

Summary
MHC class I‐related gene protein (MR1) is a non‐polymorphic MHC class IB antigen‐presenting molecule that is the restricting molecule for mucosal‐associated invariant T (MAIT) cells, a prominent population of innate‐like antibacterial T cells. The MAIT cell–MR1 axis represents a new paradigm in antigen presentation, with the MR1 ligand derived from vitamin B compounds or their metabolic precursors. Many bacteria and some fungi produce the activating ligand for MR1. In evolution, MR1 is highly conserved in most, but not all, mammals. In humans and rodents it is expressed in a broad range of cell types, both haematopoietic and non‐haematopoietic, although cell surface expression has been difficult to detect. Although MR1 trafficking shares features with both the MHC class I and MHC class II pathways, it is distinct. Several strands of evidence suggest that the intracellular location where MR1 is loaded differs for soluble ligand and for ligand derived from intact bacteria. The regulation of MR1 surface expression may also vary between different cell types. This paper will review what is currently known about the expression and trafficking of MR1 and propose a model for the loading and trafficking of MR1.
Keywords: antigen presentation/processing, bacterial, T cells
Introduction
MHC class I‐related gene protein (MR1) is an evolutionarily conserved, non‐polymorphic MHC class IB antigen‐presenting molecule that is required for the ontogeny and T‐cell receptor (TCR) ‐mediated activation of mucosal‐associated invariant T (MAIT) cells, a prominent population of anti‐bacterial innate‐like T cells.1 MR1 covalently binds unstable pyrimidine intermediates and presents them to the MAIT cell TCR.2, 3 These pyrimidines, 5‐(2‐oxoethylideneamino)6‐d‐ribitylaminouracil (5‐OE‐RU) and 5‐(2‐oxopropylideneamino)‐6‐d‐ribitylaminouracil (5‐OP‐RU), are formed by the non‐enzymatic condensation of 5‐amino‐6‐d‐ribitylaminouracil (5‐A‐RU), an intermediate in the biosynthesis of riboflavin, with glyoxal or methylglyoxal, respectively products of other metabolic pathways.3 A diverse range of bacteria and fungi, but not mammalian cells, are able to synthesize riboflavin, and hence provide the ligand for MR1.2, 4 Therefore MR1 and the MAIT cell TCR together form a pattern recognition system for riboflavin biosynthesis, and 5‐A‐RU is a pathogen‐associated molecular pattern.
MR1 is highly conserved in mammals
MR1 is highly conserved in most mammals, including humans, non‐human primates, mice, rats, hamsters, guinea pigs, cows and several marsupials, including the Tasmanian devil (Sarcophilus harrisii), grey short‐tailed opossum (Monodelphis domestica) and the tammar wallaby (Macropus eugenii).5, 6, 7, 8, 9 It is, however, lacking from carnivores, rabbits and the armadillo.10 There is 90% and 89% homology of the α1 and α2 chains between mice and humans, much higher than for other MHC class I related genes, whereas the homology of the α3 domain, which is not involved in the antigen‐binding region, is lower (73%).2, 8, 9 The similarity is also high between α1 and α2 domains in cattle and mice (81%) and humans (85%).11
Similar to MHC class I proteins, MR1 has conserved cysteine residues and β 2‐microglobulin (β 2m)and CD8 contact sites;5 in humans, interaction with CD8 appears to be important for activation of CD8+ Mycobacterium tuberculosis‐reactive MAIT cells.12 Of note, however, the cytoplasmic tail of MR1 is longer in rodents than in primates and contains a conserved motif that could be associated with endocytosis, suggesting that there may be differences in MR1 trafficking between species.13
MR1 is encoded outside the MHC region. In humans it is found on chromosome 1q25.3, in close proximity to the CD1 genes,6 whereas in mice and in rats it is found in homologous segments (chromosome 1 and chromosome 13, respectively).5, 9 In humans, MR1 has been shown to be non‐polymorphic. In a study of 10 Africans, five Asians and five Caucasians, the MR1 sequence was identical in all but one individual who had two synonymous mutations.14
MR1 mRNA is widely expressed
The expression of MR1 mRNA has been assessed in different tissues and cell lines in several different species. In rats, MR1 expression was seen in homogenates of thymus, liver, spleen, lung, kidney, testis, brain and heart.5 Similarly, MR1 mRNA has been demonstrated in liver, spleen, lung, kidney, testis, brain, heart and skeletal muscle in mice,8 and in placenta, lung, liver, kidney, spleen, thymus, prostate, testis, ovary, small intestine, colon and peripheral blood leucocytes in humans.6 It is not possible, however, to tell from these studies which cell types within tissues express MR1, nor whether expression is restricted to haematopoietic cells. One recent study has examined the expression of MR1 mRNA by cells in the mouse thymus. MR1 mRNA was found to be highly expressed in double‐positive thymocytes, with levels of expression 25‐fold higher than in single‐positive or double‐negative thymocytes or in thymic epithelium.15 Although there are no other studies that assess MR1 mRNA expression in purified primary cells, it has been examined in cell lines. MR1 mRNA expression has been demonstrated in haematopoietic cell lines of the T‐cell (Jurkat) and B‐cell (Molt‐4, Raji) lineages and in multiple non‐haematopoietic cell lines of mesenchymal (HT‐1080, SK‐LMS‐1, U‐373) and epithelial (HeLa and HT‐29) origin.8
In addition to the 1263 bp long mRNA that transcribes the 341 amino acid MR1 protein (also known as MR1A), several other MR1 transcripts have been described in multiple tissues.5, 6 All of these transcripts can also be detected in multiple human cell lines of different lineages,8 as well as multiple mouse and rat tissues.5, 8 Although the 1263‐bp transcript (MR1A) encodes the 341 amino acid MR1, three alternative transcripts (MR1B, MR1C and MR1D) encode proteins that lack the α3 domain. The MR1C (1578 bp) transcript contains a stop codon after the α2 domain, and the encoded protein is predicted to be soluble. In contrast MR1B (776 bp) and MR1D (2046 bp) retain the transmembrane domain and the encoded proteins are predicted to be membrane bound.8 Three predominant MR1 mRNA transcripts are also seen in cows: a full‐length 1100‐bp product and two splice variants (900 and 550 bp). In sheep there is no 550‐bp transcript. The 900‐bp splice variant lacks the α1 exon and the 550‐bp transcript lacks the α1 and α3 exons.11 The function of the alternative MR1 splice variants (MR1B‐D) is only beginning to be addressed and will not be discussed further in this review.16
MR1 protein expression
Several studies have assessed the expression of MR1 protein. Using anti‐MR1 antibody 26.5, which only recognizes MR1 in a folded (presumed to be ligand‐bound) form,17 surface expression of MR1 was seen at 37° on some (C1R, L721.221) but not all (JY) B‐cell‐derived cell lines, to a lesser degree on some (Jurkat and SupT1) but not all (Molt4, Peer) T‐cell lines, and was absent from myeloid (U937, HL‐60, K562, NB4) and epithelial cell lines (Caco2, T84).18 Using antibody 12.2, which also only recognizes MR1 in a folded form,17 MR1 expression was seen on the L721.221 B‐cell line and Jurkat and SupT1 T‐cell lines, and at low levels on C1R and JY B‐cell lines.19 Of note, increased surface expression of MR1 was seen at 26° in some cell lines (SupT1 and Jurkat), whereas it became detectable in others (JY, Molt4, Peer), suggesting that at lower temperature MR1 that is unloaded or bound to low‐affinity ligand may be expressed at the surface.20 MR1 has also been detected at the surface of the human bronchial epithelial cell line, BEAS‐2B.21, 22 Therefore, surface expression of MR1 seems to differ between different human cell types.
