primary and secondary antibodies. The antigen-antibody complexes were visualized using enhanced chemiluminescence (ECL) reagents (Amersham Pharmacia Biotech) according to the manufacturer's instructions.
2.4 Migration assay
Endothelial cells (50,000) were added to the upper chamber of a Transwell Permeable Supports (8 μm pore size, Corning) with growth factor and serum free media (100μL). Complete endothelial cell growth medium (500 μL) was added to the lower chamber. After 6 h, cells were fixed and stained. Cells remaining on the top of permeable surface were removed using a cotton swab; the cells that had migrated to the bottom surface were visualized microscopically (with 100× or 200× magnification), photographed, and counted.
2.5 Endothelial cell tube formation assay
Growth factor–reduced Matrigel (Corning) was used to coat the 48-well plate (150 μL/well) and endothelial cells (50,000 cells/well) were seeded. After 6 hours of incubation, capillary-like structures were scored by measuring the lengths of tubules per field in each well at 100× magnification with ImageJ software (NIH).
2.6 Matrigel plug assay
Growth factor-reduced matrigel (250 μL) containing 60 U/ml heparin (Sigma-Aldrich) was subcutaneously injected into 8-week DLC1 WT or DLC-Tek mice. After 5 days, matrigel plugs were harvested and embedded in OCT and frozen sections were processed for immunohistochemical staining using CD31 antibody.
2.7 Isolation of mouse endothelial cells
Primary lung-derived endothelial cells were isolated from lung tissues of 8 to 10-week-old mice as described [18]. Briefly, minced lungs were digested with 1.5 mg/ml collagenase A (Roche) and filtered through nylon mesh into 10% FBS–DMEM media. Cells were selected with anti-CD31-antibody (BD Biosciences) and anti-ICAM2 antibody (BD) coated Dynabeads (Invitrogen) and cultured with endothelial cell medium (ScienCell). After 3 passages, cells were immortalized using lentivirus expressing SV40 large T antigen (Genecopoeia).
2.8 Aortic ring assay
Thoracic aortas from DLC1 WT or DLC-Tek mice were excised and transferred to ice-cold PBS. Aortas were sectioned into 1-mm-long rings and individually embedded in 300μl/well growth factor–reduced Matrigel in a 48-well plate. Rings were cultured with 500 μL of medium for 6 days, and the outgrowth of endothelial tubes was visualized microscopically, photographed, and counted using ImageJ software.
3 Results
3.1 Silencing of DLC1 promotes endothelial cell migration but reduces angiogenic like tube formation of HUVECs
To examine the role of DLC1 in endothelial cells, we have silenced DLC1 in HUVEC by siRNA approaches and analyzed their cell migration and tube formation activities. The DLC1 protein level was almost completely suppressed by the siRNA treatment while RhoA activity, the major target of DLC1, was significantly increased (figure 1A). Similar to previously reported DLC2 knockdown HUVECs [13], silencing of DLC1 (siDLC1) significantly promotes endothelial cell migration (figure 1B). Nonetheless, siDLC1 HUVECs showed reduced tube formation activities (figure 1B), which is opposite to what we previously observed in DLC2 knockdown cells [13], suggesting that DLC1 and DLC2 play similar roles in endothelial cell migration but different regulatory acts in angiogenesis.
Figure 1. Silencing of DLC1 promotes migration but reduces tube formation activities of HUVECs.
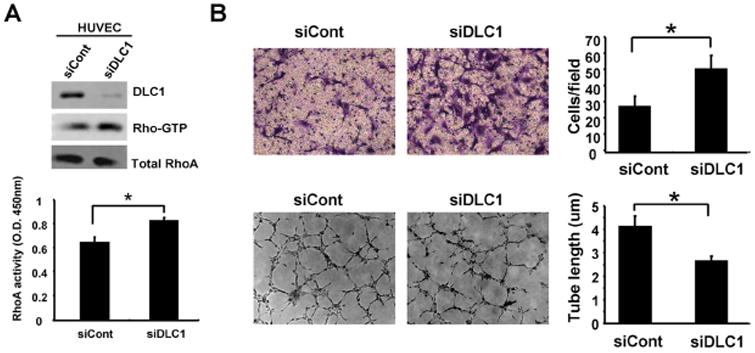
(A) HUVECs treated with control (siCont) or DLC1 (siDLC1) siRNAs were lysed for immunoblotting with indicated antibodies or for measuring RhoA activities by GST-RBD pull down assay (labeled as Rho-GTP) or by Rho G-LISA activation assay (lower panel) (N=3). (B) Representative images of transwell cell migration (upper) or tube formation (lower) of siCont or siDLC1 HUVECs cells and bar graphs of data from triplicate experiments. *, P < 0.05.
