

Summary
Cancer immunity is mediated through the effective priming and activation of tumour‐specific class I MHC molecule‐restricted CD8+ cytotoxic T lymphocytes (CTLs). DEC‐205+ dendritic cells (DCs) can cross‐present the epitope(s) of captured tumour antigens associated with class I MHC molecules alongside co‐stimulatory molecules to prime and activate tumour‐specific CD8+ CTLs. Immunosuppressive tolerogenic DCs with reduced co‐stimulatory molecules may be a cause of impaired CTL induction. Hepa1‐6‐1 cells were established from the mouse hepatoma cell line Hepa1‐6; these cells grow continuously after subcutaneous implantation into syngeneic C57BL/6 (B6) mice and do not prime CD8+ CTLs. In this study, we show that the growth of ongoing tumours was suppressed by activated CD8+ CTLs with tumour‐specific cytotoxicity through the administration of the glycolipid α‐galactosylceramide (α‐GalCer), which is a compound known to stimulate invariant natural killer T (iNKT) cells and selectively activate DEC‐205+ DCs. Moreover, we demonstrated that sequential repetitive intraperitoneal inoculation with α‐GalCer every 48 hr appeared to convert tolerogenic DEC‐205+ DCs into immunogenic DCs with a higher expression of co‐stimulatory molecules and a stronger cross‐presentation capacity, which primed CTL precursors and induced tumour‐specific CD8+ CTLs within the tumour environment without activating iNKT cells. These findings provide a new basis for cancer immunotherapy to convert tolerogenic DEC‐205+ DCs within tumours into immunogenic DCs through the sequential administration of an immuno‐potent lipid/glycolipid, and then activated immunogenic DCs with sufficient expression of co‐stimulatory molecules prime and activate tumour‐specific CD8+ CTLs within the tumour to control tumour growth.
Keywords: co‐stimulatory molecule, cytotoxic T lymphocytes, tumour‐infiltrating dendritic cells, tumour‐infiltrating lymphocytes, α‐galactosylceramide
Abbreviations
- APC
allophycocyanin
- BM
bone marrow
- CFSE
carboxyfluorescein diacetate succinimidyl ester
- CTLs
cytotoxic T lymphocytes
- DCs
dendritic cells
- FCS
fetal calf serum
- i.p.
intraperitoneal
- i.v.
intravenous
- IL‐12
interleukin‐12
- iNKT
invariant natural killer T
- KO
knockout
- mAbs
monoclonal antibodies
- PE
phycoerythrin
- s.c.
subcutaneous
- TIDCs
tumour‐infiltrating dendritic cells
- TILs
tumour‐infiltrating lymphocytes
- α‐GalCer
α‐galactosylceramide
Introduction
The most effective cells for eliminating tumour cells are CD8+ cytotoxic T lymphocytes (CTLs) that specifically recognize the tumour antigens presented in association with class I MHC molecules.1 To prime and activate CTLs in a tumour environment, dendritic cells (DCs) are key cells that uptake tumour proteins, process them into peptide fragments containing immunodominant epitope(s), and present them on their surface associated with antigen‐presenting class I MHC molecule(s) for the activation of specific T‐cell immunity.2, 3
In general, externally captured antigenic molecules are fragmented into small peptide pieces within the endosome and are presented in conjunction with class II MHC molecules, which prime CD4+ helper T cells, whereas internally synthesized proteins are broken into tiny fragments containing epitope peptides and are presented with class I MHCs, which prime CD8+ CTLs.4, 5 Recently, a unique DC subtype expressing the molecule DEC‐205 was shown to have the capacity to present an epitope of an externally captured antigenic protein in association with class I MHCs, which was called ‘cross‐presentation’,6, 7, 8, 9 and the cross‐presented antigenic epitope was specifically recognized by CD8+ CTLs for both mice and humans. Moreover, the priming and activation of antigen‐specific CD8+ CTLs required co‐stimulatory signalling via CD80 and CD86.10 Hence, CD8+ CTLs, which are tumour‐specific, can be primed and activated by tumour‐antigen‐loaded activated DEC‐205+ DCs expressing co‐stimulatory molecules.11, 12
We have established two distinct types of murine hepatoma cell lines, called Hepa1‐6‐1 and Hepa1‐6‐2, from known Hepa1‐6 cells13 and confirmed that they displayed similar antigenicity as well as an identical surface expression of class I MHC.11, 13 After subcutaneous (s.c.) implantation into syngeneic C57BL/6 (B6) mice, Hepa1‐6‐1 cells showed continuous growth and did not prime CD8+ CTLs, whereas Hepa1‐6‐2 cells failed to grow in vivo and efficiently primed the CTLs.11 Through a careful examination of the cells within these two distinct tumours, among tumour‐infiltrating DCs (TIDCs), we found that the DEC‐205+ tolerogenic DCs had reduced levels of co‐stimulatory molecules as well as impaired cross‐presenting capacities in the Hepa1‐6‐1‐derived tumour mass but not within the Hepa1‐6‐2‐derived tumour mass, and we concluded that the tolerogenic DCs may be a cause of the impaired CTL induction.11
Based on these findings, we questioned whether we could alter the conditions of the DEC‐205+ tolerogenic DCs within the Hepa1‐6‐1‐derived tumour into immunogenic DCs with higher expression levels of co‐stimulatory molecules using the external administration of α‐galactosylceramide (α‐GalCer), a potent activator of DEC‐205+ DCs.14 In this study, we show for the first time that the growth of an ongoing Hepa1‐6‐1‐derived tumour mass was profoundly suppressed by internally induced Hepa1‐6‐1‐specific CD8+ CTLs but not by the activated invariant natural killer T (iNKT) cells through a sequential repetitive intraperitoneal (i.p.) inoculation with α‐GalCer every other day. With these findings, a new notion can be proposed for cancer immunotherapy using the selective activation of immunosuppressive tolerogenic DCs within tumours by sequential administration with a known glycolipid antigen, such as α‐GalCer.
Materials and methods
Mice
Seven‐week‐old female B6 (H‐2b) mice were purchased from Charles River Japan (Kanagawa, Japan), maintained in microisolator cages under pathogen‐free conditions, and fed autoclaved laboratory chow and water. The iNKT cell knockout (Jα18 KO) mice on the B6 background were a kind gift from M. Taniguchi (RIKEN Institute, Yokohama City, Japan).15 All animal experiments were performed according to the Guidelines for the Care and Use of Laboratory Animals set by the National Institutes of Health (NIH; Bethesda, MD) and were approved by the Review Board of Nippon Medical School (Tokyo, Japan).
Culture medium
The medium used for culturing immunocompetent cells, such as CTLs, was an RPMI‐1640 (Sigma‐Aldrich, St Louis, MO) ‐based complete culture medium16 supplemented with 50 mm 2‐mercaptoethanol (Invitrogen Life Technologies, Carlsbad, CA), 2 mm l‐glutamine (Sigma‐Aldrich), 1 mm sodium pyruvate (Invitrogen), 0·1 mm non‐essential amino acids (Invitrogen), a mixture of vitamins (Invitrogen), 1 mm HEPES (Invitrogen), 100 U/ml penicillin (Invitrogen), 100 μg/ml streptomycin (Invitrogen), and 10% heated fetal calf serum (FCS; HyClone, Logan, UT). Some of the tumour cells were cultured in either Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 1 mm HEPES, 100 U/ml penicillin (Invitrogen), 100 μg/ml streptomycin and 10% heated FCS (D‐10) or with RPMI‐1640 supplemented with 1 mm HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin and 10% heated FCS (R‐10).
Establishment of Hepa1‐6‐1 and Hepa1‐6‐2 cell lines
The murine hepatoma cell line, Hepa1‐6, which was established from B6 (H‐2b) mice, was purchased from the American Type Culture Collection (ATCC, Rockville, MD). The murine hepatoma cell lines Hepa1‐6‐1 and Hepa1‐6‐2 were established from Hepa1‐6.11 Briefly, Hepa1‐6‐1 cells were obtained from the purified cells of a collagenase‐digested and surgically removed tumour mass that was grown for approximately 3 months after the s.c. implantation of Hepa1‐6 cells into syngeneic B6 mice and were maintained in D‐10 medium. Hepa1‐6‐2 is a controllable tumour line that demonstrates spontaneous regression obtained by the repetitive in vitro transfer of Hepa1‐6 cells for several months in R‐10 medium.
Tumour injections and measurement of tumour size
Ten million tumour cells with 100 μl of RPMI‐1640 were s.c. injected into the abdominal region of each mouse with a 27‐gauge needle syringe. For estimating the volume of the growing tumour mass, the diameters for both the length (a) and the width (b) were measured every day until day 24 after implantation, and the tumour volume (V) was calculated according to the formula V = ab 2/2 as previously described.11 When the longer axis of each tumour was > 20 mm in diameter, the mice were anaesthetized and killed according to the NIH guidelines.