In mice, a study using the 4E3 antibody clone, which recognizes a linear epitope in murine MR1, found no surface expression of MR1 on splenocytes, thymocytes and several murine cell lines by flow cytometry.23 Furthermore, despite being able to detect transfected MR1, MR1 could not be demonstrated in cell lysates from non‐transfected cell lines or primary tissues by Western blot with either 4E3 or antisera raised against the C‐terminal peptide of murine MR1.23, 24 However, following immunoprecipitation with the 26.5 anti‐MR1 antibody, MR1 could be detected by Western blot with antibody 4E3 in the cell lysates of a range of murine cell lines (WT3 embryonic fibroblasts, F5M and DC2.4 dendritic cells, CH27 B cells, P815 mast cells and the CMT 64.5 lung carcinoma cell line).25 Expression was also seen in a wide variety of mouse tissues. Therefore, although MR1 protein appears to be expressed in a wide range of murine tissues and cell lines, it is expressed at low levels. In cows, no MR1 could be detected in blood, thymus, spleen, colon, ileum or lymph node using an anti‐human MR1 antibody clone 20 that recognized the bovine MR1 on transfected cells.11, 17
Surface expression of MR1 has also been difficult to detect on isolated primary human cells by flow cytometry. Surface expression of MR1 was not seen on primary peripheral blood leucocytes, monocyte‐derived macrophages or dendritic cells using conformation‐dependent monoclonal antibodies, 12.2 and 26.5, or a rabbit polyclonal serum, RAMRN‐2,19 although in other studies, small amounts of MR1 were detected at the surface of dendritic cells and B cells by flow cytometry.21, 26 MR1 has also been detected on the surface of normal human bronchial epithelial cells.26
Only one study has examined expression of MR1 by different cell types in situ in tissue. Using monoclonal antibody 26.5, Gozalbo‐López et al.19 demonstrated expression of MR1 within the lamina propria of the ileum, appendix and colon by immunohistochemistry.19 MR1+ cells were located outside lymphoid follicles, were negative for myeloid markers (CD14, CD68, CD11c) and a T‐cell marker (CD3) but were positive for the B‐cell lineage marker, CD19. MR1+ lamina propria cells were also HLA‐DR+, CD38+, CD138+ and IgA+, suggesting that they were IgA‐producing plasmablasts or plasma cells. It is worth noting that only a subset of IgA+ CD38+ CD138+ cells expressed MR1. These findings are consistent with dependence on B cells for MAIT cell expansion in vivo.27, 28
In the study by Gozalbo‐López et al. MR1 expression was restricted to CD19+ cells, but it is likely that other cells also express MR1 in vivo, even if not detected by immunohistochemistry. For example, the authors were unable to detect MR1 expression in human thymus by immunohistochemistry.19 Nonetheless, MR1 must be expressed in the thymus as double‐positive thymocytes have been shown to be important for the thymic selection of MAIT cells, in an MR1‐dependent fashion.15 MR1 expression has also been detected by flow cytometry in single‐cell suspensions of human thymus, with expression restricted to CD3+ CD45+ cells, predominantly the CD4+ CD8+ subset.12 Similarly, MR1 is likely to be expressed on cell types other than B cells in vivo, as primary human and mouse myeloid cells (monocytes, dendritic cells, macrophages) and epithelial cells have been shown to activate MAIT cells in vitro in an MR1‐dependent fashion.4, 22, 29, 30
Factors affecting surface expression of MR1
Transient trafficking of endogenous MR1 to the plasma membrane was recently demonstrated using a novel monoclonal antibody (8H9.D11) that stabilized folded murine MR1 at the surface of cells.31 This allowed the activation of autoreactive mouse MAIT cell clones in the absence of exogenous ligand. Using this antibody, transient surface expression of MR1 was demonstrated in double‐positive thymocytes, macrophages and dendritic cells.31 This is consistent with the requirement of MR1 expression by double‐positive thymocytes for MAIT cell development.15
Increased surface expression of MR1 has been reported following treatment with riboflavin‐producing bacteria. Using a rabbit polyclonal anti‐MR1 antibody, infection with Escherichia coli or Salmonella Typhi was shown to induce surface expression of MR1 on an Epstein–Barr virus‐transformed lymphoblastoid B‐cell line (B‐LCL), HCT‐8 epithelial cells and primary B cells; similar staining was seen in B‐LCL with a polyclonal goat anti‐MR1 antibody and with antibody 26.5.32 In the B‐LCL, the amount of MR1 expressed on the surface correlated with the level of infection. Interestingly, MR1 surface expression was inhibited by cytochalasin D, which inhibits actin polymerization and hence phagocytosis.32 In another study MR1 could be detected at the surface of a B‐LCL with antibody 26.5, but was not increased following exposure to fixed E. coli.30 In contrast, MR1 could only be detected on the surface of THP1 cells, a myeloid cell line, after exposure to E. coli.30 Surface expression of MR1 has also been detected in M. tuberculosis‐infected, but not uninfected, A549 epithelial cells.4 Therefore, infection with ligand‐producing bacteria increases surface expression of MR1 in most, but not all, cell lines.
MR1 surface expression is also increased by ligand alone. Both stimulatory [reduced 6‐hydroxymethyl‐8‐d‐ribityllumazine (rRL‐6‐CH2OH), 5‐A‐RU ± glyoxal or methylglyoxal] and non‐stimulatory [6‐formyl pterin (6‐FP), acetyl‐6‐formyl pterin (Ac‐6‐FP)] MR1 ligands increase the surface expression of MR1 on C1R cells transduced with human MR1.2, 3, 26, 33 The non‐stimulatory ligand, 6‐FP, can also increase the basal surface expression of MR1 of the BEAS‐2B airway epithelial cell line and primary normal human bronchial epithelial cells.22
Toll‐like receptor (TLR) signalling may also play an important role in MR1 surface expression. In THP1 cells transfected with human MR1, increased surface expression was seen following treatment with heat‐killed Listeria monocytogenes, a TLR2 agonist that lacks riboflavin synthetic capacity, or with fixed Enterococcus faecalis, which also lacks the ability to synthesize riboflavin.30 Furthermore, the E. coli‐induced increase in MR1 surface expression on THP1 cells was inhibited by IKK inhibitor VII, an inhibitor of the nuclear factor‐κB (NF‐κB) signalling pathway. Interestingly, IKK inhibitor VII also inhibited basal surface expression of MR1 on both THP1 cells and B‐LCLs. Inhibition of NF‐κB signalling also inhibited subsequent MAIT cell activation.30 Supporting the importance of TLR signalling, a recent study in mice found that TLR signalling through TLR2/6 (Pam2Cys), TLR3 (poly I:C) or TLR9 (CpG oligonucleotides) was essential for the accumulation of MAIT cells in the lung in response to ligand (5‐OP‐RU).34 In contrast, in another study no MR1 up‐regulation was seen upon treatment with lipid A, the biologically active component of the TLR4 ligand lipopolysaccharide.32 Therefore, the nature of TLR stimulation may affect the surface expression of MR1 and is required for MAIT cell accumulation in vivo.
Intracellular trafficking of MR1
The intracellular trafficking of MR1 and its loading with ligand is beginning to be clarified, although most published studies pre‐date the discovery that MR1 presents a microbially derived ligand.
