

Summary
Rheumatoid arthritis (RA) is an autoimmune disease characterized by chronic inflammation, local and systemic bone loss and a lack of compensatory bone repair. Fibroblast‐like synoviocytes (FLS) are the most abundant cells of the stroma and a key population in autoimmune diseases such as RA. An increasing body of evidence suggests that these cells play not only an important role in chronic inflammation and synovial hyperplasia, but also impact bone remodelling. Under inflammatory conditions FLS release inflammatory cytokines, regulate bone destruction and formation and communicate with immune cells to control bone homeostasis. Other stromal cells, such as osteoblasts and terminally differentiated osteoblasts, termed osteocytes, are also involved in the regulation of bone homeostasis and are dysregulated during inflammation. This review highlights our current understanding of how stromal cells influence the balance between bone formation and bone destruction. Increasing our understanding of these processes is critical to enable the development of novel therapeutic strategies with which to treat bone loss in RA.
Keywords: bone remodelling, inflammatory cytokines, RA, RA‐FLS, stromal cells
Introduction
Rheumatoid arthritis (RA) is an immune‐mediated chronic inflammatory disorder of synovial joints characterized by pain, swelling and progressive joint destruction. The synovium is present in articulated joints and serves to produce and maintain synovial fluid that aids joint lubrication and movement. The synovial membrane is just one to two cells thick in a healthy joint and is formed of macrophages, which remove debris, and synovial fibroblasts that produce hyaluronan and other extracellular matrix components of the synovial fluid. In RA the synovium becomes thickened due to an expansion of synovial fibroblasts as well as an infiltration of immune cells, blood vessels and osteoclasts, all of which contribute to the swelling and stiffness characteristic of the disease. Activated RA fibroblast‐like synoviocytes (RA‐FLS) are the most abundant stromal cells in the inflamed synovium. These cells not only destroy cartilage via matrix metalloproteinase (MMP) secretion, but are also able to destroy subchondral bone via regulation of osteoclastogenesis 1, 2, 3. Once activated, RA‐FLS maintain their tumour‐like aggressive behaviour even after multiple passages in vitro 4, and to date there is no conclusive molecular explanation for this stable, persistent activation.
Besides FLS, the stroma comprises a number of matrix‐producing, structural cell types including endothelial cells, pericytes, epithelial cells and osteoblasts 5. During the last few years several studies have focused upon the influence of stromal cells on bone remodelling. Various factors released by these cells influence the balance between bone‐forming osteoblasts and bone‐resorbing osteoclasts towards bone loss. This review summarizes the direct and indirect impact of stromal cells on bone resorption in chronic inflammatory disorders (Fig. 1).
Figure 1.
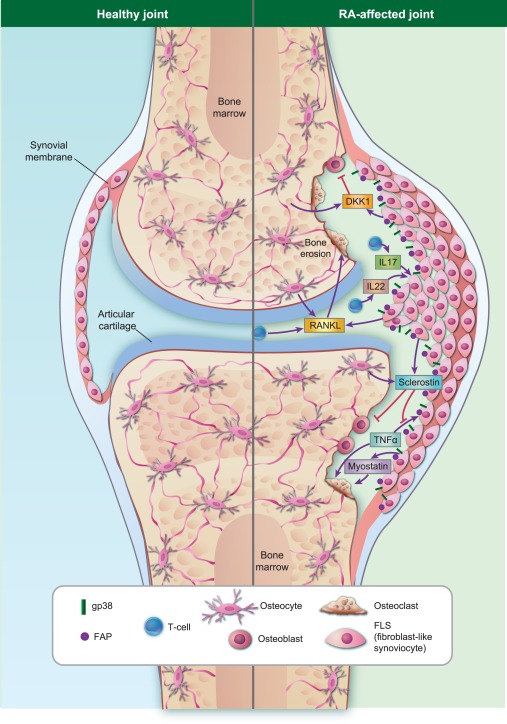
The role of fibroblast‐like synoviocytes (FLS) in inflammatory bone destruction. Under healthy conditions, there is a balance between bone formation and bone destruction to replace old bone tisssue and to repair bone defects. In rheumatoid arthritis (RA), more bone is degraded by osteoclasts than is created by osteoblasts, shifting the balance towards bone destruction. During inflammation, stromal FLS, located in the synovial membrane of the joint space, are able to influence this balance directly or indirectly. FLS release receptor‐activator of nuclear factor‐kappa B (RANKL) in response to inflammatory cytokines such as tumour necrosis factor (TNF)‐α, which subsequently stimulates osteoclastogenesis directly. They also communicate with T cells or release inhibitors of bone formation, such as sclerostin and Dickkopf‐1 (DKK1). In contrast to DKK1, sclerostin not only blocks osteoblast differentiation but also inhibits specifically TNF‐mediated bone destruction, suggesting a protective effect in TNF‐mediated bone loss. Other factors released by FLS, such as myostatin, activates bone destruction directly. Different subsets of FLS, especially gp38+ and FAP+‐expressing FLS, are highly migratory and invasive and seem to be important for cartilage and bone destruction.
Bone remodelling under resting conditions
Bone remodelling depends upon the tight coupling of bone formation and bone resorption to balance bone mass and adapt bone structure to environmental changes such as mechanical loading (ensuring that there is no net bone change). Under normal resting conditions osteoclasts are required for continuous bone remodelling via communication with osteoblasts. Osteoclasts create resorption lacunae which, in turn, activate osteoblasts that fill these lacunae with new bone matrix.
Osteoblasts arise from pluripotent mesenchymal stem cells (MSC). The differentiation of osteoblast progenitor cells into bone matrix‐producing osteoblasts requires tight control of essential signals, such as parathyroid hormone (PTH) 6, transforming growth factor (TGF)‐β and fibroblast growth factor (FGF) 7, 8. Furthermore, essential signalling pathways, including the bone morphogenic protein (BMP) pathway and the canonical Wingless and int‐1 (Wnt)/β‐catenin pathway, regulate the expression of runt‐related transcription factor 2 (Runx2) 9, alkaline phosphatase (ALP) 10 and osteoprotegerin (OPG) 11, which are all involved in osteoblast differentiation and metabolism.
Conversely, osteoclasts are multi‐nucleated cells derived from haematopoietic stem cells. Osteoclast development involves an initial differentiation step towards monocytes and macrophages, which is under the control of macrophage colony‐stimulating factor (M‐CSF). Following this there is a requirement for the receptor activator of nuclear factor kappa B (NF‐κB) (RANK) and the receptor activator of NF‐κB ligand (RANKL) 12. RANKL binds to its receptor RANK on the surface of pre‐osteoclasts, stimulating macrophage/monocyte fusion and the development of active, mature osteoclasts 13, 14. RANKL is produced predominantly by stromal cells, specifically by osteocytes, osteoblasts and fibroblasts, as well as a subset of B cells 15, 16. The RANKL decoy receptor OPG is also produced by stromal cells and blocks osteoclastogenesis by interacting with RANKL 17. This is supported by results from mouse models, revealing that RANKL deficiency causes osteopetrosis 18, while OPG‐deficient mice are osteoporotic 19. Importantly, therefore, both the main driver of osteoclastogenesis (RANKL) and the major inhibitor (OPG) are produced by stromal cells (osteoblasts and FLS) and the ratio between these two proteins is a key determinant of activation or inhibition of osteoclastogenesis during both normal, healthy bone turnover and in pathological bone destruction during inflammatory disease 17, 20.
