

Summary
Dysfunctional elimination of cell debris, and the role of opsonins such as pentraxins, is of interest regarding systemic lupus erythematosus (SLE) pathogenesis. Interferon (IFN)‐α is typically elevated during SLE flares, and inhibits hepatocyte production of the pentraxin ‘C‐reactive protein’ (CRP), partly explaining the poor correlation between CRP levels and SLE disease activity. The extrahepatically produced ‘pentraxin 3’ (PTX3) shares waste disposal functions with CRP, but has not been studied extensively in SLE. We analysed serum PTX3 in SLE, and assessed its interference with IFN‐α in vitro. Serum samples from 243 patients with SLE and 100 blood donors were analysed regarding PTX3. Patient sera were analysed for IFN‐α, and genotyped for three PTX3 single nucleotide polymorphisms reported previously to associate with PTX3 levels. Stimulated PTX3 release was assessed in the presence or absence of IFN‐α in blood donor neutrophils and peripheral blood mononuclear cells (PBMC). Serum PTX3 was 44% lower in patients with SLE compared to blood donors (P < 0·0001) and correlated with leucocyte variables. Patients with undetectable IFN‐α had 29% higher median PTX3 level than patients with detectable IFN‐α (P = 0·01). PTX3 production by PBMC was inhibited by IFN‐α, whereas neutrophil degranulation of PTX3 was increased. No differences in PTX3 levels were observed between the SNPs. In conclusion, median serum PTX3 is lower in SLE (especially when IFN‐α is detectable) compared to blood donors. In addition to its potential consumption during waste disposal, it is plausible that IFN‐α also attenuates PTX3 by inhibiting synthesis by PBMC and/or exhausting PTX3 storage in neutrophil granules.
Keywords: biomarkers, interferon‐α, leucocytes, pentraxin, systemic lupus erythematosus
Introduction
The type I interferon (IFN) system is important in the pathogenesis of systemic lupus erythematosus (SLE) 1. Many patients with SLE present elevated circulating levels of IFN‐α and/or express IFN‐inducible genes, i.e. ‘the type I IFN signature’ during periods with raised disease activity. The main IFN‐α‐producing cells are the plasmacytoid dendritic cells (pDC) 2, which respond to viral nucleic acids via endosomal Toll‐like receptors (TLR)‐7 and −9 by massive IFN‐α production. The IFN‐α/β receptor (IFNAR) is expressed by almost all cell types, and binding of IFN‐α to its receptor has consequences such as B cell proliferation, plasma cell differentiation and antibody secretion 3.
Based on results from animal models, it has been hypothesized that the short pentraxin C‐reactive protein (CRP) has a protective role in SLE 4, 5. CRP is used widely as a biomarker of inflammation in bacterial infections and chronic inflammatory diseases such as rheumatoid arthritis, whereas it is an unreliable marker of inflammation in SLE 6, 7, 8 and viral infections 9. We have shown previously that IFN‐α inhibits interleukin (IL)‐6‐induced CRP production by human hepatocytes in vitro 10 and that serum IFN‐α levels, as well as CRP genotype rs1205, affects the CRP response in patients with SLE 11. The lack of correlation between CRP levels and disease activity is thus probably explained largely by CRP gene polymorphisms and activation of the type I IFN system.
The 340 kDa protein pentraxin 3 (PTX3) is a long pentraxin that is related structurally and functionally to CRP, but its production differs both with regard to its non‐hepatic cell origin as well as to inducing stimuli 12, 13. PTX3 is composed of eight identical protomers associated through disulphide bonds 14. Monocyte‐ and macrophage‐derived production is induced by lipopolysaccharide (LPS) and IL‐1β 15, 16, and the release of stored PTX3 from neutrophils is triggered by LPS and tumour necrosis factor (TNF) 15, 17. PTX3 can also be produced by myeloid DC, but not by pDC 18. A number of single nucleotide polymorphisms (SNPs) in the PTX3 gene have been found 19. Some SNPs have been reported to associate with different blood levels of PTX3 when comparing patients with acute myocardial infarction with controls 20. Variants of the SNP rs2305619 have been associated with differences in PTX3 plasma levels both at baseline and 24 h after lung transplantation 21.
Similar to CRP, PTX3 has a role in humoral innate immunity. It is involved in waste disposal of material released from dying cells as well as in the elimination of pathogens via complement protein C1q and classical complement activation 19. Another feature of PTX3 is its ability to bind to apoptotic cells and inhibit recognition by DC in order to maintain peripheral immune tolerance 19, 22. PTX3 can also induce macrophage secretion of immunosuppressive cytokines, such as IL‐10 and transforming growth factor (TGF)‐β 19, and has been suggested to be important during tissue repair and remodelling 23, 24, as well as in female fertility 25. Considering the waste‐handling functions of PTX3, and that a dysfunctional elimination process of cell debris is believed to be a key feature of SLE pathogenesis, together with observations that monocyte PTX3 production is affected by IFN‐γ 16, 26, it is highly relevant to investigate PTX3 in SLE. The scarce previous reports have pointed both at elevated 27, 28, 29 and lowered 30, 31, 32 PTX3 levels in patients with SLE compared to control subjects.
The aims of this study were to: (i) analyse PTX3 levels in clinically well‐characterized cases with SLE; and (ii) determine whether PTX3 is influenced by IFN‐α both in vitro and in vivo.