Like MHC class I and CD1d, MR1 associates directly and non‐covalently with β 2m.19, 23, 24, 35 Consistent with this, MR1 expression is not seen in Daudi cells, which lack β 2m.18 Association of MR1 with β 2m is required for trafficking of MR1 from the endoplasmic reticulum (ER) to the Golgi35 and is also required for surface expression.16, 23 Increased surface expression of MR1 was seen when MR1 was covalently attached to β 2m.23 Similarly, replacing the α3, transmembrane and cytosolic domains of MR1 with those of Ld, a classical MHC class I molecule, increased surface expression of MR1; replacing just the transmembrane and cytosolic domains leads to a smaller increase.23 This suggests that the α3 domain of MR1 interacts more weakly with β 2m than classical MHC class I molecules and that this, along with the transmembrane and cytosolic domains, influences intracellular trafficking and surface expression. Lion et al.16 confirmed that the surface expression of native MR1 was also β 2m‐dependent. MAIT cells are absent in β 2m‐deficient mice.28, 36
MR1 is reported to be associated with the peptide loading complex (calreticulin, TAP, tapasin and ERp57), even though the MR1 α3 domain lacks some of the consensus residues that are required for loading complex interactions.23 Native MR1 co‐immunoprecipitated with members of the peptide loading complex (calreticulin, tapasin and ERp57) and other ER proteins (calnexin and protein disulphide isomerase).16 However, using knockout cell lines, surface expression of MR1 and subsequent activation of MAIT cell hybridomas by endogenous antigens was shown to be independent of TAP, tapasin, calreticulin and MHC class Ia molecules.25 Activation of primary murine MAIT cells by bacterial antigens was also shown to be TAP‐independent.29 Expression of the TAP inhibitor ICP47 in dendritic cells infected with M. tuberculosis had no effect on their ability to stimulate human MAIT cell clones.4 Furthermore, MAIT cells are present in TAP‐deficient humans and mice.28, 36 Surface expression and MR1‐mediated MAIT cell activation are also independent of the proteasome.25, 29, 32 However, similar to class I presentation, blocking protein transport past the Golgi with brefeldin A inhibited both MR1 surface expression and MR1‐mediated MAIT cell hybridoma activation.25 Therefore, although MR1 shares some features of the MHC class I pathway, its trafficking pathway is distinct.
MR1 also shares features of the MHC class II pathway. In a murine fibroblast cell line MR1 co‐immunoprecipitated with the invariant chain (Ii) and HLA‐DM, which are molecular chaperones associated with the MHC class II pathway.25 Although overexpression of Ii with or without HLA‐DM failed to increase MR1 surface expression, it enhanced the stimulation of MAIT cell hybridomas and the trafficking of MR1 to LAMP+ endosomes. Leupeptin, which inhibits proteolysis of Ii, led to the retention of MR1 in LAMP+ endosomes and inhibited surface expression. Furthermore, reduction in the amount of endogenous Ii with small interfering RNA (siRNA) in a non‐transfected B‐cell line, reduced surface expression of MR1 and inhibited activation of MAIT cell hybridomas.25 In contrast, however, Ii is not required for the ontogeny of MAIT cells28 and bone‐marrow‐derived dendritic cells from TAP−/−Ii−/− mice were equally efficient at activating MAIT cells in response to bacteria as cells from wild‐type mice,29 although cytokine‐mediated MAIT cell activation was not excluded.37 Of note, Ii has been reported to associate with MHC class I molecules in dendritic cells to mediate trafficking to endolysosomes, enabling cross‐presentation of exogenous peptides.38 In summary, whereas Ii may play a role in trafficking of MR1 to the endolysosome it is not essential.
As with MHC class II presentation, inhibition of phagolysosomal acidification with concanamycin A or bafilomycin A1 decreased surface expression of MR1 and inhibited stimulation of a MAIT cell hybridoma.25 Inhibition of endosomal acidification has also been shown to inhibit activation of primary MAIT cells in response to ligand‐producing bacteria. Treatment of murine bone‐marrow‐derived dendritic cells with chloroquine inhibited the activation of primary mouse MAIT cells in response to E. coli.29 Similarly, treatment of the human monocytic cell line THP1 with bafilomycin A reduced the activation of primary human MAIT cells in response to E. coli.30
The intracellular location(s) of MR1 in the absence of ligand is controversial. Whereas most studies have found little MR1 at the cell surface, some have found it to be restricted to the ER24, 26 and others have also found it in LAMP+ endosomes.22, 25 These differences may reflect experimental differences, in particular the presence or absence of folic acid‐derived ligands in culture media or the effects of overexpression or molecular tags on MR1 trafficking. There may also be differences in trafficking between different cell types or between humans and mice: as mentioned previously, the cytoplasmic tail of MR1 is longer in rodents than in primates and contains a conserved motif that could be associated with endocytosis.13
Two studies suggest that in the absence of ligand, MR1 is retained in the ER. In a study by Yamaguchi and Hashimoto conducted before the identification of the MR1 ligand, no MR1 could be detected at the surface of P388‐MR1 cells (a mouse lymphoblastoid cell line transfected with MR1) when surface proteins were biotinylated and immunoprecipitated;24 cells were cultured in folic‐acid‐containing media. MR1 was found to be sensitive to endoglycosidase‐H and to glycopeptidase F, suggesting that it was retained in a pre‐Golgi compartment. This sensitivity of MR1 to endoglycosidase‐H was confirmed in L‐MR1 cells, a mouse fibroblast cell line transfected with MR1.24 Therefore, in the absence of ligand MR1 did not acquire the mature state of glycosylation and was retained intracellularly. This is consistent with the findings of a recent paper by McWilliam et al.,26 where biotinylated cell surface MR1 could only be detected on C1R.hMR1 cells, a human lymphoblastoid B‐cell line transfected with MR1, after treatment with agonist (5‐OP‐RU) or antagonist (Ac‐6‐FP) ligands; cells were cultured in the absence of folic acid to prevent contamination of the media with 6‐FP. Up‐regulation of ligand‐bound MR1 at the cell surface was also demonstrated through binding of MAIT TCR tetramer (agonist ligand only) and 26.5 antibody (both agonist and antagonist ligand). In the absence of ligand, MR1 was sensitive to endoglycosidase‐H, whereas after treatment with ligand most MR1 became resistant to endoglycosidase‐H, suggesting trafficking through the Golgi; biotinylated surface MR1 was found to be endoglycosidase‐H resistant, confirming translocation to the surface of at least a proportion of ligand‐bound MR1.26 In human peripheral blood mononuclear cells (PBMCs), treatment with ligand also resulted in MR1 becoming endoglycosidase‐H resistant. Inhibiting protein egress from the ER with brefeldin A prevented the ligand‐mediated increase in surface expression and led to a gradual decline in basal surface expression. Inhibition of protein synthesis with cyclohexamide also led to a decline in basal MR1 surface expression but had less effect on ligand‐mediated increases in surface expression. Similar effects of brefeldin A and cyclohexamide on surface expression of MR1 were also seen in primary human B cells, CD14+ cells, T cells and normal human bronchial epithelial cells. There were differences in basal surface expression of MR1 in the absence of ligand, with higher levels seen in bronchial epithelial cells and B cells. There was no increase, however, in surface expression of MR1 following infection of C1R.hMR1 cells with Salmonella enterica, although they were able to activate MAIT cells. Adherent PBMCs infected with S. enterica were also able to activate MAIT cells, and this was partially inhibited when the adherent PBMCs were co‐incubated with brefeldin A or, to a lesser extent, cyclohexamide.26 Confocal microscopy confirmed that in the absence of folic acid in the cell culture medium and exogenous ligand, MR1‐GFP was retained in the ER of C1R.hMR1 cells, whereas with ligand it was found on the cell surface. In a pulse–chase experiment, endoglycosidase‐H‐sensitive MR1 slowly declined in the absence of ligand, suggesting degradation in the ER.26 Therefore, trafficking of MR1 through the ER is important for the presentation of soluble ligands and may also play a role in the presentation of ligands produced by intracellular bacteria.
