

Summary
Therapeutic blockage of cytokine signalling in autoimmune diseases has improved our understanding of the role of these cytokines in triggering, shaping and perpetuating autoimmune responses. In rheumatoid arthritis (RA), immunopathology is driven by a predominance of arthritogenic T helper cells secreting interferon‐γ [T helper type 1 (Th1)] and interleukin (IL)‐17 (Th17) over regulatory T cells (Treg). The pleiotropic cytokine IL‐6 is crucial to the differentiation of Th17 cells and the balance between pathogenic Th17 and protective Treg. Targeting the IL‐6 receptor (IL‐6R) by humanized antibodies improves signs and symptoms of RA, and has provided new insights into the mechanisms of inflammation and immune regulation. Here we review current evidence on the role of IL‐6 in the pathogenesis of RA and the molecular consequences of IL‐6R blockage in disease, with special focus on the Th17/Treg balance and plasticity.
Keywords: interleukin 6, rheumatoid arthritis, T cell plasticity, Th17/Treg balance, tocilizumab
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disorder which is characterized mainly by inflammation of the synovial tissues of the joints, but also potentially affects a variety of extra‐articular organs. An important hallmark of RA is the progressive joint damage, evidenced by radiological findings such as joint space narrowing and bone erosions, which are related to functional disability 1. Universally used disease‐modifying anti‐rheumatic drugs (DMARDs) such as methotrexate (MTX) and prednisone are able to reduce symptoms and control structural damage to some extent, especially if they are applied at early stages of the disease 2. However, new therapeutic approaches, such as monoclonal antibodies and recombinant receptors that target the action of proinflammatory cytokines, are superior to conventional treatments. Treatment with biologicals results in improved remission, as well as in the arrest of radiographic progression in RA patients, when used in combination with MTX 3. In particular, blockage of the receptor for the pleiotropic cytokine interleukin (IL)‐6 has been demonstrated to be more efficient than MTX in reducing RA activity 4. Specific blockage of proinflammatory cytokines or its receptors in autoimmune diseases, particularly in RA, is improving our understanding of the role these cytokines play in shaping and perpetuating autoimmune responses. Within this review, we discuss the impact of IL‐6 receptor (IL‐6R) blockage on CD4+ T cell polarization and function in the context of RA immunopathogenesis.
Imbalance between CD4+ T cell subpopulations in RA pathogenesis
The pathogenesis of RA is driven by an inflammatory network, in which T and B cells, autoantibodies, cytokines and other inflammatory mediators are crucial components 5. Nevertheless, the factors that lead ultimately to the breach of tolerance remain largely unknown. For years, there has been a general consensus that CD4+ T cells orchestrate the inflammatory cascade in RA, which is now supported by more recent evidence showing therapeutic efficacy of inhibitors of T cell co‐stimulation in RA patients 6. Activated CD4+ T cells exert their effect on a variety of cells that either reside in or infiltrate joints. Among others, CD4+ T cells help autoreactive B cells to differentiate into autoantibody‐producing plasma cells, and stimulate macrophages and synovial fibroblasts to secrete cytokines that are crucial to the maintenance of a chronic inflammatory state in the joints, such as tumour necrosis factor (TNF), IL‐1 and IL‐6 7. These mediators promote the expression of chemokines and their receptors, leading to the recruitment of a new wave of cells to the joints, including T and B cells, plasma cells, neutrophils, mast cells, dendritic cells and natural killer cells 8.
Traditionally, RA was considered an autoimmune process driven by CD4+ T helper (Th) cells that secrete interferon (IFN)‐γ (Th1), which predominate over the Th2 subset that secretes IL‐4, IL‐5 and IL‐13 and exerts preferably regulatory functions 9. However, since 2003 this dogma has been questioned based on experiments performed in animal models of RA, such as collagen‐induced arthritis (CIA). These studies demonstrated that a population of CD4+ T cells, characterized by the secretion of IL‐17 and dependent upon IL‐23 for its expansion, was responsible for the progressive damage observed in the joints of these animals 10. This population, referred to as Th17 cells, was shown to be involved in other models of autoimmune diseases, and has been considered a distinct lineage, mutually exclusive with Th1 11. Human Th17 cells are characterized by the expression of the lineage‐specific transcription factor retinoid acid‐related orphan receptor C2 (RORC2) 12, as well as IL‐23 receptor, chemokine receptor CCR6 13 and lectin receptor CD161 14. Besides IL‐17, Th17 cells produce IL‐21, IL‐22, IL‐26, TNF and granulocyte–macrophage colony‐stimulating factor (GM‐CSF) 15. Th17‐derived cytokines attract different cell types through the induction of other cytokines and chemokines, such as IL‐6, GM‐CSF, CC chemokine ligand (CCL)‐20 and IL‐8 16. In particular, IL‐17A and IL‐17F are key cytokines for the recruitment and/or activation of numerous cell types involved in the pathogenesis of RA, including neutrophils, monocytes, macrophages, synovial fibroblasts, chondrocytes and osteoclasts 17. Also, IL‐21 produced by Th17 cells promotes B cell differentiation to autoantibody‐producing plasma cells 18, and amplifies the Th17 response in an autocrine manner 19. These findings led to clinical trials using neutralizing antibodies to IL‐17 in patients with RA, which proved to be well tolerated and safe but failed, however, to improve clinical response compared to placebo 20, 21.
The isolation of CD4+ T cells secreting both IFN‐γ and IL‐17 from synovial tissues of RA patients has challenged the Th1/Th17 dichotomy 22. This fact has been corroborated in joints of patients with juvenile arthritis, where co‐expression of Th17 and Th1 lineage‐specific transcription factors, RORC2 and T‐bet, has been demonstrated in Th17 cells which converted to ‘non‐classical’ Th17/Th1 as a consequence of the proinflammatory environment 23, 24.
