

Summary
Mounting evidence indicates that adverse activation of the complement system plays a role in the development of diabetic vascular complications. Plasma levels of the complement proteins mannan‐binding lectin (MBL) and its associated serine proteases (MASP‐1 and MASP‐2) are elevated in diabetes. We hypothesized that single nucleotide polymorphisms (SNPs) in the MASP1 gene may contribute to altered plasma levels of the belonging gene products; MASP‐1, MASP‐3 and mannan‐binding lectin‐associated protein of 44 kDa (MAp44) in patients with type 2 diabetes. To investigate this, we compared plasma levels of MASP‐1, MASP‐3 and MAp44 in 100 patients with type 2 diabetes and 100 sex‐ and age‐matched controls. Ten carefully selected SNPs were analysed using TaqMan® genotyping assay. Additionally, we included a streptozotocin‐induced diabetes mouse model to directly examine the effect of inducing diabetes on MASP‐1 levels. MASP‐1 levels were significantly higher among patients with type 2 diabetes compared with healthy controls (P = 0·017). Five SNPs (rs874603, rs72549254, rs3774275, rs67143992, rs850312) in the MASP1 gene were associated with plasma levels of MASP‐1, MASP‐3 and MAp44. In the diabetes mouse model, diabetic mice had significantly higher MASP‐1 levels than control mice (P = 0·003). In conclusion, MASP‐1 levels were higher among patients with type 2 diabetes and diabetic mice. The mechanism behind this increase remains elusive.
Keywords: complement, mannan‐binding lectin‐associated serine proteases, single nucleotide polymorphism, type 2 diabetes mellitus
Introduction
An increasing number of studies suggest that the complement system, as a part of the innate immune system, plays an important role in the development of diabetic vascular complications 1, 2, 3, 4. The system is composed of plasma proteins produced mainly by the liver, and plays a crucial role in recognition and clearance of infectious microbes. Three distinct pathways can initiate the complement system: the classical, the alternative and the lectin pathways. All three pathways lead to formation of the C3 convertase with subsequent downstream effects that include recruitment of inflammatory cells, opsonization and generation of the membrane attack complex causing cell lyses 5.
The lectin pathway is activated when one of the five known activation molecules [mannan‐binding lectin (MBL), ficolin‐1, ficolin‐2, ficolin‐3 or collectin‐LK] binds to a target sugar molecule. The activation is mediated by the MBL‐associated serine proteases (MASPs), three of which are known: MASP‐1, MASP‐2 and MASP‐3. When MASP‐1 is activated it cleaves MASP‐2 that subsequently cleaves the complement factors C2 and C4 leading to formation of the C3 convertase 6. The role of MASP‐3 has not yet been elucidated fully 5.
MBL levels are elevated in both patients with type 1 diabetes 7 and type 2 diabetes 8, and in the last decade increased MBL and ficolin levels have, in a number of prospective studies, been associated with cardiovascular morbidity and mortality in diabetes 8, 9, 10. Circulating levels of MBL are mainly determined genetically 11, and the MBL2 genotype with the highest level of MBL is associated with mortality in type 1 diabetes 12. Compared to MBL, the role of MASPs in diabetes remains less explored. MASP‐1 and MASP‐2 levels, however, have been shown recently to be elevated in patients with type 1 diabetes 13 and polymorphisms in MASPs were found to be associated with the belonging protein products 14, 15.
The MASP1 gene is located on chromosome 3q27‐28 16, and encodes a primary transcript which is spliced by mutually exclusive splicing into three different messenger ribonucleic acids (mRNAs) coding for MASP‐1, MASP‐3 and mannan‐binding lectin‐associated protein of 44 kDa (MAp44) 17.
All three splice products share the first eight exons. The ninth exon is unique to MAp44, which makes Map44 a truncated serine protease without any cleavage function. Exon 12 and exons 13–18 encode the serine protease domain of MASP‐3 and MASP‐1, respectively 17. The locations of the polymorphisms we have investigated are listed in Table 2.
Table 2.
Distribution of MASP‐1 genotypes within diabetes and control groups
| Diabetes | Control | ||||||
|---|---|---|---|---|---|---|---|
| SNP | Localization | Gen.* | n † | n | n | P | |
| rs874603 | 3'‐UTR MASP‐3 | AA | 176 | 86 | 90 | 0·74 | |
| AG | 20 | 12 | 8 | ||||
| GG | 2 | 1 | 1 | ||||
| rs710469 | Intron 03 | TT | 46 | 18 | 28 | 0·16 | |
| TC | 100 | 56 | 44 | ||||
| CC | 46 | 22 | 24 | ||||
| rs190590338 | Promoter | GG | 197 | 98 | 99 | 0·32 | |
| AG | 1 | 1 | – | ||||
| rs35089177 | Promoter | AA | 85 | 48 | 37 | 0·16 | |
| AT | 100 | 47 | 53 | ||||
| TT | 13 | 4 | 9 | ||||
| rs7625133 | Promoter | AA | 171 | 83 | 88 | 0·14 | |
| AC | 25 | 16 | 9 | ||||
| CC | 2 | – | 2 | ||||
| rs72549254 | Intron 01 | TT | 142 | 68 | 74 | 0·58 | |
| CT | 51 | 29 | 22 | ||||
| CC | 5 | 2 | 3 | ||||
| rs3774275 | Intron 08 | AA | 28 | 12 | 16 | 0·71 | |
| AG | 85 | 43 | 42 | ||||
| GG | 85 | 44 | 41 | ||||
| rs67143992 | 3'‐UTR MASP‐3 | CC | 124 | 59 | 65 | 0·70 | |
| CT | 67 | 36 | 31 | ||||
| TT | 7 | 4 | 3 | ||||
| rs850312 | Exon 12 | AA | 87 | 40 | 47 | 0·13 | |
| AG | 89 | 51 | 38 | ||||
| GG | 22 | 8 | 14 | ||||
| rs72549154 | Exon 12 | GG | 190 | 93 | 97 | 0·12 | |
| GT | 7 | 6 | 1 | ||||
| TT | 1 | – | 1 |
*Gen. = genotypes; † n = number; MASP = mannan‐binding lectin‐associated serine protease.