3.2 RhoA regulates DLC1-mediated endothelial cell migration but not tube formation activities
Since Rho activities were up-regulated in siDLC1 HUVECs, we tested whether RhoA was responsible for the DLC1-mediated endothelial cell migration and/or tube formation phenotypes. When RhoA was further silenced in siDLC1 HUVECs, cell migration was reduced to that of control cells (figure 2A), suggesting that DLC1 regulates endothelial cell migration through RhoA. However, silencing of RhoA in siDLC1 HUVECs did not restore the tube formation activity (figure 2A), indicating that the tube formation phenotype is not a result of up-regulated RhoA activity in siDLC1 HUVECs.
Figure 2. RhoA and paxillin independently contribute to the cell migration phenotype but only paxillin is involved in tube formation defects in siDLC1 HUVECs.
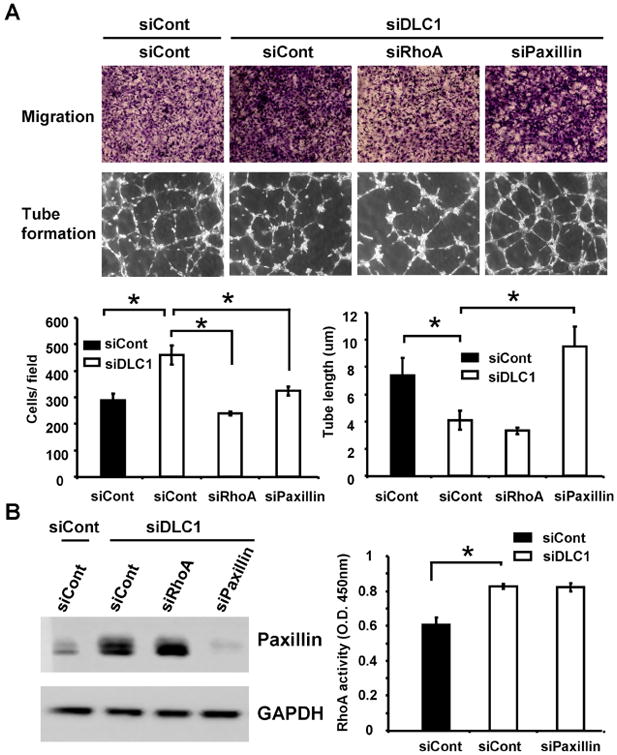
(A) Representative images of transwell cell migration (upper) or tube formation (lower) of HUVECs treated with indicated siRNAs and bar graphs of data from triplicate experiments. *, P < 0.05 (B) Cell lysates isolated from HUVECs treated with indicated siRNAs were immunoblotted with paxillin or GAPDH antibodies (left) or were used for measuring RhoA activities by Rho G-LISA activation assay (right)(N=3).
3.3 Silencing of DLC1 up-regulates paxillin expression that contributes to the tube formation and cell migration activities in HUVECs
Since the focal adhesion molecule paxillin is known to negatively regulate angiogenesis [7] and DLC1 is suggested to modulate the protein level of paxillin [8], we examined the effects of DLC1 knockdown on paxillin protein levels. Immunoblotting results showed that the paxillin level was markedly increased in siDLC1 HUVECs (figure 2B). We then investigated whether the up-regulated paxillin contributed to the decreased tube formation activity of siDLC1 HUVECs. Silencing of DLC1 and paxillin restored the tube formation (figure 2A) but not RhoA activity (figure 2C). These results demonstrate that DLC1 positively regulates angiogenesis by suppressing the level of paxillin protein in a RhoA-independent manner. Because paxillin may regulate cell migration, we further examined the effect of paxillin knockdown in siDLC1 HUVEC migration. Silencing of paxillin also reduced the increased migration of siDLC1 HUVECs (figure 2A). Since knockdown of RhoA does not affect paxillin protein levels and silencing of paxillin has no effect on RhoA activity (figure 2B), RhoA and paxillin likely contribute to DLC1-mediated endothelial cell migration independently.