Materials and methods
Patients and control subjects
A total of 243 patients (211 women, 32 men, Table 1) diagnosed with SLE were included in the study. All patients took part in the prospective, structured follow‐up programme ‘KLURING’ (Swedish acronym for Clinical LUpus Register In Northeastern Gothia) 33 at the rheumatology out‐patient clinic, Linköping University Hospital, Sweden. Of the 243 patients, 205 (84%) met at least four of the 1982 American College of Rheumatology classification criteria (ACR‐82) 34. Another 38 patients (16%) fulfilled solely the 2012 Systemic Lupus International Collaborating Clinics (SLICC) classification criteria 35; 201 patients (83%) met both ACR‐82 and SLICC‐12. The patients were recruited consecutively. Most were prevalent cases (200 patients, 82%), but 43 patients (18%) had recent‐onset disease at the time of sampling. The mean disease duration was 10 years (range = 0–45 years). The SLE Disease Activity Index 2000 (SLEDAI‐2K) 36 and the physician's global assessment (PGA; 0–4) 37 were recorded at each visit and acquired organ damage according to the SLICC/ACR damage index (SDI) score 38 was registered prospectively after inclusion in KLURING. One hundred blood donors (50 women, 50 men; mean age = 46 years; range = 22–70 years) served as healthy controls for the PTX3 analyses.
Table 1.
Characteristics of the SLE patients, n = 243
| Mean (range) or % | |
|---|---|
| Age (years) | 49 (18–88) |
| Females | 86·8% |
| Caucasian ethnicity | 90·1% |
| Disease duration (years) | 10·3 (0–45) |
| Prednisolone dosage (mg/day) | 5 (0–60) |
| SLICC/ACR damage index (score) | 1·1 (0–9) |
| SLEDAI‐2K (score) | 2·8 (0–24) |
| PGA (score) | 0·5 (0–4) |
| Patients meeting SLICC‐12 (%) | 239 (98·4) |
| Fulfilled ACR‐82 criteria (n) | 4.7 (3–9) |
| ACR‐82 criteria | n (%) |
|---|---|
| 1. Malar rash | 106 (43·6) |
| 2. Discoid rash | 39 (16·0) |
| 3. Photosensitivity | 124 (51·0) |
| 4. Oral ulcers | 28 (11·5) |
| 5. Arthritis | 184 (75·7) |
| 6. Serositis | 92 (37·9) |
| 7. Renal disorder | 62 (25·5) |
| 8. Neurological disorder | 13 (5·3) |
| 9. Haematological disorder | 139 (57·2) |
| 10. Immunological disorder | 122 (50·2) |
| 11. IF‐ANA | 240 (98·8) |
SLEDAI‐2K = systemic lupus erythematosus disease activity index 2000; SLICC = Systemic Lupus International Collaborating Clinics; ACR = American College of Rheumatology; PGA = physician's global assessment; IF‐ANA = immunofluorescence microscopy anti‐nuclear antibodies.
At all patient visits, routine laboratory analyses [leucocytes, erythrocytes, platelets, urinalysis, CRP and erythrocyte sedimentation rate (ESR)] were performed at the clinical chemistry department, Linköping University Hospital.
Peripheral venous blood was drawn from each individual at baseline. Serum was prepared and stored at −70°C until analysed.
In addition, 15 of the included patients who all met ACR‐82 were selected for consecutive analyses (two to 13 visits per patient), due to their fluctuations in disease activity (i.e. SLEDAI‐2K peak score of at least 4) over time.
Leucocyte isolation and stimulation
Polymorphonuclear neutrophil granulocytes (neutrophils) and peripheral blood mononuclear cells (PBMC) were isolated by density gradient centrifugation from heparinized healthy donor blood. The blood was layered on top of gradient media consisting of Lymphoprep® (Axis‐Shield/Alere Technologies AS, Oslo, Norway) prelayered on top of Polymorphprep® (Axis‐Shield/Alere), and centrifuged for 30 min (480 g at room temperature). Neutrophils and PBMC, respectively, were collected and washed with phosphate‐buffered saline (PBS), pH 7·4. Trace amounts of erythrocytes contaminating the neutrophil fraction were lysed by two brief (35 s) exposures to 4°C ultra‐pure water. After additional washing in PBS, neutrophils were incubated in RPMI‐1640 supplemented with 2% fetal calf serum, 2 mM L‐glutamine, 100 IU/ml penicillin and 100 μg/ml streptomycin and 20 mM HEPES (ThermoFisher Scientific, Waltham, MA, USA). PBMCs were cultured in macrophage serum‐free medium (ThermoFisher Scientific) supplemented with 20 mM HEPES, 100 IU/ml penicillin and 100 μg/ml streptomycin.
During the experiments, neutrophils and PBMC were incubated at a concentration of 2 × 106 and 4 × 106 cells/ml, respectively, at 37°C with 5% CO2. IL‐1β, LPS Escherichia coli (serotype O26:B6), TNF and mouse immunoglobulin (Ig)G2A isotype control were obtained from R&D Systems (Abingdon, UK), IFN‐α2b (IntronA®) was from Schering‐Plough (Kenilworth, NJ, USA) and neutralizing mouse anti‐human IFN‐α receptor (IFNAR) chain 2 (clone MMHAR‐2) from PBL InterferonSource (Piscataway, NJ, USA). In receptor‐blocking experiments, the cells were preincubated with blocking monoclonal antibody or isotype control, respectively, 2 h prior to cytokine addition. Cell culture supernatants were collected, centrifuged and stored for a short time at −20°C prior to analysis.
