

Summary
B‐lymphocyte hyperactivity in systemic lupus erythematosus (SLE) is T‐cell‐dependent, and CD4+ T‐cell activation is essential to SLE pathogenesis. However, the mechanism of the deregulation of CD4+ T cells in SLE is largely unknown. T‐cell immunoglobulin and ITIM domain (TIGIT) is a new inhibitory receptor preferentially expressed on activated CD4+ T cells. Here, we address the role of TIGIT in the pathogenesis of SLE. Our results showed that TIGIT expression on CD4+ T cells was significantly elevated in patients with SLE and highly correlated with the activity of the disease. TIGIT + CD4+ T cells from both healthy individuals and patients with SLE had a more activated phenotype than TIGIT − CD4+ T cells. In contrast, the activation, proliferation and cytokine production potential of TIGIT + CD4+ T cells were significantly lower than those of TIGIT − CD4+ T cells. Furthermore, activation of the TIGIT pathway by using CD155 could substantially down‐regulate the activities of CD4+ T cells from SLE patients in vitro, and in vivo administration of CD155 resulted in a delayed development of SLE in MRL/lpr mice. TIGIT is a powerful negative regulator of CD4+ T cells in SLE, which suggests that the TIGIT signalling pathway may be used as a potential therapeutic target for treating this disease.
Keywords: CD4+ T cells, systemic lupus erythematosus, therapeutic target, TIGIT
Abbreviations
- CFSE
carboxyfluorescein diacetate succinimidyl ester
- IFN‐γ
interferon‐γ
- NK
natural killer
- PBMCs
peripheral blood mononuclear cells
- PD‐1
programmed death‐1
- PE
phycoerythrin
- PerCP
peridinin chlorophyll protein
- PHA
phytohaemagglutinin
- PMA
phorbol 12‐myristate 13‐acetate
- SLE
systemic lupus erythematosus
- SLEDAI
SLE Disease Activity Index
- Tfh
follicular helper T
- TIGIT
T‐cell immunoglobulin and ITIM domain
- Treg
regulatory T
Introduction
Systemic lupus erythematosus (SLE) is characterized by the overproduction of autoantibodies, which potentially cause immune‐complex‐related inflammation in various tissues and organs.1 Therefore, this condition was traditionally thought to be a B‐cell driven disease. However, B lymphocyte hyperactivity in SLE is T‐cell‐dependent.2 It is indeed impossible for the B cells to trigger SLE‐related inflammation without the activation of CD4+ T cells.3, 4, 5 Although we know that CD4+ T‐cell deregulation contributes to SLE pathogenesis, but the mechanism is still largely unknown.
It was recently shown that a new inhibitory receptor, named T‐cell immunoglobulin and ITIM domain (TIGIT), is expressed mainly on activated T cells and natural killer (NK) cells.6 CD155 is identified as the physical ligand of TIGIT. TIGIT/CD155 engagement can inhibit T‐cell responses in a cell‐intrinsic manner by directly targeting the T‐cell receptor signalling cascade as well as via the induction of tolerogenic dendritic cells.7, 8, 9 Regulatory T (Treg) cells, also through expression of the co‐inhibitory molecule TIGIT, selectively inhibit pro‐inflammatory T helper type 1 and type 17 cell responses.10, 11 Furthermore, TIGIT‐expressing T follicular helper (Tfh) cells exhibit strong B‐cell help functions.12 Previous studies have shown that genetic ablation or antibody blockade of TIGIT enhances CD4+ T‐cell priming and exacerbates the severity of experimental autoimmune encephalitis and rheumatoid arthritis.13, 14, 15, 16 Besides, the TIGIT pathway has also been shown to play an important role in the regulation of other disease models, such as cancer and chronic viral infection.17 In addition, our previous study has shown that TIGIT expression levels on human NK cells correlate with functional heterogeneity among healthy individuals.18
Although previous evidence has indicated that the TIGIT pathway inhibits T‐cell responses in several disease models, whether TIGIT can regulate the function of CD4+ T cells in SLE is unknown. In the present study, we have demonstrated that TIGIT is a powerful negative regulator of CD4+ T cells in patients with SLE or in MRL/lpr mice, which suggests that the TIGIT signalling pathway may be used as a potential therapeutic target for treating this disease.
Material and methods
Patients
Ninety‐four patients with SLE and 64 healthy individuals were recruited from Tongji Hospital, the largest hospital in the central region of China. All of the patients fulfilled the revised criteria for SLE of the American College of Rheumatology.19 Disease activity of SLE was assessed by the SLE Disease Activity Index (SLEDAI).20 Patient demographics, clinical data, as well as laboratory test results were retrieved from the patients' medical records. The demographic characteristics and clinical presentations of the SLE patients are shown in the Supplementary material (Table S1). The study was approved by the Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China. All participants provided written informed consent.