Plasma levels of MASP‐1 and the influence of MASP1 gene polymorphisms have, to our knowledge, never been studied previously in type 2 diabetes; neither has the direct effect of diabetes on plasma level of MASP‐1 in a mouse model. The aim of this study was thus to investigate the correlation between MASP1 single nucleotide polymorphisms (SNPs) and the plasma concentrations of MASP‐1, MASP‐3 and MAp44 in 100 patients with type 2 diabetes and 100 age‐ and sex‐matched control subjects. Furthermore, we wanted to investigate the MASP‐1 concentration in a mouse model before and 18 weeks after inducing diabetes in one group of the mice.
Materials and methods
Subjects
The study cohort consisted of 100 patients with newly diagnosed type 2 diabetes and 100 age‐ and gender‐matched controls recruited, as described previously 18, 19, from the out‐patient clinics at Aarhus University Hospital, Aarhus, Denmark and by advertising in the local press. The inclusion criteria were aged more than 18 years and a diagnosis of type 2 diabetes according to World Health Organization criteria 20 with a duration of less than 5 years. Undiagnosed diabetes among the control subjects was excluded by fasting glucose and oral glucose tests.
Blood samples were not obtainable for one participant. For that reason, this participant and the belonging match were excluded in the statistical analysis.
The study complied with the Declaration of Helsinki and was approved by the Research Ethics Committee of Central Region (1‐10‐72‐349‐13) and by the Danish Data Protection Agency (1‐16‐02‐505‐13), Denmark. All patients gave their written informed consent to participate.
Protein measurements
Blood samples were obtained from our study cohort in 2008–09. The samples were stored at −80°C. Ethylenediamine tetraacetic acid (EDTA) plasma for MASP‐1, MASP‐3 and MAp44 levels were analysed using time‐resolved immunofluorometric assay with Europium (Eu)‐labelled antibodies 21, described in detail previously for MASP‐3 and Map44 assays 22. All three assays were of the conventional sandwich configuration. In brief, microtitre wells were coated overnight with monoclonal mouse anti‐human antibodies. After blocking with Tris‐buffered saline (TBS)/Tween the diluted plasma samples, standards and controls were added in duplicate and incubated overnight in 4°C; the wells were washed thrice with TBS/Tween/Ca2+ and the bound protein was detected with biotinylated antibodies. Excess antibodies were removed after washing thrice with TBS/Tween/Ca2+ and the biotinylated antibodies were detected with Eu‐streptavidin. After a final washing, enhancement buffer was added to make the Eu dissociate, and quantification of the fluorescence was measured using a dissociation‐enhanced lanthanide flouroimmunoassay reader.
Genotyping
Ten MASP1 SNPs were selected (Table 2) based on previously described phenotype associations 14, 15, non‐synonymous SNPs and synonymous SNPs located in regulatory regions, promotor regions, exons or introns in the splicing‐region of the gene. Location of the SNPs were found using Variation Viewer 23 and from previous descriptions in the literature 14, 15, 24.
The SNPs were analysed using TaqMan® genotyping assays (Applied Biosystems, Foster City, CA, USA). For all TaqMan assays deoxyribonucleic acid (DNA) amplifications were carried out in 25‐µl polymerase chain reactions (PCR) containing 20 ng DNA, 0·9 µM primers and 0·2 µM probes (final concentrations), and amplified in 96‐well plates. Reactions were performed on a GeneAmp PCR 9700 or a 7900 HT Sequence Detection System: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The genotyping was determined by measuring the end‐point fluorescence on a 7900 HT Sequence Detection System (SDS) using SDS version 2.3 software.
Animals
The animals used were 11‐week‐old female C57BL/6J wild‐type mice (Taconic, Ry, Denmark) 25.
The animals were housed three to eight per cage and had free access to tap water and standard chow (Altromin no. 1324; Altromin International, Lage, Germany). The environment was kept stable with a 12‐h light–dark cycle, a temperature of 21 ± 1°C and humidity of 55 ± 5%. The animals were randomized into a diabetic and non‐diabetic group with 11 mice in each group.
Diabetes was induced by five intraperitoneal injections of streptozotocin (STZ) dissolved in a cold 10 mM citrate buffer (doses of 55 mg/kg body weight; Sigma Aldrich, St Louis, MO, USA) on 5 consecutive days. The animals were fasting 4 h prior to injection. If the blood glucose did not rise sufficiently, two reinjections were given. Controls were injected with citrate buffer. The 18‐week experiment was initiated when the mice were diabetic (blood glucose > 15 mM). Body weight and blood glucose were measured weekly. Animals with more than 15% sustained weight loss, signs of illness or persistent ketonuria were excluded from the study. Blood glucose was measured by ContourTM (Bayer Diabetes Care, Kongens Lyngby, Denmark) and the urine was tested for ketone bodies with Combur Test D (Roche Diagnostics GmbH, Mannheim, Germany). One mouse from the diabetic group was excluded because blood glucose levels were <15 mM.
The study complied with the regulations for care and use of laboratory animals.
Statistical analysis
Comparison of clinical characteristics between the two groups was performed using a paired t‐test. Assumption of normal distributions was tested by histograms and Q–Q plots. Diabetes duration and triglyceride levels were skewed, and therefore log‐transformed before t‐tests were performed. Descriptive characteristics of the study population are presented as means ± standard deviation (s.d.) for normally distributed data or median [interquartile range (IQR)] for skewed data. All statistical results are presented as mean [95% confidence interval (CI)]. Dichotomous outcomes were tested with McNemar's test for paired data. Analysis of variance (anova) was used to investigate the association between plasma protein concentration and the covariates body mass index (BMI), haemoglobin A1c (HbA1c), diabetic status and MASP1 genotypes. In order to adjust for matching, the model was based on linear regression. The most frequent genotype was used as baseline variable. When testing diabetic and control groups separately, one‐way analysis of variance (anova) was used. If only one or two individuals had a given genotype, this genotype was not in the anova. Interactions between the effects of MASP1 SNPs and diabetic status on the protein concentration were tested with the interaction parameters incorporated into the regression models. Assumptions of normal distribution within each group and equal variances in all groups were tested with Q–Q‐plots, student's F‐test or Bartlett's test. Genotype distributions for SNPs between gender, diabetes patients and control subjects were compared using χ2 or Fisher's exact test. A P‐value < 0·05 was considered statistically significant.