In vivo activation of DEC‐205+ DCs
The in vivo activation of the DEC‐205+ DCs was performed by the injection of α‐GalCer (KRN7000; Funakoshi Co., Ltd., Tokyo, Japan).17, 18 To dissolve KRN7000, we used 0·025% Polysolvate‐20 in PBS to dissolve the material according to the manufacturer's instructions. The mice were given an i.p. injection of 2 μg α‐GalCer in 100 μl of 0·025% Polysolvate‐20 in PBS every other day from day 0 to day 18.
In vivo depletion of CD8+ T cells, CD4+ T cells and NK cells
For in vivo deletion of CD8+ T cells or CD4+ T cells, mice were given two i.p. injections (on days 1 and 3) of 400 μg/mouse anti‐Lyt2 (3.155; ATCC) or 400 μg/mouse anti‐mouse CD4 (GK1.5; BioLegend, San Diego, CA). For the deletion of NK cells, mice were intravenously (i.v.) injected twice (on days 1 and 3) with 50 μl/mouse anti‐asialo‐GM1 (poly21460; BioLegend). Flow cytometry analysis confirmed that > 95% of the CD8+ T cells, CD4+ T cells and NK cells in the spleen had been depleted.
Interleukin‐12 administration to Hepa1‐6‐1‐implanted mice
For the interleukin‐12 (IL‐12) administration into Hepa1‐6‐1‐implanted mice, 100 ng/mouse IL‐12p70 (R&D Systems, Minneapolis, MN) was injected i.p. every other day from day 0 until day 18.
Antibodies and flow cytometric analysis
Flow cytometric analyses were performed to determine the surface molecule expression of the cells using a FACSCanto II six‐colour cytometer (Becton Dickinson Immunochemical Systems, Mountain View, CA). Cell suspensions were stained with relevant antibodies for 30 min at 4° in PBS with 2% heat‐inactivated FCS and 0·1% sodium azide, washed twice and analysed. The following antibodies were used: allophycocyanin (APC)‐labelled mouse α‐GalCer‐loaded CD1d tetramer (ProImmune, Oxford, UK); phycoerythrin (PE) ‐labelled anti‐mouse CD3 (145‐2C11; BioLegend); APC‐Cy7‐labelled anti‐mouse CD45 (30‐F11; BioLegend); FITC‐labelled anti‐mouse CD69 (H1.2F3; BioLegend); PE‐ or FITC‐labelled anti‐mouse CD11c (N418; BioLegend); APC‐ or PE‐ or PE/Cy7‐labelled anti‐mouse CD205 (DEC‐205) (NLDC‐145; BioLegend); APC‐labelled anti‐mouse DC marker (33D1) (33D1; BioLegend); PE‐labelled anti‐mouse CD1d (1B1; BioLegend); FITC‐ or APC/Cy7‐labelled anti‐mouse MHC‐II (M5/114.15.2; BioLegend); APC‐ or PE‐labelled anti‐mouse CD8α (53‐6.7; BioLegend); APC‐ or PE‐labelled anti‐mouse CD80 (16‐10A1; BioLegend); APC‐ or PE‐labelled anti‐mouse CD86 (GL1; BioLegend); PE‐labelled anti‐mouse CD40 (3/23; BioLegend); and PE‐labelled anti‐mouse PD‐L1 (10F.9G2; BioLegend); PE‐labelled anti‐mouse α‐GalCer:CD1d complex (L363; Biolegend); PE‐labelled anti‐mouse NK1.1 (PK136; BioLegend). The dead cells were gated according to propidium iodide uptake. A total of 100 000 events were acquired for each sample, and the data were analysed using flowjo software (Tree Star Inc., Ashland, OR). The gating strategies for CD8+ T cells, DEC‐205+ DCs, 33D1+ DCs, iNKT cells and NK cells, as well as co‐stimulatory molecules (CD80 and CD86) and activation marker CD69, on these cells and control cells are shown in the Supplementary material (Figure S1).
Tumour‐infiltrating lymphocyte isolation
For isolation of the tumour‐infiltrating lymphocytes, the tumours were excised from the mice and digested with 1 mg/ml collagenase (Roche Diagnostics GmbH, Mannheim, Germany) for 45 min at 37°. Then, the tissue was crushed gently to homogenize it and filtered through a nylon mesh. To obtain purified tumour‐infiltrating lymphocytes (TILs), the isolated leucocytes were further purified from other haematopoietic cells with a mouse CD8+ T‐cell isolation cocktail containing a combination of biotinylated monoclonal antibodies (mAbs) directed against the cell surface antigens CD4, CD11b, CD11c, CD19, CD24, CD45R/B220, CD49b, T‐cell receptor‐γ/δ and TER119 as well as nanoparticles. The cells were negatively sorted with the immunomagnetic system (StemCell Technologies, Vancouver, BC, Canada), which yielded a population containing approximately 95% purified CD8+ TILs.
Purification of CD11c+ TIDCs
To purify the tumour‐infiltrating CD11c+ cells, the TIL suspension was incubated with PE‐labelled anti‐CD11c followed by a PE‐selection cocktail and nanoparticles and was positively sorted with the immunomagnetic system (StemCell Technologies), which yielded a population containing approximately 95% purified CD11c+ TIDCs.
Induction of bone marrow‐derived DCs
Bone marrow (BM) cells prepared from femurs and tibias of syngeneic B6 mice were depleted of red blood cells using osmotic haemolysis, as recently described.19 Next, 1 × 106 BM cells were plated on 24‐well culture plates and incubated in complete culture medium supplemented with 20 ng/ml of murine recombinant granulocyte–macrophage colony‐stimulating factor (Peprotech, Rockey Hill, NJ). On day 2 of culture, the floating cells were gently removed, and fresh medium was co‐cultured with 1 × 105 Hepa1‐6‐1 cells in the trans‐well system (Corning, Kennebunk, ME). On day 5, non‐adherent cells were collected and analysed using flow cytometry.
Re‐stimulation of Hepa1‐6‐2‐specific primed lymphocytes with CD11c+ TIDCs or BM‐derived DCs
One million Hepa1‐6‐2 cells in 200 μl of PBS were i.p. injected into each B6 mouse with a 27‐gauge needle syringe. Approximately 14 days after the Hepa1‐6‐2 inoculation, an additional administration of 1 × 106 of the original Hepa1‐6‐2 cells was performed. One week after the Hepa1‐6‐2 inoculation, 1 × 105 primed splenic CD8+ T cells were obtained by positively sorting with the immunomagnetic system (StemCell Technologies), which yielded a population containing approximately 95% purified CD8+ T cells, labelled with 5 mm carboxyfluorescein diacetate succinimidyl ester (CFSE) and further co‐cultured with either 5 × 104 CD11c+ TIDCs or BM‐derived DCs pulsed with hepa1‐6‐1 lysate obtained from 5 × 103 Hepa1‐6‐1 cells overnight and completely washed out to remove free antigen in a 200 μl round‐bottom 96‐well cell culture plate for 4 days. Next, the re‐stimulated cells were harvested and analysed using flow cytometry to determine the number of cell divisions, which correlated with the ability of the TIDCs to prime T cells.
In vitro induction of Hepa1‐6‐1‐specific CTLs
Four million spleen cells obtained from sequentially α‐GalCer‐treated Hepa1‐6‐1‐implanted mice were re‐stimulated with 1 × 105/well Hepa1‐6‐1 cells treated with mitomycin‐C (Kyowa Hakko‐Kirin, Tokyo, Japan) for 5 days in 24‐well culture plates with 1·0 ml of complete culture medium containing 10% rat concanavalin A supernatant‐containing medium (Rat T‐STIM; Corning, Bedford, MA).
CTL assay
A standard chromium‐51‐release assay was performed to determine the cytotoxicity of the purified CD8+ TILs, or generated Hepa1‐6‐specific immune cells, as previously described.16 Briefly, various numbers of effector cells were incubated with 5 × 103 chromium‐51‐labelled targets for 4 hr at 37° in 200 μl of RPMI‐1640 containing 10% warmed FCS in a round‐bottom 96‐well cell culture plate. After incubation, the plate was centrifuged for 10 min at 330 g, and 100 μl of the cell‐free supernatant was collected to measure the radioactivity with a Packard Auto‐Gamma 5650 counter (Hewlett‐Packard, Tokyo, Japan). The maximum release was determined from the supernatant of the cells that had been lysed by the addition of 5% Triton X‐100, and the spontaneous release was determined from the target cells incubated without the added effector cells. The percentage of specific lysis was calculated as 100 × (experimental release – spontaneous release)/ (maximum release – spontaneous release).