3.4 Endothelial DLC1 is not required for mouse embryo and tissue development
The above finding drove us to investigate the role of endothelial DLC1 in mice. Tek-Cre (Cre recombinase driven by the receptor tyrosine kinase Tek (aka Tie2) promoter/enhancer) from Jackson laboratory and DLC1loxP/loxP mice established in our laboratory (manuscript in preparation) were used to produce endothelial specific DLC1 knockout Tek-Cre:DLC1loxP/loxP (named DLC1-Tek) mice. In DLC1loxP/loxP mice, wild type DLC1 exon 3 regions are replaced with loxP-flanked exon 3 sequences. In the presence of Cre, the loxP-flanked exon 3 (82 base pairs encoding 26 amino acids: 38LLFPIDIALVKREHDFLDRDAIEALC) is expected to be excised, which leads to a frame-shift and premature termination. Although DLC1 total knockout led to embryonic lethality and showed defects in vascularization [4, 16], DLC1-Tek mice were apparently normal and healthy (not shown). This finding indicated that endothelial DLC1 is not required for vasculogenesis during embryo development and for mouse survival.
3.5 Loss of DLC1 in endothelial cells reduces angiogenic responses in mice
Since silencing of DLC1 decreased HUVEC tube formation, we investigated whether angiogenic processes were compromised in DLC1-Tek mice by using gel plug and aorta ring assays. Matrigel was injected subcutaneously into mice to induce neovascularization. Five days post-injection, matrigel plugs were isolated and analyzed by CD31 immunohistochemistry staining. The staining results showed that CD31-positive cells were markedly reduced in gel plugs from DLC1-Tek mice (figure 3A). In addition, ex vivo aorta ring assays showed significantly reduced sprouting in samples from DLC1-Tek (figure 3B). Both results suggest that the angiogenic response is compromised in DLC1-Tek mice. To confirm the efficiency of DLC1 deletion, primary endothelial cells isolated from the lungs of DLC1-Tek or wild-type control (DLC1loxP/loxP) mice were subjected to immunoblot analysis. The level of DLC1 protein was diminished in DLC1-Tek endothelial cells and the Rho-GTP level was up-regulated as expected (figure 4A). In agreement with our findings using siDLC1 HUVECs, DLC1-Tek cells migrated faster than DLC1loxP/loxP control cells and the tube formation activity was reduced in DLC1-Tek cells (figure 4B). Furthermore, paxillin protein levels were also upregulated in DLC1-Tek cells (figure 5A). Further silencing of paxillin rescued DLC1-Tek cell migration and tube formation phenotypes (figure 5B), although less completely than those in siDLC1 HUVECs (figure 2A). This probably was due to lower paxillin knockdown efficiency in primary mouse endothelial cells as evident by immunoblotting (figure 5B). On the other hand, silencing of RhoA reduced DLC1-Tek cell migration but had no effect on the tube formation activity (figure 5B). These data indicate that endothelial DLC1 contributes to angiogenesis in mice and mechanistically, DLC1 regulates endothelial cell migration through RhoA and paxillin independently and tube formation mainly through paxillin signaling (figure 5C).
Figure 3. Lack of endothelial DLC1 reduces in vivo and ex vivo angiogenesis.