The data were analysed using Stata software version 13.1 (StataCorp, College Station, TX, USA).
Results
Characteristics of the study population are presented in Table 1. The median diabetes duration of the patients was less than 2 years and the glycaemic control was excellent, but they were generally overweight. There was no difference in smoking habits, 24‐h ambulatory systolic or diastolic blood pressure between the two groups. However, significantly more patients with diabetes than controls were receiving anti‐hypertensive and cholesterol‐lowering treatment. Consequently, the diabetic group had significantly lower cholesterol levels.
Table 1.
Characteristics of the study population
| Patients with diabetes | Control subjects | P‐value | |
|---|---|---|---|
| Sex (men/women), n * | 51/48 | 51/48 | – |
| Age (years) ± s.d.† | 58 ± 10 | 58 ± 10 | – |
| Diabetes duration, median (IQR‡) | 1·8 (0·7; 3·2) | – | – |
| HbA1c, % ± s.d. | 6·5 ±0·6 | 5·6 ± 0·3 | < 0·0001 |
| BMI, kg/m2 ± s.d. | 30 ± 5 | 26 ± 4 | < 0·0001 |
| Smoking, n (ever/never) | 57/42 | 53/46 | 0·68 |
| 24‐h ABPM§ systolic BP (mmHg) ± s.d. | 126 ± 11 | 125 ± 13 | 0·68 |
| 24‐h ABPM diastolic BP (mmHg) ± s.d. | 74 ± 7 | 76 ± 8 | 0·14 |
| 24‐h HR (bpm) ± s.d. | 74 ± 10 | 68 ± 9 | 0·008 |
| Statin use, n (yes/no) | 75/23 | 18/81 | < 0·0001 |
| Anti‐hypertensive treatment, n (yes/no) | 63/35 | 25/74 | < 0·0001 |
| Total cholesterol (mmol/l) ± s.d. | 4·4 ± 0·8 | 5·7 ± 1·0 | < 0·0001 |
| LDL‐cholesterol (mmol/l) ± s.d. | 2·3 ± 0·8 | 3·4 ± 1·0 | < 0·0001 |
| HDL‐cholesterol (mmol/l) ± s.d. | 1·4 ± 0·3 | 1·7 ± 0·6 | < 0·0001 |
| Triglycerides (mmol/l), median (IQR) | 1·4 (1·1; 1·9) | 1·2 (0·9; 1·6) | 0·011 |
*n, = number; †s.d. = standard deviation; BP = blood pressure; ‡IQR = interquartile range; §ABPM = ambulatory blood pressure monitoring; BMI = body mass index; HbA1c = haemoglobin A1c; HR = heart rate; LDL = low‐density lipoprotein; HDL = high‐density lipoprotein.
Plasma levels of MASPs and MAp44
MASP‐1 levels were significantly higher in patients with type 2 diabetes (mean = 10·17 µg/ml, 95% CI = 9·68, 10·66) than in the control subjects (mean = 9·39 µg/ml, 95% CI = 9·03, 9·75, P = 0·017). This remained significant when adjusting for BMI (P = 0·039). MAp44 levels were not significantly higher among the patients (mean = 2·59 µg/ml, 95% CI = 2·46, 2·73) versus mean = 2·41 µg/ml, 95% CI = 2·30, 2·53, P = 0·055), nor did MASP‐3 levels differ between patients (mean = 6·10 µg/ml, 95% CI = 5·78, 6·41) and controls (mean = 5·87 µg/ml, 95% CI = 5·60, 6·15, P = 0·30 (Fig. 1).
Figure 1.
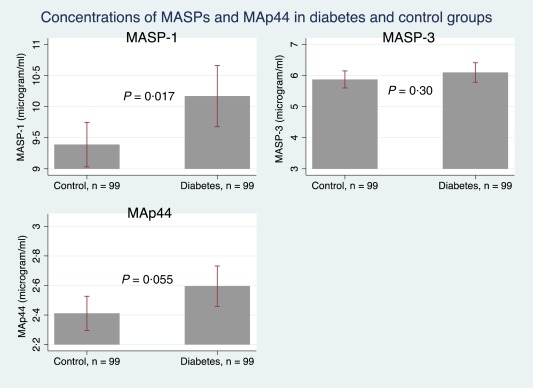
The mean plasma levels of mannan‐binding lectin‐associated serine protease (MASPs) and mannan‐binding lectin‐associated protein of 44 kDa (MAp44) in patients with type 2 diabetes compared to control subjects. The bars illustrate mean and the whiskers illustrate 95% confidence interval. [Colour figure can be viewed at wileyonlinelibrary.com].
Effects of body mass index and glycaemic control
A significant correlation between MASP‐3 levels (P = 0·008, r 2 = 0·03), MAp44 levels (P = 0·002, r 2 = 0·05) and body mass index (BMI) was observed (Fig. 2), but not between BMI and MASP‐1 levels (P = 0·23). These associations remained significant when adjusting for diabetes, MAp44 (P = 0·01) and MASP‐3 (P = 0·02), respectively. Moreover, a significant association of the plasma concentration of MASP‐1 and MAp44 (P = 0·02 for both proteins) with glycated haemoglobin (HbA1c) was observed.
Figure 2.