Bone remodelling in RA
In addition to the pain, swelling and associated loss of function caused by synovial inflammation, resorption of bone tissue is a classical characteristic of RA. Erosion starts very early in disease, is irreversible and accompanied by permanent functional impairment 21. This process occurs predominantly at the regions where the articular cartilage, bone and inflamed synovium (pannus) meet with subchondral bone destroyed from the outside inwards by cells within the invading pannus tissue. The predominant bone‐degrading cell within the pannus is the osteoclast 22, 23.
In RA, inflammation and bone loss are closely linked processes. In this context inflammatory cytokines, including tumour necrosis factor (TNF)‐α, interleukin (IL)‐1β, IL‐6 and IL‐17, are produced by infiltrating macrophages and T cells. These cytokines induce RA‐FLS, osteoblasts and bone marrow stromal cells to express RANKL, thus promoting osteoclastogenesis 24, 25, 26, 27. RA synovial tissues express elevated RANKL mRNA and protein compared to patients with osteoarthritis (OA) or to RA patients with less active disease. This elevated level of RANKL is accompanied by diminished OPG expression 28, 29, 30. Thus, the uneven quantities of OPG and RANKL within the RA microenvironment results in an imbalance in the osteoblast–osteoclast axis leading to an overall bias towards bone resorption.
In addition to RANKL, OPG and cytokines, stromal cells of the inflamed synovial tissue also express other factors, which influence osteoclastogenesis directly. One of these factors is myostatin (also known as GDF‐8), which belongs to the TGF‐β family and is expressed mainly in skeletal muscle. Deletion of myostatin leads to muscle hypertrophy 31. In addition to its role as a negative regulator of muscle growth, several studies have revealed a new role for myostatin in bone homeostasis. Genetic deletion or antibody‐mediated blockade of myostatin leads to high bone density and volume in a mouse model of osteotomy 32, 33, 34, 35. Recently, Dankbar and colleagues showed for the first time that myostatin is expressed highly in stromal cells of RA synovial tissues and that deficiency or antibody‐mediated inhibition leads to an amelioration of arthritis in mouse models of arthritis. Functional in vitro studies revealed that myostatin enhances RANKL‐mediated osteoclast formation by promoting the fusion of pre‐osteoclasts, leading to enhanced bone loss 36.
In addition to encouraging osteoclast activity, inflammatory conditions inhibit the reparative activity of osteoblasts. Gilbert et al. 37, 38 identified that, when TNF‐α was included in pre‐osteoblast cultures, osteoblast differentiation and maturation in vitro was arrested. Others have found similarly that osteoblast maturation markers such as collagen type I, alkaline phosphatase and osteocalcin are all reduced in the presence of TNF‐α 24, 39, 40, leaving these osteoblasts unable to up‐regulate matrix mineralization 41. Osteoblasts cultured with serum from RA patients on infliximab therapy show reduced expression of IL‐6, a cytokine linked to arthritis‐related bone loss 42. IL‐6 binds the IL‐6 receptor, an interaction which induces prostaglandin E2 synthesis, reducing in turn the ratio of OPG/RANKL expression by osteoblasts, thereby favouring osteoclastogenesis 43. In addition, osteoblasts cultured with serum from patients treated with infliximab show reduced expression of IL‐1β 42, known to inhibit bone formation in vitro 44 and in vivo 45, as well as impaired osteoblast migration 46. Presumably because of the factors described above, it has been reported that TNF blockade in RA patients corrects bone metabolism imbalance seen in RA 47.
In RA the local microenvironment is changed profoundly due to the influx of immune cells and proliferation of FLS at affected joints. This produces localized hypoxia and a reduced pH, both of which are capable of influencing the behaviour of osteoblasts within the joint. Hypoxia inhibits Wnt signalling (discussed in more detail below) in osteoblasts by sequestering β‐catenin to inhibit transcriptional activity 48. By up‐regulating Dickkopf‐1 (DKK1) 49, low pH causes the down‐regulation of alkaline phosphatase synthesis in osteoblasts which prevents mineralization 50.
Hypoxia and acidosis also increase affect osteoclastogenesis and resorptive capacity. Arnett et al. (2003) identified that in vitro differentiation of monocytes to osteoclasts (in the presence of M‐CSF and RANKL) was fourfold more efficient at 2% oxygen compared to 20% oxygen. Importantly, however, the experimental protocol in this instance in fact subjected the cells to repeated periods of hypoxia and normoxia 51. The requirement for periods of reoxygenation was confirmed in another study by Knowles and Athanasou (2009), who demonstrated that repeated hypoxia/reoxygenation cycles, such as those expected to occur during period of inflammation, enhanced osteoclast differentiation; however, when cells were subjected to continued hypoxia at 2% oxygen osteoclastogenesis was inhibited dramatically 52. In addition, the resorption activity of the osteoclasts formed at 2% oxygen was two‐ to fourfold higher than osteoclasts formed at higher oxygen tensions 51, 52 (reviewed in 53). This combination of increased osteoclast formation and activity, combined with a dramatic decrease in osteoblast function in response to hypoxia and low pH, combine to drive bone destruction during inflammation.
Cytokine‐mediated bone destruction
Pannus tissue contains large numbers of activated macrophages, leucocytes and FLS, which release proinflammatory cytokines, including TNF‐α, IL‐1β, IL‐6 and IL‐17. TNF‐α, expressed mainly by monocytes and macrophages but also by T cells, B cells and FLS, plays a pivotal role in the inflamed synovial microenvironment in RA. TNF‐α is considered to be at the top of the inflammatory cascade, based on observations that TNF‐α induces the expression of other cytokines (e.g. IL‐1β, IL‐6, IL‐8) and that anti‐TNF‐α treatment in RA patients reduces IL‐1β release by FLS significantly 54, 55, 56. Moreover, in FLS, TNF‐α induces the production of adhesion molecules to attract leucocytes into the affected joints 57, as well as MMPs that destroy cartilage 58.
During the last few years the impact of TNF‐α on bone remodelling has been addressed in several studies. Classically, it has been shown that this proinflammatory cytokine increases the number of osteoclast precursors 59 and activates osteoclastogenesis indirectly by triggering RANKL release from lymphocytes 60, 61 and endothelial cells 62, as well as increasing RANKL and M‐CSF production by stromal cells 63, 64. The effect of TNF‐α on bone erosion has been studied in detail using the human TNF transgenic (hTNFtg) 65 and the mouse TNF‐driven spondyloarthritis (TNFΔARE) 66 mouse models of RA. Both these models over‐express TNF‐α and result in osteoclast‐mediated bone destruction in joints. TNF is known to enhance osteoclast activity directly, by promoting maturation of bone marrow macrophages into mature osteoclasts in the presence of RANK‐ligand 60, 67.
Stromal cells promote bone loss via interaction with immune cells
Recent studies have shown that the interaction of RA‐FLS with infiltrating immune cells plays a key role in both chronic inflammation and bone destruction. In particular, CD4+ T helper cells (Th cells), the prominent T cell subset in the sublining of rheumatoid synovium, express RANKL and also cytokines which have stimulatory, as well as inhibitory effects on osteoclastogenesis. Th1 cells release IL‐4 and IL‐10, which blocks osteoclastogenesis, whereas Th17 cells release IL‐17, IL‐22, RANKL, IL‐1, IL‐6 and TNF‐α 60, 67, which activate osteoclastogenesis directly on osteoclast precursors. IL‐17 and IL‐22 also stimulate RANKL expression in RA‐FLS to activate osteoclastogenesis, suggesting an indirect role of T cells in bone loss via cross‐talk with RA‐FLS 68, 69, 70, 71.