Priming of CD8+ CTLs from naive T cells by α‐GalCer‐activated DCs
To examine the effect of α‐GalCer‐activated DCs on CTL priming, TIDCs from Hepa1‐6‐1‐implanted B6 mice with an i.p. injection of 2 μg/mouse α‐GalCer every other day for 14 days were co‐cultured with syngeneic splenic naive T cells obtained by passing them through a nylon wool column as previously described.20 TIDCs (1 × 105/well) and purified naive splenic T cells (5 × 105/well) were co‐cultured in a flat‐bottom 96‐well cell‐culture plate. After 4 days of culture, cells were collected and the percentages of naive T cells (CD3+ CD8+ CD44− CD62L+) and memory T cells (CD3+ CD8+ CD44+ CD62L−) were evaluated by flow cytometry. After a week, the primed T cells were further incubated with mitomycin‐C‐treated Hepa1‐6‐1 cells for 48 hr and the amount of interferon‐γ secreted in the culture supernatant was measured by ELISA.
Cytokine detection
The amount of mouse serum IL‐12p40 and the amount of interferon‐γ in the culture supernatant were measured with the IL‐12p40 detection Kit (R&D Systems) and the IFN‐γ detection Kit (BioLegend), respectively.
Statistical analysis
The results were analysed using a Student's t‐test, and the results are presented as the mean ± SEM. Differences at P < 0·05 were considered significant. The statistical analysis was performed using prism software (GraphPad Software, La Jolla, CA).
Results
Effect of the administration of Hepa1‐6‐1‐specific CD8+ CTLs on Hepa1‐6‐1 tumour growth in vivo
We previously reported that we have established two distinct types of murine hepatoma cell lines, Hepa1‐6‐1 and Hepa1‐6‐2,11 from the known hepatoma cell line Hepa1‐6.13 We confirmed that these cells showed similar antigenicity as well as identical surface class I MHC molecule restriction.21 The original Hepa1‐6‐1 cells showed continuous growth after s.c. implantation into syngeneic B6 mice and did not prime CD8+ CTLs, whereas the Hepa1‐6‐2 cells failed to grow in vivo by Hepa1‐6‐2‐specific CD8+ CTLs, which also efficiently eliminated Hepa1‐6‐1 cells in vitro.11 Therefore, we first established Hepa1‐6‐2‐specific CD8+ CTLs through the repetitive stimulation of Hepa1‐6‐2‐primed spleen cells with Hepa1‐6‐2 tumour cells in vitro and confirmed that these cells specifically killed both Hepa1‐6‐1 and Hepa1‐6‐2 cells in vitro (Fig. 1a). In addition, we confirmed that there was no difference in H‐2Db class‐I MHC molecule expression between Hepa1‐6‐1 and Hepa1‐6‐2 cells (Fig. 1b). These results clearly indicated that both the Hepa1‐6‐1 and Hepa1‐6‐2 tumour cell lines express the same epitopes and the same levels of MHC class I molecules. Then, we applied 1 × 106 of the Hepa1‐6‐2‐specific CD8+ CTLs s.c. into the tumour mass of B6 mice that were implanted with Hepa1‐6‐1 cells 7 days before the CTL administration, when the implanted Hepa1‐6‐1 cells had established a solid tumour mass.11 However, the growth of the implanted Hepa1‐6‐1 cells was not inhibited at all (Fig. 1c). We also examined the effects of the i.v. administration of 1 × 106 Hepa1‐6‐2‐specific CD8+ CTLs/mouse to the mice implanted with Hepa1‐6‐1 cells. Similar to the findings obtained by i.v. administration of 1 × 106 OVA‐specific CD8+ CTLs/mouse21 the growth of the tumour was not at all inhibited (Fig. 1d). These findings indicate that adoptive immunotherapy using tumour‐specific CTLs does not work in this system.
Figure 1.
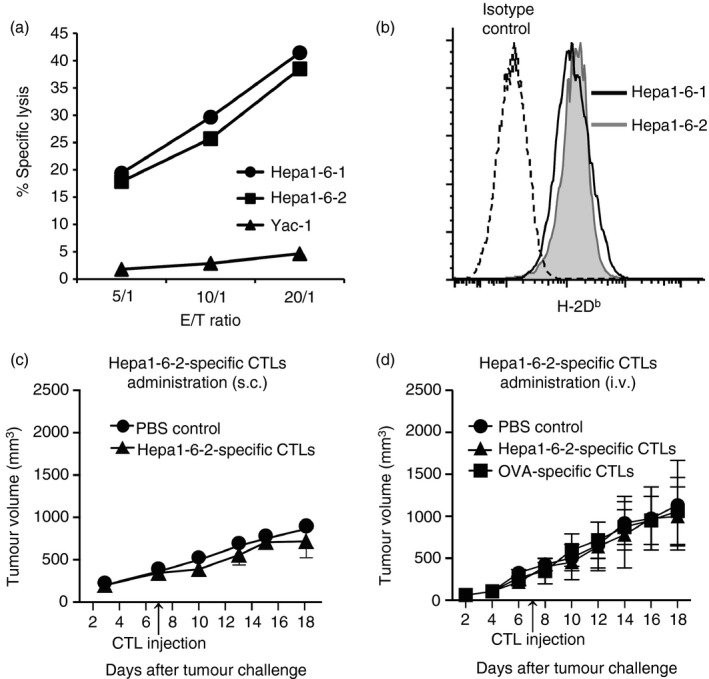
Effect of the external administration of Hepa1‐6‐2‐specific CD8+ cytotoxic T lymphocytes (CTLs) on Hepa1‐6‐1 tumour growth in vivo. (a) To determine whether Hepa1‐6‐1 or Hepa1‐6‐2 tumour cells could be eliminated by Hepa1‐6‐2‐specific CD8+ CTLs in vitro, a standard chromium‐51 release assay was performed. (b) H‐2Db expression on the Hepa1‐6‐1 and Hepa1‐6‐2 cells evaluated by flow cytometry. (c) Effects of subcutaneous (s.c.) administration of Hepa1‐6‐2‐specific CD8+ CTLs (1 × 106) on a Hepa1‐6‐1 tumour growth were examined. (d) Effects of intravenous (i.v.) administration of Hepa1‐6‐2‐specific CD8+ CTLs (1 × 106) on a Hepa1‐6‐1 tumour growth were examined. Hepa1‐6‐2‐specific CD8+ CTLs were obtained from B6 mice immunized with Hepa1‐6‐2. Ovalbumin (OVA) ‐specific CD8+ CTLs were also examined as a control for i.v. administration. The diameter of the length (a) and the width (b) were measured every day until day 18 after the implantation, and the tumour volume (V) was calculated according to the formula V = ab 2/2. The data are shown as the mean ± SEM of the mice per group (n = 3).
Activation of DEC‐205+ DCs in vivo by i.p. injection of α‐GalCer in B6 mice
We recently reported that CD11c+ DEC‐205+ DCs are selectively activated in vivo through the i.p. injection of α‐GalCer in BALB/c mice.14 Based on this finding, we examined whether α‐GalCer also selectively stimulated DEC‐205+ DCs in the spleen cells of B6 mice. We prepared spleen cells from B6 mice at 6, 12, 24 and 48 hr after the i.p. injection of α‐GalCer at 2 μg/mouse and analysed the kinetics in terms of the percentage and the number of DEC‐205+ or 33D1+ cells among the CD11c+ DCs using flow cytometry and a haemocytometer. We found that among the CD11c+ DC subsets, the percentage (Fig. 2a; left panel) as well as the actual number (Fig. 2a; right panel) of DEC‐205+ DCs significantly increased to their peak value at approximately 12 hr after injection, whereas the percentage (Fig. 2a; left panel) and number (Fig. 2a; right panel) of 33D1+ DCs profoundly declined at approximately 24 hr after inoculation. We also examined the expression of CD80, CD86, CD40 and PD‐L1 on either the DEC‐205+, 33D1+ or DEC‐205− 33D1− DCs using flow cytometry. The expression of co‐stimulatory molecules, such as CD80, CD86 and CD40, on the DEC‐205+ DCs but not on the 33D1+ or the DEC‐205− 33D1− DCs was up‐regulated and reached an optimal value at approximately 24 hr before the expression began to decline 48 hr after stimulation with α‐GalCer (Fig. 2b). Moreover, IL‐12 production in the sera similarly increased and reached its maximum at approximately 6 hr and then declined 48 hr after the administration of 2 μg/mouse α‐GalCer (Fig. 2c). These data suggest that an i.p. injection of α‐GalCer selectively activated DEC‐205+ DCs to secrete IL‐12, and the expression of co‐stimulatory molecules on these cells was up‐regulated in vivo over 48 hr.
Figure 2.