Conversely, it has been postulated that autoimmune processes triggering RA are caused by an imbalance between CD4+ effector T cells and regulatory T cells (Treg), with a pre‐eminence of the former 25, 26. Treg from peripheral blood of RA patients have been described to exhibit an impaired ability to suppress proinflammatory cytokine production by effector T cells and monocytes, a defect that has been attributed to epigenetic modifications and to the effect of TNF on Treg, among other mechanisms 27, 28. This view has been challenged by other groups, which have shown that the impairment is due to the resistance of RA effector T cells to be suppressed by Treg 29, 30. Similarly, there is contradictory evidence regarding the functional proficiency of Treg from RA synovial fluid, where Treg are enriched compared to peripheral blood 30.
The pleiotropic cytokine IL‐6 and its role in RA
IL‐6 is secreted by a large number of cell types, including T and B cells, monocytes, fibroblasts and synoviocytes. IL‐6 exerts its effects once it has bound to a receptor complex formed by the ligand‐binding IL‐6Rα chain (CD126) and the signal‐transducing β‐subunit gp130 (CD130) 31. Binding of IL‐6 to its receptor induces homodimerization of gp130 32, leading to phosphorylation of tyrosine kinases of the Janus kinase (JAK) family, as well as the recruitment and activation of signal transducers and activators of transcription (STAT)‐1 and STAT‐3 33. Importantly, IL‐6R as a membrane‐bound receptor, is restricted to hepatocytes, leucocytes and megakaryocytes, and as a soluble form (sIL‐6R) is present in circulation and at sites of inflammation 34. Accordingly, there are two mechanisms through which IL‐6 exerts its biological effects: classical IL‐6R signal transduction via the membrane‐bound IL‐6R and IL‐6 ‘trans‐signalling’ 34, a process in which sIL‐6R binds IL‐6 and thereby prolongs its circulating half‐life and bioavailability 35. The gp130 subunit is expressed ubiquitously, so many cells that lack IL‐6R can still respond to IL‐6 through trans‐signalling, accounting for the pleiotropic effects of IL‐6 36.
While classical IL‐6R signalling controls central homeostatic processes and immunological outcomes, including acute‐phase response, glucose metabolism, haematopoiesis and regulation of the neuroendocrine system, IL‐6 trans‐signalling plays a role in the recruitment and apoptosis of leucocytes, the maintenance of T cell effector functions and activation of stromal tissues 37. sIL‐6R is released upon activation from T cells, monocytes and neutrophils via IL‐6R ectodomain shedding through adamalysin proteases ADAM metallopeptidase domain 17 (ADAM17) and ADAM10 38, 39, 40. Therefore, it is not surprising that serum concentrations of sIL‐6R are elevated during inflammation 37. Additionally, there are soluble forms of gp130 (sgp130) that are released by differential splicing 41 and bind to the IL‐6‐sIL‐6R complex to block trans‐signalling 42. However, high serum concentrations of sgp130 remain largely unaltered during inflammation, suggesting a buffering effect on IL‐6 trans‐signalling 37, 42.
Serum concentrations of IL‐6 are elevated rapidly in the context of inflammation in response to IL‐1β and TNF (major activators of IL‐6 expression), Toll‐like receptors, prostaglandins and mediators of stress response 37. IL‐6 expression is also induced by IL‐32, a cytokine that is over‐expressed in RA patients and associated highly with synovial inflammation 43, 44.
In RA, IL‐6 signalling triggers a variety of degenerative and inflammatory processes involving multiple mechanisms (Fig. 1). A relevant effect of IL‐6 in the pathogenesis of RA is its capacity to trigger systemic inflammatory events, such as increase in thermogenesis and synthesis of acute‐phase proteins (C‐reactive protein, fibrinogen, haptoglobin and serum amyloid A), resulting in manifestations such as fever, asthenia and anaemia 45. IL‐6 also induces synovial neovascularization, infiltration of inflammatory cells such as neutrophils and synovial hyperplasia, leading to RA joint destruction 46. Moreover, IL‐6 promotes the generation of osteoclasts and bone resorption by stimulating the expression of receptor activator of nuclear factor kappa B (NF‐κB) ligand (RANKL), among other mechanisms 47. In addition, IL‐6 promotes survival of B cells and their differentiation into autoantibody‐secreting long‐lived plasma cells through the STAT‐3‐mediated up‐regulation of PR domain zinc finger protein 1 (Blimp‐1) 48, and drives the commitment of CD4+ T cells to follicular Th cells that promote immunoglobulin class‐switching in B cell follicles 49. Direct evidence of a relationship between IL‐6 and chronic joint inflammation in RA arises from the findings that IL‐6 levels are elevated in serum and synovial fluid of RA patients, and that IL‐6 serum concentration correlates positively with disease activity 50, 51. In accordance, it has been reported that IL‐6‐deficient mice are resistant to the induction of arthritis 52, and that blocking the IL‐6R reduces the incidence of arthritis in mice 53.
Figure 1.
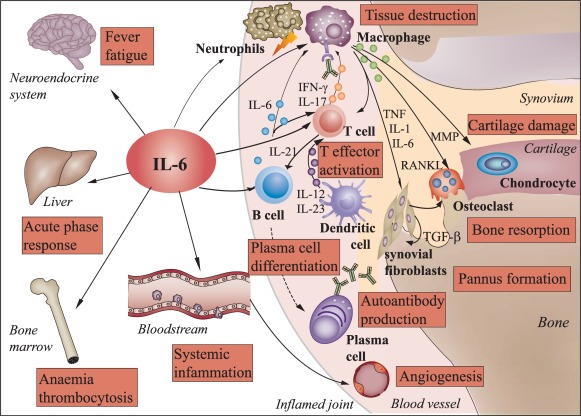
Pleiotropic effects of interleukin‐6 in rheumatoid arthritis. Interleukin (IL)‐6 exerts systemic effects on multiple tissues and cells of the immune system. In rheumatoid arthritis (RA), IL‐6 triggers systemic inflammatory processes, such as increase in thermogenesis and synthesis of acute phase proteins by hepatocytes, resulting in manifestations such as fever, fatigue and anaemia. In the synovial tissue of the joints, IL‐6 also induces vascularization, infiltration of inflammatory cells, such as neutrophils and monocytes/macrophages, and expansion of synovial fibroblasts leading to RA tissue destruction and pannus formation, respectively. IL‐6 also promotes cartilage damage, the differentiation of osteoclasts and bone resorption by stimulating the expression of matrix metalloproteinases (MMPs) and receptor activator of nuclear factor kappa B (NK‐κB) ligand (RANKL). Furthermore, IL‐6 promotes the expansion of CD4+ T cells and, together with cytokines secreted by inflammatory dendritic cells, induces the differentiation into interferon (IFN)‐γ and/or IL‐17‐producing T effector cells which, in turn, activate macrophages and attract neutrophils. IL‐6 drives also the differentiation of B cells to autoantibody‐producing plasma cells. Binding of autoantibodies to Fc receptors further triggers activation and release of inflammatory mediators by macrophages. TGF = transforming growth factor; TNF = tumour necrosis factor.