The first evidence that activated T cells play an important role in bone destruction was shown by Kong and colleagues in 1999 72. They could demonstrate that CD4+ T cells produce a sufficient amount of soluble RANKL to promote osteoclastogenesis, which subsequently induces bone loss in a model of adjuvant arthritis.
The importance of Th17 cells and IL‐17 in bone destruction is also supported by studies using the collagen‐induced arthritis (CIA) mouse model. IL‐17A‐deficient mice developed a markedly reduced severity of CIA accompanied by less bone erosion and less synovial hyperplasia 73. Therapeutic treatment with neutralizing anti‐IL‐17A antibodies reduces significantly the severity of inflammation and bone erosion in various RA mouse models, including CIA 74, antigen‐induced arthritis 75 and glucose‐6‐phosphate isomerase (G6PI)‐induced arthritis 76.
IL‐17 is considered to be a potential osteoclastic cytokine, because it increases RANKL expression in osteoblasts, RA‐FLS and IL‐1 and TNF‐α expression in synovial macrophages, which activates osteoclastogenesis and subsequently drives bone destruction. In an in vitro co‐culture model with murine bone marrow cells and osteoblasts, treatment with IL‐17 derived from synovial fluid of RA patients results in an increased osteoclastogenesis by an up‐regulation of prostaglandin E2 in osteoblasts 77. Higher IL‐17A concentrations were found in synovial fluid and sera from RA patients compared to OA patients or healthy controls 78, 79; therefore, targeting IL‐17 is suggested as an attractive therapeutic target in RA. IL‐17 blockers have been evaluated and are currently being tested in clinical trials for human RA. So far, however, inhibition of IL‐17 has not led to complete disease remission, and monoclonal antibodies against IL‐17 receptor seems to be ineffective in RA 80.
Wnt signalling is critical to bone homeostasis
The Wnt signalling pathway not only controls developmental processes such as skeletal patterning 81, 82, but is also crucial for maintaining bone homeostasis. Three major branches of Wnt signalling exist: the canonical, the Ca2+‐dependent non‐canonical and the planar cell polarity signalling pathway. Of these, the canonical Wnt/β‐catenin pathway is known to be the predominant component that impacts upon bone remodelling. In the absence of Wnt, β‐catenin forms a destruction complex composed of axin, casein kinase 1 (CK1), adenomatous polyposis coli (APC) and glycogen synthase kinase 3β (GSK3β). This complex facilitates phosphorylation, ubiquitination and subsequently degradation of β‐catenin by the proteasome. Activation of signalling occurs upon binding of Wnt proteins to the low‐density lipoprotein receptor‐related protein 5/6 (LRP5/6) receptors and Frizzled (Fz) co‐receptors on the cell surface. Dishevelled (DSH) and the destruction complex are recruited to the cell membrane, allowing β‐catenin accumulation within the cytoplasm and subsequent translocation into the nucleus, where it activates transcription of specific target genes 83.
A strong link between the canonical Wnt/β‐catenin pathway and bone homeostasis has been demonstrated by studying mutations of several members of the pathway. Mutations in Wnt proteins such as Wnt3, Wnt3a and Wnt7A/Wnt7a lead to skeletal malformations in humans 84, 85 and in mice 86, 87. Moreover, loss of function mutations in the human LRP5 gene as well as LRP5 knock‐out mice are associated with low bone density and skeletal fragility 88, 89, whereas gain of function mutations in the LRP5 gene in humans and in mice leads to an increased bone density 90, 91. In previous studies it has been shown that the high bone mass caused by LRP5 mutations is associated with decreased binding of the Wnt inhibitors sclerostin 92, 93 and DKK1 94. An explanation for these observed phenotypes could be the influence of Wnt/β‐catenin signalling on the regulation of the OPG/RANKL ratio. Recent findings have shown that the Wnt/β‐catenin pathway in osteoblasts inhibits osteoclastogenesis through down‐regulation of RANKL expression and up‐regulation of OPG expression, leading to an altered OPG/RANKL ratio 11, 95. Therefore, the Wnt/β‐catenin pathway not only affects cell differentiation into mature osteoblasts and bone renewal, but also arrests bone degradation by blockade of RANK‐RANKL interaction through OPG.
Wnt signalling in RA
The role of Wnt signalling in RA is not yet understood fully. β‐catenin expression was found to be elevated in synovial tissues and FLS from RA patients compared to those from OA patients 96. Several years ago, Sen and colleagues revealed that Wnt1‐mediated Wnt/β‐catenin signalling is constitutively active in RA‐FLS leading to pro‐matrix metalloproteinase 3 (MMP3) secretion and fibronectin expression. However, Wnt1 not only activates the canonical Wnt pathway but also the non‐canonical Wnt/Ca2+ (β‐catenin and LRP5/6 independent) pathway. Wnt1 and Wnt5a initiate the non‐canonical signalling cascade by binding to Fz co‐receptor causing intracellular Ca2+ release, activation of the calcium sensitive enzymes calmodulin kinase II (CamKII) and protein kinase C (PKC). These kinases activate several transcription factors such as nuclear factor of activated T cells (NFAT), NF‐κB and activator protein 1 (AP‐1) 97. The same authors demonstrated that Wnt5a/Fz‐5 non‐canonical signalling increased IL‐6, IL‐8, IL‐15 and RANKL release, indicating that the non‐canonical Wnt pathway might also be important for RA‐FLS activation 98, 99. These observations suggest that constitutive activation of canonical and/or non‐canonical Wnt signalling in RA‐FLS may promote synovial inflammation, pannus formation and bone/cartilage erosion during RA.
Wnt antagonists released by stromal cells under inflammatory conditions control bone remodelling
A number of extracellular Wnt antagonists provide fine‐tuning of the Wnt signalling cascade. Secreted glycoproteins such as sclerostin and DKK1 bind to LRP5 and LRP6 to antagonize canonical Wnt signalling, leading to inhibition of bone formation. Loss of Wnt inhibitor sclerostin expression results in high bone mass and strength in patients with sclerosteosis 100 and Van Buchem disease 101, as well as in sclerostin‐deficient mice 102.
Knowledge that the Wnt/β‐catenin pathway regulates bone formation and degradation has sparked tremendous interest during the last decade; in particular, the use of anti‐sclerostin antibodies in osteoporosis, in which loss of sclerostin enhances bone mineral density seems to be extremely promising. As Wnt signalling is required for bone formation, it was assumed that the enhanced production of Wnt‐antagonists in the inflamed joints was responsible for the lack of bone repair seen in RA joints; hence, blocking Wnt‐antagonists could be a promising approach to reactivate the Wnt pathway and counteract bone destruction. However, in reality the situation in vivo is more complicated.