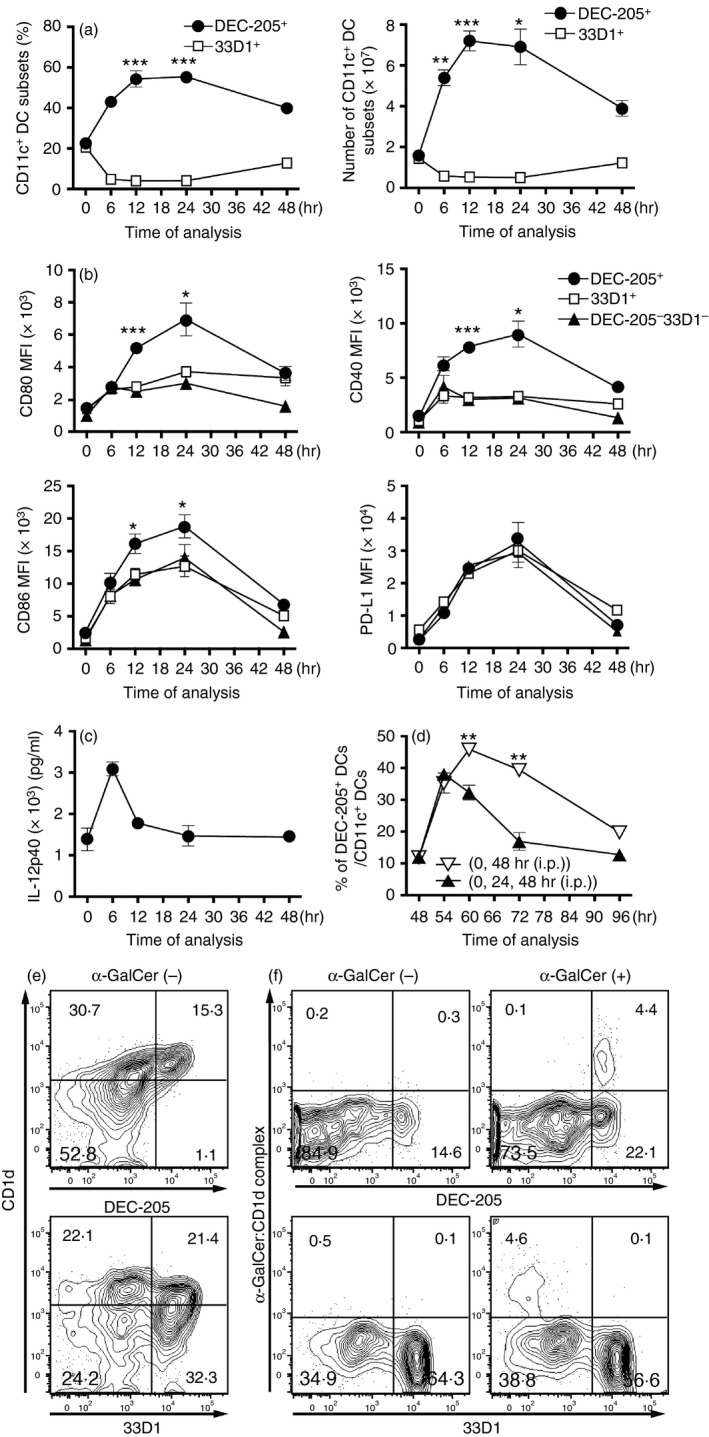
Activation of DEC‐205+ dendritic cells (DCs) in vivo by an intraperitoneal (i.p.) injection of α‐GalCer in B6 mice. (a) Spleen cells were prepared from normal B6 mice 6, 12, 24 and 48 hr after an i.p. injection with 2 μg/mouse α‐GalCer, and the kinetics of the percentage (left panel) and the number (right panel) of the DEC‐205+ or 33D1+ cells among the CD11c+ DCs were analysed by flow cytometry and haemocytometer. The data are shown as the mean ± SEM of the mice per group (n = 3), *P < 0·05, **P < 0·01 and ***P < 0·005, Student's t‐test. (b) Similarly, spleen cells were prepared from the B6 mice 6, 12, 24 and 48 hr after an i.p. injection with 2 μg/mouse α‐GalCer, and the expression of CD80, CD86, CD40 and PD‐L1 on either the DEC‐205+, the 33D1+, or the DEC‐205−33D1− DCs was analysed by flow cytometry. The data are shown as the mean ± SEM of the mice per group (n = 3), *P < 0·05, **P < 0·01 and ***P < 0·005, Student's t‐test, which compared the expression of co‐stimulatory molecules on DEC‐205+ DCs with that of 33D1+ DCs. (c) The amount of interleukin‐12p40 (IL‐12p40) secretion in the serum from B6 mice 6, 12, 24 and 48 hr after an i.p. injection with 2 μg/mouse α‐GalCer was determined by ELISA. The data are shown as the mean ± SEM of the mice per group (n = 5). (d) Spleen cells were prepared from B6 mice 6, 12, 24 and 48 hr after i.p. administration with 2 μg/mouse α‐GalCer at 0 hr, 24 hr and 48 hr (every 24 hr; closed triangle) or at 0 hr and 48 hr (every 48 hr; open triangle), and the kinetics in the percentages of DEC‐205+ cells among the CD11c+ DCs was analysed by flow cytometry. The data are shown as the mean ± SEM of the mice in each group (n = 3), **P < 0·05, Student's t‐test. (e) CD1d expression on splenic DEC‐205+ DCs and 33D1+ DCs obtained from B6 mice. (f) α‐GalCer:CD1d complex expression on splenic DEC‐205+ DCs and 33D1+ DCs obtained from B6 mice 6 hr after i.p. injection with 2 μg/mouse α‐GalCer.
We further examined the effect of boosting expression with α‐GalCer on the percentage of DEC‐205+ DCs in α‐GalCer‐immunized B6 mice. When the immunized mice were boosted with α‐GalCer 48 hr after the initial i.p. injection, the percentage of DEC‐205+ DCs similarly increased from 20% to 50% and reached its maximum at approximately 12 hr before it rapidly declined (Fig. 2d; open inverse triangle). However, the percentage of DEC‐205+ DCs in the primed mice showed a weak response at approximately 6 hr when it was boosted with α‐GalCer at both 24 and 48 hr (Fig. 2d; closed triangle). These data suggest that innate immunity, especially DEC‐205+ DCs, can be activated by stimulation at a 48‐hr, but not a 24‐hr, interval to achieve the maximum response with lipid/glycolipid α‐GalCer.
Next, we investigated the expression of the CD1d molecule and the α‐GalCer–CD1d complex on DEC‐205+ DCs as well as 33D1+ DCs by using a unique mAb (L363) that responds predominantly to the α‐GalCer–CD1d complex. The CD1d expression on DEC‐205+ DCs was far higher than that on 33D1+ DCs (Fig. 2e). To our surprise, the α‐GalCer–CD1d complex was predominantly observed on DEC‐205+ DCs (Fig. 2f). Therefore, the DEC‐205+ DCs seem to be selectively activated by α‐GalCer through the CD1d molecules on them.
Effects of the selective activation of TIDCs within the Hepa1‐6‐1 tumour mass on tumour growth by the external administration of α‐GalCer
We then investigated whether the activation of tolerogenic TIDCs within the Hepa1‐6‐1 tumour mass could suppress tumour growth in vivo through a single administration of high‐dose α‐GalCer. Hepa1‐6‐1 cells (1 × 107) were implanted s.c. into the right abdominal region of syngeneic B6 mice, and 2 hr later (day 0), the tumour‐implanted mice were treated i.p. once with either 20 μg α‐GalCer or 0·025% Polysolvate‐20 in 100 μl of PBS as a vehicle control. The growth kinetics of the Hepa1‐6‐1 tumour volume was evaluated by measuring the diameter of the length and the width daily until day 24 after the implantation. The Hepa1‐6‐1‐implanted control mice, which were given 0·025% Polysolvate‐20 in 100 μl of PBS alone, established a solid tumour mass after approximately 7 days, whereas the tumour growth in mice that received 20 μg α‐GalCer was weakly suppressed, despite the high dose of α‐GalCer (Fig. 3a).
Figure 3.

Effects of the selective activation of tumour‐infiltrating dendritic cells (TIDCs) within the Hepa1‐6‐1 tumour mass from sequential administration of α‐GalCer every other day on tumour regression. (a) To determine the effect of the selective activation of TIDCs within the Hepa1‐6‐1 tumour mass from a Day 0 single intraperitoneal (i.p.) administration of 20 μg/mouse α‐GalCer on tumour regression, the diameters of the length (a) and the width (b) of the tumours was measured every day until day 20 after the implantation, and the tumour volume (V) was calculated according to the formula V = ab 2/2. The data are shown as the mean ± SEM of the mice per group (n = 3). (b) To estimate the effect of the selective activation of TIDCs within the Hepa1‐6‐1 tumour mass from an i.p. administration of 2 μg/mouse α‐GalCer every other day on tumour regression, the diameter of the length (a) and the width (b) of the tumour was measured daily until day 24 after the implantation, and the tumour volume (V) was calculated according to the formula V = ab 2/2 . The data are shown as the mean ± SEM of the mice per group (n = 21). (c) Observation of the growth kinetics of the treated or untreated mice until day 24 after the Hepa1‐6‐1 cells implantation. Scale bars, 10 mm. (d) We measured and plotted the volume of each tumour 24 days after the implantation (n = 21). The data are shown as the mean ± SEM of mice per group (n = 21), ***P < 0·005, Student's t‐test. (e) Tumour volume in CD8+ T cell, CD4+ T cell, and natural killer (NK) cell deletion mice with an i.p. administration of 2 μg/mouse α‐GalCer every other day. For interleukin‐12 (IL‐12) treatment of mice, 100 ng of IL‐12p70 was injected i.p. every other day from day 0 to day 18. The data are shown as the mean ± SEM of the mice per group (n = 6 to n = 8). (f) Serum IL‐12 level in α‐GalCer‐administered mice. The data are shown as the mean ± SEM of the mice per group (n = 6).