IL‐6 as critical factor in the Th17/Treg balance
Experiments performed with murine naive CD4+ T cells have revealed that Treg and Th17 cells share a common pathway for their generation: both lineages require TGF‐β to differentiate (Fig. 2) 54. However, addition of IL‐6 represses Treg differentiation 55 while favouring the development of Th17 cells, an effect that is potentiated by other proinflammatory cytokines, including IL‐1β, IL‐21, IL‐23 and TNF 12, 54, 56. The full acquisition of pathogenic functions by effector Th17 cells is mediated by IL‐23 rather than IL‐6 and TGF‐β, because apart from their role in driving the initial lineage commitment, IL‐6 and TGF‐β are capable of restraining the inflammatory response of activated Th17 cells by inducing IL‐10 production 57. However, a shift in the balance between proinflammatory and regulatory signals, for example through decreased numbers of Treg, might lead to a reduced IL‐10 production, concomitant with an up‐regulation of proinflammatory mediators produced by unrestrained Th17 cells. It has been shown that IL‐6‐deficient mice fail to develop a Th17 response, while their peripheral repertoire is dominated by forkhead box protein 3 (FoxP3+) Treg 58. Interestingly, deletion of Treg in this model leads to the reappearance of Th17 cells, suggesting an additional pathway of Th17 generation in vivo, which involves IL‐21 and TGF‐β 58. In addition, IL‐6 over‐expression in vivo was shown to inhibit the generation of induced Treg (iTreg), but had no impact on thymus‐derived natural Treg (nTreg) 59.
Figure 2.

The effect of interleukin (IL)‐6 and the cytokine milieu on CD4+ T cell commitment and plasticity. IL‐6 is involved in the commitment of naive CD4+ T cells to various effector cell lineages, which are characterized by the expression of specific transcription factors and transcriptional activators such as signal transducer and activators of transcription‐3 (STAT‐3) and a particular cytokine secretion profile. In combination with transforming growth factor (TGF)‐β, IL‐6 promotes the differentiation of IL‐17‐secreting T helper type 17 (Th17) cells. Conversely, IL‐6 blocks the generation of regulatory T cells (Treg). In combination, IL‐6 and IL‐21 induce the polarization into follicular T helper (Tfh) cells. Both Th1 and Th17 cells are involved in the pathogenesis of autoimmune diseases such as rheumatoid arthritis. The balance between Treg and Th17 cells is crucial to the maintenance of self‐tolerance. Plasticity between effector T cell lineages allows adaptation to the requirements of any immune response. Under the influence of IL‐12 and interferon (IFN)‐γ or IL‐4, Th17 cells acquire a Th1‐ or Th2‐like phenotype, respectively. In the absence of TGF‐β, IL‐6 converts induced (i)Treg into pathogenic Th17 cells. BCL‐6 = B cell CLL/lymphoma 6; FoxP3 = forkhead box protein 3; RORC2 = Retinoic acid‐related orphan nuclear receptor C2; TNF = tumour necrosis factor.
There is also a certain plasticity between Th17 and Treg lineages. It has been demonstrated that stimulating Treg with IL‐6 destabilizes the transcription factor FoxP3, while increasing IL‐17 and RORγt expression 60, 61. Furthermore, CD4+ T cells that co‐express IL‐17 and FoxP3 have been found in the synovium of RA patients, suggesting that Treg instability contributes to the pathogenesis of RA 62. There is evidence that IL‐6‐driven CD4+ T cell activation via STAT‐3 occurs as an early event in the pathogenesis of RA, especially in seronegative patients 63. Thus, a proinflammatory environment enriched with IL‐6 could favour the de‐novo generation of highly pathogenic Th17 lymphocytes, and also suppress iTreg generation and function, amplifying the inflammatory process further.
Blockage of IL‐6R for the treatment of rheumatoid arthritis
Based on the important role that IL‐6 plays in triggering systemic inflammation, it is not surprising that IL‐6 antagonists have been tested for the treatment of RA and other inflammatory diseases. Tocilizumab (TCZ) is a humanized monoclonal antibody that blocks the IL‐6 binding site of the IL‐6R 37. TCZ, used as monotherapy as well as in combination with MTX, showed an acceptable safety profile, and was proved effective in reducing disease activity, delaying joint destruction and improving physical function, especially in RA patients who were refractory to other anti‐rheumatic therapies, including TNF inhibitors 4, 64, 65, 66, 67, 68. Moreover, clinical studies have demonstrated a similar efficacy of TCZ compared to other biological therapies 69, 70. Furthermore, it has been demonstrated recently that TCZ is effective in treating early‐diagnosed RA patients who had not been treated previously with DMARDs 71. These results led to the approval of TCZ for RA treatment and the successful use of TCZ on other rheumatic diseases such as giant cell arteritis 72, and also encouraged the development of more biological drugs targeting IL‐6 or its receptor.
Sarilumab, a monoclonal human antibody directed against anti‐IL‐6Rα, has shown efficacy and safety profiles similar to TCZ as monotherapy as well as in combination with MTX 73, 74. Like TCZ, sarilumab has been demonstrated to be superior to adalimumab when used as monotherapy 75. Monoclonal antibodies directed against IL‐6, such as clazakizumab, olokizumab, and sirukumab, have been shown in Phase II clinical trials to be well tolerated and to reduce disease activity in RA patients with inadequate response to MTX or TNF inhibitors 76, 77, 78.