Immunoassays
An enzyme‐linked immunosorbent assay (ELISA) kit was used to analyse PTX3 levels in SLE and control sera (Quantikine®; R&D Systems, Minneapolis, MN, USA). This plasma‐validated kit showed excellent correlation between plasma and serum (r = 0·972, P = 0·001, n = 6). For analysis of PTX3 in cell culture supernatants, a DuoSet ELISA was used (R&D Systems). Assays were performed according to the manufacturers’ instructions. Briefly, Costar (Corning, NY, USA) half‐area plates were coated with 2 µg/ml of mouse anti‐human PTX3 and incubated overnight. Plates were blocked by 1% bovine serum albumin in PBS for 1 h and incubated thereafter with samples and standards for 2 h. Biotinylated goat anti‐human PTX3 (360 ng/ml) was added and incubated for 2 h, followed by addition of streptavidin horseradish peroxidase (R&D Systems) diluted 1 : 200 and another 20 min of incubation. Plates were developed with tetramethylbenzidine substrate and the reaction was stopped by adding 1 M H2SO4. All incubations were performed at room temperature. Plate reader Sunrise (Tecan, Männedorf, Switzerland) and software Magellan version 7.1 (Tecan) were used.
IFN‐α levels were measured in sera from patients with SLE by a dissociation‐enhanced lanthanide fluoroimmunoassay (detection limit 1 unit/ml) at Uppsala University, described elsewhere 39.
IL‐1β and TNF were analysed by a high‐sensitivity multiplex magnetic bead assay (Milliplex, Millipore, Solna, Sweden).
Cell viability
A tetrazolium‐based assay was used to determine the relative number of viable cells (CellTiter 96 Aqueous One Solution Cell Proliferation Assay; Promega, Madison, WI, USA). Optical density was measured at 490 nm (Sunrise, Tecan) and the number of viable cells was expressed as the percentage of unstimulated control cells.
Genotyping
Genomic DNA was obtained from whole blood samples using a QIAamp DNA Blood Midi kit (Qiagen, Hilden, Germany). The SNPs rs3816527, rs3845978 and rs2305619, selected based on their associations with PTX3 blood levels 19, 20, 40, were genotyped using the Infinium ImmunoChip (Illumina Inc., San Diego, CA, USA). Genotyping was performed at the SNP&SEQ Technology Platform at the National Genomics Infrastructure (NGI) hosted by the Science for Life Laboratory in Uppsala, Sweden.
Statistics
Wilcoxon's matched‐pairs signed‐rank test was used to evaluate differences in the neutrophil and PBMC experiments. The Mann–Whitney U‐test was used to evaluate differences in PTX3 levels between patients and controls and between SLE cases with and without detectable IFN‐α. Spearman's correlation was used to determine the association between PTX3 and disease variables. The Kruskal–Wallis test was used to evaluate differences in PTX3 levels between the genetic variants of the SNPs. Two‐tailed P‐values < 0·05 were considered significant. Statistical analyses were performed with spss statistics version 22 (IBM, Armonk, NY, USA) or GraphPad Prism version 5.04 (GraphPad Software, La Jolla, CA, USA).
Ethics
Oral and written informed consent was obtained from all subjects. The study protocol was approved by the Regional Ethics Review Board in Linköping (Dnr: M75‐08/2008).
Results
PTX3 levels in SLE and healthy controls
Levels of PTX3 were 44% lower (P < 0·0001) among the patients with SLE (median 2·5 ng/ml) compared to the healthy controls (median 4·5 ng/ml) (Fig. 1). A less pronounced, but statistically significant, inverse correlation was found between IFN‐α and PTX3 (r = –0·154, P = 0·017) for all patients. Hence, we compared patients with and without detectable IFN‐α levels. Patients without detectable IFN‐α (IFN‐α < 1 U/ml) showed a 29% higher median serum level (2·7 ng/ml) of PTX3 compared to patients with detectable IFN‐α (IFN‐α > 1 U/ml) (median 2·1 ng/ml), P = 0.01 (Fig. 1). There were no significant differences in PTX3 levels between men and women, either among the controls or among the patients. We found no significant correlation between PTX3 and the inducing cytokines IL‐1β and TNF in patients with SLE, nor between PTX3 and CRP (data not shown).
Figure 1.
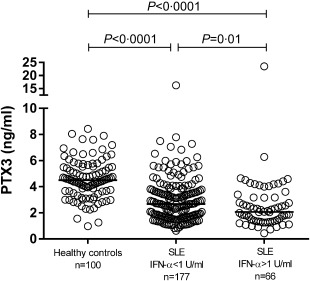
Serum pentraxin 3 (PTX3) levels determined by enzyme‐linked immunosorbent assay (ELISA) in healthy controls and patients with systemic lupus erythematosus (SLE). Serum levels of PTX3 were significantly lower in the patients with SLE (median 2·5 ng/ml) compared to the healthy controls (median 4·5 ng/ml). Patients without detectable interferon (IFN)‐α (< 1 U/ml) showed significantly higher levels (median 2·7 ng/ml) of PTX3 compared with patients with detectable IFN‐α (> 1 U/ml) (median 2·1 ng/ml). Solid lines represent median values. Note axis break.