Surprisingly, antibody‐mediated blockade of sclerostin in the hTNFtg mouse model of RA caused an unexpected acceleration of bone erosion. Moreover, loss of sclerostin in the partially TNF‐dependent glucose‐6‐phosphate‐isomerase (G6PI) mouse model of arthritis had no effect on the progression of RA. Disease severity was ameliorated with loss of sclerostin in the K/BxN serum transfer model, which is TNF receptor‐independent. Combined, these data suggest a specific role for sclerostin in TNF‐α signalling‐induced bone erosions. Sclerostin has a protective function in TNF‐dependent but not TNF‐independent inflammatory arthritis: the more inflammation is driven by TNF, the higher the protective effect of sclerostin 103. In line with these data, several publications have shown that inhibition of sclerostin has either no effect or a destructive effect on cartilage and bone: in the CIA model of RA, sclerostin inhibition had no effect on the improvement of focal bone destruction 104, and pharmacological inhibition of sclerostin in a rat model of osteoarthritis showed no effect on inflammatory cartilage remodelling 105. Of note, one study reported that increased chondrocyte sclerostin is chondroprotective in a sheep model of osteoarthritis 106, and Bouzis et al. found that loss of sclerostin promotes OA in mice 107. However, there has been a report that anti‐sclerostin therapy is protective in TNF‐driven arthritis 108. The protective effect was seen largely when arthritic mice were co‐treated with blocking TNF antibodies, which is in line with the notion that TNF triggers bone loss. Clearly, there is complexity in the function of sclerostin, as uncovered by the animal models of RA described above, and this should be considered carefully when using anti‐sclerostin antibodies in patients with RA or other TNF‐dependent immune‐mediated inflammatory diseases.
Although sclerostin and DKK1 are both Wnt‐inhibitors that bind LRP receptors and are up‐regulated in response to TNF‐α, they exhibit very different effects on bone under inflammatory conditions compared to non‐inflammatory conditions (as seen in most osteoporosis situations). Diarra and co‐workers have demonstrated that anti‐DKK1 treatment in arthritic mice is able to reverse the pattern of bone destruction to promote activation of bone repair, resulting in new bone and osteophyte formation 109. To explore the role of DKK1 in RA patients, Juarez et al. took synovial fibroblasts from treatment‐naive patients with undifferentiated inflammatory arthritis of less than 3 months duration. Fibroblasts from patients that would be diagnosed subsequently with RA expressed significantly higher levels of DKK1 messenger RNA and protein compared to fibroblasts from patients whose arthritis resolved. In co‐cultures with lymphocytes, more DKK1 was secreted by RA fibroblasts than by fibroblasts from non‐inflamed joints or resolving arthritis, and the levels of DKK1 secretion during co‐culture correlated positively with lymphocyte adhesion 110. Recently, Seror et al. 111 found increased DKK1 levels in a cohort of early RA patients with enhanced bone destruction. Therefore, together with findings from those of the RA mouse model in which anti‐DKK1 antibodies were used successfully to enhance bone formation, blocking DKK1 could provide a new therapeutic target for treating bone loss.
Not all stromal cells are created equal!
Fibroblasts, despite being the most ubiquitous stromal cells in the synovium, have proved difficult to characterize in molecular terms, and it is only relatively recently that fibroblast‐specific markers have been identified to allow the identification of fibroblast subsets. Clearly, as has been described above, there are differences between the phenotypes of FLS and RA‐FLS; however, it is becoming apparent that greater complexity exists than simply between disease and healthy synovial fibroblasts. Key fibroblast‐specific markers identified so far include fibroblast activation protein 1 (FAP1), endosialin (CD248), vascular cell adhesion molecule 1 (VCAM‐1) and podoplanin (GP38). The identification of these markers has allowed us to begin to differentiate between RA‐FLS subsets and investigate their function.
In 2016 Croft et al. assessed the functional differences between two of these disease subsets: podoplanin+ fibroblasts, which predominate in the RA synovial lining layer, and endosialin+ fibroblasts that are restricted to the sublining. Using a human cartilage and RA‐FLS graft in severe combined immunodeficient (SCID) mice they showed that it is the podoplanin+ RA‐FLS subset that is migratory and invasive 112. A recent publication has also confirmed the assumption that FAP plays a crucial role in inflammatory destructive arthritis. FAP deletion in a mouse model of RA ameliorates cartilage degradation and isolated FLS from these mice show a lower cartilage adhesion capacity. These findings point to a previously unknown function of FAP in the attachment of FLS to cartilage during RA 113. Taken together, these data match with similar findings of podoplanin+ cancer‐associated fibroblasts (CAFs) promoting metastasis 114 and FAP+ fibroblasts promoting tumour growth in a mouse xenograft model 115 and suggest that targeting a specific fraction of the stromal cells may be an appropriate therapeutic strategy in inflammation as well as cancer.
The importance of osteocytes in bone remodelling
Other predominant cell types in the synovial joint are the osteoblasts and the bone‐embedded osteoblasts termed the osteocytes. Until relatively recently osteocytes were ignored due to difficulties isolating them from tissue and maintaining their phenotype ex vivo. However, in the last few years improvements in techniques have allowed researchers to interrogate their function during bone disease. Osteocytes are descendants of matrix‐producing osteoblasts. They are embedded in the bone matrix but are not passive cells in bone behaviour; rather, they act upon bone remodelling through regulation of both osteoclast and osteoblast activity. It has been reported that osteocytes are able to release RANKL as well as M‐CSF to recruit osteoclast progenitors to sites of remodelling, supporting the generation of functional resorbing osteoclasts 116. It has generally been believed that osteoblasts and stromal cells are the main source of RANKL; however, co‐culture studies from Nakashima et al. 117 demonstrated that purified osteocytes have a greater capacity to support osteoclastogenesis than osteoblasts and bone marrow stromal cells. Osteocytes not only communicate with osteoclasts, but also with osteoblasts through the release of the canonical Wnt/β‐catenin sclerostin, which negatively regulates osteoblast differentiation 118, 119.
Although osteocytes communicate with both osteoblasts and osteoclasts to produce RANKL and sclerostin, the question remains as to how RANKL and sclerostin reach the bone surface from deep within the bone. During their embedding phase, osteocytes form dendritic extensions (40–100 per cell) 120 to build a lacuna–canalicular network, maintaining connections with the bone surface and the vascular space 118. Recently, Honma and co‐workers 121 developed a novel co‐culture system using osteoclast precursors together with osteocytes, embedded in a collagen gel. On the basis of this system, they demonstrated clearly that the osteocytic, membrane‐bound form of RANKL communicates directly with osteoclast precursors through osteocyte dendritic extensions.
The importance of osteocytes has been reported in many musculoskeletal diseases. Decreased connectivity between the osteocytes occurs in osteoporotic and osteoarthritic bones. Moreover, bones taken from patients with osteoporosis also display disorientation of the dendrites 122. Xiong et al. 123 reported that mice with osteocyte‐specific RANKL deletion develop postnatal osteopetrosis. Recently it has been reported that osteocytes are associated with bone loss in inflammatory bowel disease (IBD) in rodents 124. Another study revealed that patients with Crohn's disease possess increased osteocyte apoptosis and reduced bone mass and bone formation 125.
Currently, the function of osteocytes and the control of osteocytogenesis under inflammatory conditions in RA is not well understood. Recently, Pathak et al. reported that in vitro stimulation of osteocytes with serum from RA patients results in enhanced osteocyte‐to‐osteoclast communication. They found that RA serum containing inflammatory cytokines enhances the RANKL/OPG ratio in osteocytes, which leads subsequently to enhanced osteoclastogenesis and bone destruction 126.
However, nothing is known so far about changes in connectivity and orientation of osteocytes under inflammatory conditions. It is well known that chronic inflammation is a major risk factor for systemic bone loss leading to osteoporosis. Even in chronic inflammatory diseases such as RA, local bone erosions are associated typically with systemic bone loss. As already discussed above, osteoporosis patients seem to have a decreased connectivity and disorientation of dendritic extensions in their osteocyte network. Therefore, it can be assumed that the osteocyte network in bones of RA patients might be altered, which could influence signalling molecules involved in bone remodelling processes. This is a relatively new area of research; however, the influences of inflammatory factors in RA on osteocyte‐mediated systemic bone loss has yet to be investigated thoroughly.