Mice
Six‐week‐old female MRL/lpr mice were purchased from The Jackson Laboratory (Bar Harbor, ME), housed under specific pathogen‐free conditions at the animal facilities of Tongji Medical College animal facility, Huazhong University of Science and Technology, Wuhan, China. All of the mice involved in the current study were approved by the Huazhong University of Science and Technology Animal Care and Use Committee. After 3 days of adaptive feeding, the mice were injected intraperitoneally with 50 µg of recombinant mouse CD155 protein (6909‐CD‐050, R&D Systems, Minneapolis, MN) once a week for 4 weeks. Administration of the same amount of mouse IgG (Sigma‐Aldrich, St Louis, MO) diluted in PBS was served as control. The mice were examined daily, and the date was recorded when remarkably enlarged lymph nodes were observed and touched. In some experiments, mice were killed after 1 month of treatment, and spleen and kidney were harvested as needed.
Cell preparation and activation
Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized blood of patients with SLE and healthy individuals by using Ficoll‐Hypaque density gradients (Sigma‐Aldrich). All cells were cultured in RPMI‐1640 medium (GIBCO, Grand Island, NY) supplemented with 10% fetal bovine serum and maintained at 37° in a 5% CO2 humidified atmosphere. Splenic lymphocytes were isolated from MRL/lpr mice by density gradient centrifugation of splenocytes on lymphocyte separation medium (Dakewe Biotech Company Ltd, Shenzhen, China). For cell stimulation studies, PBMCs or splenic lymphocytes were cultured with phytohaemagglutinin (PHA) (5 µg/ml, Sigma‐Aldrich) for 24 hr in the presence of recombinant human CellExp™ CD155 protein (0·5 µg/ml; Biovision, Milpitas, CA), recombinant mouse CD155 protein (0·5 µg/ml; R&D Systems), or IgG control, respectively. To detect the intracellular interferon‐γ (IFN‐γ) production, PMA (50 ng/ml; Sigma‐Aldrich) was also used for stimulation, and monensin (1 μm; eBioscience, San Diego, CA) was added to cultures for the last 6 hr of incubation. After culture, the cells were collected and analysed by flow cytometry.
Flow cytometric analysis
Cell surface staining was performed on PBMCs or splenic lymphocytes using the following anti‐human or anti‐mouse monoclonal antibodies: anti‐CD3‐peridinin chlorophyll protein (PerCP) Cy5.5 (HIT3a), anti‐CD4‐FITC (RM4‐5), anti‐CD4‐PerCP Cy5.5 (A161A1), anti‐CD25‐FITC (M‐A251) and anti‐CD69‐FITC (FN50) (Biolegend, San Diego, CA); anti‐CD8‐phycoerythrin (PE) (RPA‐T8), anti‐CD56‐PE (B159), anti‐CD69‐PerCP Cy5.5 (FN50), anti‐Ki‐67‐FITC (B56), anti‐HLA‐DR‐FITC (G46‐6), anti‐CD45RA‐FITC (5H9), anti‐CXCR5‐PE (RF8B2), anti‐CD127‐PE (HIL‐7R‐M21), anti‐CD154‐PE (TRAP1) and anti‐CD226‐PE (11A8) (BD Biosciences, San Jose, CA); anti‐PD‐1‐FITC (J116), anti‐TIGIT‐allophycocyanin (MBSA43), and anti‐CD25‐PE (PC61.5) (eBioscience). Isotype controls with irrelevant specificities were included as negative controls. All of these cell suspensions were incubated for 30 min on ice. For intracellular staining, the cells were fixed and permeabilized with Fixation and Permeabilization Buffer (BD Biosciences) and stained with anti‐IFN‐γ‐allophycocyanin (4S.B3) (BD Biosciences) and anti‐IFN‐γ‐PE (XMG1.2) (eBioscience) for 30 min in the dark. After washing, the pellets were resuspended in 500 μl cold staining buffer and analysed with FACSCalibur or FACSCanto II flow cytometers (BD Biosciences). Data analysis was performed using flowjo version 7.6.1 software (TreeStar, Ashland, OR).