Correlation analyses between PTX3 and different disease activity variables revealed weak significant correlations with leucocyte‐associated variables; leucocyte count (r = 0·293, P < 0·0001), monocytes (r = 0·143, P = 0·027), neutrophils (r = 0·262, P < 0·0001) and urinary leucocytes (r = –0·242, P = 0·045). No association was found between PTX3 and disease activity defined as SLEDAI‐2K (data not shown).
PTX3 levels were also analysed in the consecutive samples from 15 cases (Supporting information, Fig. S1 shows longitudinal data of each patient). PTX3 levels at the highest and lowest disease activity (defined by SLEDAI‐2K and PGA) were compared, but no significant differences were found (data not shown).
No influence of PTX3 genetic variants on PTX3 serum levels
Patients with SLE were genotyped for three PTX3 SNPs, rs3816527, rs3845978 and rs2305619, and the effects on PTX3 serum levels was examined. No significant differences in PTX3 serum levels were observed between the genetic variants (Fig. 2).
Figure 2.
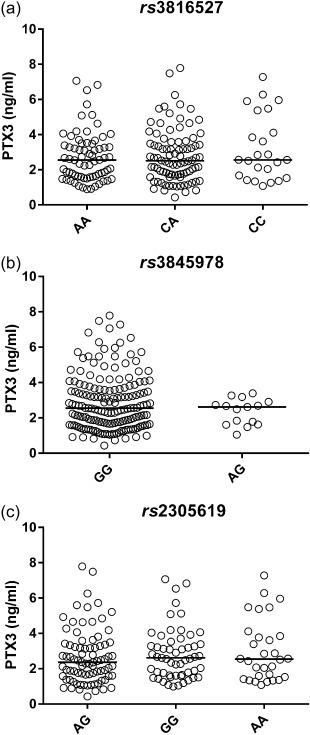
Influence of pentraxin 3 (PTX3) genetic variants on PTX3 serum levels. Patients with systemic lupus erythematosus (SLE) were genotyped for three PTX3 single nucleotide polymorphisms (SNPs), rs3816527, rs3845978 and rs2305619. No significant differences were observed based on gene variants.
Effects of IFN‐α on PTX3 release in neutrophils and PBMC
In order to investigate a possible mechanistic connection to the inverse correlation between IFN‐α and PTX3, the influence of IFN‐α on PTX3 release from neutrophils and PBMC was analysed. Production of PTX3 in PBMC was induced by IL‐1β and LPS and to some extent by TNF (Fig. 3a–c). After 3 h of stimulation, no statistically significant differences were seen in PMBC production of PTX3 (Fig. 3a). IL‐1β‐induced PTX3 production in PBMC was inhibited significantly by IFN‐α with a 17% median decrease at 6 h and a 78% median decrease at 24 h (Fig. 3b,c). The PTX3 production induced by LPS was inhibited significantly with a 29% decrease at 6 h and a 54% decrease at 24 h (Fig. 3b,c). TNF‐induced PTX3 production was inhibited significantly with a 30% decrease at 6 h (Fig. 3b).
Figure 3.
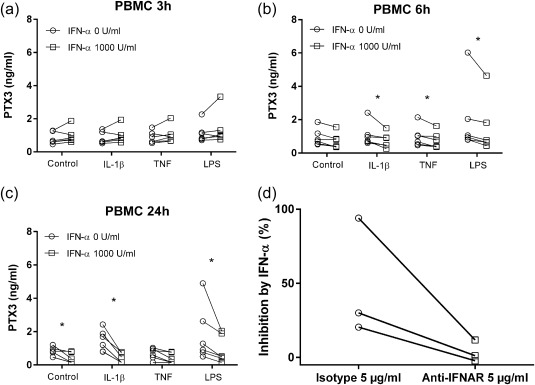
Pentraxin 3 (PTX3) production from peripheral blood mononuclear cells (PBMC). The effect of interferon (IFN)‐α on PTX3 production induced by interleukin (IL)‐1β (20 ng/ml), tumour necrosis factor (TNF) (25 ng/ml) or lipopolysaccharide (LPS) (10 ng/ml) in PBMC stimulated for (a) 3 h, (b) 6 h and (c) 24 h (n = 6). (d) Percentage inhibition of PTX3 by IFN‐α. Effect of a neutralizing antibody to type I interferon receptor (IFNAR). PBMC were preincubated with 5 µg of antibody for 2 h and then stimulated with IFN‐α and IL‐1β, n = 3. *P < 0·05.
We observed no reduced cell viability due to IFN‐α exposure in the cell viability assay (n = 3, data not shown). To ensure that the IFN‐α‐mediated suppression of PBMC PTX3 was mediated by receptor‐dependent signalling, we used a neutralizing antibody to the type I IFN receptor (IFNAR). PBMC were preincubated with 5 µg of antibody for 2 h and then stimulated with IFN‐α and IL‐1β. Presence of this receptor‐blocking antibody reversed the IFN‐α‐dependent inhibition (Fig. 3d).
Neutrophil release of PTX3 was induced by TNF and LPS, but not by IL‐1β (Fig. 4a–d), and the stimulated PTX3 release was similar at all sampling time‐points. LPS‐induced neutrophil release of PTX3 was increased significantly by IFN‐α with a 30% median increase at 1·5 h, 19% at 3 h and 18% at 6 h (Fig. 4a–c). Furthermore, TNF‐induced release of PTX3 was amplified significantly by IFN‐α at 3 and 16 h, with a median increase of 22 and 34%, respectively (Fig. 4b,d).