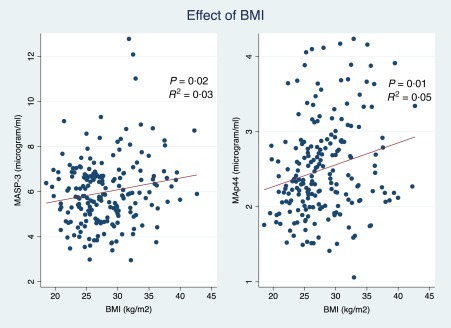
The relationship between body mass index (BMI), mannan‐binding lectin‐ associated serine protease (MASP)‐3 and mannan‐binding lectin‐associated protein of 44 kDa (MAp44) concentrations. The red lines illustrate the regression lines of MASP‐3 and MAp44 as function of BMI. R 2 is the correlation coefficient of the model and P is the P‐value of the correlation adjusted for diabetes. [Colour figure can be viewed at wileyonlinelibrary.com].
When adjusting for HbA1c the difference of MASP‐1 concentration between patients with type 2 diabetes and control subjects did not remain significant (P = 0·26).
MASP1 genotypes
The genotypes were distributed equally between diabetic patients and control subjects (Table 2). Men and women were also tested separately, but no significant differences were observed (data not shown). Rs3774275 was associated with BMI, but only in the diabetic group, and when adjusting for this in the genetic analysis no difference was observed. For these reasons we did not adjust for BMI in further analyses.
Association between SNPs and plasma levels
Association between SNPs and plasma concentration of MASP‐1, MASP‐3 and MAp44 are presented in Tables 3, 4, 5, 6. When only one or two individual had a given genotype the variance from this genotype was not tested, and no P‐value is therefore presented.
Table 3.
The effect of SNPs on the concentration of MASP‐1, MASP‐3 and MAp44 for the entire study population
| MASP‐1 | MASP‐3 | MAp44 | ||||||
|---|---|---|---|---|---|---|---|---|
| SNP | Gen.* | n † | Concentration µg/ml (95% CI ‡ ) | P | Concentration µg/ml (95% CI) | P | Concentration µg/ml (95% CI) | P |
| rs874603 | AA | 176 | 9·79 (9·45; 10·11) | 5·87 (5·66; 6·08) | 2·48 (2·39; 2·58) | |||
| GA | 20 | 9·70 (8·85; 10·55) | 0·86 | 6·91 (6·04; 7·78) | 0·011 | 2·68 (2·32; 3·03) | 0·23 | |
| rs710469 | TT | 46 | 9·81 (9·27; 10·35) | 0·73 | 6·33 (5·87; 6·79) | 0·11 | 2·41 (2·23; 2·58) | 0·32 |
| CT | 100 | 9·68 (9·22; 10·15) | 5·89 (5·61; 6·17) | 2·52 (2·39; 2·65) | ||||
| CC | 46 | 10·06 (9·40; 10·71) | 0·38 | 5·99 (5·53; 6·45) | 0·72 | 2·63 (2·42; 2·87) | 0·39 | |
| rs190590338 | GG | 197 | 9·77 (9·46; 10·08) | – | 5·99 (5·78; 6·19) | – | 2·50 (2·41; 2·59) | – |
| AG | 1 | 11·99 | 5·92 | 3·09 | ||||
| rs35089177 | AA | 85 | 9·85 (9·41; 10·30) | 0·66 | 6·01 (5·67; 6·35) | 1·00 | 2·60 (2·46; 2·74) | 0·08 |
| AT | 100 | 9·71 (9·23; 10·18) | 6·01 (5·72; 6·30) | 2·44 (2·32; 2·58) | ||||
| TT | 13 | 9·86 (9·11; 10·61) | 0·71 | 5·61 (4·98; 6·23) | 0·19 | 2·31 (2·04; 2·59) | 0·34 | |
| rs7625133 | AA | 171 | 9·83 (9·50; 10·16) | 5·92 (5·70; 6·14) | 2·53 (2·43; 2·62) | |||
| AC | 25 | 9·59 (8·62; 10·55) | 0·63 | 6·42 (5·70; 7·15) | 0·17 | 2·39 (2·12; 2·66) | 0·34 | |
| CC | 2 | 7·97 | 6·24 | 1·79 | ||||
| rs72549254 | TT | 142 | 9·86 (9·59; 10·22) | 5·87 (5·64; 6·10) | 2·55 (2·44; 2·65) | |||
| CT | 51 | 9·65 (9·03; 10·26) | 0·56 | 6·23 (5·77; 6·69) | 0·17 | 2·43 (2·26; 2·61) | 0·28 | |
| CC | 5 | 8·85 (7·01; 10·69) | 0·11 | 6·82 (4·04; 9·59) | 0·30 | 1·93 (1·36; 2·51) | 0·002 | |
| rs3774275 | AA | 28 | 9·00 (8·11; 9·89) | 0·03 | 6·22 (5·70; 6·75) | 0·28 | 2·32 (2·04; 2·60) | 0·03 |
| AG | 85 | 9·63 (9·19; 10·07) | 0·10 | 6·02 (5·74; 6·31) | 0·49 | 2·43 (2·29; 2·56) | 0·04 | |
| GG | 85 | 10·19 (9·71; 10·67) | 5·87 (5·51; 6·23) | 2·64 (2·51; 2·77) | ||||
| rs67143992 | CC | 124 | 9·59 (9·20; 9·98) | 6·24 (5·96; 6·52) | 2·43 (2·32; 2·55) | |||
| CT | 67 | 10·09 (9·55; 10·63) | 0·12 | 5·60 (5·31; 5·90) | 0·001 | 2·61 (2·46; 2·76) | 0·49 | |
| TT | 7 | 10·22 (8·60; 11·86) | 0·32 | 5·11 (3·88; 6·34) | 0·02 | 2·76 (2·16; 3·37) | 0·17 | |
| rs850312 | AA | 87 | 9·53 (9·10; 9·96) | 6·42 (6·07; 6·77) | 2·32 (2·19; 2·45) | |||
| AG | 89 | 10·04 (9·54; 10·54) | 0·56 | 5·68 (5·42; 5·93) | 0·001 | 2·61 (2·49; 2·74) | 0·003 | |
| GG | 22 | 9·71 (8·82; 10·61) | 0·81 | 5·50 (4·90; 6·10) | 0·005 | 2·78 (2·47; 3·10) | 0·005 | |
| rs72549154 | GG | 190 | 9·77 (9·46; 10·09) | 5·97 (5·76; 6·18) | 2·50 (2·41; 2·59) | |||
| GT | 7 | 9·80 (7·95; 11·65) | 0·98 | 6·36 (5·09; 7·62) | 0·43 | 2·67 (2·11; 3·22) | 0·45 | |
| TT | 1 | 10·57 | – | 6·38 | – | 1·76 | – |
*Gen. = genotypes; † n = number. ‡CI = confidence interval; MASP = mannan‐binding lectin‐associated serine protease; SNPs = single nucleotide polymorphisms; MAp44 = mannan‐binding lectin‐associated protein of 44 kDa.