Proliferation assay
CD4+ T cells were purified from human PBMCs or mouse splenic lymphocytes by immunomagnetic negative selection using the human or mouse CD4+ T‐cell isolation Kit (Miltenyi Biotec, Auburn, CA), respectively. Purified CD4+ T cells were labelled with 2·5 μm carboxyfluorescein diacetate succinimidyl ester (CFSE) (Sigma‐Aldrich) at 37° for 15 min. The unconjugated CFSE was eliminated by washing the cells with RPMI‐1640 containing 10% fetal bovine serum. The labelled cells were resuspended in culture medium and co‐cultured with human or mouse anti‐CD3 and anti‐CD28 antibodies in 96‐well flat‐bottom plates, respectively. In some experiments, recombinant CD155 protein or IgG control was added to the medium. After 4 days of culture, cells from 96‐well plates were harvested and analysed for CFSE intensities.
Apoptosis analysis
The PBMCs or splenic lymphocytes were cultured with CD155 or IgG control for 24 hrs. After culture, cells were harvested and apoptosis rates were measured using an FITC Annexin V Apoptosis Detection Kit (BD Biosciences) according to the manufacturer's instructions.
Histological analysis
Samples of kidney from MRL/lpr mice were collected after 1 month of recombinant mouse CD155 protein treatment. The samples were fixed in 10% paraformaldehyde and embedded in paraffin, and 4‐μm‐thick sections were prepared. Sections were stained with haematoxylin & eosin and examined by light microscopy.
Statistical analysis
Data are expressed as the mean ± standard deviation (SD). Statistical analysis differences between groups were analysed using the Mann–Whitney U‐test. Spearman's rank correlation test for non‐parametric data was employed to analyse the relationship between two factors. Rates of mice with no enlarged lymph nodes were studied with Kaplan–Meier analysis. Comparison between curves was performed by the log‐rank method. graphpad prism (version 5.01, GraphPad, La Jolla, CA) software was used for statistical analysis. Statistical significance was determined as P < 0·05.
Results
TIGIT expression on CD4+ T cells is significantly elevated in patients with SLE
To determine whether TIGIT is involved in the pathogenesis of SLE, we used flow cytometry to assess the expression of TIGIT on peripheral blood cells. Given that our previous study has shown that TIGIT is not expressed on B cells, monocytes, dendritic cells and neutrophils,18 the present study only focused on the expression of TIGIT on CD4+ and CD8+ T cells and NK cells. We observed that TIGIT was indeed expressed on CD4+ and CD8+ T cells and NK cells in both healthy individuals and patients with SLE. However, the expression of TIGIT was significantly elevated on CD4+ T cells but decreased on CD8+ T cells and NK cells in patients with SLE compared with in healthy individuals (Fig. 1a, b). It suggests that TIGIT expression on CD4+ T cells may play a more important role in the pathogenesis of SLE.
Figure 1.
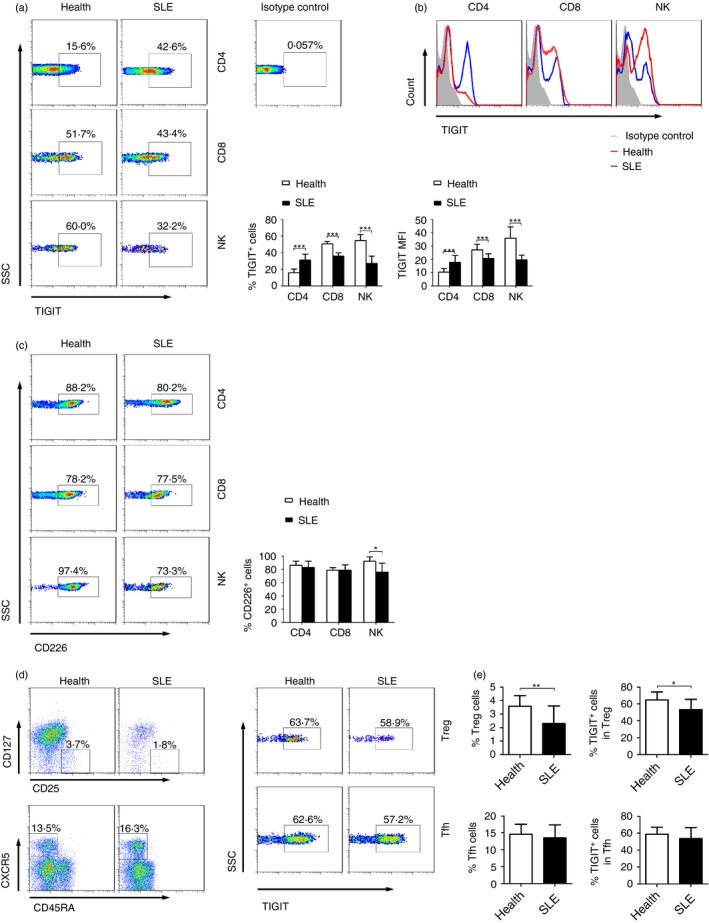
TIGIT expression on CD4+ T cells is elevated in patients with systemic lupus erythematosus (SLE). (a) Representative FACS plots or (b) representative histograms showing the expression of TIGIT on peripheral blood CD4+ T cells, CD8+ T cells, and natural killer (NK) cells in patients with SLE (n = 54) and healthy individuals (n = 45). The percentages of TIGIT + cells or the MFI of TIGIT in different groups are shown as the mean ± SD and are pooled from three to five independent experiments. (c) Representative FACS plots showing the expression of CD226 on peripheral blood CD4+ T cells, CD8+ T cells, and NK cells in patients with SLE (n = 16) and healthy individuals (n = 