Figure 4.
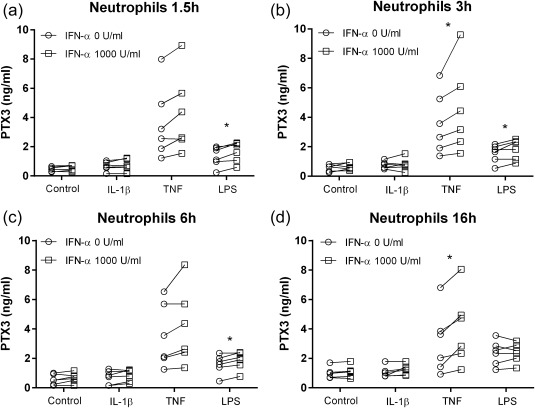
Pentraxin 3 (PTX3) release from neutrophils. The effect of interferon (IFN)‐α on PTX3 release induced by interleukin (IL)‐1β (20 ng/ml), tumour necrosis factor (TNF) (25 ng/ml) or lipopolysaccharide (LPS) (10 ng/ml) in neutrophils stimulated for (a) 1·5 h, (b) 3 h, (c) 6 h and (d) 16 h (n = 6).
Discussion
Accumulation of cellular debris due to insufficient elimination is considered a key feature of lupus pathogenesis. Pentraxins such as CRP and PTX3 have biological properties that contribute to clearance of dying cells, and low levels of these proteins could thus result in the accumulation of cell debris and subsequent inflammation and autoimmunity 41, 42. Similarly, CRP supplementation to lupus model mice leads to decreased levels of autoantibodies, fewer autoimmune manifestations and enhanced survival 43. Conversely, a recent study demonstrated that immunization with PTX3 in a murine model led to anti‐PTX3 antibody production which delayed lupus‐like nephritis and prolonged survival 44. Together with our findings, this points towards a complex biological regulation and role of PTX3.
In the present study we found that serum levels of PTX3 were markedly lower among patients with SLE compared to healthy controls, due perhaps to circulating IFN‐α. Based on results from our in‐vitro experiments, we draw the conclusion that circulating IFN‐α cause reduction in PTX3 production from PBMC. This theory is strengthened by the fact that patients with detectable IFN‐α had lower PTX3 levels than patients without detectable IFN‐α. Consequently, it is likely that IFN‐α, together with the potential PTX3 consumption during waste disposal of dying cells, is a major cause of lowered systemic levels of PTX3 in SLE.
The biological roles of PTX3 in SLE are far from proved, but results from animal models of lupus suggest a protective role of pentraxins in SLE 4, 5. As both CRP and PTX3 contribute to the clearance of apoptotic cells and inhibits self‐recognition by DC, a hampered production, or other exhaustion, of CRP and PTX3 could enhance further the problems of deficient waste disposal in SLE. Conversely, both circulating and tissue levels of PTX3 were reported recently to associate with lupus nephritis, and PTX3 was suggested as a biomarker of tubulointerstitial injury 45. Moreover, PTX3 plays part in angiogenesis and remodelling of the extracellular matrix 24, 46.
Circulating PTX3 has been reported previously to be both elevated 27, 28 and lowered 30, 31, 32 in SLE. The reasons to the discrepancies between the studies remain unknown, but may be due to differences in study design, e.g. selection of study population (sex and age may influence PTX3 levels 47), and definition of disease activity, ethnicity, detection methodologies and genetics. For the latter, some PTX3 gene variants have been associated with differences in PTX3 levels 20, 21. However, genotyping of three PTX3 SNPs in the present study revealed no significant differences in PTX3 serum levels based on genetic variants. To our knowledge, the influence of SNPs on PTX3 blood levels in SLE has not been investigated previously. Differences in absolute PTX3 levels between studies may be related to the use of serum versus plasma.
To pursue the inverse relation between IFN‐α and PTX3 mechanistically, in vitro studies on PBMC and neutrophils were performed. PTX3 production by PBMC increased with time, especially the IL‐1β induced production, and IFN‐α inhibited both IL‐1β‐ and LPS‐stimulated PTX3 production at 6 and 24 h of incubation. Furthermore, PTX3 was inhibited in control PBMC at 24 h, implying that IFN‐α also inhibits the baseline synthesis. Doni et al. examined the effect of IFN‐α on PTX3 production 26, albeit in purified myeloid DC. However, in their study IFN‐α had no suppressive effect, but rather amplified PTX3 production in response to LPS. PTX3 is stored in the specific granules of neutrophils, and becomes exocytosed upon stimulation 17, 46, e.g. following IFN‐α activation 48. Accordingly, in the present study, stimulated release of neutrophil PTX3 was increased significantly by IFN‐α in LPS‐stimulated cells. Furthermore, the TNF‐induced neutrophil release of PTX3 was increased by IFN‐α. Given that PTX3 levels were relatively stable in cell culture supernatants over time, our results support that it is rather a quick release/degranulation, but not de‐novo synthesis of the protein by the neutrophils. In accordance, neutrophils have been described as a reservoir of ‘ready‐to‐use’ PTX3 17. Neutrophils exposed to IFN‐α are primed to become activated by immune complexes and subsequent induction of neutrophil extracellular trap (NET) formation 49, allowing co‐localization of PTX3 17. Speculatively, raised IFN‐α may provoke tissue‐recruited neutrophils to release and deposit PTX3 by degranulation. Moreover, neutrophil degranulation has been suggested as a major source of local elevation of PTX3 in rheumatoid arthritis 50.