JAK inhibitors, such as tofacitinib (JAK1/3 inhibitor) and baricitinib (JAK1/2 inhibitor) which, as a consequence of their mechanism of action, interfere with IL‐6 secretion and downstream pathway activation, have also demonstrated efficacy in reducing signs and symptoms of RA in combination with MTX as well as monotherapy in Phase III clinical studies with patients who did not respond to MTX or TNF inhibitors 79, 80, 81.
The mechanisms by which IL‐6 inhibitors achieve this symptomatic improvement in RA patients have not yet been clarified entirely. However, some effects which are related directly to the known functions of IL‐6 have been described, such as normalization of bone formation/resorption markers and cartilage replacement, reduction of chemokine concentrations in serum and decrease of plasma cells and circulating autoantibodies 82, 83, 84. It has also been suggested that CD56+ natural killer (NK) cells play a protective role in RA and that high levels of NK cells are associated with RA remission upon treatment with TCZ 85. It is likely that new data on immunological parameters able to predict an adequate response to IL‐6 inhibitors begin to appear 86, 87. Evidence regarding the immunological impact of IL‐6R blockage has been obtained mainly through application of TCZ in clinical practice. Because sarilumab and anti‐IL‐6 monoclonal antibodies show a clinical efficacy similar to TCZ, it is conceivable that they may trigger similar immunological effects; however, this assert must be tested.
Effects of IL‐6R blockage on T cell subpopulations
Recent discoveries regarding the role that IL‐6 plays in the generation and expansion of Th17 responses, while affecting Treg adversely, suggest another angle addressed by TCZ in rheumatological diseases which, unlike the aforementioned described effects, involve initial events of the inflammatory cascade. Years ago, precursor experiments in mice showed that IL‐6R blockage affects the capacity of T cells to respond to antigenic stimuli 88. This hypothesis has been proved in the murine model of CIA by inoculating an anti‐IL‐6R antibody at the time of disease induction 89. The authors of this study observed that treated mice did not develop pathogenic Th17 activity without affecting the percentage of Treg, thus shifting the balance between these populations towards a protective response and, consequently, reducing the severity of arthritis 89. However, this effect could not be reproduced in mice with already established disease 89. Similar results have been obtained in other animal models of arthritis, both in spontaneous and in induced models 90, 91.
Regarding clinical settings, data describing the behaviour of Th17 and Treg subpopulations during TCZ treatment are somewhat conflicting (Table 1). A study on RA patients described a decrease in Th17 frequency and an increase of Tregs after 3 months of TCZ treatment 25. As the authors did not observe changes in IL‐6 serum levels upon TCZ treatment, they hypothesized that TCZ rather acts on restoration of the Th17/Treg balance than on the reduction of IL‐6‐induced inflammation 25. In contrast, our group has shown that the percentages of peripheral Treg and Th17/Th1 cells increased, while the frequency of Th1 or Th17 cells remained unaltered in RA patients who received TCZ for 6 months 26. This is in accordance with another study by Thiolat and co‐workers reporting an expansion of Treg expressing the adenosine triphosphate (ATP)‐hydrolysing ectonucleotidase CD39, but stable levels of Th17 cells after 3 months of TCZ treatment 93. The stability of the Th17 subpopulation upon TCZ treatment, together with data from IL‐6R blockage in murine arthritis, suggests that IL‐6 is necessary for Th17 induction, but might be dispensable for the maintenance of Th17 responses 94. It is likely that recovering Treg frequency and function is key in achieving a positive clinical outcome, as an increase in the Treg population has also been described after therapeutic TNF blockage 95. Anti‐TNF‐induced Treg can mediate suppression via IL‐10 and TGF‐β and compensate for the defective natural Treg in RA patients 96. Interestingly, differentiation of T cells into Treg instead of Th17 cells might be due partially to decreased IL‐6 levels in response to anti‐TNF treatment 97.
Table 1.
Effect of interleukin (IL)‐6R blockage by tocilizumab (TCZ) on CD4+ T cell subsets in treated rheumatoid arthritis (RA) patients (update of Tanaka 2013 92)
| RA patients | Treatment (months) | CD4+ T cell stimulus | Th1 | Th17 | Th17/1 | Th2 | Treg | Treg/Th17 ratio | Ref |
|---|---|---|---|---|---|---|---|---|---|
| MTX‐IR, BioDrug‐IR | 4 | PMA + ionomycin 8 h | = | ↓ | n.d. | n.d. | ↑ | ↑ | 25 |
| MTX‐IR | 6 | PMA + ionomycin 5 h | = | = | ↑ | n.d. | ↑ | ↑ | 26 |
| MTX‐IR, BioDrug‐IR | 3 | anti‐CD3 + anti‐CD28 24 h | n.d. | = | n.d. | n.d. | ↑ | ↑ | 92 |
|
Early RA MTX‐IR, Anti‐TNF‐IR |
3 | PMA + ionomycin 24 h | ↓ | ↓ | n.d. | ↑ | ↓ | n.d. | 97 |
↑ = Increase, ↓ = decrease, = no change with respect to baseline; BioDrug = biological drugs; IR = inadequate response; MTX = methotrexate; n.d. = not determined; TNF = anti‐tumour necrosis factor; Treg = regulatory T cells.
Unlike the studies described above, Guggino and colleagues reported that both 3‐month treatment of early‐diagnosed RA patients with TCZ and in‐vitro exposure of patient‐derived peripheral blood mononuclear cells to TCZ induced a depletion of Th1, Th17 and Treg and their related cytokines IL‐12, IL‐17 and IL‐10, while increasing the Th2 subset and IL‐4 secretion 98. The authors suggest that blockage of IL‐6 signalling reverts T cell resistance to Fas‐mediated apoptosis 99, and that distinct sensitivities of activated Th17, Th1 and Th2 cells to Fas‐mediated apoptosis might result in the depletion of Th1 and Th17, but not Th2, cells 100. Nevertheless, this discrepancy might be due to the differences in duration of T cell stimulation between the studies.