McWilliam et al.26 found that MR1 needed to bind to ligand before it could bind β 2m and exit the ER. In contrast with previous papers, which used folate‐containing cell culture media,19, 23, 24, 35 McWilliam et al.26 found little or no β 2m associated with MR1 in C1R.hMR1 cells in the absence of ligand, suggesting that it was not fully folded. In human PBMCs, however, a small proportion of MR1 appeared to be associated with β 2m, which may reflect binding of an endogenous ligand in vivo. Overall, this suggests that folding of MR1 and association with β 2m is required for trafficking to the Golgi. This is consistent with an earlier study by Huang et al.,17 performed before the identification of the MR1 ligand, where only folded MR1 was found at the cell surface. Huang et al. used a mutated MR1 that had been modified to contain the 64‐3‐7 epitope (R46Q for human MR1 and K46Q for mouse MR1), which is normally found in open forms of Ld molecules.17, 23 MR1 modified to contain the 64‐3‐7 epitope could be detected in either an open conformation (with the 64‐3‐7 monoclonal antibody) or in a folded conformation (with 26.5 or 12.2 antibodies). Significantly more of the folded conformation of MR1 was seen at the cell surface and only folded MR1 could activate MAIT cell hybridomas. Mutations of the ligand binding groove significantly reduced surface expression of folded MR1 and stimulation of MAIT cell hybridomas, suggesting that loading of MR1 with an (endogenous) ligand is required for efficient surface expression.17
The requirement for ligand binding for MR1 to fold and reach the cell surface was confirmed by McWilliam et al.26 When C1R.hMR1 cells were cultured in folate‐deficient media and in the absence of exogenous ligand, only a small amount of MR1 could be immunoprecipitated with a conformation‐dependent anti‐MR1 antibody (8F2.F9). In contrast, large amounts of MR1 could be immunoprecipitated following addition of ligand Ac‐6‐FP, and, in HeLa.hMR1 cells, MR1 could be visualized at the plasma membrane. When ER–Golgi transport was disrupted with brefeldin A, folded MR1 accumulated in the ER of cells treated with Ac‐6‐FP. They found that the formation of a Schiff base between the MR1 ligand and residue K43 of MR1 was required for folding of MR1 and exit from the ER. If the basically charged lysine at residue 43 (K43) was mutated to an alanine (K43A), neutralizing the basic charge, the K43A mutant MR1 associated with β 2m, became endoglycosidase‐H resistant, and was expressed at the cell surface of C1R cells in the absence of ligand. In contrast, the K43R mutant, which retained the positive charge and was unable to form a Schiff bass with Ac‐6‐FP, could not fold, and was unable to leave the ER.26 Therefore the K43 residue of MR1 prevents complete folding, including association with β 2m, and causes retention in ER until ligand is bound; binding of ligand to K43 neutralizes the basic charge, allowing folding, association with β 2m, and trafficking to the cell surface. These experiments also suggest that Ac‐6‐FP can access the ER by a yet to be defined mechanism.
Other studies, however, which have used folic‐acid‐containing cell culture media, have found MR1 in endosomal compartments in the absence of exogenous ligand.22, 25 Harriff et al.22 found that in the BEAS‐2B.MR1‐GFP airway epithelial cell line, MR1 with a C‐terminal GFP localized not only to the ER but also to the ER–Golgi intermediate compartment (ERGIC‐53), the trans‐Golgi network (TGN‐46), and late endosomes/lysosomes (Rab7, LAMP1); MR1‐GFP was not found in Rab5+ early endosomes. The majority of MR1‐GFP co‐localized with β 2m.22 This is consistent with another study that used a mouse B‐cell line (CH27) overexpressing mMR1.eGFP, where MR1 was found predominantly to reside in LAMP1+ endolysosomes and in the ER.25 The addition of a C‐terminal GFP tag did not appear to affect the function of MR1 in either study. Harriff et al. found that similar to endogenous MR1, MR1‐GFP was detectable at the cell surface and, in BEAS‐2B cells infected with mycobacteria, was able to stimulate a MAIT cell clone. Up‐regulation of surface expression of both MR1 and MR1‐GFP was seen following treatment with 6‐FP.22 Similarly, Huang et al.25 demonstrated that both CH27.mMR1.eGFP cells and CH27.mMR1 cells expressed similar levels of MR1 at the cell surface and were equally able to stimulate MAIT cell hybridomas in the absence of external ligand. It is still possible, however, that the C‐terminal GFP tag alters the intracellular trafficking of MR1. Aldemir found that MR1‐GFP co‐immunoprecipitated with β 2m in L721.221 cells but not in 293 or HeLa cells, yet in 293 and HeLa cells MR1‐GFP partially co‐localized with LAMP‐1.35 Future studies should determine the intracellular localization of untagged endogenous MR1 in various cell types; however, it should be noted that many of the antibodies available (12.2, 26.5 and 8F2.F9) only recognize folded MR1.17, 31
Loading of MR1 with soluble ligand can occur in endosomal compartments. Harriff et al.22 found that 6‐FP increased the surface expression of MR1 on both BEAS‐2B cells and normal human bronchial epithelial cells, even though folate was present in the cell culture media, and that up‐regulation of surface expression was markedly decreased by brefeldin A. Incubation of cells with 6‐FP also resulted in a decrease in the number of MR1‐GFP+ vesicles per cell and a decrease in co‐localization with β 2m+ endosomal compartments. A further reduction in MR1‐GFP+ vesicles was seen when cells were pre‐treated with cyclohexamide before 6‐FP.22 Together, this suggests that 6‐FP can displace endogenous ligand in MR1+ β 2m+ endosomal compartments, leading to surface expression. McWilliam et al. also demonstrated that reloading of MR1 could occur in endosomal compartments. They found that ~50% of surface MR1 was internalized within 2–4 hr, and trafficked to either early (EEA1+) or late (LAMP+) endosomal compartments.26 Only a small amount (~4%) recycled to the surface. The Schiff base formed between ligand and K43 is labile at the pH present in late endosomes, suggesting the possibility of exchange of covalently bound ligand during recycling. Indeed, MR1 pre‐loaded with 6‐FP could be re‐loaded with 5‐OP‐RU, with 40% of MR1 at the cell surface binding 5‐OP‐RU.26
MR1 can also be loaded at the cell surface. Le Bourhis et al.29 demonstrated that glutaraldehyde‐fixed WT3 cells, a fibroblast cell line, overexpressing MR1 that were co‐incubated with live bacteria were able to activate primary MAIT cells. Consistent with this, McWilliam et al.26 showed that MR1 could be loaded at the surface of C1R.hMR1 cells if they were cultured in the absence of vitamin B antigens but not if pre‐treated with 6‐FP. C1R.hMR1 cells were pre‐incubated or not with 6‐FP at 37°, then incubated on ice with 5‐OP‐RU for 20 min; surface‐loaded MR1‐5‐OP‐RU was detected with the TCR tetramer. This suggests that small amounts of empty MR1 or MR1 bound to non‐covalently bound ligand must be expressed at cell surface, even when cultured in the absence of folic acid.26 Interestingly, when ligand was removed from the cell culture media, ligand‐bound MR1 was rapidly lost from the cell surface, with ~50% lost in 8 hr.26 Therefore, in C1R cells at least, MR1 only remains at the cell surface as long as vitamin B antigens are present in the environment. There may, however, be differences in trafficking between cell types. In THP1.hMR1 cells, surface expression of MR1 increased with TLR stimulation, even in the absence of exogenous ligand, and subsequently declined, regardless of whether ligand was present or not.30