8). The percentages of CD226+ cells in different groups are shown as the mean ± SD and are pooled from two independent experiments. (d) Regulatory T (Treg) cells (CD4+ CD25+ CD127−) and follicular helper T (Tfh) cells (CD4+ CD45RA − CXCR5+) were gated for analysis of TIGIT expression. (e) The percentages of Treg cells and Tfh cells in CD4+ T cells and the expression of TIGIT + cells are shown as the mean ± SD, n = 11 to n = 16 subjects per group. Data are pooled from two or three independent experiments. *P < 0·05, **P < 0·01, ***P < 0·001 (Mann–Whitney U‐test). [Colour figure can be viewed at wileyonlinelibrary.com]
We also measured the expression of CD226 on T cells and NK cells because TIGIT exerts its function by competing with CD226 for the same ligand, CD155.21 We observed that not only CD4+, CD8+ T cells but also NK cells had a high level of CD226 expression in both healthy individuals and patients with SLE (Fig. 1c). Moreover, both Treg and Tfh cells had a relatively high level of TIGIT expression in SLE patients (Fig. 1d, e). The percentage of TIGIT+ cells in Treg cells was significantly decreased in SLE patients compared with healthy individuals (Fig. 1e).
TIGIT expression on CD4+ T cells is highly correlated with the SLEDAI
We further observed that the expression of TIGIT on CD4+ T cells in patients with SLEDAI > 10 was significantly higher than that in those with SLEDAI < 10 (Fig. 2a, b). Further correlation analysis showed that TIGIT expression on CD4+ T cells was highly correlated with the SLEDAI (Fig. 2c). In addition, our results showed that a significantly higher TIGIT expression on CD4+ T cells was found in patients with SLE with a positive high titre of anti‐dsDNA antibodies or proteinuria compared with those patients without these conditions (Fig. 2d–f). The expression of TIGIT on CD4+ T cells was also significantly correlated with erythrocyte sedimentation rate and C‐reaction protein levels in patients with SLE (Fig. 2g, h). These data demonstrate that the expression of TIGIT on CD4+ T cells is associated with the SLEDAI, indicating that TIGIT may be a biomarker for monitoring disease activity in patients with SLE.
Figure 2.
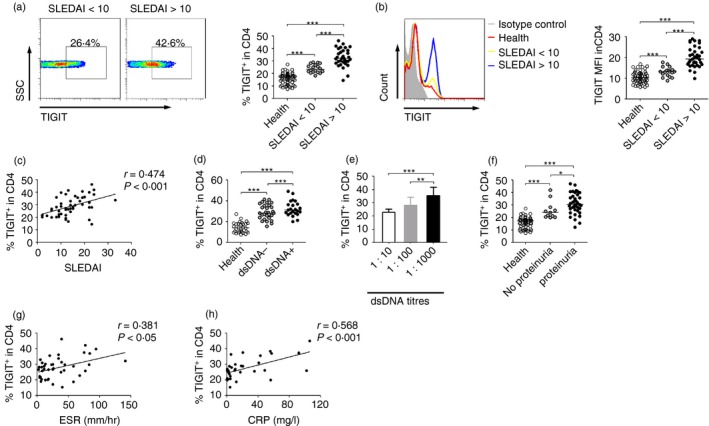
TIGIT expression on CD4+ T cells is correlated with the systemic lupus erythematosus activity index (SLEDAI). (a) Representative FACS plots or (b) representative histograms showing the expression of TIGIT on peripheral blood CD4+ T cells in patients with SLE with different groups of SLEDAI. The percentages of TIGIT + cells or the MFI of TIGIT in CD4+ T cells from healthy individuals (n = 45), patients with SLE with SLEDAI < 10 (n = 19), and patients with SLE with SLEDAI > 10 (n = 36) are shown. (c) Correlation between TIGIT expression on CD4+ T cells and SLEDAI is shown (Spearman's rank correlation test). (d) The percentages of TIGIT + cells in CD4+ T cells from healthy individuals (n = 30) and SLE patients with negative (n = 23) or positive (n = 29) anti‐dsDNA antibody are shown. (e) The percentages of TIGIT + cells in CD4+ T cells in patients with SLE with different titres of anti‐dsDNA antibodies (1 : 10, n = 7; 1 : 100, n = 7; 1 : 1000, n = 15) are shown as the mean ± SD and are pooled from two independent experiments. (f) The percentages of TIGIT + cells in CD4+ T cells in healthy individuals (n = 45) and patients with SLE with (n = 38) or without (n = 11) proteinuria are shown. (g) Correlation between TIGIT expression on CD4+ T cells and erythrocyte sedimentation rate (ESR) or (h) between TIGIT expression on CD4+ T cells and C‐reactive protein (CRP) is shown (Spearman's rank correlation test). Each symbol represents an individual donor, and horizontal bars indicate the median. *P < 0·05, **P < 0·01, ***P < 0·001 (Mann–Whitney U‐test). [Colour figure can be viewed at wileyonlinelibrary.com]