Table 4.
The effect of rs3774275 on MASP‐1 levels in diabetes and control groups separately
| SNP | Gen.* | n † dm ‡ | Concentration µg/ml (95% CI § ) Diabetes group | P | n Con. ¶ | Concentration µg/ml (95% CI § ) Control group | P |
|---|---|---|---|---|---|---|---|
| rs3774275 | AA | 12 | 8·66 (6·71; 10·61) | 0·01 | 16 | 9·26 (8·42; 10·10) | 0.90 |
| AG | 43 | 9·90 (9·27; 10·53) | 42 | 9·35 (8·72; 9·97) | |||
| GG | 44 | 10·84 (10·08; 11·60) | 41 | 9·48 (8·95; 10·01) |
*Gen. = genotypes; † n = number. ‡dm = diabetes group; §CI = confidence interval; ¶con. = control group; MASP = mannan‐binding lectin‐associated serine protease.
Table 5.
The effect of SNPs on MASP‐3 levels in diabetes and control groups separately
| SNP | Gen.* | n † dm ‡ | Concentration µg/ml (95% CI § ) Diabetes group | P | n Con. ¶ | Concentration µg/ml (95% CI § ) Control group | P |
|---|---|---|---|---|---|---|---|
| rs874603 | AA | 86 | 5·94 (5·64; 6·23) | 0·02 | 90 | 5·81 (5·52; 6·10) | 0·07 |
| GA | 12 | 7·04 (5·57; 8·51) | 8 | 6·71 (5·87; 7·56) | |||
| rs67143992 | CC | 59 | 6·45 (6·02; 6·88) | 0·02 | 65 | 6·05 (5·69; 6·41) | 0·07 |
| CT | 36 | 5·57 (5·12; 6·02) | 31 | 5·64 (5·26; 6·03) | |||
| TT | 4 | 5·64 (4·21; 7·06) | 3 | 4·40 (0·24; 8·55) | |||
| rs850312 | AA | 40 | 6·66 (6·10; 7·22) | 0·01 | 47 | 6·22 (5·77; 6·67) | 0·04 |
| AG | 51 | 5·70 (5·34; 6·06) | 38 | 5·65 (5·27; 6·02) | |||
| GG | 8 | 5·82 (4·40; 7·23) | 14 | 5·32 (4·65; 5·99) |
*Gen. = genotypes; † n = number; ‡dm = diabetes group; = §CI = confidence interval = ¶con. = control group; MASP = mannan‐binding lectin‐associated serine protease; SNPs = single nucleotide polymorphisms.
Table 6.
The effect of SNPs on MAp44 levels in diabetes and control groups separately
| SNP | Gen.* | n † dm ‡ | Concentration µg/ml (95% CI § ) Diabetes group | P | n Con. ¶ | Concentration µg/ml (95% CI § ) Control group | P |
|---|---|---|---|---|---|---|---|
| rs72549254 | TT | 68 | 2·68 (2·52; 2·84) | 0·07 | 74 | 2·43 (2·29; 2·57) | 0·58 |
| CT | 29 | 2·46 (2·19; 2·73) | 22 | 2·40 (2·17; 2·62) | |||
| CC | 2 | 1·72 | 3 | 2·07 | |||
| rs3774275 | AA | 12 | 2·46 (1·97; 2·95) | 0·23 | 16 | 2·21 (1·85; 2·57) | 0·10 |
| AG | 43 | 2·50 (2·29; 2·70) | 42 | 2·35 (2·18; 2·53) | |||
| GG | 44 | 2·73 (2·52; 2·93) | 41 | 2·55 (2·37; 2·72) | |||
| rs850312 | AA | 40 | 2·46 (2·24; 2·68) | 0·06 | 47 | 2·20 (2·05; 2·35) | 0·002 |
| AG | 51 | 2·62 (2·46; 2·79) | 38 | 2·60 (2·39; 2·80) | |||
| GG | 8 | 3·08 (2·26; 3·91) | 14 | 2·61 (2·35; 2·87) |
*Gen. = genotypes; † n = number; ‡dm = diabetes group; §CI = confidence interval; ¶con. = control group; MAp44 = mannan‐binding lectin‐associated protein of 44 kDa; SNPs = single nucleotide polymorphisms.
For the MASP‐1 protein, the only SNP found to be associated significantly with the plasma concentration was rs3774275. The less frequent genotype (AA, n = 28) was associated with lower MASP‐1 levels (mean = 9·00 µg/ml, 95% CI = 8·11, 9·89) compared to the more frequent homozygote GG (n = 85) (mean = 10·19 µg/ml, 95% CI = 9·71, 10·67, P = 0·03). Interactions between the effects of genotypes in the MASP1 SNPs and diabetic status were tested, and a significant interaction parameter was found only for this SNP (rs3774275), GG × diabetic status (P = 0·045). The association of this SNP was present only in the diabetes group (P = 0·01) and not in the control group (P = 0·90) when analysing the two groups separately (Table 4). When adjusting for diabetic status, no significantly different associations between genotypes and MASP‐1 levels were obtained (data not shown).
Three SNPs (rs874603, rs67143992, rs850312) were associated with plasma concentrations of MASP‐3. Data for the entire study population are shown in Table 3 and for the groups separately in Table 5. In separate analyses, the associations remained statistically significant only in the diabetes group for rs87460 and rs67143992, but were attenuated in the control group (P = 0·07 for both SNPs).