As indicated in Fig. 2(a–f), we confirmed that innate DEC‐205+ DCs could be selectively activated in a 48‐hr rather than a 24‐hr cycle stimulation with 2 μg of α‐GalCer in vivo. Based on these findings, we attempted to determine the effect of 10 sequential injections of α‐GalCer at 2 μg/mouse at 48‐hr intervals beginning 2 hr after the Hepa1‐6‐1 implantation until day 18. We compared the results with those of Hepa1‐6‐1‐implanted mice that were injected with 20 μg/mouse α‐GalCer once. Although the total amount of α‐GalCer was the same, the effect of the sequential repetitive activation of the DEC‐205+ DCs with 2 μg/mouse of α‐GalCer every other day was potent (Fig 3b,c). To confirm the findings, we plotted the volume of each tumour for 24 days after implantation and confirmed the stability of the suppressive effect (Fig. 3d). These results indicate that sequential administration with a smaller amount of α‐GalCer every other day induced a more stable and potent suppressive effect on tumour growth.
Moreover, we examined the effect of depletion of either CD8+ T cells, CD4+ T cells, or NK cells on tumour growth in the sequentially α‐GalCer‐administered Hepa1‐6‐1 tumour‐implanted mice. As demonstrated in Fig. 3(e), the inhibitory effect of tumour growth by α‐GalCer‐administration was apparently abolished by CD8+ T‐cell depletion but not by CD4+ T‐cell or NK cell depletion. Therefore, the inhibitory effect of α‐GalCer‐administration requires CD8+ T cells but not CD4+ T cells or NK cells. Additionally, the effect of sequential i.p. administration of 100 ng/mouse IL‐12p70 into tumour‐implanted mice was examined and found that the tumour growth was moderately inhibited. Indeed, we detected approximately 3000 pg/ml of IL‐12 in the blood of α‐GalCer‐administered tumour‐growth‐suppressed mice (Fig. 3f) and concluded that the very small amount of IL‐12 secreted in vivo appeared to inhibit tumour growth.
Effects of sequential i.p. administration of α‐GalCer into tumour‐bearing mice every other day on the activation of CD8+ TILs and TIDCs
Next, we examined the characteristics of the TILs in the Hepa1‐6‐1 tumour mass in the α‐GalCer‐administered B6 mouse to determine whether the Hepa1‐6‐1‐specific CD8+ CTLs were primed and activated. We examined the expression of an activation marker, CD69, on the TILs using flow cytometry 7, 10 and 14 days after Hepa1‐6‐1 implantation in both α‐GalCer‐administered and control vehicle‐injected B6 mice. The CD3+ TILs in the α‐GalCer‐administered B6 mice were significantly activated with enhanced CD69 expression (Fig. 4a; panels in the first lane). In particular, the percentage of CD8+ TILs significantly increased by α‐GalCer administration (Fig. 4a; panels in the second lane). Moreover, CD8+ T cells bearing enhanced CD69 expression in the TILs were also strongly activated (Fig. 4a; panels in the third lane). Conversely, the CD69 expression on NK1.1+ NK cells in the TILs was almost unchanged by the α‐GalCer injection (Fig. 4a; panels in the fourth lane). These findings suggest that ongoing tumour growth can be suppressed by activated CD8+ CTLs with tumour‐specific cytotoxicity through sequential i.p. administration of α‐GalCer every other day.
Figure 4.

Effect of the sequential intraperitoneal (i.p.) administration of α‐GalCer into the Hepa1‐6‐1 tumour‐bearing mice on the activation of CD8+ tumour‐infiltrating lymphocytes (TILs) and tumour‐infiltrating dendritic cells (TIDCs). (a) CD69 expression on the TILs of the α‐GalCer‐treated B6 mice was analysed by flow cytometry 7, 10 and 14 days after the Hepa1‐6‐1 cell implantation. CD69 expression on TILs (first lane), the percentage of CD8+ T cells (second lane), CD69 expression on CD8+ T cells (third lane), and the CD69 expression on NK1.1+ NK cells among TILs (fourth lane) were examined. The data are representative of n = 4 mice per group. (b and c) Administration of α‐GalCer to the tumour‐bearing mice altered the level of co‐stimulatory molecule expression on the TIDCs. The phenotype and expression of the co‐stimulatory molecules on the TIDCs in the tumour‐bearing mice treated with α‐GalCer were examined. The expression of the co‐stimulatory molecules CD80 and CD86 on the DEC205+ CD11c+ MHC‐II+ TIDCs or 33D1+ CD11c+ MHC‐II+ TIDCs from the B6 mice injected with/without α‐GalCer 10 days after the Hepa1‐6‐1 cell implantation was analysed by flow cytometry. The data are shown as the mean + SEM of the mice per group (n = 4), *P < 0·05, Student's t‐test. n.s., not significant.
We then confirmed that the selective activation of DEC‐205+ DCs in vivo could be achieved by an i.p. injection of α‐GalCer in B6 mice (Fig. 2b). Hence, we examined the phenotype and expression of co‐stimulatory molecules on the TIDCs in the sequentially α‐GalCer‐administered B6 mice 10 days after Hepa1‐6‐1 implantation by flow cytometry. Enhanced expression of CD80 and CD86 on the CD11c+ class II+ DEC‐205+ TIDCs in the α‐GalCer‐treated mice (Fig. 4b) but not in the CD11c+ 33D1+ TIDCs was apparently observed (Fig. 4c). These findings suggest the possibility that sequential i.p. administration of α‐GalCer prevents the induction of tolerogenic DEC‐205+ DCs or converts them into immunogenic DCs expressing higher amounts of CD80/86 that may specifically prime CTL precursors.
Comparison of the number of iNKT cells between α‐GalCer‐treated mice and untreated mice
α‐GalCer is also known as a compound that specifically stimulates iNKT cells via CD1d on the DEC‐205+ DCs. To confirm whether the iNKT cells were actually activated within the TILs, the number and activation of iNKT cells in the spleen, as well as in the TILs, of the Hepa1‐6‐1‐implanted mice treated repetitively with α‐GalCer, were examined 10 days after tumour implantation. Compared with that in the mice not administered α‐GalCer, no significant differences in the number of α‐GalCer‐specific CD1d‐restricted iNKT cells (Fig. 5a) and CD69+‐activated iNKT cells (Fig. 5b) were observed. Moreover, we examined whether the α‐GalCer treatment could suppress growth of tumoir in iNKT KO mice. As demonstrated in Fig. 5(c), tumour growth was also markedly inhibited in iNKT‐deficient Jα18–/– (Jα18 KO) mice19 with sequential i.p. administration of 2 μg/mouse α‐GalCer every other day, indicating that the α‐GalCer‐activated CD8+ T cells but not iNKT cells may be the actual effector cells for the anti‐tumour reaction iNKT KO mice. These findings suggest that the major effectors for suppressing Hepa1‐6‐1 growth were not the activated iNKT cells but rather the activated tumour‐specific CD8+ CTLs in the tumour‐bearing mice treated with α‐GalCer.
Figure 5.