TIGIT is expressed on activated CD4+ T cells in patients with SLE
Next, we tried to characterize the phenotype of TIGIT+ CD4+ T cells in patients with SLE. We found that the expression of immune activation markers CD25 and HLA‐DR as well as intracellular proliferation marker Ki‐67 on CD4+ T cells was significantly increased in patients with SLE compared with healthy individuals (Fig. 3a–c). The expression of CD25 on TIGIT+ CD4+ T cells was significantly higher than that on TIGIT− CD4+ T cells in patients with SLE with low SLEDAI (Fig. 3a). Furthermore, the TIGIT+ CD4+ T cells from patients with SLE with both low and high SLEDAI had a significantly higher expression of HLA‐DR and Ki‐67 than TIGIT− CD4+ T cells from those patients (Fig. 3b, c). We also determined the expression of programmed death 1 (PD‐1), which was discovered as a death receptor in programmed cell death and is up‐regulated on CD4+ T cells upon activation.22 We observed that TIGIT+ CD4+ T cells also showed a significantly higher expression of PD‐1 than TIGIT− CD4+ T cells (Fig. 3d). However, the apoptosis of CD4+ T cells had no difference between TIGIT− and TIGIT+ subsets, in both SLE patients and healthy individuals (see Supplementary material; Fig. S1a). These data suggest that TIGIT is expressed on activated CD4+ T cells in patients with SLE.
Figure 3.
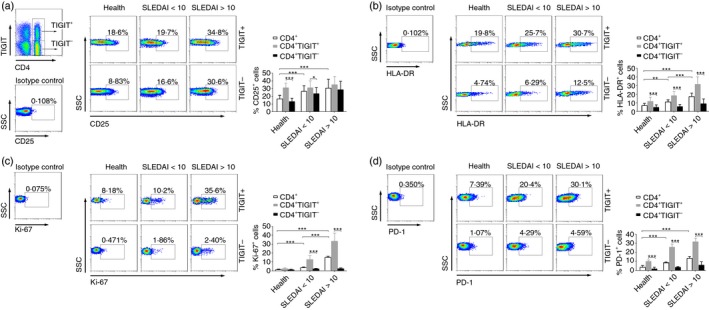
TIGIT expression on activated CD4+ T cells in patients with systemic lupus erythematosus (SLE). Peripheral blood of healthy individuals and patients with SLE with different groups of SLE activity index (SLEDAI) was collected for analysis of the phenotypic characteristics of TIGIT + or TIGIT − CD4+ T cells. (a) Representative FACS plots showing the expression of CD25, (b) HLA‐DR, (c) Ki‐67, and (d) PD‐1 on TIGIT + or TIGIT − CD4+ T cells in different groups. The percentages of CD25+, HLA‐DR +, Ki‐67+ and PD‐1+ cells in TIGIT + or TIGIT − CD4+ T cells from healthy individuals, patients with SLE with SLEDAI < 10, and patients with SLE with SLEDAI > 10 are shown as the mean ± SD, n = 10 to n = 17 subjects per group. Data are pooled from three or four independent experiments. *P < 0·05, **P < 0·01, ***P < 0·001 (Mann–Whitney U‐test). [Colour figure can be viewed at wileyonlinelibrary.com]
TIGIT as a negative regulator of CD4+ T‐cell function in patients with SLE
We further evaluated the relationship between TIGIT and the function of CD4+ T cells. After PHA stimulation, there was a significantly decreased expression of activation markers CD25 and CD69 on CD4+ T cells from patients with SLE compared with healthy individuals (Fig. 4a, b). In contrast, CD4+ T cells from patients with SLE showed a higher IFN‐γ production than those from healthy individuals after the same stimulation (Fig. 4c). More importantly, we observed that, for patients with SLE, the expression of CD25, CD69 and intracellular IFN‐γ in TIGIT− CD4+ T cells was all significantly higher than that in TIGIT+ CD4+ T cells after stimulation (Fig. 4a–c). Cell proliferation analysis also showed that the proliferation of CD4+ T cells was significantly decreased in patients with SLE compared with healthy individuals. Similarly, the proliferation of TIGIT− CD4+ T cells was significantly higher than that of TIGIT+ CD4+ T cells from both patients with SLE and healthy individuals (Fig. 4d). These data suggest that TIGIT serves as a negative regulator of CD4+ T‐cell function in patients with SLE.