The loading of ligand from intact bacteria onto MR1 may differ from soluble ligand. Harriff et al. found that the trafficking molecules involved in loading of MR1 in M. tuberculosis‐infected epithelial cells differ from those required for loading soluble ligand. To identify trafficking molecules in airway epithelial cells that are uniquely required for MR1‐dependent MAIT cell activation following intracellular infection with M. tuberculosis, Harriff et al.22 used short hairpin RNA to knock down the expression of various trafficking molecules in BEAS‐2B airway epithelial cells expressing only endogenous MR1. BEAS‐2B cells were then infected with M. tuberculosis and production of interferon‐γ by various T‐cell clones was assessed. Twenty‐seven candidate genes were identified that resulted in decreased responses by the MAIT cell clone but not by the HLA‐E or HLA‐B45‐restricted clones. Of these, 10 were further evaluated by siRNA knockdown and eight (Rab3a, Rab3c, Rab3GAP2, Rab6, Rab34, Sec22b, syntaxin‐18 and Vamp4) were validated.22 Rab proteins are GTPases that regulate membrane trafficking: Rab3a is involved in exocytosis;39 Rab3c is found in recycling vesicles and is involved in recycling of MHC class I to the plasma membrane and in cross‐presentation;40 Rab6 is a Golgi‐resident protein that regulates both retrograde and anterograde Golgi transport;41 Rab34 is a Golgi‐resident protein involved in intra‐Golgi transport, the secretory pathway, the repositioning of lysosomes, and fusion of phagosomes with lysosomes;42, 43 Rab3GAP2 is a subunit of a GTPase‐activating protein that is involved in the activation of the Rab3 subfamily of proteins.44 Sec22b, syntaxin‐18 (Stx18) and Vamp4 are SNARE proteins involved in intracellular membrane trafficking: Sec22b is involved in both anterograde and retrograde transport between the Golgi and the ER, in formation of the autophagosome, and in cross‐presentation via transport of proteins directly from the ER–Golgi intermediate compartment to the phagosome;45, 46 Stx18 is localized to the ER and involved in retrograde Golgi–ER transport and ER‐mediated phagocytosis,47, 48 and possibly post‐Golgi‐trafficking pathways;22, 49 Vamp4 is involved in Golgi–endosome transport and retrograde endosome–Golgi transport and may also play a role in regulated exocytosis.50, 51, 52 Of note, both Rab6 and Sec22b have been found in Mycobacterium bovis bacillus Calmette–Guérin‐containing phagosomes,53 Rab34 accumulates in Staphylococcus aureus‐containing phagosomes and is associated with delivery of cathepsin D to M. tuberculosis‐containing phagosomes,54 and transcription of Rab34 is up‐regulated following infection with Mycobacterium smegmatis in an NF‐κB‐dependent manner.55
Although knockdown of Stx18, Vamp4 or Rab6 inhibited MAIT cell activation following intracellular infection with M. tuberculosis, only knockdown of Stx18 inhibited (partially) 6‐FP‐dependent translocation of MR1 to the cell surface; this was seen in both BEAS‐2B‐MR1‐GFP cells and the parental BEAS‐2B cell line.22 Knockdown of Stx18, Vamp4 or Rab6 had no effect on basal cell surface expression of MR1 in either cell line; the effect of knockdown on MR1 surface expression following infection with M. tuberculosis was not assessed. This suggests that there are distinct pathways for the presentation of soluble exogenous ligand and ligand from intracellular bacteria. Supporting this hypothesis, incubation of BEAS‐2B cells with M. smegmatis supernatant led to MAIT cell activation but this could be inhibited by pre‐incubation with 6‐FP. In contrast, pre‐incubation with 6‐FP had little effect on MAIT cell activation in response to intracellular M. tuberculosis infection. Stx18, which appeared to be involved in both pathways, co‐localized with MR1 in the ER, whereas Vamp4, which appeared only to be involved with presentation of ligand from intracellular bacteria, co‐localized with MR1 in endosomal compartments.22
Harriff et al. suggested that the role of Stx18 in MR1 presentation may be independent of its role in retrograde Golgi–ER transport and ER‐mediated phagocytosis.22 Stx18 forms a complex with additional SNAREs (Sec22b, BNIP1 and Use1) to regulate these processes.48 The siRNA knockdown of Sec22b and BNIP1 also inhibited MAIT cell activation, but knockdown of Use1 had no effect, although the efficiency of silencing was not reported.22 Knockdown of Stx18, Sec22b and BNIP1 has previously been shown to disrupt the structure of the ER and/or Golgi,56, 57, 58 whereas knockdown of the murine homologue of Use1, D12, had no effect on Golgi structure or the distribution of the KDEL receptor, which cycles between the Golgi and the ER.49 Although this complex is involved in retrograde transport, knockdown of Stx18, Sec22b, BNIP1 or Use1 have also been reported to inhibit the constitutive secretory pathway, with cargo blocked in the ER/Golgi.58 Stx18, Sec22b and Use1 have also been found in phagosomes;46, 48, 59 Sec22b was shown to mediate fusion of the ER–Golgi intermediate compartment with the phagosome through interactions with Stx4, a plasma membrane syntaxin, and this was shown to be necessary for cross‐presentation in dendritic cells.46 Overall, this suggests that even though no effect was seen with knockdown of Use1, ER–Golgi transport, either retrograde or anterograde, plays an important role in antigen presentation by MR1; ER‐mediated phagocytosis may also be relevant.
In BEAS‐2B‐MR1‐GFP cells, knockdown of Stx18, Vamp4 or Rab6 led to an increase in MR1‐GFP+ endosomal compartments and the amount of GFP per endosome.22 This could represent fragmentation of the ER and/or Golgi or expansion of multi‐vesicular bodies/autophagosomal compartments.57, 60, 61 Using TGN46 as a marker of the trans‐Golgi, disruption of the trans‐Golgi was seen with knockdown of Stx18 or Vamp4, whereas knockdown of Rab6 made the trans‐Golgi more compact.22 Expansion of multi‐vesicular bodies/autophagosomal compartments has been reported following Rab6 knockdown.60