Conclusions
Despite some differences, the studies presented herein support the hypothesis that anti‐IL‐6R treatment restores the physiological Treg/Th17 balance and promotes the expansion of protective Treg, which may be important for the clinical response to anti‐IL‐6R observed in most RA patients. Although further studies are required to unravel the complete spectrum of effects that IL‐6R blockage exerts in the human organism, we postulate that its activity upstream of the inflammatory cascade, re‐establishing the Treg/Th17 equilibrium, is crucial to prevent downstream inflammatory effects of IL‐6, induced by a storm of proinflammatory cytokines and chemokines attracting and activating multiple cell populations, and therefore there lies a substantial proportion of its clinical efficacy.
Disclosure
L. S. has received consultant fees from Roche Laboratories. The other authors have no competing interests to declare.
Acknowledgements
We are very grateful to Dr Madhvi Menon for critically reading this review. This study was supported by FONDECYT grants no. 1121100 and no. 11121497.
Contributor Information
D. Catalán, Email: dfcatalan@med.uchile.cl.
L. Soto, Email: sotolian@gmail.com
References
- 1. Fuchs HA, Kaye JJ, Callahan LF, Nance EP, Pincus T. Evidence of significant radiographic damage in rheumatoid arthritis within the first 2 years of disease. J Rheumatol 1989; 16:585–91. [PubMed] [Google Scholar]
- 2. Goekoop‐Ruiterman YP, de Vries‐Bouwstra JK, Allaart CF et al Patient preferences for treatment: report from a randomised comparison of treatment strategies in early rheumatoid arthritis (BeSt trial). Ann Rheum Dis 2007; 66:1227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smolen JS, Landewe R, Breedveld FC et al EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease‐modifying antirheumatic drugs: 2013 update. Ann Rheum Dis 2014; 73:492–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dougados M, Kissel K, Sheeran T et al Adding tocilizumab or switching to tocilizumab monotherapy in methotrexate inadequate responders: 24‐week symptomatic and structural results of a 2‐year randomised controlled strategy trial in rheumatoid arthritis (ACT‐RAY). Ann Rheum Dis 2013; 72:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van der Woude D, Huizinga TW. Translating basic research into clinical rheumatology. Best Pract Res Clin Rheumatol 2008; 22:299–310. [DOI] [PubMed] [Google Scholar]
- 6. Linsley PS, Nadler SG. The clinical utility of inhibiting CD28‐mediated costimulation. Immunol Rev 2009; 229:307–21. [DOI] [PubMed] [Google Scholar]
- 7. Choy E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxf) 2012; 51 (Suppl 5):v3–11. [DOI] [PubMed] [Google Scholar]
- 8. Iwamoto T, Okamoto H, Toyama Y, Momohara S. Molecular aspects of rheumatoid arthritis: chemokines in the joints of patients. FEBS J 2008; 275:4448–55. [DOI] [PubMed] [Google Scholar]
- 9. Schulze‐Koops H, Kalden JR. The balance of Th1/Th2 cytokines in rheumatoid arthritis. Best Pract Res Clin Rheumatol 2001; 15:677–91. [DOI] [PubMed] [Google Scholar]
- 10. Murphy CA, Langrish CL, Chen Y et al Divergent pro‐ and antiinflammatory roles for IL‐23 and IL‐12 in joint autoimmune inflammation. J Exp Med 2003; 198:1951–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T‐cell repertoire: the Th17 lineage. Curr Opin Immunol 2006; 18:349–56. [DOI] [PubMed] [Google Scholar]
- 12. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)‐17 cells requires transforming growth factor‐beta and induction of the nuclear receptor RORgammat. Nat Immunol 2008; 9:641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Acosta‐Rodriguez EV, Rivino L, Geginat J et al Surface phenotype and antigenic specificity of human interleukin 17‐producing T helper memory cells. Nat Immunol 2007; 8:639–46. [DOI] [PubMed] [Google Scholar]
- 14. Cosmi L, De Palma R, Santarlasci V et al Human interleukin 17‐producing cells originate from a CD161+CD4+ T cell precursor. J Exp Med 2008; 205:1903–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. Main features of human T helper 17 cells. Ann NY Acad Sci 2013; 1284:66–70. [DOI] [PubMed] [Google Scholar]
- 16. Romagnani S. Human Th17 cells. Arthritis Res Ther 2008; 10:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miossec P. Interleukin‐17 in rheumatoid arthritis: if T cells were to contribute to inflammation and destruction through synergy. Arthritis Rheum 2003; 48:594–601. [DOI] [PubMed] [Google Scholar]
- 18. Sakuraba K, Oyamada A, Fujimura K et al Interleukin‐21 signaling in B cells, but not in T cells, is indispensable for the development of collagen‐induced arthritis in mice. Arthritis Res Ther 2016; 18:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nurieva R, Yang XO, Martinez G et al Essential autocrine regulation by IL‐21 in the generation of inflammatory T cells. Nature 2007; 448:480–3. [DOI] [PubMed] [Google Scholar]
- 20. Genovese MC, Greenwald M, Cho CS et al A phase II randomized study of subcutaneous ixekizumab, an anti‐interleukin‐17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheumatol 2014; 66:1693–704. [DOI] [PubMed] [Google Scholar]
- 21. Burmester GR, Durez P, Shestakova G et al Association of HLA‐DRB1 alleles with clinical responses to the anti‐interleukin‐17A monoclonal antibody secukinumab in active rheumatoid arthritis. Rheumatology (Oxford) 2016; 55:49–55. [DOI] [PubMed] [Google Scholar]
- 22. Aarvak T, Chabaud M, Miossec P, Natvig JB. IL‐17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol 1999; 162:1246–51. [PubMed] [Google Scholar]
- 23. Nistala K, Adams S, Cambrook H et al Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci USA 2010; 107:14751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cosmi L, Cimaz R, Maggi L et al Evidence of the transient nature of the Th17 phenotype of CD4+CD161+ T cells in the synovial fluid of patients with juvenile idiopathic arthritis. Arthritis Rheum 2011; 63:2504–15. [DOI] [PubMed] [Google Scholar]