Figure 4.
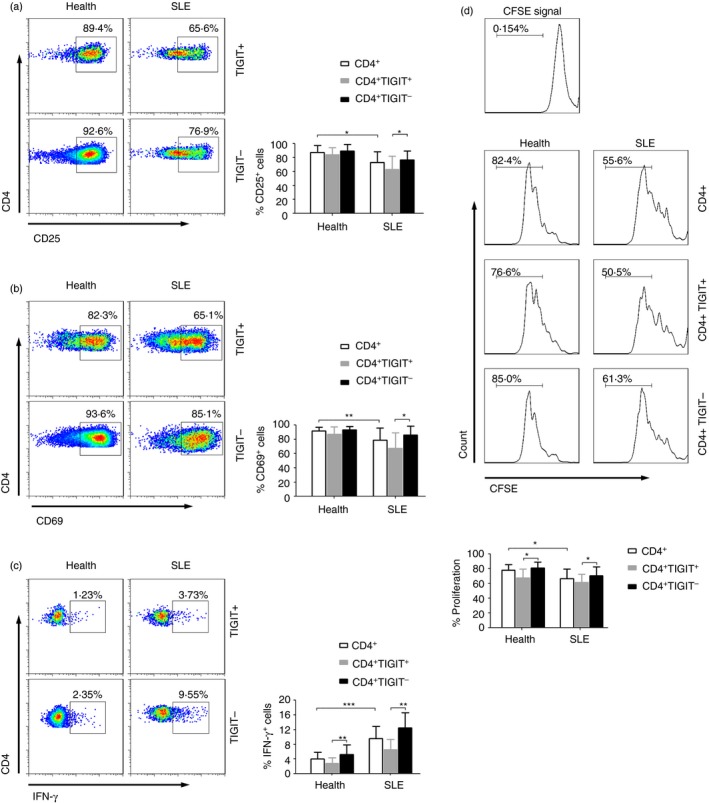
TIGIT as a negative regulator of CD4+ T‐cell function in systemic lupus erythematosus (SLE) patients. Peripheral blood mononuclear cells (PBMCs) isolated from healthy individuals and patients with SLE were stimulated with phytohaemagglutinin (PHA) for 24 hr. (a) Representative FACS plots showing the expression of CD25, (b) CD69, and (c) interferon‐γ (IFN‐γ) in TIGIT + or TIGIT − CD4+ T cells after stimulation. The percentages of CD25+, CD69+ and IFN‐γ + cells in TIGIT + or TIGIT − CD4+ T cells in different groups are shown as the mean ± SD, n = 9 to n = 13 subjects per group. (d) Purified CD4+ T cells from healthy individuals and patients with SLE were labelled with CFSE and stimulated with anti‐CD3 and anti‐CD28 antibodies for 4 days. Representative FACS histograms showing the proliferation of TIGIT + or TIGIT − CD4+ T cells in healthy individuals and patients with SLE. The percentages of TIGIT + or TIGIT − CD4+ T‐cell proliferation in different groups are shown as the mean ± SD, n = 8 to n = 10 subjects per group. Data are pooled from two or three independent experiments. *P < 0·05, **P < 0·01, ***P < 0·001 (Mann–Whitney U test). [Colour figure can be viewed at wileyonlinelibrary.com]
Activation of TIGIT pathway down‐regulates the function of CD4+ T cells from patients with SLE
We next determined whether activation of the TIGIT pathway by using recombinant human CD155 protein would affect the function of CD4+ T cells. Our results showed that CD155 had no effect on CD25 expression but significantly decreased the expression of CD69 on CD4+ T cells (Fig. 5a, b). Furthermore, CD155 could substantially decrease the production of PMA‐ or PHA‐stimulated IFN‐γ in CD4+ T cells from both patients with SLE and healthy individuals compared with the IgG control (Fig. 5c, d). We also observed that the proliferation of CD4+ T cells from both patients with SLE and healthy individuals was substantially decreased by using recombinant CD155 protein (Fig. 5e). However, activation of the TIGIT pathway had no effect on the apoptosis of CD4+ T cells (see Supplementary material, Fig. S1b). These data suggest that activation of the TIGIT pathway can down‐regulate the function of CD4+ T cells from patients with SLE.