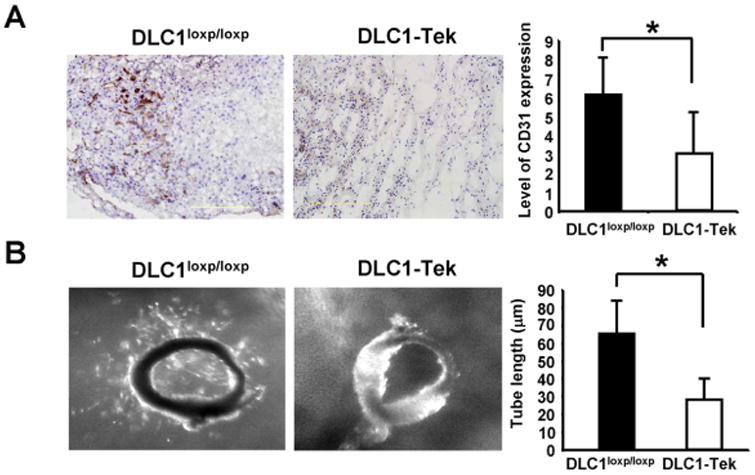
(A) Representative images of angiogenic response to Matrigel in DLC1-Tek and DLC1loxP/loxP mice. Gel plugs from DLC1-Tek showed significantly lower CD31-positive vessels than DLC1loxP/loxP samples. Bar graph shows quantification of vessel formation per sample (N=3). *P < 0.05 (B) Representative images of aortic ring explants placed in Matrigel. Bar graph shows quantification of tube-like structure. Sprouting of aortic explants is significantly reduced in DLC1-Tek samples (N=5). P value was calculated by paired Student's t-test. *, P < 0.05
Figure 4. Loss of DLC1 promotes endothelial cell migration but suppresses tube formation activities.
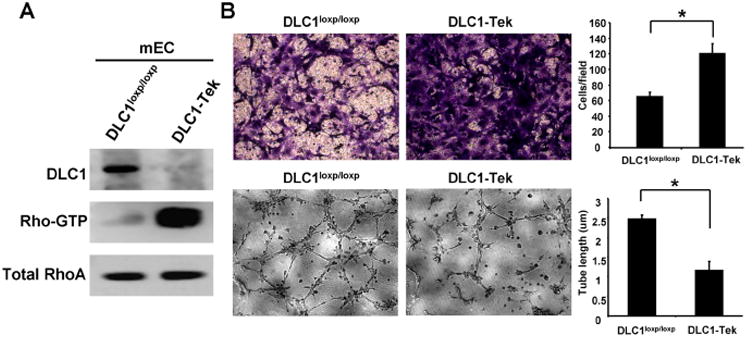
(A) Cell lysates prepared from DLC-Tek or DLC1loxP/loxP endothelial cells were subjected to immunoblotting or RhoA activity assays. (B) Representative images of transwell cell migration (upper) or tube formation (lower) of indicated cells and bar graphs of data from triplicate experiments. *, P < 0.05.
Figure 5. Rescue DLC1-Tek cell phenotypes by silencing of RhoA or paxillin.
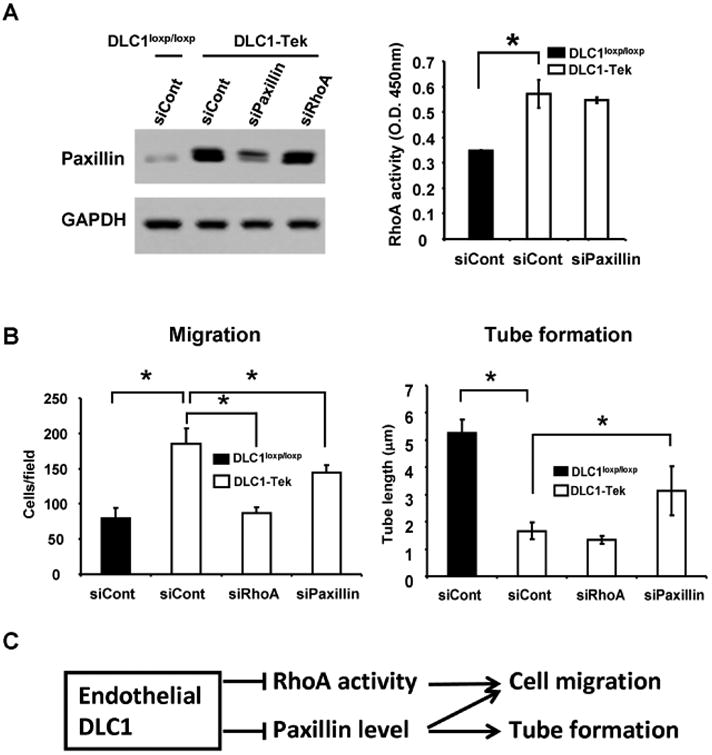
(A) Cell lysates isolated from DLC1loxp/loxp or DLC1-Tek treated with indicated siRNAs were immunoblotted with paxillin or GAPDH antibodies (left) or were used for measuring RhoA activities by Rho-GTP G-LISA activation assay (right) (N=3). (B) Bar graphs of cell migration (left) and tube formation (right) of indicated cells from triplicate experiments. *, P < 0.05. (C) Schematic representation of DLC1 regulating endothelial cell migration and tube formation through RhoA and/or paxillin.