Besides association with the MASP‐3 concentration, rs850312 was also associated with the plasma concentration of MAp44 in the entire study population, as were rs72549254 and rs3774275. While the genotype GG in rs850312 was associated with lower MASP‐3 levels (mean = 5·50 µg/ml, 95% CI = 4·90, 6·10) compared to the genotype AA (mean = 6·42 µg/ml, 95% CI = 6·07, 6·77, P = 0·005), GG had the opposite effect on MAp44 levels (GG, mean = 2·78 µg/ml, 95% CI = 2·47, 3·10 versus AA, mean = 2·32 µg/ml, 95% CI = 2·19, 2·45, P = 0·005). The effect of rs850312 on MAp44 plasma levels remained significant in the control group (P = 0·002) but not in the diabetes group (P = 0·06). The effect of the two other SNPs (rs72549254 and rs3774275) did not remain significant when analysing the groups separately (Table 6). When incorporating the interaction parameters, genotypes and diabetic status to plasma levels of MASP‐3 and MAp44, respectively, no interactions were found, nor were any significantly different associations between genotypes and protein levels of the two proteins found when adjusting for diabetic status (data not shown).
Diabetes mouse model
MASP‐1 levels were identical in the two groups at baseline (P = 0·66, data not shown), and a fall in MASP‐1 levels was observed from the study start (mean = 6·04 units/ml, 95% CI = 5·18, 6·90) to study end (mean = 2·14 units/ml, 95% CI = 1·76, 2·52) in all mice, P < 0·001, indicating that MASP‐1 levels in general decline with age in mice. However, at study end MASP‐1 levels were significantly higher in the diabetic mice (mean = 2·6 units/ml, 95% CI = 2·0, 3·3) compared to the non‐diabetic mice (mean = 1·6 units/ml, 95% CI = 1·5, 1·8), P = 0·003 (Fig. 3).
Figure 3.
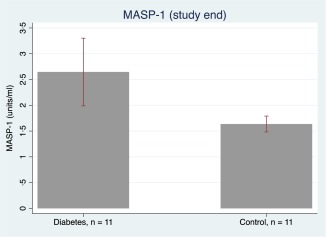
Mean mannan‐binding lectin‐associated serine protease (MASP)‐1 concentrations in the diabetic and control group. The bars illustrate mean MASP‐1 concentration and the whiskers indicate 95% confidence interval. [Colour figure can be viewed at wileyonlinelibrary.com].
Discussion
In the present study, we investigated the association between MASP1 SNPs and plasma concentrations of the corresponding gene products, MASP‐1, MASP‐3 and MAp44, in patients with type 2 diabetes and control subjects. In line with a previous study in patients with type 1 diabetes 13, we found significantly higher MASP‐1 levels, but not MASP‐3 or MAp44 levels, in type 2 diabetic patients compared to control subjects. In our genetic results, we did not find any statistically significant differences between genotype distribution in diabetes and control groups. As far as we know, the MASP1 gene has not been observed as a candidate gene in any type 2 diabetes genomewide association studies. Only one SNP was associated with the MASP‐1 level. From our study, genetics alone do not seem to be the reason for higher MASP‐1 plasma levels in patients with diabetes.
We saw a positive correlation between MASP‐1 and HbA1c, and as it has been speculated previously that hyperglycaemia causes elevated levels of MBL, MASP‐1 and MASP‐2 we included a diabetes mouse model to investigate the causality of diabetes inducing increased MASP‐1 levels. Diabetes induction led to higher MASP‐1 levels among the diabetic mice compared to the control mice, suggesting a higher MASP‐1 level as a consequence of diabetes. In accordance with this finding, it has been shown previously for MBL‐C in a similar mouse model 26. A possible link between hyperglycaemia and complement activation has been suggested to be via MBL binding with high affinity to the glycation product fructoseamine 27. Whether the underlying mechanism of elevated MASP‐1 level in diabetes is secondary to hyperglycaemia‐induced MBL increase remains to be studied.
MASP‐3 and MAp44 levels did not differ between the diabetes and control groups when adjusting for BMI. However, we found a weak positive correlation between BMI, MASP‐3 and MAp44 levels. To our knowledge, this finding has not been described previously. Even though our finding does not show a strong correlation, it can be speculated that as white adipose tissue is an endocrine and secretory organ found to secrete other inflammatory cytokines 28, it might also be involved in secretion and regulation of complement components such as MASP‐3 and MAp44. In contrast, association of weight loss and serum MBL concentration has been studied, but no correlation was found 29.
In the genetic analysis we found three SNPs to be associated with MASP‐3 levels (Table 5) and three SNPs to be associated with MAp44 levels (Table 6). The effect of the three SNPs on the MASP‐3 levels was consistent in the diabetes group, but not in the control group. For MAp44, only one SNP remained associated significantly with the plasma level when analysing the two groups separately. However, as we did not find any significant interaction parameters between genotypes and diabetic status to the plasma levels of MASP‐3, we cannot conclude that the association between MASP‐3 and the genotypes are significantly different in the two groups. The association of rs67143992 and rs850312 with the plasma level of MASP‐3 is in accordance with previous findings 14, 15.
Our study provides important new information about components in the lectin pathway in diabetes. We are the first to investigate polymorphisms in the MASP1 gene in relation to its associated gene products in type 2 diabetes. The fact that MASP‐1 levels are elevated in type 2 diabetes emphasizes the importance of the lectin pathway in diabetes. As we included a diabetes mouse model showing higher levels of MASP‐1 in the diabetic mice, we suggest a causal link between diabetes and the higher MASP‐1 levels. The relevance of investigating the role of MASP‐1 in diabetes and diabetic vascular complications is in accordance with its pathophysiological role in blood coagulation (reviewed in 30). An in‐vitro study found MASP‐1 to promote fibrin formation, influence the fibrin structure, activate endothelial cells and hence be involved in blood clot formation 31. Of clinical relevance, a pilot study exploring plasma levels of MASPs in cardio‐ and cerebrovascular disease found elevated MASP‐1 levels in patients with subacute phase of myocardial infarction, but decreased levels of MASP‐1 in patients with acute ischaemic stroke, whereas MASP‐3 and MAp44 did not differ between the groups 32. These studies suggest MASP‐1 to be involved in blood clot formation, and hence may be of importance of the increased prothrombotic state found in patients with diabetes.