Conclusion
Bone loss is a common feature of a variety of musculoskeletal disorders. Under inflammatory conditions such as RA the main trigger of articular bone erosion is synovitis, including the production of inflammatory cytokines and RANKL, leading to activation of osteoclastogenesis. Both activation of bone‐destroying osteoclasts and a lack of compensatory bone repair mechanisms contribute to a progressive loss of joint structure in RA patients.
In this review we have explored the role of stromal cells and their influence in bone remodelling. Emerging data obtained have provided evidence that stromal cells are more than just structural cells. Under chronic inflammation they acquire novel features which are important for the development of pathological processes (Fig. 1). Knowledge of stromal cells and their influence in bone formation and bone destruction will facilitate the development of future therapeutic strategies for repair of bone erosion.
Disclosure
The authors have no conflicts of interest.
References
- 1. Tak PP, Bresnihan B. The pathogenesis and prevention of joint damage in rheumatoid arthritis: advances from synovial biopsy and tissue analysis. Arthritis Rheum 2000; 43:2619–33. [DOI] [PubMed] [Google Scholar]
- 2. Korb‐Pap A, Bertrand J, Sherwood J, Pap T. Stable activation of fibroblasts in rheumatic arthritis – causes and consequences. Rheumatology (Oxford) 2016; 55:ii64–7. [DOI] [PubMed] [Google Scholar]
- 3. Gravallese EM. Bone destruction in arthritis. Ann Rheum Dis 2002; 61 Suppl 2:ii84–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lefevre S, Knedla A, Tennie C et al Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med 2009; 15:1414–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Naylor AJ, Filer A, Buckley CD. The role of stromal cells in the persistence of chronic inflammation. Clin Exp Immunol 2013; 171:30–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qin L, Qiu P, Wang L et al Gene expression profiles and transcription factors involved in parathyroid hormone signaling in osteoblasts revealed by microarray and bioinformatics. J Biol Chem 2003; 278:19723–31. [DOI] [PubMed] [Google Scholar]
- 7. Globus RK, Patterson‐Buckendahl P, Gospodarowicz D. Regulation of bovine bone cell proliferation by fibroblast growth factor and transforming growth factor beta. Endocrinology 1988; 123:98–105. [DOI] [PubMed] [Google Scholar]
- 8. Wrana JL, Maeno M, Hawrylyshyn B, Yao KL, Domenicucci C, Sodek J. Differential effects of transforming growth factor‐beta on the synthesis of extracellular matrix proteins by normal fetal rat calvarial bone cell populations. J Cell Biol 1988; 106:915–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gaur T, Lengner CJ, Hovhannisyan H et al Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem 2005; 280:33132–40. [DOI] [PubMed] [Google Scholar]
- 10. Rawadi G, Vayssiere B, Dunn F, Baron R, Roman‐Roman S. BMP‐2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J Bone Miner Res 2003; 18:1842–53. [DOI] [PubMed] [Google Scholar]
- 11. Glass DA II, Bialek P, Ahn JD et al Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell 2005; 8:751–64. [DOI] [PubMed] [Google Scholar]
- 12. Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone 2007; 40:251–64. [DOI] [PubMed] [Google Scholar]
- 13. Armstrong AP, Tometsko ME, Glaccum M, Sutherland CL, Cosman D, Dougall WC. A RANK/TRAF6‐dependent signal transduction pathway is essential for osteoclast cytoskeletal organization and resorptive function. J Biol Chem 2002; 277:44347–56. [DOI] [PubMed] [Google Scholar]
- 14. Takayanagi H, Kim S, Koga T et al Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 2002; 3:889–901. [DOI] [PubMed] [Google Scholar]
- 15. Yeo L, Toellner KM, Salmon M et al Cytokine mRNA profiling identifies B cells as a major source of RANKL in rheumatoid arthritis. Ann Rheum Dis 2011; 70:2022–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yeo L, Lom H, Juarez M et al Expression of FcRL4 defines a pro‐inflammatory, RANKL‐producing B cell subset in rheumatoid arthritis. Ann Rheum Dis 2015; 74:928–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lacey DL, Timms E, Tan HL et al Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998; 93:165–76. [DOI] [PubMed] [Google Scholar]
- 18. Kong YY, Yoshida H, Sarosi I et al OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph‐node organogenesis. Nature 1999; 397:315–23. [DOI] [PubMed] [Google Scholar]
- 19. Bucay N, Sarosi I, Dunstan CR et al Osteoprotegerin‐deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 1998; 12:1260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kostenuik PJ. Osteoprotegerin and RANKL regulate bone resorption, density, geometry and strength. Curr Opin Pharmacol 2005; 5:618–25. [DOI] [PubMed] [Google Scholar]
- 21. Scott DL, Pugner K, Kaarela K et al The links between joint damage and disability in rheumatoid arthritis. Rheumatology 2000; 39:122–32. [DOI] [PubMed] [Google Scholar]
- 22. Gravallese EM, Harada Y, Wang JT, Gorn AH, Thornhill TS, Goldring SR. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am J Pathol 1998; 152:943–51. [PMC free article] [PubMed] [Google Scholar]
- 23. Tsuboi H, Matsui Y, Hayashida K et al Tartrate resistant acid phosphatase (TRAP) positive cells in rheumatoid synovium induce the destruction of articular cartilage. Arthritis Rheum 2002; 46:S618–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bertolini DR, Nedwin GE, Bringman TS, Smith DD, Mundy GR. Stimulation of bone resorption and inhibition of bone formation in vitro by human tumour necrosis factors. Nature 1986; 319:516–8. [DOI] [PubMed] [Google Scholar]
- 25. Redlich K, Hayer S, Ricci R et al Osteoclasts are essential for TNF‐alpha mediated joint destruction. Arthritis Rheum 2002; 46:S624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hashizume M, Hayakawa N, Mihara M. IL‐6 trans‐signalling directly induces RANKL on fibroblast‐like synovial cells and is involved in RANKL induction by TNF‐alpha and IL‐17. Rheumatology (Oxford) 2008; 47:1635–40. [DOI] [PubMed] [Google Scholar]
- 27. Kotake S, Udagawa N, Takahashi N et al IL‐17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest 1999; 103:1345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Haynes DR, Crotti TN, Loric M, Bain GI, Atkins GJ, Findlay DM. Osteoprotegerin and receptor activator of nuclear factor kappaB ligand (RANKL) regulate osteoclast formation by cells in the human rheumatoid arthritic joint. Rheumatology (Oxford) 2001; 40:623–30. [DOI] [PubMed] [Google Scholar]
- 29. Crotti TN, Smith MD, Weedon H et al Receptor activator NF‐kappaB ligand (RANKL) expression in synovial tissue from patients with rheumatoid arthritis, spondyloarthropathy, osteoarthritis, and from normal patients: semiquantitative and quantitative analysis. Ann Rheum Dis 2002; 61:1047–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haynes DR, Barg E, Crotti TN et al Osteoprotegerin expression in synovial tissue from patients with rheumatoid arthritis, spondyloarthropathies and osteoarthritis and normal controls. Rheumatology (Oxford) 2003; 42:123–34. [DOI] [PubMed] [Google Scholar]