4. Discussion
Previous studies have demonstrated that DLC1 total knockout in mice leads to embryonic lethality with severe defects in the neural tube, heart, and vasculature of placenta [4, 16]. Our current studies on DLC1-Tek endothelial-specific knockout mice have shown that endothelial DLC1 is not required for appropriate vasculogenesis and have suggested that abnormalities from several cell types and microenvironments may coordinately contribute to vascular defects in total knockouts. On the other hand, DLC2 total knockout mice are apparently normal [13]. It would be interesting for future studies to generate DLC1/DLC2 double endothelial specific knockout mice to reveal the function of DLC in vasculogenesis.
The role of DLC1 in cell migration is intriguing. Our previous studies showed a positive role of DLC1 in epithelial cell migration because silencing of DLC1 in prostate epithelial cells reduces cell migration mainly due to upregulation of plasminogen activator inhibitor 1 (PAI-1) [19]. In the endothelial cells, knockout or knockdown of DLC1 promotes migration through two independent routes - the RhoA pathway and paxillin signaling. These findings suggest that DLC1 may regulate cellular activities through different mechanisms that are cell type dependent.
The finding that lack of endothelial DLC1 reduces angiogenesis is somewhat surprising, because our previous report [13] showed that total knockout of DLC2 in mice enhances angiogenic responses and silencing of DLC2 in HUVECs increases tube formation in a RhoA-dependent manner. Although down-regulation of DLC1 or DLC2 both lead to up-regulation of RhoA activities, it appears that RhoA activity is more relevant to DLC2-mediated angiogenesis since knockdown of RhoA does not change the tube formation phenotype in DLC1-Tek or siDLC1 cells. RhoA is a known positive regulator of tube formation [21] and is upregulated in DLC1-Tek and siDLC1 cells, indicating that lack of DLC1 elicits a negative regulation that overcomes or bypasses the positive effect of RhoA. To this end, we found that paxillin is up-regulated in the absence of DLC1. Up-regulated paxillin appears to be responsible for the observed tube formation reduction because silencing of paxillin restores the tube formation activity without altering RhoA activity in DLC1-Tek or siDLC1 cells. This result is in agreement with the finding that paxillin negatively controls angiogenesis by altering neurophilin 2 (NRP2) expression [7]. NRP2 is a non-kinase receptor that binds to VEGFR-2 and regulates endothelial cell functions, such as survival and migration [6]. Our studies have placed DLC1 at the upstream of the paxillin-NRP2 pathway in regulating angiogenesis. Now the question is how DLC1 suppresses paxillin protein levels. One possibility is that paxillin protein is more stable in the absence of DLC1. This is supported by the report that DLC1 down-regulates paxillin in spread fibroblasts and that this process does not require DLC1's RhoGAP activity [8], which is consistent with our finding that silencing of RhoA does not affect the up-regulation of paxllin in DLC1-Tek or siDLC1 cells. In addition, it is shown that the LD motif of DLC1 binds to the talin R8 domain, which also interacts with the paxillin LD1 and LD2 motifs [23]. Therefore, DLC1 and paxillin are competing for the same binding site on talin. It is likely that talin-bound paxillin is more stable in the absence of DLC1 in a cell. On the other hand, DLC1 may suppress paxillin expression at the transcriptional stage. These possibilities require further investigation in the future.