Figure 5.
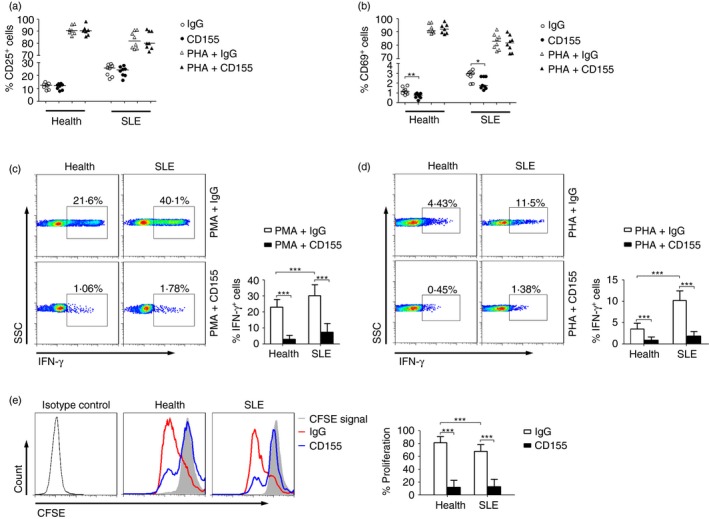
Activation of TIGIT pathway down‐regulates the function of CD4+ T cells from systemic lupus erythematosus (SLE) patients. Peripheral blood mononuclear cells (PBMCs) isolated from healthy individuals and patients with SLE were stimulated with PMA or PHA in the presence of recombinant human CD155 protein or IgG control for 24 hr. (a) The percentages of CD25+ or (b) CD69+ cells in CD4+ T cells from healthy individuals (n = 7) and patients with SLE (n = 8) are shown. Each symbol represents an individual donor, and horizontal bars indicate the median. (c) Representative FACS plots showing the expression of interferon‐γ (IFN‐γ) in CD4+ T cells after PMA or (d) phytohaemagglutinin (PHA) stimulation. The percentages of IFN‐γ + cells in CD4+ T cells from healthy individuals and SLE patients are shown as the mean ± SD, n = 8 to n = 12 subjects per group, pooled from three independent experiments. (e) Representative FACS histograms showing the proliferation of CD4+ T cells from healthy individuals and SLE patients by using CD155 or IgG control. The percentages of CD4+ T‐cell proliferation in different groups are shown as the mean ± SD, n = 8 to n = 10 subjects per group, pooled from three independent experiments. *P < 0·05, **P < 0·01, ***P < 0·001 (Mann–Whitney U‐test). [Colour figure can be viewed at wileyonlinelibrary.com]
Recombinant CD155 protein treatment delays the development of SLE in MRL/lpr mice
To further test the effect of activation of the TIGIT pathway on the progression of SLE, we used recombinant mouse CD155 protein to treat the MRL/lpr mice. An enlarged cervical lymph node was obviously observed in 9‐ to 10‐week‐old MRL/lpr mice after treatment with IgG control, but this was not observed in most mice treated with recombinant CD155 protein during that period (Fig. 6a). Statistical analysis showed that administration of CD155 significantly delayed the time of appearance of lymph node enlargement in MRL/lpr mice (Fig. 6a). Histopathological analysis confirmed that treatment of MRL/lpr mice with CD155 reduced the damage of kidney tissue (Fig. 6b).
Figure 6.