In addition to IFN‐α‐dependent inhibition of PTX3 production in PBMCs, autoantibodies directed towards PTX3 51 and tissue deposition of PTX3 could possibly explain low circulating levels in patients in general 45. Although we found no significant association between PTX3 and CRP levels, it is interesting to compare with our previous finding of circulating anti‐CRP antibodies in SLE 52 as well as co‐localization of glomerular IgG‐, CRP‐ and C1q‐deposits in lupus nephritis 53. The typically low levels of circulating CRP in SLE are not explained by circulating anti‐CRP antibodies. It would be interesting to investigate if this also holds true for anti‐PTX3 in the present study group. Furthermore, many patients presenting with increased expression of IFN inducible genes (the type I IFN signature) lack measurable IFN‐α in serum 54, 55, indicating that IFN‐α can exert its biological effects locally and/or in concentrations that could not be measured properly. Such effects could explain why patients without detectable IFN‐α have lower levels compared to healthy controls.
In conclusion, we have shown that IFN‐α exerts diverse effects on neutrophils and PBMCs, leading to release of PTX3 from neutrophils and attenuated synthesis of PTX3 by PBMCs in vitro. The net effect is probably reduced circulating levels of PTX3, as lupus patients with raised circulating IFN‐α have reduced levels of circulating PTX3. Given the important role of PTX3 in the clearance of dying cells and the prominent activation of the type I IFN system in SLE, our results suggest that the suppressed PTX3 levels in SLE contribute to the autoimmune disease process by providing autoantigens to B cells and endogenous IFN inducers to pDC, all of which further sustain the disease. This has implications regarding waste disposal and peripheral immune tolerance in SLE.
Disclosure
No disclosures.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site
Fig. S1. Individual systemic lupus erythematosus (SLE) manifestations and longitudinal variations of pentraxin 3 (PTX3) and interferon (IFN)‐α. The graphs illustrate individual variations in PTX3 and IFN‐α over time for the 15 patients who were followed consecutively (A–O). Observe that axes and scales are different in the graphs.
Acknowledgements
This study was supported by the Swedish Society for Medical Research, Region Östergötland, the Swedish Research Council, the Swedish Rheumatism Association, the Swedish Society of Medicine, the Professor Nanna Svartz Foundation and King Gustaf V's 80‐year foundation.
We thank Marianne Peterson for biobank administration, Johanna Sandling for performing the genotyping and Andrei Alexsson for quality control of the genotyping.
References
- 1. Bengtsson AA, Ronnblom L. Systemic lupus erythematosus: still a challenge for physicians. J Intern Med 2017; 281:52–64. [DOI] [PubMed] [Google Scholar]
- 2. Ronnblom L, Alm GV. The natural interferon‐alpha producing cells in systemic lupus erythematosus. Hum Immunol 2002; 63:1181–93. [DOI] [PubMed] [Google Scholar]
- 3. Eloranta ML, Alm GV, Ronnblom L. Disease mechanisms in rheumatology – tools and pathways: plasmacytoid dendritic cells and their role in autoimmune rheumatic diseases. Arthritis Rheum 2013; 65:853–63. [DOI] [PubMed] [Google Scholar]
- 4. Rodriguez W, Mold C, Marnell LL et al Prevention and reversal of nephritis in MRL/lpr mice with a single injection of C‐reactive protein. Arthritis Rheum 2006; 54:325–35. [DOI] [PubMed] [Google Scholar]
- 5. Szalai AJ, Weaver CT, McCrory MA et al Delayed lupus onset in (NZB x NZW)F1 mice expressing a human C‐reactive protein transgene. Arthritis Rheum 2003; 48:1602–11. [DOI] [PubMed] [Google Scholar]
- 6. Barnes EV, Narain S, Naranjo A et al High sensitivity C‐reactive protein in systemic lupus erythematosus: relation to disease activity, clinical presentation and implications for cardiovascular risk. Lupus 2005; 14:576–82. [DOI] [PubMed] [Google Scholar]
- 7. Gabay C, Roux‐Lombard P, de Moerloose P, Dayer JM, Vischer T, Guerne PA. Absence of correlation between interleukin 6 and C‐reactive protein blood levels in systemic lupus erythematosus compared with rheumatoid arthritis. J Rheumatol 1993; 20:815–21. [PubMed] [Google Scholar]
- 8. Rezaieyazdi Z, Sahebari M, Hatef MR et al Is there any correlation between high sensitive CRP and disease activity in systemic lupus erythematosus? Lupus 2011; 20:1494–500. [DOI] [PubMed] [Google Scholar]
- 9. Lubell Y, Blacksell SD, Dunachie S et al Performance of C‐reactive protein and procalcitonin to distinguish viral from bacterial and malarial causes of fever in Southeast Asia. BMC Infect Dis 2015; 15:511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Enocsson H, Sjowall C, Skogh T, Eloranta ML, Ronnblom L, Wettero J. Interferon‐alpha mediates suppression of C‐reactive protein: explanation for muted C‐reactive protein response in lupus flares?. Arthritis Rheum 2009; 60:3755–60. [DOI] [PubMed] [Google Scholar]