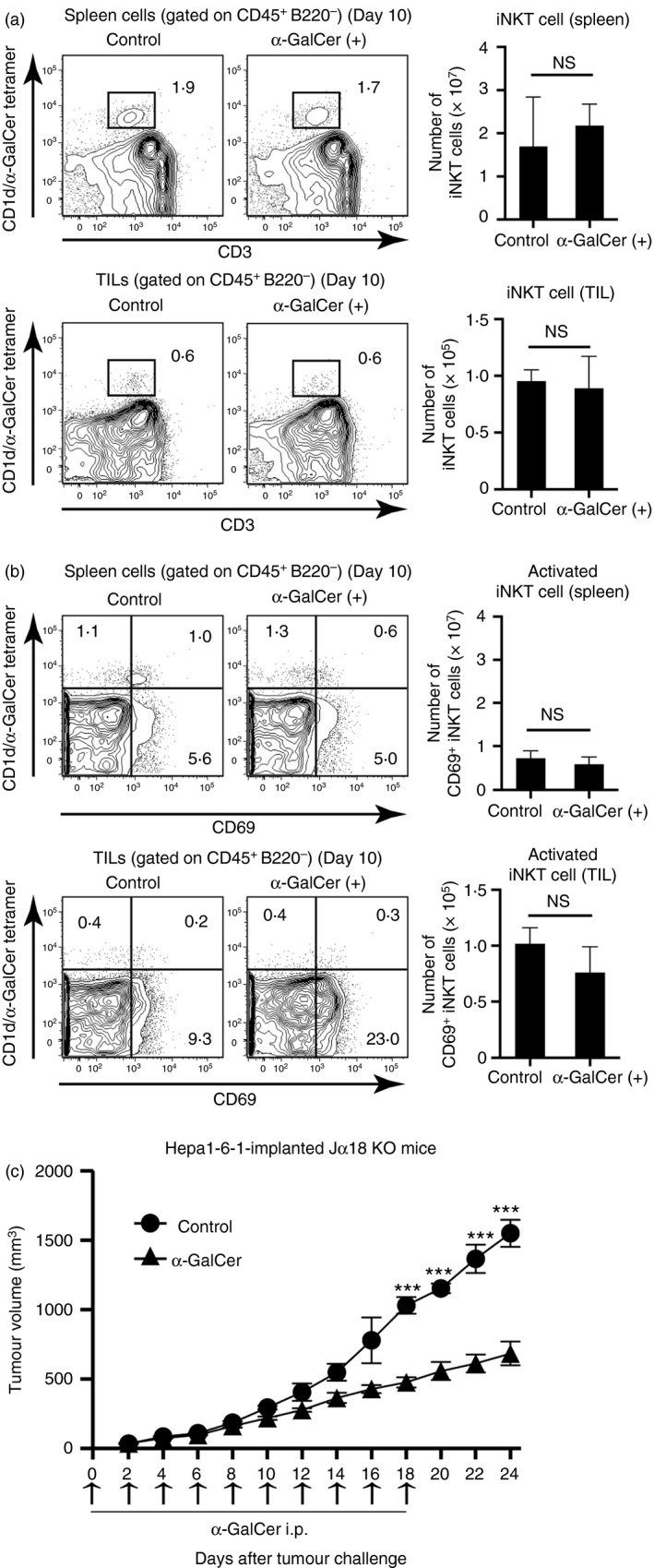
Comparison of the number and activation of invariant natural killer T (iNKT) cells between the α‐GalCer‐treated and untreated mice. (a) The percentage of CD1d/α‐GalCer tetramer+ CD3+ cells in the spleen and the tumour‐infiltrating lymphocytes (TILs) from B6 mice injected with/without α‐GalCer 10 days after Hepa1‐6‐1 cell implantation were analysed by flow cytometry. The data are shown as the mean + SEM of the mice per group (n = 4), Student's t‐test. (b) The percentage of CD69+ iNKT cells in the spleen or in the TILs of the mice treated with α‐GalCer was analysed by flow cytometry. The data are shown as the mean + SEM of the mice per group (n = 4), Student's t‐test. n.s., not significant. (c) Tumour volume in iNKT‐deficient Jα18(–/–) (Jα18 KO) mice15 with sequential intraperitoneal (i.p.) administration of 2 μg/mouse α‐GalCer every other day. Hepa1‐6‐1 tumour cells were implanted on day 0. The data are shown as the mean ± SEM of the mice per group (n = 5), ***P < 0·005, Student's t‐test.
Sequential administration of α‐GalCer in vivo appears to induce priming and activation of Hepa1‐6‐1‐specific CD8+ CTLs
As indicated above, the sequential administration of α‐GalCer every 48 hr in vivo appeared to convert tolerogenic DEC‐205+ DCs into immunogenic DCs that will prime tumour‐specific CTL precursors, which may be activated by neighbouring immunogenic DCs that express higher amounts of CD80/86 within the Hepa‐1‐6‐1 tumour mass. To examine this possibility, we performed a standard chromium‐51‐release assay to determine whether cytotoxicity of the primed CD8+ CTLs within the spleen of the α‐GalCer‐treated Hepa‐1‐6‐1‐bearing mice was observed. We observed specific cytotoxicity against Hepa1‐6‐1 tumour cells when the primed spleen cells were re‐stimulated in vitro with Hepa1‐6‐1 cells in the Hepa1‐6‐1‐implanted B6 mice treated with repetitive α‐GalCer but not in the untreated mice (Fig. 6a). The cytotoxicity may be initiated through the cross‐presenting capacity of the converted immunogenic DEC‐205+ DCs that presented captured tumour antigen(s) in association with class I MHC molecules and co‐stimulation molecules for priming virgin splenic T cells.
Figure 6.

Sequential administration of α‐GalCer in vivo every other day induced the priming of Hepa1‐6‐1‐specific CD8+ cytotoxic T lymphocytes (CTLs) and tumour‐infiltrating lymphocytes (TILs). (a) Four million spleen cells from sequentially α‐GalCer‐treated mice implanted with Hepa‐1‐6‐1 cells 25 days before were re‐stimulated in vitro, and a standard chromium‐51‐release assay was performed. The data are shown as the mean ± SEM of the cells per group (n = 4). (b) CFSE‐labelled splenic CD8+ T cells from the primed mice were stimulated for 4 days with complete culture medium alone, with tumour‐infiltrating dendritic cells (TIDCs) from untreated mice 20 days after Hepa1‐6‐1 cell implantation, or with TIDCs from α‐GalCer‐treated mice 20 days after Hepa1‐6‐1 cell implantation. The co‐cultured cells were then harvested and analysed to determine the number of cell divisions using flow cytometry. The data are representative of n = 3 mice per group. (c) The ex vivo cytotoxic activity of the CD8+ TILs within the Hepa1‐6‐1‐derived tumour mass of the α‐GalCer‐treated mice implanted with Hepa1‐6‐1 cells 24 days before was determined by a standard chromium‐51‐release assay. The data are shown as the mean + SEM of the number of cells per group (n = 3).
To confirm this possibility, we examined the cross‐presenting capacity of the DEC‐205+ TIDCs in the Hepa‐1‐6‐1‐bearing mice that were repetitively treated with 2 μg/mouse α‐GalCer every other day until day 18. The CD11c+ TIDCs were enriched from a Hepa1‐6‐1 tumour mass using a magnetic separation kit and were co‐cultured with CFSE‐labelled primed splenic T cells from mice pre‐immunized i.p. with Hepa1‐6‐2 cells 4 days previously. Then, the co‐cultured cells were harvested and analysed by flow cytometry. The Hepa1‐6‐2‐primed splenic T cells were apparently activated when the cells were stimulated with the TIDCs from the α‐GalCer‐treated mice implanted with Hepa1‐6‐1. In contrast, the primed T cells were not significantly activated when the cells were stimulated with the control TIDCs from mice treated with vehicle alone (Fig. 6b). Hence, we confirmed that the TIDCs from the Hepa1‐6‐1 tumour‐bearing mice that were repetitively treated with α‐GalCer gained the ability to specifically activate primed splenic T cells. These findings suggest that the tolerogenic TIDCs within the Hepa1‐6‐1 tumour mass might be converted into immunogenic DCs, which had the ability to prime virgin T cells.
We showed here that α‐GalCer initiated the internal activation of tolerogenic DCs within the tumours and the activated tolerogenic DCs gained a suppressive ability against tumour growth through priming and activation of tumour‐specific CD8+ CTLs. Therefore, we examined whether the TILs within the tumour mass showed Hepa1‐6‐1 tumour cell‐specific cytotoxicity ex vivo when α‐GalCer was sequentially administered to the mice. The specific cytotoxicity against the Hepa1‐6‐1 tumour cells was detected in the TILs. However, the TILs in the vehicle‐treated control mice bearing Hepa1‐6‐1 showed no cytotoxicity (Fig. 6c). These findings suggest that the sequential administration of α‐GalCer to the Hepa1‐6‐1‐bearing mice induced tumour‐specific CD8+ CTLs within the tumour mass through converting tolerogenic DEC‐205+ TIDCs into immunogenic DCs that gained the ability to cross‐present and had augmented co‐stimulatory molecules.