DLC1 is known as a tumor suppressor and is down-regulated in numerous types of cancer, including hepatocellular carcinoma (HCC). Our previous findings demonstrate that down-regulation of DLC1 in epithelial cells promotes angiogenesis by releasing more VEGF [17]. This is not observed in current studies since endothelial cells are generally not known to express VEGF and we fail to detect it in HUVECs and our mouse endothelial cells (not shown). Nonetheless, our current studies show that reduction of DLC1 in endothelial cells impairs angiogenesis, which is not favorable for tumor development. These seem contradictory to each other in terms of promoting tumorigenesis. Interestingly, a recent report analyzing DLC1 expression and localization in HCC showed that although DLC1 is often lost in cancer cells, it remains within the stromal components and is concentrated within a close proximity to endothelial (CD34 positive) cells [22]. It will be worthwhile to investigate DLC1 expression not only in cancer cells but also within tumor microenvironments in the future.
Highlight.
Silencing of DLC1 enhances endothelial cell migration
Down-regulation of DLC1 reduces tube formation activity
DLC1 knockdown increases RhoA activity and paxillin protein level.
RhoA and paxillin independently contribute to DLC1-medilated cell migration.
DLC1-medilated endothelial tube formation is mainly controlled by paxillin.
Endothelial specific DLC1 knockout mice show compromised angiogenic responses
Acknowledgments
This study was supported in part by NIH grants DK097291 to YPS and CA151366 to SHL.
Footnotes
The authors disclose no potential conflicts of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barras D, Widmann C. GAP-independent functions of DLC1 in metastasis. Cancer metastasis reviews. 2014;33:87–100. doi: 10.1007/s10555-013-9458-0. [DOI] [PubMed] [Google Scholar]
- 2.Braun AC, Olayioye MA. Rho regulation: DLC proteins in space and time. Cell Signal. 2015;27:1643–1651. doi: 10.1016/j.cellsig.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Cao X, Voss C, Zhao B, Kaneko T, Li SS. Differential regulation of the activity of deleted in liver cancer 1 (DLC1) by tensins controls cell migration and transformation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1455–1460. doi: 10.1073/pnas.1114368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Durkin ME, Avner MR, Huh CG, Yuan BZ, Thorgeirsson SS, Popescu NC. DLC-1, a Rho GTPase-activating protein with tumor suppressor function, is essential for embryonic development. FEBS letters. 2005;579:1191–1196. doi: 10.1016/j.febslet.2004.12.090. [DOI] [PubMed] [Google Scholar]
- 5.Durkin ME, Yuan BZ, Zhou X, Zimonjic DB, Lowy DR, Thorgeirsson SS, Popescu NC. DLC-1:a Rho GTPase-activating protein and tumour suppressor. J Cell Mol Med. 2007;11:1185–1207. doi: 10.1111/j.1582-4934.2007.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Favier B, Alam A, Barron P, Bonnin J, Laboudie P, Fons P, Mandron M, Herault JP, Neufeld G, Savi P, Herbert JM, Bono F. Neuropilin-2 interacts with VEGFR-2 and VEGFR-3 and promotes human endothelial cell survival and migration. Blood. 2006;108:1243–1250. doi: 10.1182/blood-2005-11-4447. [DOI] [PubMed] [Google Scholar]
- 7.German AE, Mammoto T, Jiang E, Ingber DE, Mammoto A. Paxillin controls endothelial cell migration and tumor angiogenesis by altering neuropilin 2 expression. Journal of cell science. 2014;127:1672–1683. doi: 10.1242/jcs.132316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaushik S, Ravi A, Hameed FM, Low BC. Concerted modulation of paxillin dynamics at focal adhesions by Deleted in Liver Cancer-1 and focal adhesion kinase during early cell spreading. Cytoskeleton. 2014;71:677–694. doi: 10.1002/cm.21201. [DOI] [PubMed] [Google Scholar]