A first limitation of our study is that, although we found a significantly higher MASP‐1 level in type 2 diabetes compared to control subjects, our cross‐sectional study design does not provide hard evidence for causal involvement. Another limitation that needs to be considered is our matching that is only performed on age and gender, leaving the risk of confounding from other risk factors. Nevertheless, in the genetic analysis we did not find any association between BMI and investigated genotypes, and one must consider external confounding to be limited for two reasons: (1) genes are assorted randomly during gamete formation and (2) genes should not be susceptible to external risk factors. Conversely, in SNP association studies it is difficult to exclude that the analysis of genotype and gene products could be confounded by SNPs in linkage disequilibrium with the SNPs analysed. The size of our study population and number of SNPs analysed also need to be taken into account. The 10 SNPs are selected cautiously to cover exons, introns and untranslated regions and are based on previously described associations with the protein levels. However, we do not intend to study all genetic variations in the MASP1 gene, and hence, we cannot exclude the importance of any other SNPs in relation to the MASP1 gene products in type 2 diabetes. Conversely, the preselective procedure reduces the risk of false associations found by statistical coincidence and for that reason is considered a more effective study design 33. Statistical coincidence must be considered in our genetic analysis with multiple testing. A limitation of the size of our study population concerns the risk of missing associations that might exist, but are too small to be significant in our relatively small sample size. This limitation is also present in the mouse model, where the difference of the MASP‐1 level between the two groups actually might be of greater significance.
Although our findings in our mouse model support the observation of elevated MASP1 levels in diabetic patients, the use of STZ is a limitation. STZ is a pancreatic β cell toxin that destroys the β cells, and we cannot be sure that the higher plasma level of MASP‐1 seen in the diabetic mice is caused by STZ itself rather than the higher glycaemic level caused by STZ. Our mouse model imitates a type 1 diabetes condition that is different to the type 2 diabetic condition in the patients of our study cohort. However, the MASP‐1 concentration remained significantly higher in the diabetes group when adjusting for BMI, and we found a significant correlation between HbA1c and plasma levels of MASP‐1. This may suggest that the reason of the higher MASP‐1 level in the patients of our study population is not caused by an underlying inflammatory condition in the adipose tissue alone, but rather the higher glycaemic level.
In conclusion, this study demonstrates higher MASP‐1 levels in patients with type 2 diabetes compared to control subjects. Surprisingly, we also found a positive correlation between MASP‐3, MAp44 and BMI. In the genetic analysis, we found several SNPs to be associated with the plasma levels of MASP‐1, MASP‐3 and MAp44 with different significance in the diabetes and control group. In our mouse model, we are the first to show that after inducing diabetes the plasma level of MASP‐1 is significantly higher compared to the level in the control mice.
Author contributions
S. S. K. measured the protein levels, made the analysis and wrote the paper. T. K. H., S. T. and S. S. K. designed the study. The genotyping was conducted under the supervision of R. S. C. H. was responsible for the mouse experiment and E. L. and P. H. recruited and characterized the study cohort. T. K. H., K. F., E. L. and P. L. P. were persistent co‐supervisors during the research process. Finally, the Aase and Ejnar Danielsens Foundation was helpful with financial support. All authors revised the manuscript.
Disclosure
We have no financial or commercial conflicts of interest to declare.
Acknowledgement
This paper is based on work done from Department of Endocrinology and Internal Medicine, Aarhus University Hospital, Department of Biomedicine, Aarhus University and Department of Immunology, Aalborg University Hospital in Denmark.
References
- 1. Ghosh P, Sahoo R, Vaidya A, Chorev M, Halperin JA. Role of complement and complement regulatory proteins in the complications of diabetes. Endocr Rev 2015; 36:272–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Flyvbjerg A. Diabetic angiopathy, the complement system and the tumor necrosis factor superfamily. Nat Rev Endocrinol 2010; 6:94–101. [DOI] [PubMed] [Google Scholar]
- 3. Ostergaard J, Hansen TK, Thiel S, Flyvbjerg A. Complement activation and diabetic vascular complications. Clin Chim Acta 2005; 361:10–9. [DOI] [PubMed] [Google Scholar]
- 4. Hansen TK, Tarnow L, Thiel S et al Association between mannose‐binding lectin and vascular complications in type 1 diabetes. Diabetes 2004; 53:1570–6. [DOI] [PubMed] [Google Scholar]
- 5. Bajic G, Degn SE, Thiel S, Andersen GR. Complement activation, regulation, and molecular basis for complement‐related diseases. EMBO J 2015; 34:2735–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Degn SE, Jensen L, Hansen AG et al Mannan‐binding lectin‐associated serine protease (MASP)‐1 is crucial for lectin pathway activation in human serum, whereas neither MASP‐1 nor MASP‐3 is required for alternative pathway function. J Immunol 2012; 189:3957–69. [DOI] [PubMed] [Google Scholar]
- 7. Hansen TK, Thiel S, Knudsen ST et al Elevated levels of mannan‐binding lectin in patients with type 1 diabetes. J Clin Endocrinol Metab 2003; 88:4857–61. [DOI] [PubMed] [Google Scholar]