Effects of α‐GalCer on converting tolerogenic DEC‐205+ DCs induced by Hepa1‐6‐1 tumour cells into immunogenic DCs in vitro
Finally, we wished to determine whether tolerogenic DEC‐205+ DCs expressing down‐regulated co‐stimulation molecules could be converted into immunogenic cells by treating them with α‐GalCer. BM‐derived DCs were incubated with Hepa‐1‐6‐1 cells in the trans‐well system, which is a system in which the DCs can only be reached by humoral factors secreted from Hepa‐1‐6‐1 tumours. When the DCs were incubated in the culture supernatant of Hepa1‐6‐1 cells for 5 days, the expression of CD86 co‐stimulatory molecules on the DEC‐205+ DCs was markedly down‐regulated, indicating that the cells became tolerogenic (Fig. 7a). However, when the tolerated DCs were treated with 100 ng/ml of α‐GalCer in the presence of Hepa1‐6‐1 cells using a trans‐well system for an additional 24 hr, CD86 expression on the DEC‐205+ DCs was almost completely recovered to a normal or slightly enhanced level (Fig. 7b). Moreover, these recovered DCs treated with α‐GalCer gained the ability to stimulate Hepa1‐6‐2‐primed splenic T cells (Fig. 7c). Furthermore, we examined whether α‐GalCer treatment could induce priming of naive T cells. Here, we found that, when splenic naive T cells from normal B6 mice were co‐cultured with TIDCs from Hepa1‐6‐1‐bearing mice given i.p. injections of 2 μg/mouse α‐GalCer every other day for 14 days, the CD44− CD62L+ naive T cells were converted into the CD44+ CD62L− memory T cells (Fig. 7d) and the converted memory T cells secreted interferon‐γ when stimulated with Hepa1‐6‐1 cells for a further 48 hr (Fig. 7e). Collectively, sequential administration of α‐GalCer into the tumour‐implanted mice not only converted the tolerogenic DCs into immunogenic cells but also induced the priming of naive T cells into tumour‐specific CD8+ CTLs together with providing DCs with the cross‐presenting ability of captured tumour antigens.
Figure 7.
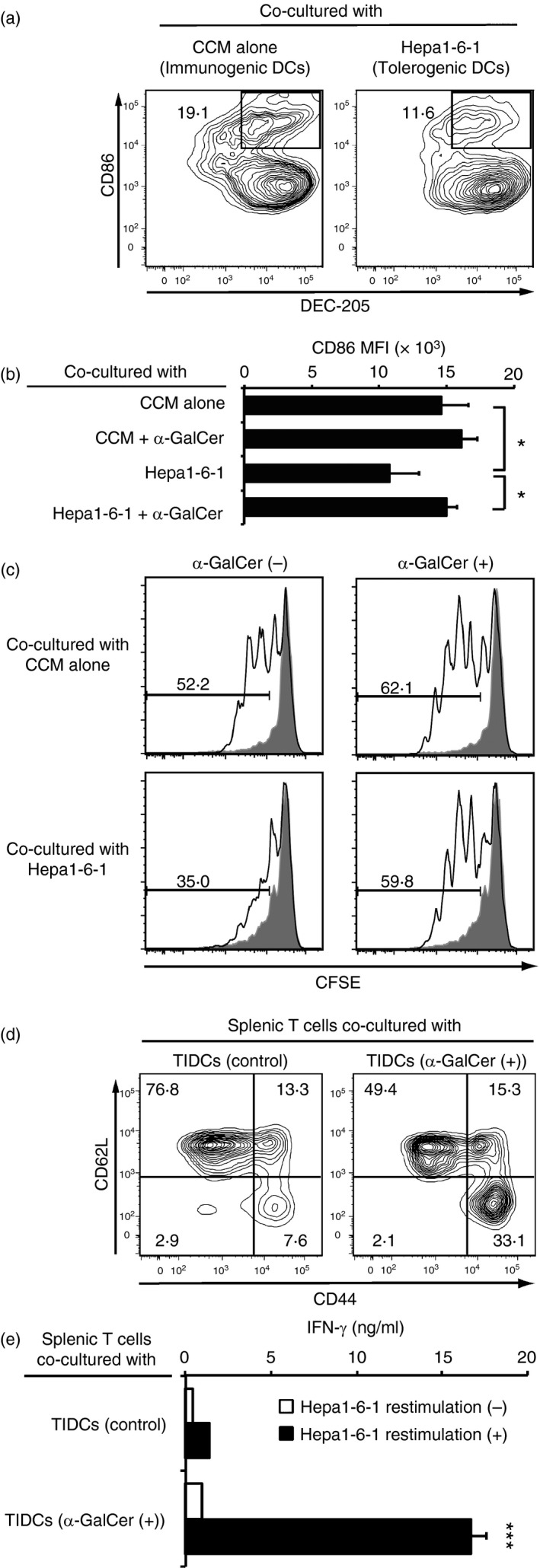
Effects of α‐GalCer on converting tolerogenic DEC‐205+ DCs+ induced by Hepa1‐6‐1 tumour cells into immunogenic dendritic cells (DCs) in vitro. (a) The DCs were incubated with Hepa1‐6‐1 cells for 5 days in the trans‐well system and their expression of CD86 was analysed by flow cytometry. The data are representative of n = 3 mice per group. (b) Those tolerated DCs were treated with complete culture medium (CCM) containing 100 ng/ml α‐GalCer in the presence of Hepa1‐6‐1 cells using a trans‐well system for an additional 24 hr, and their expression of CD86 was analysed by flow cytometry. The data are shown as the mean + SEM of the cells per group (n = 3), *P < 0·05, Student's t‐test. (c) 5 × 104 of α‐GalCer‐treated recovered DCs were re‐stimulated with the lysate of 5 × 103 Hepa1‐6‐1 cells and 100 ng/ml α‐GalCer for an additional 48 hr and the antigen‐loaded DCs were washed extensively to remove the free tumour lysate. Then the cells were co‐cultured with 1 × 105 Hepa1‐6‐2‐primed splenic CD8+ T cells labelled with CFSE for 4 days after and analysed by flow cytometry. Shaded portions indicate the number of division of primed T cells co‐cultured with antigen unloaded DCs. The data are representative of n = 3 mice per group. (d) 5 × 105 splenic naive T cells from normal B6 mice were co‐cultured with 1 × 105 TIDCs from Hepa1‐6‐1‐bearing mice injected intraperitoneally (i.p.) with 2 μg/mouse α‐GalCer every other day for 14 days, and their surface expression of CD44 and CD62L was examined by flow cytometry. (e) The amount of secreted interferon‐γ (IFN‐γ) after stimulation with Hepa1‐6‐1 cells for a further 48 hr was measured by ELISA. The data are shown as the mean + SEM of the cells per group (n = 3), ***P < 0·005, Student's t‐test.
Discussion
In the present study, we showed that sequential repetitive i.p. administration of α‐GalCer every 48 hr in hepatoma‐cell‐bearing mice initiated tumour‐specific CD8+ CTLs among the TILs in vivo. α‐GalCer is a glycolipid associated with CD1d molecules that can activate iNKT cells.17, 18 Indeed, in cases of human myeloma or glioma cells, α‐GalCer‐activated iNKT cells recognize endogenous glycolipids in these tumour cells and eliminate them.22 Additionally, the targeted delivery of α‐GalCer to DEC‐205+ DCs optimized the Type I NKT cell‐based anti‐tumour response.23 However, as shown in this study, the number of CD69+‐activated iNKT cells was not increased in mice that were repeatedly treated with α‐GalCer (Fig. 5a). This outcome may have occurred because the frequent stimulation of innate iNKT cells with α‐GalCer drives them into anergy rather than activation. Indeed, it was reported that sequential administration of α‐GalCer resulted in iNKT cells that were unresponsive and drove them into an anergic state.24 Therefore, the suppressive effect of sequential administration with α‐GalCer on Hepa1‐6‐1 tumour growth was not correlated with iNKT cells but rather with other immune effectors activated by α‐GalCer. In addition, as demonstrated here, sequential administration with α‐GalCer inhibited Hepa1‐6‐1 tumour growth in vivo even in the iNKT‐deficient mice. Additionally, in vivo suppression of Hepa1‐6‐1 growth appears to be mediated by CD8+ CTLs. Moreover, It should be noted that only a small amount of IL‐12 (approximately 3000 pg/ml) was detected in the sera of α‐GalCer‐administered tumour‐regressed mice, indicating that a small amount of IL‐12 seems to be sufficient in the DEC‐205+ DC‐activated state for tumour‐growth suppression.
In the tumour microenvironment, cancer cells secrete a variety of immunosuppressive factors. As we previously reported,11 Hepa1‐6‐1 cells secrete various humoral factors, such as α‐fetoprotein, transforming growth factor‐β and vascular endothelial growth factor, which down‐regulate the co‐stimulatory molecules on DCs. Hence, the TIDCs near Hepa1‐6‐1 cells might become immunosuppressive tolerogenic DCs with down‐regulated co‐stimulatory molecules. We speculated whether those tolerogenic TIDCs within the Hepa1‐6‐1 tumour mass might be able to cancel the effect of externally administered CD8+ CTLs that have the capacity to specifically eliminate Hepa1‐6‐1 cells. We confirmed that the cytotoxicity of CTLs and the effect of TIDC2 were completely abrogated. Taking all of the data into consideration, we hypothesized that the internal activation of immunosuppressive tolerogenic DCs within the tumours would inhibit tumour growth.