Other data support the existence of separate pathways for loading MR1. In a recent study, inhibition of endosomal acidification (with bafilomycin A) or phagocytosis (with cytochalasin D) partially inhibited the ability of THP1 cells, a human monocytic cell line, to activate primary human MAIT cells in response to fixed intact E. coli, but had no effect on the response to E. coli supernatant.30 This is consistent with a previous report that found that treatment of mouse bone‐marrow‐derived dendritic cells with inhibitors of endocytosis and/or phagocytosis (Dynasore, cytochalasin D) or of endosomal acidification (chloroquine) inhibited activation of primary mouse MAIT cells in response to E. coli.29 THP1 cells treated with fixed intact bacteria (E. coli or S. enterica) stimulated more MAIT cells to produce interferon‐γ than THP1 cells treated with the equivalent culture volume of bacterial supernatant; treating cells with both E. coli supernatant and fixed intact Enterococcus faecalis, which does not produce ligand, did not enhance MAIT cell activation.30 This suggests that uptake and loading of soluble ligand differs to bacteria‐associated ligand. The ability of ligand‐producing bacteria with differing infectious cycles to stimulate MAIT cells also supports loading of MR1 in an endolysosomal compartment. Using a non‐phagocytic epithelial cell line (HeLa) as the antigen‐presenting cell, Le Bourhis et al.62 assessed their ability to activate MAIT cells following infection with E. coli, which does not invade the cell, Salmonella Typhimurium, which invades the cell but resides in endosomes by preventing fusion with lysosomes, or Shigella flexneri, which invades the cell and resides in the cytoplasm. HeLa cells infected with E. coli or Salmonella Typhimurium failed to significantly activate MAIT cells, even at high multiplicity of infection, whereas those infected with Shigella flexneri readily activated MAIT cells, with efficient killing of infected HeLa cells. In contrast, HeLa cells overexpressing hMR1 (HeLa.hMR1) readily activated MAIT cells following infection with E. coli or Salmonella Typhimurium; HeLa.hMR1 express high levels of MR1 at the cell surface, suggesting that in addition to the ER, loading of soluble ligand may have occurred at the cell surface. Interestingly, abrogating Shigella flexneri's ability to invade HeLa cells by deleting MixD, a virulence protein required for entry into cells, reduced MAIT cell activation to the minimal levels seen with E. coli and Salmonella Typhimurium. In contrast to HeLa cells, primary human monocytes infected with formaldehyde‐fixed E. coli, Salmonella Typhimurium, or Shigella flexneri readily activated MAIT cells. These differences suggest that bacteria need to access the endolysosome for ligand to be efficiently loaded onto MR1. When bacteria fail to reach the endolysosome, ligand is not available to MR1. Bacteria residing in the cytoplasm, such as Shigella flexneri, may be targeted to the lysosomal compartment by autophagy (xenophagy).63
Finally, several lines of evidence support the existence of an endogenous MR1 ligand(s). First, MR1‐dependent development of MAIT cells occurs in the thymus in utero; MAIT cells can be detected in multiple human fetal tissues.15, 64 Second, surface expression of folded MR1 can be detected on a variety of non‐transfected cell lines and primary human PBMCs, albeit at low levels, in the absence of exogenous ligand and even in the absence of folic acid in the cell culture medium.19, 21, 22, 26, 30 Third, mutations in the ligand‐binding groove of MR1 reduced surface expression and inhibited the ability of WT3 cells overexpressing MR1 to stimulate MAIT cell hybridomas.17 Endogenous ligand(s) may be derived from folic acid or unrelated molecules; it is unknown whether 6‐FP occurs in vivo. The existence of non‐folic acid‐derived ligands is suggested by the detection of low levels of the folded form of MR1 at the cell surface in the absence of folic acid or an exogenous ligand.26 An endogenous ligand could covalently bind to MR1 Lys43 through the formation of a Schiff base, as reported for 6‐FP,2 or could bind non‐covalently to MR1 and neutralize Lys43, as reported for some ribityllumazines, diclofenac and 5′‐hydroxylated diclofenac.18, 65 The existence of a non‐covalently bound ligand would be consistent with the observation that MR1 can be loaded at the cell surface;26, 29 alternatively, surface loading may be explained by the surface expression of small amounts of unloaded, open conformation MR1, which has been reported in cells overexpressing MR1.17
In summary, we propose the following model of MR1 loading and trafficking (Fig. 1). In the absence of any ligand, including that supplied by folic acid supplementation of media, most MR1 accumulates in the ER in an unfolded state. In the ER MR1 binds to an endogenous ligand, allowing folding and association with β 2m. MR1 bound to β 2m then trafficks through the Golgi to an endolysosomal compartment, either directly or via the cell surface. The invariant chain may assist in this trafficking but is not necessary. Loading of MR1 with activating ligands can occur in four ways. First, soluble ligand can access the ER via an unknown mechanism, resulting in loading and trafficking to the cell surface, where it can interact with the MAIT cell TCR; this is the dominant pathway for loading of soluble ligand. Second, soluble ligand can replace non‐covalently bound endogenous ligand on MR1 transiently expressed at the cell surface or covalently bound or non‐covalently bound endogenous ligand on MR1 in endolysosomal compartments. Alternatively, ligand may be able to bind to small amounts of un‐liganded, open conformation MR1 that reach the cell surface. Given the lack of MR1 at the cell surface, loading by either mechanism is likely to be a minor pathway. Third, phagocytosed bacteria can access the MR1‐containing endolysosomal compartment where ligand is released and exchanges with the endogenous ligand. Finally, cytoplasmic bacteria, such as Shigella, may be delivered to the MR1‐containing endolysosomal compartment by xenophagy (autophagy). Ligand‐loaded MR1 then trafficks to the cell surface, where it is transiently expressed, before recycling to the endolysosomal compartment, where it is eventually degraded. Ligand exchange could also potentially occur upon recycling. Trafficking of MR1 is also influenced by TLR signalling, at least in some cell types, with increased cell surface expression seen even in the absence of exogenous ligand.
Figure 1.
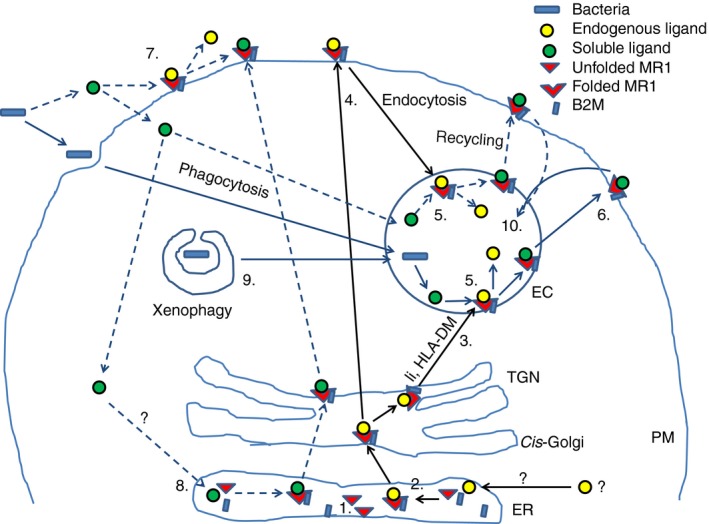
Model of trafficking of MHC‐related protein 1 (MR1) and loading with ligand. (1) In the absence of ligand, empty MR1 molecules accumulate in the endoplasmic reticulum (ER). (2) Binding of an endogenous ligand, such as 6‐FP, to MR1 leads to folding and stable association with β 2‐microglobulin (β 2m), allowing exit from the ER. (3) MR1 bound to endogenous ligand translocates through the Golgi and reaches an endolysosomal compartment, either directly, potentially with the help of the invariant chain (li), or (4) indirectly via cell surface expression and subsequent endocytosis. (5) The low pH environment of the endolysosomal compartment favours exchange of covalently bound ligand with exogenous ligand (5‐OP‐RU or 5‐OE‐RU) derived from either phagocytosed bacteria or acquired directly from the extracellular environment by an unknown mechanism. (6) From the endolysosomal compartment, ligand‐bound MR1 recycles to the cell surface where it can interact with the MAIT cell T‐cell receptor (TCR). (7) Extracellular soluble exogenous ligand can be loaded onto MR1 at cell surface, either by displacing non‐covalently bound endogenous ligand or by binding to unliganded MR1 (not shown), or (8) can access the ER via an unknown mechanism. In the ER, exogenous ligand is loaded directly onto empty MR1, allowing its trafficking to the cell surface. (9) Xenophagy may deliver cytoplasmic bacteria to the endolysosomal compartment, where loading of bacterial‐derived ligand can occur. (10) Following surface expression MR1 may either undergo further rounds of recycling or may traffic to the lysosome for degradation.
Disclosures
The authors have no competing interests to declare.