- 31. McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF‐beta superfamily member. Nature 1997; 387:83–90. [DOI] [PubMed] [Google Scholar]
- 32. Kellum E, Starr H, Arounleut P et al Myostatin (GDF‐8) deficiency increases fracture callus size, Sox‐5 expression, and callus bone volume. Bone 2009; 44:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hamrick MW, Shi X, Zhang W et al Loss of myostatin (GDF8) function increases osteogenic differentiation of bone marrow‐derived mesenchymal stem cells but the osteogenic effect is ablated with unloading. Bone 2007; 40:1544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hamrick MW. Increased bone mineral density in the femora of GDF8 knockout mice. Anat Rec A Discov Mol Cell Evol Biol 2003; 272:388–91. [DOI] [PubMed] [Google Scholar]
- 35. Hamrick MW, Pennington C, Byron CD. Bone architecture and disc degeneration in the lumbar spine of mice lacking GDF‐8 (myostatin). J Orthop Res 2003; 21:1025–32. [DOI] [PubMed] [Google Scholar]
- 36. Dankbar B, Fennen M, Brunert D et al Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat Med 2015; 21:1085–90. [DOI] [PubMed] [Google Scholar]
- 37. Gilbert L et al Inhibition of osteoblast differentiation by tumor necrosis factor‐alpha. Endocrinology 2000; 141:3956–64. [DOI] [PubMed] [Google Scholar]
- 38. Gilbert L, He X, Farmer P et al Expression of the osteoblast differentiation factor RUNX2 (Cbfa1/AML3/Pebp2alpha A) is inhibited by tumor necrosis factor‐alpha. J Biol Chem 2002; 277:2695–701. [DOI] [PubMed] [Google Scholar]
- 39. Centrella M, McCarthy TL, Canalis E. Tumor necrosis factor‐alpha inhibits collagen synthesis and alkaline phosphatase activity independently of its effect on deoxyribonucleic acid synthesis in osteoblast‐enriched bone cell cultures. Endocrinology 1988; 123:1442–8. [DOI] [PubMed] [Google Scholar]
- 40. Li YP, Stashenko P. Proinflammatory cytokines tumor necrosis factor‐alpha and IL‐6, but not IL‐1, down‐regulate the osteocalcin gene promoter. J Immunol 1992; 148:788–94. [PubMed] [Google Scholar]
- 41. Panagakos FS, Fernandez C, Kumar S. Ultrastructural analysis of mineralized matrix from human osteoblastic cells: effect of tumor necrosis factor‐alpha. Mol Cell Biochem 1996; 158:81–9. [DOI] [PubMed] [Google Scholar]
- 42. Musacchio E, Valvason C, Botsios C et al The tumor necrosis factor‐{alpha}‐blocking agent infliximab inhibits interleukin 1beta (IL‐1beta) and IL‐6 gene expression in human osteoblastic cells. J Rheumatol 2009; 36:1575–9. [DOI] [PubMed] [Google Scholar]
- 43. Liu XH, Kirschenbaum A, Yao S, Levine AC. Cross‐talk between the interleukin‐6 and prostaglandin E(2) signaling systems results in enhancement of osteoclastogenesis through effects on the osteoprotegerin/receptor activator of nuclear factor‐{kappa}B (RANK) ligand/RANK system. Endocrinology 2005; 146:1991–8. [DOI] [PubMed] [Google Scholar]
- 44. Stashenko P, Dewhirst FE, Rooney ML, Desjardins LA, Heeley JD. Interleukin‐1 beta is a potent inhibitor of bone formation in vitro. J Bone Miner Res 1987; 2:559–65. [DOI] [PubMed] [Google Scholar]
- 45. Nguyen L, Dewhirst FE, Hauschka PV, Stashenko P. Interleukin‐1 beta stimulates bone resorption and inhibits bone formation in vivo. Lymphokine Cytokine Res 1991; 10:15–21. [PubMed] [Google Scholar]
- 46. Hengartner NE, Fiedler J, Ignatius A, Brenner RE. IL‐1beta inhibits human osteoblast migration. Mol Med 2013; 19:36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zwerina J, Tuerk B, Redlich K, Smolen JS, Schett G. Imbalance of local bone metabolism in inflammatory arthritis and its reversal upon tumor necrosis factor blockade: direct analysis of bone turnover in murine arthritis. Arthritis Res Ther 2006; 8:R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Almeida M, Han L, Martin‐Millan M, O'Brien CA, Manolagas SC. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta‐catenin from T cell factor‐ to forkhead box O‐mediated transcription. J Biol Chem 2007; 282:27298–305. [DOI] [PubMed] [Google Scholar]
- 49. Colla S, Zhan F, Xiong W et al The oxidative stress response regulates DKK1 expression through the JNK signaling cascade in multiple myeloma plasma cells. Blood 2007; 109:4470–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brandao‐Burch A, Utting JC, Orriss IR, Arnett TR. Acidosis inhibits bone formation by osteoblasts in vitro by preventing mineralization. Calcif Tissue Int 2005; 77:167–74. [DOI] [PubMed] [Google Scholar]
- 51. Arnett TR, Gibbons DC, Utting JC et al Hypoxia is a major stimulator of osteoclast formation and bone resorption. J Cell Physiol 2003; 196:2–8. [DOI] [PubMed] [Google Scholar]
- 52. Knowles HJ, Athanasou NA. Acute hypoxia and osteoclast activity: a balance between enhanced resorption and increased apoptosis. J Pathol 2009; 218:256–64. [DOI] [PubMed] [Google Scholar]
- 53. Arnett TR. Acidosis, hypoxia and bone. Arch Biochem Biophys 2010; 503:103–9. [DOI] [PubMed] [Google Scholar]
- 54. Butler DM, Maini RN, Feldmann M, Brennan FM. Modulation of proinflammatory cytokine release in rheumatoid synovial membrane cell cultures. Comparison of monoclonal anti TNF‐alpha antibody with the interleukin‐1 receptor antagonist. Eur Cytokine Netw 1995; 6:225–30. [PubMed] [Google Scholar]
- 55. Alvaro‐Gracia JM, Zvaifler NJ, Brown CB, Kaushansky K, Firestein GS. Cytokines in chronic inflammatory arthritis. VI. Analysis of the synovial cells involved in granulocyte‐macrophage colony‐stimulating factor production and gene expression in rheumatoid arthritis and its regulation by IL‐1 and tumor necrosis factor‐alpha. J Immunol 1991; 146:3365–71. [PubMed] [Google Scholar]
- 56. Buchan G, Barrett K, Turner M, Chantry D, Maini RN, Feldmann M. Interleukin‐1 and tumour necrosis factor mRNA expression in rheumatoid arthritis: prolonged production of IL‐1 alpha. Clin Exp Immunol 1988; 73:449–55. [PMC free article] [PubMed] [Google Scholar]
- 57. Chin JE, Winterrowd GE, Krzesicki RF, Sanders ME. Role of cytokines in inflammatory synovitis. The coordinate regulation of intercellular adhesion molecule 1 and HLA class I and class II antigens in rheumatoid synovial fibroblasts. Arthritis Rheum 1990; 33:1776–86. [DOI] [PubMed] [Google Scholar]
- 58. Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci 2006; 11:529–43. [DOI] [PubMed] [Google Scholar]
- 59. Li P, Schwarz EM, O'Keefe RJ, Ma L, Boyce BF, Xing L. RANK signaling is not required for TNFalpha‐mediated increase in CD11(hi) osteoclast precursors but is essential for mature osteoclast formation in TNFalpha‐mediated inflammatory arthritis. J Bone Miner Res 2004; 19:207–13. [DOI] [PubMed] [Google Scholar]