In our previous study, we speculated that tolerogenic DCs within the tumour mass could be converted into functional immunogenic DCs through the activation of DEC‐205+ DCs.11 Additionally, as we recently demonstrated, the selective activation of the DEC‐205+ DC subset might activate tumour‐specific CD8+ CTLs without additional tumour–antigen stimulation in vivo.19, 25 In this study, we demonstrate for the first time that the sequential administration of α‐GalCer every 48 hr might convert tolerogenic DCs within the tumour mass into immunogenic DEC‐205+ DCs, which have an enhanced expression of co‐stimulation molecules and a cross‐presenting capacity that might prime and activate naive T cells within the tumour mass.
The findings presented here may provide a new basis for cancer immunotherapy to convert immunosuppressive tolerogenic DCs within tumours into immunogenic DCs through sequential external administration of an immunopotent lipid/glycolipid, such as α‐GalCer, and activated immunogenic DCs with sufficient expression of co‐stimulatory molecules can then prime and activate tumour‐specific CD8+ CTLs within the tumour mass to control tumour growth.
Disclosure
The authors declare no financial conflicts of interest.
Supporting information
Figure S1 Representative gating strategies for DEC‐205+ dendritic cells (DCs), 33D1+ DCs, CD3+ T cells, natural killer (NK) cells, and invariant natural killer T (iNKT) cells as well as co‐stimulatory molecules (CD80 and CD86) and activation marker CD69 on these cells with controls in (a) tumour tissue and (b) spleen.
Acknowledgements
H.K., M.S. and Y.N. performed the experiments, E.U. and H.T. assisted with the experiments, H.T. designed the study, and H.K. and H.T. wrote the paper. This study was supported in part by grants from the Ministry of Education, Science, Sport, and Culture, the Ministry of Health and Labor and Welfare, Japan (25461715 to H.T.); the Japanese Health Sciences Foundation; the Promotion and Mutual Aid Corporation for Private Schools of Japan; and from a MEXT‐supported Programme for the Strategic Research Foundation at Private Universities, Japan.
References
- 1. Buller RM, Holmes KL, Hugin A, Frederickson TN, Morse HC 3rd. Induction of cytotoxic T‐cell responses in vivo in the absence of CD4 helper cells. Nature 1987; 328:77–9. [DOI] [PubMed] [Google Scholar]
- 2. Germain RN. MHC‐dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell 1994; 76:287–99. [DOI] [PubMed] [Google Scholar]
- 3. Takahashi H. Antigen presentation in vaccine development. Comp Immunol Microbiol Infect Dis 2003; 26:309–28. [DOI] [PubMed] [Google Scholar]
- 4. Takahashi H, Takeshita T, Morein B, Putney S, Germain RN, Berzofsky JA. Induction of CD8+ cytotoxic T cells by immunization with purified HIV‐1 envelope protein in ISCOMs. Nature 1990; 344:873–5. [DOI] [PubMed] [Google Scholar]
- 5. Takahashi H, Nakagawa Y, Yokomuro K, Berzofsky JA. Induction of CD8+ cytotoxic T lymphocytes by immunization with syngeneic irradiated HIV‐1 envelope derived peptide‐pulsed dendritic cells. Int Immunol 1993; 5:849–57. [DOI] [PubMed] [Google Scholar]
- 6. Steinman RM. Dendritic cells and the control of immunity: enhancing the efficiency of antigen presentation. Mt Sinai J Med 2001; 68:160–6. [PubMed] [Google Scholar]
- 7. Bozzacco L, Trumpfheller C, Siegal FP, Mehandru S, Markowitz M, Carrington M et al DEC‐205 receptor on dendritic cells mediates presentation of HIV gag protein to CD8+ T cells in a spectrum of human MHC I haplotypes. Proc Natl Acad Sci USA 2007; 104:1289–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S et al Differential antigen processing by dendritic cell subsets in vivo . Science 2007; 315:107–11. [DOI] [PubMed] [Google Scholar]
- 9. Bozzacco L, Trumpfheller C, Huang Y, Longhi MP, Shimeliovich I, Schauer JD et al HIV gag protein is efficiently cross‐presented when targeted with an antibody towards the DEC‐205 receptor in Flt3 ligand‐mobilized murine DC. Eur J Immunol 2010; 40:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li YL, Wu YG, Wang YQ, Li Z, Wang RC, Wang L et al Bone marrow‐derived dendritic cells pulsed with tumor lysates induce anti‐tumor immunity against gastric cancer ex vivo . World J Gastroenterol 2008; 14:7127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harimoto H, Shimizu M, Nakagawa Y, Nakatsuka K, Wakabayashi A, Sakamoto C et al Inactivation of tumor‐specific CD8+ CTLs by tumor‐infiltrating tolerogenic dendritic cells. Immunol Cell Biol 2013; 91:545–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ochsenbein AF, Sierro S, Odermatt B, Pericin M, Karrer U, Hermans J et al Roles of tumour localization, second signals and cross priming in cytotoxic T‐cell induction. Nature 2001; 411:1058–64. [DOI] [PubMed] [Google Scholar]
- 13. Nakatsuka K, Sugiyama H, Nakagawa Y, Takahashi H. Purification of antigenic peptide from murine hepatoma cells recognized by Class‐I major histocompatibility complex molecule‐restricted cytotoxic T‐lymphocytes induced with B7‐1‐gene‐transfected hepatoma cells. J Hepatol 1999; 30:1119–29. [DOI] [PubMed] [Google Scholar]
- 14. Murakami R, Nakagawa Y, Shimizu M, Wakabayashi A, Negishi Y, Hiroi T et al Effects of dendritic cell subset manipulation on airway allergy in a mouse model. Int Arch Allergy Immunol 2015; 168:219–32. [DOI] [PubMed] [Google Scholar]
- 15. Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I et al Requirement for Vα14 NKT cells in IL‐12‐mediated rejection of tumors. Science 1997; 278:1623–6. [DOI] [PubMed] [Google Scholar]
- 16. Takahashi H, Nakagawa Y, Leggatt GR, Ishida Y, Saito T, Yokomuro K et al Inactivation of human immunodeficiency virus (HIV)‐1 envelope‐specific CD8+ cytotoxic T lymphocytes by free antigenic peptide: a self‐veto mechanism? J Exp Med 1996; 183:879–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Taniguchi M, Harada M, Kojo S, Nakayama T, Wakao H. The regulatory role of Vα14 NKT cells in innate and acquired immune response. Annu Rev Immunol 2003; 21:483–513. [DOI] [PubMed] [Google Scholar]
- 18. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K et al CD1d‐restricted and TCR‐mediated activation of vα14 NKT cells by glycosylceramides. Science 1997; 278:1626–9. [DOI] [PubMed] [Google Scholar]
- 19. Ichikawa T, Negishi Y, Shimizu M, Takeshita T, Takahashi H. α‐Galactosylceramide‐activated murine NK1.1+ invariant‐NKT cells in the myometrium induce miscarriages in mice. Eur J Immunol 2016; 46:1867–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takahashi H, Cease KB, Berzofsky JA. Identification of proteases that process distinct epitopes on the same protein. J Immunol 1989; 142:2221–9. [PubMed] [Google Scholar]
- 21. Ishii R, Shimizu M, Nakagawa Y, Shimizu K, Tanaka S, Takahashi H. In vivo priming of natural killer T cells by dendritic cells pulsed with hepatoma‐derived acid‐eluted substances. Cancer Immunol Immunother 2004; 53:383–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dhodapkar KM, Cirignano B, Chamian F, Zagzag D, Miller DC, Finlay JL et al Invariant natural killer T cells are preserved in patients with glioma and exhibit antitumor lytic activity following dendritic cell‐mediated expansion. Int J Cancer 2004; 109:893–9. [DOI] [PubMed] [Google Scholar]
- 23. Macho‐Fernandez E, Cruz LJ, Ghinnagow R, Fontaine J, Bialecki E, Frisch B et al Targeted delivery of α‐galactosylceramide to CD8α + dendritic cells optimizes type I NKT cell‐based antitumor responses. J Immunol 2014; 193:961–9. [DOI] [PubMed] [Google Scholar]
- 24. Sullivan BA, Kronenberg M. Activation or anergy: NKT cells are stunned by α‐galactosylceramide. J Clin Invest 2005; 115:2328–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moriya K, Wakabayashi A, Shimizu M, Tamura H, Dan K, Takahashi H. Induction of tumor‐specific acquired immunity against already established tumors by selective stimulation of innate DEC‐205+ dendritic cells. Cancer Immunol Immunother 2010; 59:1083–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Representative gating strategies for DEC‐205+ dendritic cells (DCs), 33D1+ DCs, CD3+ T cells, natural killer (NK) cells, and invariant natural killer T (iNKT) cells as well as co‐stimulatory molecules (CD80 and CD86) and activation marker CD69 on these cells with controls in (a) tumour tissue and (b) spleen.