References
- 1. Ussher JE, Klenerman P, Willberg CB. Mucosal‐associated invariant T (MAIT) cells: new players in anti‐bacterial immunity. Front Immunol 2014; 5:450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kjer‐Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L et al MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 2012; 491:717–23. [DOI] [PubMed] [Google Scholar]
- 3. Corbett AJ, Eckle SBG, Birkinshaw RW, Liu L, Patel O, Mahony J et al T‐cell activation by transitory neo‐antigens derived from distinct microbial pathways. Nature 2014; 509:361–5. [DOI] [PubMed] [Google Scholar]
- 4. Gold MC, Cerri S, Smyk‐Pearson S, Cansler ME, Vogt TM, Delepine J et al Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol 2010; 8:e1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Walter L, Günther E. Isolation and molecular characterization of the rat MR1 homologue, a non‐MHC‐linked class I‐related gene. Immunogenetics 1998; 47:477–82. [DOI] [PubMed] [Google Scholar]
- 6. Hashimoto K, Hirai M, Kurosawa Y. A gene outside the human MHC related to classical HLA class I genes. Science 1995; 269:693–5. [DOI] [PubMed] [Google Scholar]
- 7. Cheng Y, Belov K. Characterisation of non‐classical MHC class I genes in the Tasmanian devil (Sarcophilus harrisii). Immunogenetics 2014; 66:727–35. [DOI] [PubMed] [Google Scholar]
- 8. Riegert P, Wanner V, Bahram S. Genomics, isoforms, expression, and phylogeny of the MHC class I‐related MR1 gene. J Immunol 1998; 161:4066–77. [PubMed] [Google Scholar]
- 9. Yamaguchi H, Hirai M, Kurosawa Y, Hashimoto K. A highly conserved major histocompatibility complex class I‐related gene in mammals. Biochem Biophys Res Commun 1997; 238:697–702. [DOI] [PubMed] [Google Scholar]
- 10. Boudinot P, Mondot S, Jouneau L, Teyton L, Lefranc M‐P, Lantz O. Restricting nonclassical MHC genes coevolve with TRAV genes used by innate‐like T cells in mammals. Proc Natl Acad Sci U S A 2016; 113:E2983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goldfinch N, Reinink P, Connelley T, Koets A, Morrison I, Van Rhijn I. Conservation of mucosal associated invariant T (MAIT) cells and the MR1 restriction element in ruminants, and abundance of MAIT cells in spleen. Vet Res 2010; 41:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gold MC, Eid T, Smyk‐Pearson S, Eberling Y, Swarbrick GM, Langley SM et al Human thymic MR1‐restricted MAIT cells are innate pathogen‐reactive effectors that adapt following thymic egress. Mucosal Immunol 2013; 6:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parra‐Cuadrado JF, del Moral M, García‐Pavía P, Setién F, Martínez‐Naves E. Characterization of the MHC class I‐related MR1 locus in nonhuman primates. Immunogenetics 2001; 53:643–8. [DOI] [PubMed] [Google Scholar]
- 14. Parra‐Cuadrado JF, Navarro P, Mirones I, Setién F, Oteo M, Martínez‐Naves E. A study on the polymorphism of human MHC class I‐related MR1 gene and identification of an MR1‐like pseudogene. Tissue Antigens 2000; 56:170–2. [DOI] [PubMed] [Google Scholar]
- 15. Seach N, Guerri L, Le Bourhis L, Mburu Y, Cui Y, Bessoles S et al Double positive thymocytes select mucosal‐associated invariant T cells. J Immunol 2013; 191:6002–9. [DOI] [PubMed] [Google Scholar]
- 16. Lion J, Debuysscher V, Wlodarczyk A, Hodroge A, Serriari N‐E, Choteau L et al MR1B, a natural spliced isoform of the MHC‐related 1 protein, is expressed as homodimers at the cell surface and activates MAIT cells. Eur J Immunol 2013; 43:1363–73. [DOI] [PubMed] [Google Scholar]
- 17. Huang S, Gilfillan S, Cella M, Miley MJ, Lantz O, Lybarger L et al Evidence for MR1 antigen presentation to mucosal‐associated invariant T cells. J Biol Chem 2005; 280:21183–93. [DOI] [PubMed] [Google Scholar]
- 18. Abós B, Gómez del Moral M, Gozalbo‐López B, López‐Relaño J, Viana V, Martínez‐Naves E. Human MR1 expression on the cell surface is acid sensitive, proteasome independent and increases after culturing at 26°C. Biochem Biophys Res Commun 2011; 411:632–6. [DOI] [PubMed] [Google Scholar]
- 19. Gozalbo‐López B, del Moral MG, Campos‐Martín Y, Setién F, Martín P, Bellas C et al The MHC‐related protein 1 (MR1) is expressed by a subpopulation of CD38+, IgA+ cells in the human intestinal mucosa. Histol Histopathol 2009; 24:1439–49. [DOI] [PubMed] [Google Scholar]
- 20. Ljunggren H‐G, Stam NJ, Ohlen C, Neefjes JJ, Hoglund P, Heemels M‐T et al Empty MHC class I molecules come out in the cold. Nature 1990; 346:476–80. [DOI] [PubMed] [Google Scholar]
- 21. Harriff MJ, Cansler ME, Toren KG, Canfield ET, Kwak S, Gold MC et al Human lung epithelial cells contain Mycobacterium tuberculosis in a late endosomal vacuole and are efficiently recognized by CD8+ T cells. PLoS ONE 2014; 9:e97515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harriff MJ, Karamooz E, Burr A, Grant WF, Canfield ET, Sorensen ML et al Endosomal MR1 trafficking plays a key role in presentation of Mycobacterium tuberculosis ligands to MAIT Cells. PLoS Pathog 2016; 12:e1005524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miley MJ, Truscott SM, Yu YYL, Gilfillan S, Fremont DH, Hansen TH et al Biochemical features of the MHC‐related protein 1 consistent with an immunological function. J Immunol 2003; 170:6090–8. [DOI] [PubMed] [Google Scholar]
- 24. Yamaguchi H, Hashimoto K. Association of MR1 protein, an MHC Class I‐related molecule, with β2‐microglobulin. Biochem Biophys Res Commun 2002; 290:722–9. [DOI] [PubMed] [Google Scholar]
- 25. Huang S, Gilfillan S, Kim S, Thompson B, Wang X, Sant AJ et al MR1 uses an endocytic pathway to activate mucosal‐associated invariant T cells. J Exp Med 2008; 205:1201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McWilliam HEG, Eckle SBG, Theodossis A, Liu L, Chen Z, Wubben JM et al The intracellular pathway for the presentation of vitamin B‐related antigens by the antigen‐presenting molecule MR1. Nat Immunol 2016; 17:531–7. [DOI] [PubMed] [Google Scholar]
- 27. Martin E, Treiner E, Duban L, Guerri L, Laude H, Toly C et al Stepwise development of MAIT cells in mouse and human. PLoS Biol 2009; 7:e1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F et al Selection of evolutionarily conserved mucosal‐associated invariant T cells by MR1. Nature 2003; 422:164–9. [DOI] [PubMed] [Google Scholar]
- 29. Le Bourhis L, Martin E, Péguillet I, Guihot A, Froux N, Coré M et al Antimicrobial activity of mucosal‐associated invariant T cells. Nat Immunol 2010; 11:701–8. [DOI] [PubMed] [Google Scholar]
- 30. Ussher JE, van Wilgenburg B, Hannaway RF, Ruustal K, Phalora P, Kurioka A et al TLR signaling in human antigen‐presenting cells regulates MR1‐dependent activation of MAIT cells. Eur J Immunol 2016; 46:1600–14. https://doi.org/10.1002/eji.201545969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chua W‐J, Kim S, Myers N, Huang S, Yu L, Fremont DH et al Endogenous MHC‐related protein 1 is transiently expressed on the plasma membrane in a conformation that activates mucosal‐associated invariant T cells. J Immunol 2011; 186:4744–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Salerno‐Goncalves R, Rezwan T, Sztein MB. B cells modulate mucosal associated invariant T cell (MAIT) immune responses. Front Immunol 2014; 4:511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eckle SBG, Birkinshaw RW, Kostenko L, Corbett AJ, McWilliam HEG, Reantragoon R et al A molecular basis underpinning the T cell receptor heterogeneity of mucosal‐associated invariant T cells. J Exp Med 2014; 211:1585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen Z, Wang H, D'Souza C, Sun S, Kostenko L, Eckle SBG et al Mucosal‐associated invariant T‐cell activation and accumulation after in vivo infection depends on microbial riboflavin synthesis and co‐stimulatory signals. Mucosal Immunol 2017; 10:58–68. [DOI] [PubMed] [Google Scholar]