- 25. Samson M, Audia S, Janikashvili N et al Inhibition of IL‐6 function corrects Th17/Treg imbalance in rheumatoid arthritis patients. Arthritis Rheum 2012; 64:2499–503. [DOI] [PubMed] [Google Scholar]
- 26. Pesce B, Soto L, Sabugo F et al Effect of interleukin‐6 receptor blockade on the balance between regulatory T cells and T helper type 17 cells in rheumatoid arthritis patients. Clin Exp Immunol 2013; 171:237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ehrenstein MR, Evans JG, Singh A et al Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti‐TNFalpha therapy. J Exp Med 2004; 200:277–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cribbs AP, Kennedy A, Penn H et al Treg cell function in rheumatoid arthritis is compromised by CTLA‐4 promoter methylation resulting in a failure to activate the indoleamine 2,3‐dioxygenase pathway. Arthritis Rheumatol 2014; 66:2344–54. [DOI] [PubMed] [Google Scholar]
- 29. Walter GJ, Fleskens V, Frederiksen KS et al Phenotypic, functional, and gene expression profiling of peripheral CD45RA+ and CD45RO+ CD4+CD25+CD127(low) Treg cells in patients with chronic rheumatoid arthritis. Arthritis Rheumatol 2016; 68:103–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum 2004; 50:2775–85. [DOI] [PubMed] [Google Scholar]
- 31. Kishimoto T. Interleukin‐6: from basic science to medicine – 40 years in immunology. Annu Rev Immunol 2005; 23:1–21. [DOI] [PubMed] [Google Scholar]
- 32. Murakami M, Hibi M, Nakagawa N et al IL‐6‐induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science 1993; 260:1808–10. [DOI] [PubMed] [Google Scholar]
- 33. Kimura A, Kishimoto T. IL‐6: regulator of Treg/Th17 balance. Eur J Immunol 2010; 40:1830–5. [DOI] [PubMed] [Google Scholar]
- 34. Jones SA, Scheller J, Rose‐John S. Therapeutic strategies for the clinical blockade of IL‐6/gp130 signaling. J Clin Invest 2011; 121:3375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peters M, Jacobs S, Ehlers M et al The function of the soluble interleukin 6 (IL‐6) receptor in vivo: sensitization of human soluble IL‐6 receptor transgenic mice towards IL‐6 and prolongation of the plasma half‐life of IL‐6. J Exp Med 1996; 183:1399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taga T, Kishimoto T. Gp130 and the interleukin‐6 family of cytokines. Annu Rev Immunol 1997; 15:797–819. [DOI] [PubMed] [Google Scholar]
- 37. Hunter CA, Jones SA. IL‐6 as a keystone cytokine in health and disease. Nat Immunol 2015; 16:448–57. [DOI] [PubMed] [Google Scholar]
- 38. Briso EM, Dienz O, Rincon M. Cutting edge: soluble IL‐6R is produced by IL‐6R ectodomain shedding in activated CD4 T cells. J Immunol 2008; 180:7102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jones GW, McLoughlin RM, Hammond VJ et al Loss of CD4+ T cell IL‐6R expression during inflammation underlines a role for IL‐6 trans signaling in the local maintenance of Th17 cells. J Immunol 2010; 184:2130–9. [DOI] [PubMed] [Google Scholar]
- 40. Matthews V, Schuster B, Schutze S et al Cellular cholesterol depletion triggers shedding of the human interleukin‐6 receptor by ADAM10 and ADAM17 (TACE). J Biol Chem 2003; 278:38829–39. [DOI] [PubMed] [Google Scholar]
- 41. Richards PJ, Nowell MA, Horiuchi S et al Functional characterization of a soluble gp130 isoform and its therapeutic capacity in an experimental model of inflammatory arthritis. Arthritis Rheum 2006; 54:1662–72. [DOI] [PubMed] [Google Scholar]
- 42. Narazaki M, Yasukawa K, Saito T et al Soluble forms of the interleukin‐6 signal‐transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane‐anchored gp130. Blood 1993; 82:1120–6. [PubMed] [Google Scholar]
- 43. Netea MG, Azam T, Ferwerda G et al IL‐32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 ligands for IL‐1beta and IL‐6 production through a caspase 1‐dependent mechanism. Proc Natl Acad Sci USA 2005; 102:16309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Joosten LA, Netea MG, Kim SH et al IL‐32, a proinflammatory cytokine in rheumatoid arthritis. Proc Natl Acad Sci USA 2006; 103:3298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Castell JV, Gomez‐Lechon MJ, David M, Hirano T, Kishimoto T, Heinrich PC. Recombinant human interleukin‐6 (IL‐6/BSF‐2/HSF) regulates the synthesis of acute phase proteins in human hepatocytes. FEBS Lett 1988; 232:347–50. [DOI] [PubMed] [Google Scholar]
- 46. Lally F, Smith E, Filer A et al A novel mechanism of neutrophil recruitment in a coculture model of the rheumatoid synovium. Arthritis Rheum 2005; 52:3460–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Palmqvist P, Persson E, Conaway HH, Lerner UH. IL‐6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF‐kappa B ligand, osteoprotegerin, and receptor activator of NF‐kappa B in mouse calvariae. J Immunol 2002; 169:3353–62. [DOI] [PubMed] [Google Scholar]
- 48. Diehl SA, Schmidlin H, Nagasawa M et al STAT3‐mediated up‐regulation of BLIMP1 Is coordinated with BCL6 down‐regulation to control human plasma cell differentiation. J Immunol 2008; 180:4805–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nurieva RI, Chung Y, Martinez GJ et al Bcl6 mediates the development of T follicular helper cells. Science 2009; 325:1001–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Madhok R, Crilly A, Watson J, Capell HA. Serum interleukin 6 levels in rheumatoid arthritis: correlations with clinical and laboratory indices of disease activity. Ann Rheum Dis 1993; 52:232–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sack U, Kinne RW, Marx T, Heppt P, Bender S, Emmrich F. Interleukin‐6 in synovial fluid is closely associated with chronic synovitis in rheumatoid arthritis. Rheumatol Int 1993; 13:45–51. [DOI] [PubMed] [Google Scholar]