- 9.Kim TY, Vigil D, Der CJ, Juliano RL. Role of DLC-1, a tumor suppressor protein with RhoGAP activity, in regulation of the cytoskeleton and cell motility. Cancer metastasis reviews. 2009;28:77–83. doi: 10.1007/s10555-008-9167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liao YC, Lo SH. Deleted in liver cancer-1 (DLC-1): a tumor suppressor not just for liver. The international journal of biochemistry & cell biology. 2008;40:843–847. doi: 10.1016/j.biocel.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liao YC, Shih YP, Lo SH. Mutations in the focal adhesion targeting region of deleted inliver cancer-1 attenuate their expression and function. Cancer research. 2008;68:7718–7722. doi: 10.1158/0008-5472.CAN-08-2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liao YC, Si L, Devere White RW, Lo SH. The phosphotyrosine-independent interaction of DLC-1 and the SH2 domain of cten regulates focal adhesion localization and growth suppression activity of DLC-1. The Journal of cell biology. 2007;176:43–49. doi: 10.1083/jcb.200608015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Y, Chen NT, Shih YP, Liao YC, Xue L, Lo SH. DLC2 modulates angiogenic responses in vascular endothelial cells by regulating cell attachment and migration. Oncogene. 2010;29:3010–3016. doi: 10.1038/onc.2010.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qian X, Li G, Asmussen HK, Asnaghi L, Vass WC, Braverman R, Yamada KM, Popescu NC, Papageorge AG, Lowy DR. Oncogenic inhibition by a deleted in liver cancer gene requires cooperation between tensin binding and Rho-specific GTPase-activating protein activities. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:9012–9017. doi: 10.1073/pnas.0703033104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ravi A, Kaushik S, Ravichandran A, Pan CQ, Low BC. Epidermal growth factor activates the Rho GTPase-activating protein (GAP) Deleted in Liver Cancer 1 via focal adhesion kinase and protein phosphatase 2A. J Biol Chem. 2015;290:4149–4162. doi: 10.1074/jbc.M114.616839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sabbir MG, Wigle N, Loewen S, Gu Y, Buse C, Hicks GG, Mowat MR. Identification and characterization of Dlc1 isoforms in the mouse and study of the biological function of a single gene trapped isoform. BMC biology. 2010;8:17. doi: 10.1186/1741-7007-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shih YP, Liao YC, Lin Y, Lo SH. DLC1 negatively regulates angiogenesis in a paracrine fashion. Cancer research. 2010;70:8270–8275. doi: 10.1158/0008-5472.CAN-10-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shih YP, Sun P, Wang A, Lo SH. Tensin1 positively regulates RhoA activity through its interaction with DLC1. Biochimica et biophysica acta. 2015;1853:3258–3265. doi: 10.1016/j.bbamcr.2015.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shih YP, Takada Y, Lo SH. Silencing of DLC1 upregulates PAI-1 expression and reduces migration in normal prostate cells. Molecular cancer research : MCR. 2012;10:34–39. doi: 10.1158/1541-7786.MCR-11-0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tripathi BK, Qian X, Mertins P, Wang D, Papageorge AG, Carr SA, Lowy DR. CDK5 is a major regulator of the tumor suppressor DLC1. The Journal of cell biology. 2014;207:627–642. doi: 10.1083/jcb.201405105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Nieuw Amerongen GP, Koolwijk P, Versteilen A, van Hinsbergh VW. Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:211–217. doi: 10.1161/01.atv.0000054198.68894.88. [DOI] [PubMed] [Google Scholar]
- 22.Wolosz D, Walczak A, Wilczynski GM, Szparecki G, Wilczek E, Gornicka B. Deleted in liver cancer 1 expression and localization in hepatocellular carcinoma tissue sections. Oncology letters. 2014;8:785–788. doi: 10.3892/ol.2014.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zacharchenko T, Qian X, Goult BT, Jethwa D, Almeida TB, Ballestrem C, Critchley DR, Lowy DR, Barsukov IL. LD Motif Recognition by Talin: Structure of the Talin-DLC1 Complex. Structure. 2016;24:1130–1141. doi: 10.1016/j.str.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]