- 60. Cenci S, Weitzmann MN, Roggia C et al Estrogen deficiency induces bone loss by enhancing T‐cell production of TNF‐alpha. J Clin Invest 2000; 106:1229–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kanematsu M, Sato T, Takai H, Watanabe K, Ikeda K, Yamada Y. Prostaglandin E2 induces expression of receptor activator of nuclear factor‐kappa B ligand/osteoprotegrin ligand on pre‐B cells: implications for accelerated osteoclastogenesis in estrogen deficiency. J Bone Miner Res 2000; 15:1321–9. [DOI] [PubMed] [Google Scholar]
- 62. Collin‐Osdoby P, Rothe L, Anderson F, Nelson M, Maloney W, Osdoby P. Receptor activator of NF‐kappa B and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflammatory cytokines, and role in human osteoclastogenesis. J Biol Chem 2001; 276:20659–72. [DOI] [PubMed] [Google Scholar]
- 63. Hofbauer LC, Lacey DL, Dunstan CR, Spelsberg TC, Riggs BL, Khosla S. Interleukin‐1beta and tumor necrosis factor‐alpha, but not interleukin‐6, stimulate osteoprotegerin ligand gene expression in human osteoblastic cells. Bone 1999; 25:255–9. [DOI] [PubMed] [Google Scholar]
- 64. Kimble RB, Srivastava S, Ross FP, Matayoshi A, Pacifici R. Estrogen deficiency increases the ability of stromal cells to support murine osteoclastogenesis via an interleukin‐1and tumor necrosis factor‐mediated stimulation of macrophage colony‐stimulating factor production. J Biol Chem 1996; 271:28890–7. [DOI] [PubMed] [Google Scholar]
- 65. Keffer J, Probert L, Cazlaris H et al Transgenic mice expressing human tumor‐necrosis‐factor – a predictive genetic model of arthritis. EMBO J 1991; 10:4025–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU‐rich elements: implications for joint and gut‐associated immunopathologies. Immunity 1999; 10:387–98. [DOI] [PubMed] [Google Scholar]
- 67. Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF‐alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest 2000; 106:1481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kwok SK, Cho ML, Park MK et al Interleukin‐21 promotes osteoclastogenesis in humans with rheumatoid arthritis and in mice with collagen‐induced arthritis. Arthritis Rheum 2012; 64:740–51. [DOI] [PubMed] [Google Scholar]
- 69. Kim KW, Kim HR, Park JY et al Interleukin‐22 promotes osteoclastogenesis in rheumatoid arthritis through induction of RANKL in human synovial fibroblasts. Arthritis Rheum 2012; 64:1015–23. [DOI] [PubMed] [Google Scholar]
- 70. Komatsu N, Takayanagi H. Inflammation and bone destruction in arthritis: synergistic activity of immune and mesenchymal cells in joints. Front Immunol 2012; 3:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim KW, Kim HR, Kim BM, Cho ML, Lee SH. Th17 cytokines regulate osteoclastogenesis in rheumatoid arthritis. Am J Pathol 2015; 185:3011–24. [DOI] [PubMed] [Google Scholar]
- 72. Kong YY, Feige U, Sarosi I et al Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 1999; 402:304–9. [DOI] [PubMed] [Google Scholar]
- 73. Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen‐induced arthritis in IL‐17‐deficient mice. J Immunol 2003; 171:6173–7. [DOI] [PubMed] [Google Scholar]
- 74. Lubberts E, Koenders MI, Oppers‐Walgreen B et al Treatment with a neutralizing anti‐murine interleukin‐17 antibody after the onset of collagen‐induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum 2004; 50:650–9. [DOI] [PubMed] [Google Scholar]
- 75. Koenders MI, Lubberts E, Oppers‐Walgreen B et al Blocking of interleukin‐17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin‐1. Am J Pathol 2005; 167:141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ishiguro A, Akiyama T, Adachi H, Inoue J, Nakamura Y. Therapeutic potential of anti‐interleukin‐17A aptamer: suppression of interleukin‐17A signaling and attenuation of autoimmunity in two mouse models. Arthritis Rheum 2011; 63:455–66. [DOI] [PubMed] [Google Scholar]
- 77. Kotake S, Udagawa N, Takahashi N et al IL‐17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest 1999; 103:1345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ziolkowska M, Koc A, Luszczykiewicz G et al High levels of IL‐17 in rheumatoid arthritis patients: IL‐15 triggers in vitro IL‐17 production via cyclosporin A‐sensitive mechanism. J Immunol 2000; 164:2832–8. [DOI] [PubMed] [Google Scholar]
- 79. Metawi SA, Abbas D, Kamal MM, Ibrahim MK. Serum and synovial fluid levels of interleukin‐17 in correlation with disease activity in patients with RA. Clin Rheumatol 2011; 30:1201–7. [DOI] [PubMed] [Google Scholar]
- 80. Kugyelka R, Kohl Z, Olasz K et al Enigma of IL‐17 and Th17 cells in rheumatoid arthritis and in autoimmune animal models of arthritis. Mediators Inflamm 2016; 2016:6145810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nusslein‐Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature 1980; 287:795–801. [DOI] [PubMed] [Google Scholar]
- 82. Cabrera CV, Alonso MC, Johnston P, Phillips RG, Lawrence PA. Phenocopies induced with antisense RNA identify the wingless gene. Cell 1987; 50:659–63. [DOI] [PubMed] [Google Scholar]
- 83. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med 2013; 19:179–92. [DOI] [PubMed] [Google Scholar]
- 84. Niemann S, Zhao C, Pascu F et al Homozygous WNT3 mutation causes tetra‐amelia in a large consanguineous family. Am J Hum Genet 2004; 74:558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Woods CG, Stricker S, Seemann P et al Mutations in WNT7A cause a range of limb malformations, including Fuhrmann syndrome and Al‐Awadi/Raas‐Rothschild/Schinzel phocomelia syndrome. Am J Hum Genet 2006; 79:402–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Galceran J, Farinas I, Depew MJ, Clevers H, Grosschedl R. Wnt3a–/– like phenotype and limb deficiency in Lef1(–/–)Tcf1(–/–) mice. Genes Dev 1999; 13:709–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Parr BA, McMahon AP. Dorsalizing signal Wnt‐7a required for normal polarity of D‐V and A‐P axes of mouse limb. Nature 1995; 374:350–3. [DOI] [PubMed] [Google Scholar]
- 88. Gong Y, Slee RB, Fukai N et al LDL receptor‐related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001; 107:513–23. [DOI] [PubMed] [Google Scholar]
- 89. Kato M, Patel MS, Levasseur R et al Cbfa1‐independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol 2002; 157:303–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Little RD, Carulli JP, Del Mastro RG et al A mutation in the LDL receptor‐related protein 5 gene results in the autosomal dominant high‐bone‐mass trait. Am J Hum Genet 2002; 70:11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Babij P, Zhao W, Small C et al High bone mass in mice expressing a mutant LRP5 gene. J Bone Miner Res 2003; 18:960–74. [DOI] [PubMed] [Google Scholar]
- 92. Balemans W, Cleiren E, Ai M, Van Wesenbeeck L, Warman ML, Van Hul W. The binding between sclerostin and LRP5 is altered by DKK1 and by high‐bone mass LRP5 mutations. Calcif Tissue Int 2008; 82:445–53. [DOI] [PubMed] [Google Scholar]