- 52. Hata H, Sakaguchi N, Yoshitomi H et al Distinct contribution of IL‐6, TNF‐alpha, IL‐1, and IL‐10 to T cell‐mediated spontaneous autoimmune arthritis in mice. J Clin Invest 2004; 114:582–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Takagi N, Mihara M, Moriya Y et al Blockage of interleukin‐6 receptor ameliorates joint disease in murine collagen‐induced arthritis. Arthritis Rheum 1998; 41:2117–21. [DOI] [PubMed] [Google Scholar]
- 54. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL‐17‐producing T cells. Immunity 2006; 24:179–89. [DOI] [PubMed] [Google Scholar]
- 55. Bettelli E, Carrier Y, Gao W et al Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006; 441:235–8. [DOI] [PubMed] [Google Scholar]
- 56. Volpe E, Servant N, Zollinger R et al A critical function for transforming growth factor‐beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)‐17 responses. Nat Immunol 2008; 9:650–7. [DOI] [PubMed] [Google Scholar]
- 57. McGeachy MJ, Bak‐Jensen KS, Chen Y et al TGF‐beta and IL‐6 drive the production of IL‐17 and IL‐10 by T cells and restrain T(H)‐17 cell‐mediated pathology. Nat Immunol 2007; 8:1390–7. [DOI] [PubMed] [Google Scholar]
- 58. Korn T, Bettelli E, Gao W et al IL‐21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 2007; 448:484–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fujimoto M, Nakano M, Terabe F et al The influence of excessive IL‐6 production in vivo on the development and function of Foxp3+ regulatory T cells. J Immunol 2011; 186:32–40. [DOI] [PubMed] [Google Scholar]
- 60. Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25–Foxp3– T cells or are self‐induced to become Th17 cells in the absence of exogenous TGF‐beta. J Immunol 2007; 178:6725–9. [DOI] [PubMed] [Google Scholar]
- 61. Yang XO, Nurieva R, Martinez GJ et al Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 2008; 29:44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Komatsu N, Okamoto K, Sawa S et al Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 2014; 20:62–8. [DOI] [PubMed] [Google Scholar]
- 63. Anderson AE, Pratt AG, Sedhom MA et al IL‐6‐driven STAT signalling in circulating CD4+ lymphocytes is a marker for early anticitrullinated peptide antibody‐negative rheumatoid arthritis. Ann Rheum Dis 2016; 75:466–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kremer JL, Blanco R, Brzosko M et al Tocilizumab inhibits structural joint damage in rheumatoid arthritis patients with inadequate responses to methotrexate at 1 year: the LITHE study. Arthritis Rheum 2011; 63:609–21. [DOI] [PubMed] [Google Scholar]
- 65. Nishimoto N, Hashimoto J, Miyasaka N et al Study of active controlled monotherapy used for rheumatoid arthritis, an IL‐6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an X‐ray reader‐blinded randomised controlled trial of tocilizumab. Ann Rheum Dis 2007; 66:1162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Emery P, Keystone E, Tony HP et al IL‐6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti‐tumour necrosis factor biologicals: results from a 24‐week multicentre randomised placebo‐controlled trial. Ann Rheum Dis 2008; 67:1516–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Burmester GR, Feist E, Kellner H, Braun J, Iking‐Konert C, Rubbert‐Roth A. Effectiveness and safety of the interleukin 6‐receptor antagonist tocilizumab after 4 and 24 weeks in patients with active rheumatoid arthritis: the first phase IIIb real‐life study (TAMARA). Ann Rheum Dis 2011; 70:755–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gabay C, Emery P, van Vollenhoven R et al Tocilizumab monotherapy versus adalimumab monotherapy for treatment of rheumatoid arthritis (ADACTA): a randomised, double‐blind, controlled phase 4 trial. Lancet 2013; 381:1541–50. [DOI] [PubMed] [Google Scholar]
- 69. Smolen JS, Beaulieu A, Rubbert‐Roth A et al Effect of interleukin‐6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double‐blind, placebo‐controlled, randomised trial. Lancet 2008; 371:987–97. [DOI] [PubMed] [Google Scholar]
- 70. Genovese MC, McKay JD, Nasonov EL et al Interleukin‐6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease‐modifying antirheumatic drugs: the tocilizumab in combination with traditional disease‐modifying antirheumatic drug therapy study. Arthritis Rheum 2008; 58:2968–80. [DOI] [PubMed] [Google Scholar]
- 71. Bijlsma JW, Welsing PM, Woodworth TG et al Early rheumatoid arthritis treated with tocilizumab, methotrexate, or their combination (U‐Act‐Early): a multicentre, randomised, double‐blind, double‐dummy, strategy trial. Lancet 2016; 388:343–55. [DOI] [PubMed] [Google Scholar]
- 72. Villiger PM, Adler S, Kuchen S et al Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double‐blind, placebo‐controlled trial. Lancet 2016; 387:1921–7. [DOI] [PubMed] [Google Scholar]
- 73. Genovese MC, Fleischmann R, Kivitz AJ et al Sarilumab plus methotrexate in patients with active rheumatoid arthritis and inadequate response to methotrexate: results of a phase III study. Arthritis Rheumatol 2015; 67:1424–37. [DOI] [PubMed] [Google Scholar]
- 74. Fleischmann R, van Adelsberg J, Lin Y et al Sarilumab and nonbiologic disease‐modifying antirheumatic drugs in patients with active rheumatoid arthritis and inadequate response or intolerance to tumor necrosis factor inhibitors. Arthritis Rheumatol 2017; 69:277–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Burmester GR, Lin Y, Patel R et al Efficacy and safety of sarilumab monotherapy versus adalimumab monotherapy for the treatment of patients with active rheumatoid arthritis (MONARCH): a randomised, double‐blind, parallel‐group phase III trial. Ann Rheum Dis 2016. doi: 10.1136/annrheumdis-2016-210310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Weinblatt ME, Mease P, Mysler E et al The efficacy and safety of subcutaneous clazakizumab in patients with moderate‐to‐severe rheumatoid arthritis and an inadequate response to methotrexate: results from a multinational, phase IIb, randomized, double‐blind, placebo/active‐controlled, dose‐ranging study. Arthritis Rheumatol 2015; 67:2591–600. [DOI] [PubMed] [Google Scholar]