- 35. Aldemir H. Novel MHC class I‐related molecule MR1 affects MHC class I expression in 293T cells. Biochem Biophys Res Commun 2008; 366:328–34. [DOI] [PubMed] [Google Scholar]
- 36. Tilloy F, Treiner E, Park S‐H, Garcia C, Lemonnier F, de la Salle H et al An invariant T cell receptor α chain defines a novel TAP‐independent major histocompatibility complex class Ib–restricted α/β T cell subpopulation in mammals. J Exp Med 1999; 189:1907–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ussher JE, Bilton M, Attwod E, Shadwell J, Richardson R, Lara C et al CD161++ CD8+ T cells, including the MAIT cell subset, are specifically activated by IL‐12 + IL‐18 in a TCR‐independent manner. Eur J Immunol 2014; 44:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Basha G, Omilusik K, Chavez‐Steenbock A, Reinicke AT, Lack N, Choi KB et al A CD74‐dependent MHC class I endolysosomal cross‐presentation pathway. Nat Immunol 2012; 13:237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schlüter OM, Khvotchev M, Jahn R, Südhof TC. Localization versus function of Rab3 proteins: evidence for a common regulatory role in controlling fusion. J Biol Chem 2002; 277:40919–29. [DOI] [PubMed] [Google Scholar]
- 40. Zou L, Zhou J, Zhang J, Li J, Liu N, Chai L et al The GTPase Rab3b/3c‐positive recycling vesicles are involved in cross‐presentation in dendritic cells. Proc Natl Acad Sci U S A 2009; 106:15801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu S, Storrie B. Are Rab proteins the link between Golgi organization and membrane trafficking? Cell Mol Life Sci 2012; 69:4093–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goldenberg NM, Grinstein S, Silverman M. Golgi‐bound Rab34 Is a novel member of the secretory pathway. Mol Biol Cell 2007; 18:4762–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gutierrez MG. Functional role(s) of phagosomal Rab GTPases. Small GTPases 2013; 4:148–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barr F, Lambright DG. Rab GEFs and GAPs. Curr Opin Cell Biol 2010; 22:461–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Daste F, Galli T, Tareste D. Structure and function of longin SNAREs. J Cell Sci 2015; 128:4263–72. [DOI] [PubMed] [Google Scholar]
- 46. Cebrian I, Visentin G, Blanchard N, Jouve M, Bobard A, Moita C et al Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell 2011; 147:1355–68. [DOI] [PubMed] [Google Scholar]
- 47. Aoki T, Kojima M, Tani K, Tagaya M. Sec22b‐dependent assembly of endoplasmic reticulum Q‐SNARE proteins. Biochem J 2008; 410:93–100. [DOI] [PubMed] [Google Scholar]
- 48. Hatsuzawa K, Tamura T, Hashimoto H, Hashimoto H, Yokoya S, Miura M et al Involvement of syntaxin 18, an endoplasmic reticulum (ER)‐localized SNARE protein, in ER‐mediated phagocytosis. Mol Biol Cell 2006; 17:3964–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Okumura AJ, Hatsuzawa K, Tamura T, Nagaya H, Saeki K, Okumura F et al Involvement of a novel Q‐SNARE, D12, in quality control of the endomembrane system. J Biol Chem 2006; 281:4495–506. [DOI] [PubMed] [Google Scholar]
- 50. Krzewski K, Gil‐Krzewska A, Watts J, Stern JNH, Strominger JL. VAMP4‐ and VAMP7‐expressing vesicles are both required for cytotoxic granule exocytosis in NK cells. Eur J Immunol 2011; 41:3323–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Steegmaier M, Klumperman J, Foletti DL, Yoo J‐S, Scheller RH. Vesicle‐associated membrane protein 4 is implicated in trans‐Golgi network vesicle trafficking. Mol Biol Cell 1999; 10:1957–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hirata T, Fujita M, Nakamura S, Gotoh K, Motooka D, Murakami Y et al Post‐Golgi anterograde transport requires GARP‐dependent endosome‐to‐TGN retrograde transport. Mol Biol Cell 2015; 26:3071–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee B‐Y, Jethwaney D, Schilling B, Clemens DL, Gibson BW, Horwitz MA. The Mycobacterium bovis Bacille Calmette–Guérin phagosome proteome. Mol Cel Proteomics 2010; 9:32–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Seto S, Tsujimura K, Koide Y. Rab GTPases regulating phagosome maturation are differentially recruited to mycobacterial phagosomes. Traffic 2011; 12:407–20. [DOI] [PubMed] [Google Scholar]
- 55. Gutierrez MG, Mishra BB, Jordao L, Elliott E, Anes E, Griffiths G. NF‐κB activation controls phagolysosome fusion‐mediated killing of mycobacteria by macrophages. J Immunol 2008; 181:2651–63. [DOI] [PubMed] [Google Scholar]
- 56. Nakajima K, Hirose H, Taniguchi M, Kurashina H, Arasaki K, Nagahama M et al Involvement of BNIP1 in apoptosis and endoplasmic reticulum membrane fusion. EMBO J 2004; 23:3216–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Iinuma T, Aoki T, Arasaki K, Hirose H, Yamamoto A, Samata R et al Role of syntaxin 18 in the organization of endoplasmic reticulum subdomains. J Cell Sci 2009; 122:1680–90. [DOI] [PubMed] [Google Scholar]
- 58. Gordon DE, Bond LM, Sahlender DA, Peden AA. A targeted siRNA screen to identify SNAREs required for constitutive secretion in mammalian cells. Traffic 2010; 11:1191–204. [DOI] [PubMed] [Google Scholar]
- 59. Campbell‐Valois F‐X, Trost M, Chemali M, Dill BD, Laplante A, Duclos S et al Quantitative proteomics reveals that only a subset of the endoplasmic reticulum contributes to the phagosome. Mol Cell Proteomics 2012; 11(M111):016378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Storrie B, Micaroni M, Morgan GP, Jones N, Kamykowski JA, Wilkins N et al Electron tomography reveals Rab6 is essential to the trafficking of trans‐Golgi clathrin and COPI‐coated vesicles and the maintenance of Golgi cisternal number. Traffic 2012; 13:727–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shitara A, Shibui T, Okayama M, Arakawa T, Mizoguchi I, Shakakura Y et al VAMP4 is required to maintain the ribbon structure of the Golgi apparatus. Mol Cell Biochem 2013; 380:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Le Bourhis L, Dusseaux M, Bohineust A, Bessoles S, Martin E, Premel V et al MAIT cells detect and efficiently lyse bacterially‐infected epithelial cells. PLoS Pathog 2013; 9:e1003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Knodler LA, Celli J. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol 2011; 13:1319–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Leeansyah E, Loh L, Nixon DF, Sandberg JK. Acquisition of innate‐like microbial reactivity in mucosal tissues during human fetal MAIT‐cell development. Nat Commun 2014; 5:3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Keller AN, Eckle SBG, Xu W, Liu L, Hughes VA, Mak JYW et al Drugs and drug‐like molecules can modulate the function of mucosal‐associated invariant T cells. Nat Immunol 2017; 18:402–11. [DOI] [PubMed] [Google Scholar]
- 66. Patel O, Kjer‐Nielsen L, Le Nours J, Eckle SBG, Birkinshaw RW, Beddoe T et al Recognition of vitamin B metabolites by mucosal‐associated invariant T cells. Nat Commun 2013; 4:2142. [DOI] [PubMed] [Google Scholar]