- 93. Ellies DL, Viviano B, McCarthy J et al Bone density ligand, sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J Bone Miner Res 2006; 21:1738–1749. [DOI] [PubMed] [Google Scholar]
- 94. Ai M, Holmen SL, Van Hul W, Williams BO, Warman ML. Reduced affinity to and inhibition by DKK1 form a common mechanism by which high bone mass‐associated missense mutations in LRP5 affect canonical Wnt signaling. Mol Cell Biol 2005; 25:4946–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Spencer GJ, Utting JC, Etheridge SL, Arnett TR, Genever PG. Wnt signalling in osteoblasts regulates expression of the receptor activator of NFkappaB ligand and inhibits osteoclastogenesis in vitro . J Cell Sci 2006; 119:1283–96. [DOI] [PubMed] [Google Scholar]
- 96. Xiao CY, Pan YF, Guo XH, Wu YQ, Gu JR, Cai DZ. Expression of beta‐catenin in rheumatoid arthritis fibroblast‐like synoviocytes. Scand J Rheumatol 2011; 40:26–33. [DOI] [PubMed] [Google Scholar]
- 97. Sen M, Reifert J, Lauterbach K et al Regulation of fibronectin and metalloproteinase expression by Wnt signaling in rheumatoid arthritis synoviocytes. Arthritis Rheum 2002; 46:2867–77. [DOI] [PubMed] [Google Scholar]
- 98. Sen M, Lauterbach K, El‐Gabalawy H, Firestein GS, Corr M, Carson DA. Expression and function of wingless and frizzled homologs in rheumatoid arthritis. Proc Natl Acad Sci USA 2000; 97:2791–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sen M, Chamorro M, Reifert J, Corr M, Carson DA. Blockade of Wnt‐5A/frizzled 5 signaling inhibits rheumatoid synoviocyte activation. Arthritis Rheum 2001; 44:772–81. [DOI] [PubMed] [Google Scholar]
- 100. Brunkow ME, Gardner JC, Van Ness J et al Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot‐containing protein. Am J Hum Genet 2001; 68:577–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Balemans W, Patel N, Ebeling M et al Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 2002; 39:91–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Li X, Ominsky MS, Niu QT et al Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res 2008; 23:860–9. [DOI] [PubMed] [Google Scholar]
- 103. Wehmeyer C, Frank S, Beckmann D et al Sclerostin inhibition promotes TNF‐dependent inflammatory joint destruction. Sci Transl Med 2016; 8:330ra35. [DOI] [PubMed] [Google Scholar]
- 104. Marenzana M, Vugler A, Moore A, Robinson M. Effect of sclerostin‐neutralising antibody on periarticular and systemic bone in a murine model of rheumatoid arthritis: a microCT study. Arthritis Res Ther 2013; 15:R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Roudier M, Li X, Niu QT et al Sclerostin is expressed in articular cartilage but loss or inhibition does not affect cartilage remodeling during aging or following mechanical injury. Arthritis Rheum 2013; 65:721–31. [DOI] [PubMed] [Google Scholar]
- 106. Chan BY, Fuller ES, Russell AK et al Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthritis Cartilage 2011; 19:874–85. [DOI] [PubMed] [Google Scholar]
- 107. Bouaziz W, Funck‐Brentano T, Lin H et al Loss of sclerostin promotes osteoarthritis in mice via β‐catenin‐dependent and ‐independent Wnt pathways. Arthritis Res Ther 2015; 17:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chen XX, Baum W, Dwyer D et al Sclerostin inhibition reverses systemic, periarticular and local bone loss in arthritis. Ann Rheum Dis 2013; 72:1732–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Diarra D, Stolina M, Polzer K et al Dickkopf‐1 is a master regulator of joint remodeling. Nat Med 2007; 13:156–63. [DOI] [PubMed] [Google Scholar]
- 110. Juarez M, McGettrick HM, Scheel‐Toellner D et al DKK1 expression by synovial fibroblasts in very early rheumatoid arthritis associates with lymphocyte adhesion in an in vitro flow co‐culture system. Arthritis Res Ther 2016; 18:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Seror R, Boudaoud S, Pavy S et al Increased Dickkopf‐1 in recent‐onset rheumatoid arthritis is a new biomarker of structural severity. Data from the ESPOIR cohort. Sci Rep 2016; 6:18421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Croft AP, Naylor AJ, Marshall JL et al Rheumatoid synovial fibroblasts differentiate into distinct subsets in the presence of cytokines and cartilage. Arthritis Res Ther 2016; 18:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Waldele S, Koers‐Wunrau C, Beckmann D et al Deficiency of fibroblast activation protein alpha ameliorates cartilage destruction in inflammatory destructive arthritis. Arthritis Res Ther 2015; 17:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Dang Q, Liu J, Li J, Sun Y. Podoplanin: a novel regulator of tumor invasion and metastasis. Med Oncol 2014; 31:24. [DOI] [PubMed] [Google Scholar]
- 115. Cheng JD, Dunbrack RL, Jr., Valianou M, Rogatko A, Alpaugh RK, Weiner LM. Promotion of tumor growth by murine fibroblast activation protein, a serine protease, in an animal model. Cancer Res 2002; 62:4767–72. [PubMed] [Google Scholar]
- 116. Zhao S, Kato Y, Zhang Y, Harris S, Ahuja SS, Bonewald LF. MLO‐Y4 osteocyte‐like cells support osteoclast formation and activation. J Bone Miner Res 2002; 17:2068–2079. [DOI] [PubMed] [Google Scholar]
- 117. Nakashima T, Hayashi M, Fukunaga T et al Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 2011; 17:1231–4. [DOI] [PubMed] [Google Scholar]
- 118. Bonewald LF. The amazing osteocyte. J Bone Miner Res 2011; 26:229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL‐dependent pathway. PLOS ONE 2011; 6:e25900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Beno T, Yoon YJ, Cowin SC, Fritton SP. Estimation of bone permeability using accurate microstructural measurements. J Biomech 2006; 39:2378–87. [DOI] [PubMed] [Google Scholar]
- 121. Honma M, Ikebuchi Y, Kariya Y et al RANKL subcellular trafficking and regulatory mechanisms in osteocytes. J Bone Miner Res 2013; 28:1936–1949. [DOI] [PubMed] [Google Scholar]
- 122. Knothe Tate ML, Adamson JR, Tami AE, Bauer TW. The osteocyte. Int J Biochem Cell Biol 2004; 36:1–8. [DOI] [PubMed] [Google Scholar]
- 123. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix‐embedded cells control osteoclast formation. Nat Med 2011; 17:1235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Metzger CE, Narayanan A, Zawieja DC, Bloomfield SA. Inflammatory bowel disease in a rodent model alters osteocyte protein levels controlling bone turnover. J Bone Miner Res 2017; 32:802–13. [DOI] [PubMed] [Google Scholar]
- 125. Oostlander AE, Bravenboer N, Sohl E et al Histomorphometric analysis reveals reduced bone mass and bone formation in patients with quiescent Crohn's disease. Gastroenterology 2011; 140:116–23. [DOI] [PubMed] [Google Scholar]
- 126. Pathak JL, Bravenboer N, Luyten FP et al Mechanical loading reduces inflammation‐induced human osteocyte‐to‐osteoclast communication. Calcif Tissue Int 2015; 97:169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]