- 8. Mellbin LG, Hamsten A, Malmberg K et al Mannose‐binding lectin genotype and phenotype in patients with type 2 diabetes and myocardial infarction: a report from the DIGAMI 2 trial. Diabetes Care 2010; 33:2451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hansen TK, Gall MA, Tarnow L et al Mannose‐binding lectin and mortality in type 2 diabetes. Arch Intern Med 2006; 166:2007–13. [DOI] [PubMed] [Google Scholar]
- 10. Ostergaard JA, Thiel S, Hovind P et al Association of the pattern recognition molecule H‐ficolin with incident microalbuminuria in an inception cohort of newly diagnosed type 1 diabetic patients: an 18 year follow‐up study. Diabetologia 2014; 57:2201–7. [DOI] [PubMed] [Google Scholar]
- 11. Steffensen R, Thiel S, Varming K, Jersild C, Jensenius JC. Detection of structural gene mutations and promoter polymorphisms in the mannan‐binding lectin (MBL) gene by polymerase chain reaction with sequence‐specific primers. J Immunol Methods 2000; 241:33–42. [DOI] [PubMed] [Google Scholar]
- 12. Ostergaard JA, Thiel S, Lajer M et al Increased all‐cause mortality in patients with type 1 diabetes and high‐expression mannan‐binding lectin genotypes: a 12‐year follow‐up study. Diabetes Care 2015; 38:1898–903. [DOI] [PubMed] [Google Scholar]
- 13. Jenny L, Ajjan R, King R, Thiel S, Schroeder V. Plasma levels of mannan‐binding lectin‐associated serine proteases MASP‐1 and MASP‐2 are elevated in type 1 diabetes and correlate with glycaemic control. Clin Exp Immunol 2015; 180:227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ammitzboll CG, Steffensen R, Jorgen Nielsen H et al Polymorphisms in the MASP1 gene are associated with serum levels of MASP‐1, MASP‐3, and MAp44. PLOS ONE 2013; 8:e73317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ingels C, Vanhorebeek I, Steffensen R et al Lectin pathway of complement activation and relation with clinical complications in critically ill children. Pediatr Res 2014; 75:99–108. [DOI] [PubMed] [Google Scholar]
- 16. Takada F, Seki N, Matsuda Y, Takayama Y, Kawakami M. Localization of the genes for the 100‐kDa complement‐activating components of Ra‐reactive factor (CRARF and Crarf) to human 3q27‐q28 and mouse 16B2‐B3. Genomics 1995; 25:757–9. [DOI] [PubMed] [Google Scholar]
- 17. Degn SE, Hansen AG, Steffensen R, Jacobsen C, Jensenius JC, Thiel S. MAp44, a human protein associated with pattern recognition molecules of the complement system and regulating the lectin pathway of complement activation. J Immunol 2009; 183:7371–8. [DOI] [PubMed] [Google Scholar]
- 18. Cichosz SL, Fleischer J, Hoeyem P et al Objective measurements of activity patterns in people with newly diagnosed Type 2 diabetes demonstrate a sedentary lifestyle. Diabet Med 2013; 30:1063–6. [DOI] [PubMed] [Google Scholar]
- 19. Laugesen E, Hoyem P, Stausbol‐Gron B et al Carotid‐femoral pulse wave velocity is associated with cerebral white matter lesions in type 2 diabetes. Diabetes Care 2013; 36:722–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Surveillance. WHODoND. Definition, Diagnosis and Classification of Diabetes Mellitus and its Complications . Report of a WHO Consultation. Available at: http://apps.who.int/iris/bitstream/10665/66040/1/WHO_NCD_NCS_99.2.pdf (accessed 8 January 2016).
- 21. Hemmila I, Dakubu S, Mukkala VM, Siitari H, Lovgren T. Europium as a label in time‐resolved immunofluorometric assays. Anal Biochem 1984; 137:335–43. [DOI] [PubMed] [Google Scholar]
- 22. Degn SE, Jensen L, Gal P et al Biological variations of MASP‐3 and MAp44, two splice products of the MASP1 gene involved in regulation of the complement system. J Immunol Methods 2010; 361:37–50. [DOI] [PubMed] [Google Scholar]
- 23. Information TNCfB . Variation Viewer. Available at: http://www.ncbi.nlm.nih.gov/variation/view/?q=rs874603&filters=source:dbsnp&assm=GCF_000001405.28 (accessed 20 October 2015).
- 24. Beltrame MH, Boldt AB, Catarino SJ et al MBL‐associated serine proteases (MASPs) and infectious diseases. Mol Immunol 2015; 67:85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Holt CB, Ostergaard JA, Axelgaard E et al Ficolin B in diabetic kidney disease in a mouse model of type 1 diabetes. Mediators Inflamm 2015; 2015:653260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ostergaard JA, Bjerre M, Dagnaes‐Hansen F, Hansen TK, Thiel S, Flyvbjerg A. Diabetes‐induced changes in mannan‐binding lectin levels and complement activation in a mouse model of type 1 diabetes. Scand J Immunol 2013; 77:187–94. [DOI] [PubMed] [Google Scholar]
- 27. Fortpied J, Vertommen D, Van Schaftingen E. Binding of mannose‐binding lectin to fructosamines: a potential link between hyperglycaemia and complement activation in diabetes. Diabetes Metab Res Rev 2010; 26:254–60. [DOI] [PubMed] [Google Scholar]
- 28. Trayhurn P, Beattie JH. Physiological role of adipose tissue: white adipose tissue as an endocrine and secretory organ. Proc Nutr Soc 2001; 60:329–39. [DOI] [PubMed] [Google Scholar]
- 29. Hoyem PH, Bruun JM, Pedersen SB et al The effect of weight loss on serum mannose‐binding lectin levels. Clin Dev Immunol 2012; 2012:354894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dobo J, Schroeder V, Jenny L, Cervenak L, Zavodszky P, Gal P. Multiple roles of complement MASP‐1 at the interface of innate immune response and coagulation. Mol Immunol 2014; 61:69–78. [DOI] [PubMed] [Google Scholar]
- 31. Gulla KC, Gupta K, Krarup A et al Activation of mannan‐binding lectin‐associated serine proteases leads to generation of a fibrin clot. Immunology 2010; 129:482–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frauenknecht V, Thiel S, Storm L et al Plasma levels of mannan‐binding lectin (MBL)‐associated serine proteases (MASPs) and MBL‐associated protein in cardio‐ and cerebrovascular diseases. Clin Exp Immunol 2013; 173:112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brookes AJ. The essence of SNPs. Gene 1999; 234:177–86. [DOI] [PubMed] [Google Scholar]