- 11. Enocsson H, Sjowall C, Kastbom A et al Association of serum C‐reactive protein levels with lupus disease activity in the absence of measurable interferon‐alpha and a C‐reactive protein gene variant. Arthritis Rheumatol 2014; 66:1568–73. [DOI] [PubMed] [Google Scholar]
- 12. Manfredi AA, Rovere‐Querini P, Bottazzi B, Garlanda C, Mantovani A. Pentraxins, humoral innate immunity and tissue injury. Curr Opin Immunol 2008; 20:538–44. [DOI] [PubMed] [Google Scholar]
- 13. Inforzato A, Rivieccio V, Morreale AP et al Structural characterization of PTX3 disulfide bond network and its multimeric status in cumulus matrix organization. J Biol Chem 2008; 283:10147–61. [DOI] [PubMed] [Google Scholar]
- 14. Inforzato A, Baldock C, Jowitt TA et al The angiogenic inhibitor long pentraxin PTX3 forms an asymmetric octamer with two binding sites for FGF2. J Biol Chem 2010; 285:17681–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Imamura M, Kawasaki T, Savchenko AS et al Lipopolysaccharide induced expression of pentraxin 3 in human neutrophils and monocyte‐derived macrophages. Cell Immunol 2007; 248:86–94. [DOI] [PubMed] [Google Scholar]
- 16. Polentarutti N, Picardi G, Basile A et al Interferon‐gamma inhibits expression of the long pentraxin PTX3 in human monocytes. Eur J Immunol 1998; 28:496–501. [DOI] [PubMed] [Google Scholar]
- 17. Jaillon S, Peri G, Delneste Y et al The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J Exp Med 2007; 204:793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doni A, Peri G, Chieppa M et al Production of the soluble pattern recognition receptor PTX3 by myeloid, but not plasmacytoid, dendritic cells. Eur J Immunol 2003; 33:2886–93. [DOI] [PubMed] [Google Scholar]
- 19. Ortega‐Hernandez OD, Bassi N, Shoenfeld Y, Anaya JM. The long pentraxin 3 and its role in autoimmunity. Semin Arthritis Rheumat 2009; 39:38–54. [DOI] [PubMed] [Google Scholar]
- 20. Barbati E, Specchia C, Villella M et al Influence of pentraxin 3 (PTX3) genetic variants on myocardial infarction risk and PTX3 plasma levels. PLoS One 2012; 7:e53030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Diamond JM, Meyer NJ, Feng R et al Variation in PTX3 is associated with primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med 2012; 186:546–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vezzoli M, Sciorati C, Campana L et al The clearance of cell remnants and the regeneration of the injured muscle depend on soluble pattern recognition receptor PTX3. Mol Med 2016; 22:809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Doni A, Musso T, Morone D et al An acidic microenvironment sets the humoral pattern recognition molecule PTX3 in a tissue repair mode. J Exp Med 2015; 212:905–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leali D, Inforzato A, Ronca R et al Long pentraxin 3/tumor necrosis factor‐stimulated gene‐6 interaction: a biological rheostat for fibroblast growth factor 2‐mediated angiogenesis. Arterioscler Thromb Vasc Biol 2012; 32:696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Salustri A, Garlanda C, Hirsch E et al PTX3 plays a key role in the organization of the cumulus oophorus extracellular matrix and in in vivo fertilization. Development 2004; 131:1577–86. [DOI] [PubMed] [Google Scholar]
- 26. Doni A, Michela M, Bottazzi B et al Regulation of PTX3, a key component of humoral innate immunity in human dendritic cells: stimulation by IL‐10 and inhibition by IFN‐gamma. J Leukocyte Biol 2006; 79:797–802. [DOI] [PubMed] [Google Scholar]
- 27. Shimada Y, Asanuma YF, Yokota K et al Pentraxin 3 is associated with disease activity but not atherosclerosis in patients with systemic lupus erythematosus. Mod Rheumatol 2014; 24:78–85. [DOI] [PubMed] [Google Scholar]
- 28. Cieslik P, Hrycek A. Pentraxin 3 as a biomarker of local inflammatory response to vascular injury in systemic lupus erythematosus. Autoimmunity 2015; 48:242–50. [DOI] [PubMed] [Google Scholar]
- 29. Assandri R, Monari M, Colombo A, Dossi A, Montanelli A. Pentraxin 3 plasma levels and disease activity in systemic lupus erythematosus. Autoimmune Dis 2015; 2015:354014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bassi N, Del Prete D, Ghirardello A et al PTX3, anti‐PTX3, and anti‐C1q autoantibodies in lupus glomerulonephritis. Clin Rev Allergy Immunol 2015; 49:217–26. [DOI] [PubMed] [Google Scholar]
- 31. Fazzini F, Peri G, Doni A et al PTX3 in small‐vessel vasculitides: an independent indicator of disease activity produced at sites of inflammation. Arthritis Rheum 2001; 44:2841–50. [DOI] [PubMed] [Google Scholar]
- 32. Hollan I, Bottazzi B, Cuccovillo I et al Increased levels of serum pentraxin 3, a novel cardiovascular biomarker, in patients with inflammatory rheumatic disease. Arthritis Care Res 2010; 62:378–85. [DOI] [PubMed] [Google Scholar]