- 77. Genovese MC, Fleischmann R, Furst D et al Efficacy and safety of olokizumab in patients with rheumatoid arthritis with an inadequate response to TNF inhibitor therapy: outcomes of a randomised Phase IIb study. Ann Rheum Dis 2014; 73:1607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Smolen JS, Weinblatt ME, Sheng S, Zhuang Y, Hsu B. Sirukumab, a human anti‐interleukin‐6 monoclonal antibody: a randomised, 2‐part (proof‐of‐concept and dose‐finding), phase II study in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis 2014; 73:1616–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fleischmann R, Kremer J, Cush J et al Placebo‐controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med 2012; 367:495–507. [DOI] [PubMed] [Google Scholar]
- 80. Charles‐Schoeman C, Burmester G, Nash P et al Efficacy and safety of tofacitinib following inadequate response to conventional synthetic or biological disease‐modifying antirheumatic drugs. Ann Rheum Dis 2016; 75:1293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dougados M, van der Heijde D, Chen YC et al Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: results from the RA‐BUILD study. Ann Rheum Dis 2017; 76:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Garnero P, Thompson E, Woodworth T, Smolen JS. Rapid and sustained improvement in bone and cartilage turnover markers with the anti‐interleukin‐6 receptor inhibitor tocilizumab plus methotrexate in rheumatoid arthritis patients with an inadequate response to methotrexate: results from a substudy of the multicenter double‐blind, placebo‐controlled trial of tocilizumab in inadequate responders to methotrexate alone. Arthritis Rheum 2010; 62:33–43. [DOI] [PubMed] [Google Scholar]
- 83. Kawashiri SY, Kawakami A, Iwamoto N et al Proinflammatory cytokines synergistically enhance the production of chemokine ligand 20 (CCL20) from rheumatoid fibroblast‐like synovial cells in vitro and serum CCL20 is reduced in vivo by biologic disease‐modifying antirheumatic drugs. J Rheumatol 2009; 36:2397–402. [DOI] [PubMed] [Google Scholar]
- 84. Illei GG, Shirota Y, Yarboro CH et al Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open‐label phase I dosage‐escalation study. Arthritis Rheum 2010; 62:542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Daien CI, Gailhac S, Audo R et al High levels of natural killer cells are associated with response to tocilizumab in patients with severe rheumatoid arthritis. Rheumatology (Oxf) 2015; 54:601–8. [DOI] [PubMed] [Google Scholar]
- 86. Pers YM, Fortunet C, Constant E et al Predictors of response and remission in a large cohort of rheumatoid arthritis patients treated with tocilizumab in clinical practice. Rheumatology (Oxf) 2014; 53:76–84. [DOI] [PubMed] [Google Scholar]
- 87. Boyapati A, Msihid J, Fiore S, van Adelsberg J, Graham NM, Hamilton JD. Sarilumab plus methotrexate suppresses circulating biomarkers of bone resorption and synovial damage in patients with rheumatoid arthritis and inadequate response to methotrexate: a biomarker study of MOBILITY. Arthritis Res Ther 2016; 18:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mihara M, Nishimoto N, Yoshizaki K, Suzuki T. Influences of anti‐mouse interleukin‐6 receptor antibody on immune responses in mice. Immunol Lett 2002; 84:223–9. [DOI] [PubMed] [Google Scholar]
- 89. Fujimoto M, Serada S, Mihara M et al Interleukin‐6 blockade suppresses autoimmune arthritis in mice by the inhibition of inflammatory Th17 responses. Arthritis Rheum 2008; 58:3710–9. [DOI] [PubMed] [Google Scholar]
- 90. Hirota K, Hashimoto M, Yoshitomi H et al T cell self‐reactivity forms a cytokine milieu for spontaneous development of IL‐17+ Th cells that cause autoimmune arthritis. J Exp Med 2007; 204:41–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Iwanami K, Matsumoto I, Tanaka‐Watanabe Y et al Crucial role of the interleukin‐6/interleukin‐17 cytokine axis in the induction of arthritis by glucose‐6‐phosphate isomerase. Arthritis Rheum 2008; 58:754–63. [DOI] [PubMed] [Google Scholar]
- 92. Tanaka T. Can IL‐6 blockade rectify imbalance between Tregs and Th17 cells? Immunotherapy 2013; 5:695–7. [DOI] [PubMed] [Google Scholar]
- 93. Thiolat A, Semerano L, Pers YM et al Interleukin‐6 receptor blockade enhances CD39+ regulatory T cell development in rheumatoid arthritis and in experimental arthritis. Arthritis Rheumatol 2014; 66:273–83. [DOI] [PubMed] [Google Scholar]
- 94. Liu X, Leung S, Wang C et al Crucial role of interleukin‐7 in T helper type 17 survival and expansion in autoimmune disease. Nat Med 2010; 16:191–7. [DOI] [PubMed] [Google Scholar]
- 95. Biton J, Semerano L, Delavallee L et al Interplay between TNF and regulatory T cells in a TNF‐driven murine model of arthritis. J Immunol 2011; 186:3899–910. [DOI] [PubMed] [Google Scholar]
- 96. Nadkarni S, Mauri C, Ehrenstein MR. Anti‐TNF‐alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF‐beta. J Exp Med 2007; 204:33–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Charles P, Elliott MJ, Davis D et al Regulation of cytokines, cytokine inhibitors, and acute‐phase proteins following anti‐TNF‐alpha therapy in rheumatoid arthritis. J Immunol 1999; 163:1521–8. [PubMed] [Google Scholar]
- 98. Guggino G, Giardina AR, Raimondo S et al Targeting IL‐6 signalling in early rheumatoid arthritis is followed by Th1 and Th17 suppression and Th2 expansion. Clin Exp Rheumatol 2014; 32:77–81. [PubMed] [Google Scholar]
- 99. Atreya R, Mudter J, Finotto S et al Blockade of interleukin 6 trans signaling suppresses T‐cell resistance against apoptosis in chronic intestinal inflammation: evidence in Crohn disease and experimental colitis in vivo . Nat Med 2000; 6:583–8. [DOI] [PubMed] [Google Scholar]
- 100. Fang Y, Yu S, Ellis JS, Sharav T, Braley‐Mullen H. Comparison of sensitivity of Th1, Th2, and Th17 cells to Fas‐mediated apoptosis. J Leukoc Biol 2010; 87:1019–28. [DOI] [PMC free article] [PubMed] [Google Scholar]