- 33. Ighe A, Dahlstrom O, Skogh T, Sjowall C. Application of the 2012 Systemic Lupus International Collaborating Clinics classification criteria to patients in a regional Swedish systemic lupus erythematosus register. Arthritis Res Ther 2015; 17:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tan EM, Cohen AS, Fries JF et al The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25:1271–7. [ [DOI] [PubMed] [Google Scholar]
- 35. Petri M, Orbai AM, Alarcon GS et al Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 2012; 64:2677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gladman DD, Ibanez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol 2002; 29:288–91. [PubMed] [Google Scholar]
- 37. Scott DL. A simple index to assess disease activity in rheumatoid arthritis. J Rheumatol 1993; 20:582–4. [PubMed] [Google Scholar]
- 38. Gladman D, Ginzler E, Goldsmith C et al The development and initial validation of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis Rheum 1996; 39:363–9. [DOI] [PubMed] [Google Scholar]
- 39. Cederblad B, Blomberg S, Vallin H, Perers A, Alm GV, Ronnblom L. Patients with systemic lupus erythematosus have reduced numbers of circulating natural interferon‐alpha‐ producing cells. J Autoimmun 1998; 11:465–70. [DOI] [PubMed] [Google Scholar]
- 40. Chiarini M, Sabelli C, Melotti P et al PTX3 genetic variations affect the risk of Pseudomonas aeruginosa airway colonization in cystic fibrosis patients. Genes Immun 2010; 11:665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rhodes B, Furnrohr BG, Vyse TJ. C‐reactive protein in rheumatology: biology and genetics. Nat Rev Rheumatol 2011; 7:282–9. [DOI] [PubMed] [Google Scholar]
- 42. Russell AI, Cunninghame Graham DS, Shepherd C et al Polymorphism at the C‐reactive protein locus influences gene expression and predisposes to systemic lupus erythematosus. Hum Mol Genet 2004; 13:137–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Du Clos TW, Zlock LT, Hicks PS, Mold C. Decreased autoantibody levels and enhanced survival of (NZB x NZW) F1 mice treated with C‐reactive protein. Clin Immunol Immunopathol 1994; 70:22–7. [DOI] [PubMed] [Google Scholar]
- 44. Gatto M, Ghirardello A, Luisetto R et al Immunization with pentraxin 3 (PTX3) leads to anti‐PTX3 antibody production and delayed lupus‐like nephritis in NZB/NZW F1 mice. J Autoimmun 2016; 74:208–16. [DOI] [PubMed] [Google Scholar]
- 45. Pang Y, Tan Y, Li Y et al Pentraxin 3 is closely associated with tubulointerstitial injury in lupus nephritis: a large multicenter cross‐sectional study. Medicine (Baltimore) 2016; 95:e2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maina V, Cotena A, Doni A et al Coregulation in human leukocytes of the long pentraxin PTX3 and TSG‐6. J Leukocyte Biol 2009; 86:123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yamasaki K, Kurimura M, Kasai T, Sagara M, Kodama T, Inoue K. Determination of physiological plasma pentraxin 3 (PTX3) levels in healthy populations. Clin Chem Lab Med 2009; 47:471–7. [DOI] [PubMed] [Google Scholar]
- 48. Corssmit EP, Heijligenberg R, Hack CE, Endert E, Sauerwein HP, Romijn JA. Effects of interferon‐alpha (IFN‐alpha) administration on leucocytes in healthy humans. Clin Exp Immunol 1997; 107:359–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Garcia‐Romo GS, Caielli S, Vega B et al Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 2011; 3:73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weitoft T, Larsson A, Saxne T et al Pentraxin 3 in serum and synovial fluid of patients with rheumatoid arthritis with and without autoantibodies. Scand J Rheumatol 2016; doi: 10.1080/03009742.2016.1244288. [DOI] [PubMed] [Google Scholar]
- 51. Bassi N, Ghirardello A, Blank M et al IgG anti‐pentraxin 3 antibodies in systemic lupus erythematosus. Ann Rheumat Dis 2010; 69:1704–10. [DOI] [PubMed] [Google Scholar]
- 52. Sjowall C, Eriksson P, Almer S, Skogh T. Autoantibodies to C‐reactive protein is a common finding in SLE, but not in primary Sjogren's syndrome, rheumatoid arthritis or inflammatory bowel disease. J Autoimmun 2002; 19:155–60. [DOI] [PubMed] [Google Scholar]
- 53. Sjowall C, Olin AI, Skogh T et al C‐reactive protein, immunoglobulin G and complement co‐localize in renal immune deposits of proliferative lupus nephritis. Autoimmunity 2013; 46:205–14. [DOI] [PubMed] [Google Scholar]
- 54. Bengtsson AA, Sturfelt G, Truedsson L et al Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus 2000; 9:664–71. [DOI] [PubMed] [Google Scholar]
- 55. Baechler EC, Batliwalla FM, Karypis G et al Interferon‐inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA 2003; 100:2610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site
Fig. S1. Individual systemic lupus erythematosus (SLE) manifestations and longitudinal variations of pentraxin 3 (PTX3) and interferon (IFN)‐α. The graphs illustrate individual variations in PTX3 and IFN‐α over time for the 15 patients who were followed consecutively (A–O). Observe that axes and scales are different in the graphs.