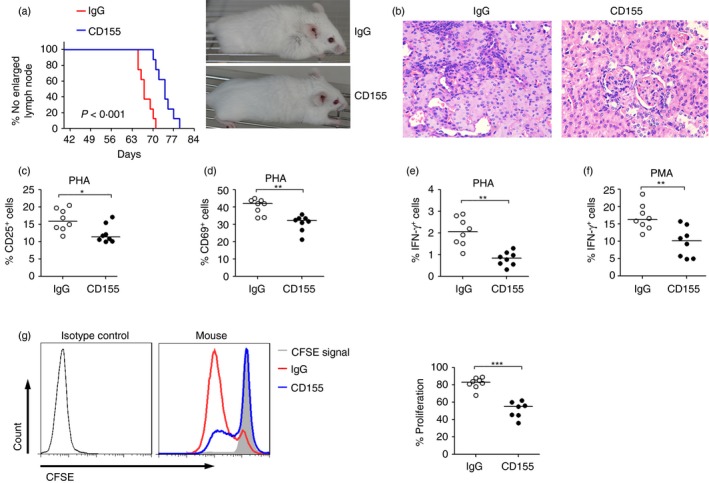
Recombinant CD155 protein treatment delays the development of systemic lupus erythematosus (SLE) in MRL/lpr mice. (a) The 6‐week‐old MRL/lpr mice were injected intraperitoneally with recombinant mouse CD155 protein or IgG control once a week for 4 weeks. Representative photographs were taken in MRL/lpr mice after 1 month of the treatment. The date was recorded when an enlarged lymph node was obviously observed and plotted as indicated (P < 0·001, log‐rank test). There were eight mice in each group. (b) Samples of kidney from mice in the experiment were harvested after 1 month of recombinant mouse CD155 protein or IgG control treatment. Pathological damage of the kidney was detected by haematoxylin & eosin staining. Photographs are representative results of 6‐week‐old MRL/lpr mice after 1 month of treatment. (c) The 6‐week‐old MRL/lpr mice were injected intraperitoneally with recombinant mouse CD155 protein or IgG control once a week for 4 weeks. After that, splenic lymphocytes were isolated and stimulated with phytohaemagglutinin (PHA) or PMA for 24 hr. After culture, the cells were collected and analysed by flow cytometry. The percentages of CD25+ cells, (d) CD69+ cells, and (e, f) IFN‐γ + cells (PHA or PMA stimulation) in CD4+ T cells from CD155‐ or IgG control‐treated mice are shown. (g) Purified mouse splenic CD4+ T cells labelled with CFSE were stimulated with anti‐CD3 and anti‐CD28 antibodies for 4 days. Representative FACS histograms showing the proliferation of CD4+ T cells from CD155‐ or IgG control‐treated mice. The percentages of CD4+ T‐cell proliferation in different groups are shown. Each symbol represents an individual mouse, and horizontal bars indicate the median. *P < 0·05, **P < 0·01, ***P < 0·001 (Mann–Whitney U‐test). [Colour figure can be viewed at wileyonlinelibrary.com]
We further investigated the mechanism by which activation of the TIGIT pathway delays the development of SLE in mice. The activities of CD4+ T cells were determined in MRL/lpr mice after 1 month of CD155 or IgG control treatment. We found that, after stimulation, the expression of CD25, CD69 and intracellular IFN‐γ was significantly decreased in splenic CD4+ T cells from mice treated with CD155 compared with those treated with IgG control (Fig. 6c–f). In addition, the proliferation of splenic CD4+ T cells from CD155‐treated mice was markedly inhibited compared with those from mice receiving IgG control treatment (Fig. 6g). The apoptosis of CD4+ T cells also had no difference between CD155‐treated and IgG‐treated mice (see Supplementary material, Fig. S1). These data suggest that in vivo administration of CD155 can down‐regulate the activities of CD4+ T cells, which may result in a delayed development of SLE in MRL/lpr mice.
Discussion
Systemic lupus erythematosus is a multisystem autoimmune disorder and displays a broad spectrum of clinical and immunological manifestations.23 Although previously considered a rare disease, SLE now appears to be relatively common in certain groups of the population.24 Between 1955 and 1974, the incidence of SLE in the USA increased from 1·0 to 7·6 cases per 100,000 population.25 The key issue of the SLE pathogenesis is B‐cell hyperactivity with high titre production of autoantibodies. Hence, besides using corticosteroids, new therapeutic strategies were aimed to deplete or inhibit the highly activated B cells, such as by using CD20 monoclonal antibody.26 However, B‐lymphocyte hyperactivity in SLE is T‐cell‐dependent.2 A highly specific role of T cells is involved in the activation of B cells producing anti‐native DNA antibodies. Although patients with SLE have a reduced functional capacity of T cells, it actually results from a post‐activation/refractory state.2 Further elucidation of the mechanism of CD4+ T‐cell deregulation in SLE will contribute to finding new therapeutic targets for the disease. The present study has revealed another mechanism by which TIGIT regulates CD4+ T‐cell function in SLE. These data suggest that the TIGIT signalling pathway may be used as a new therapeutic target for the treatment of SLE.
Although many researchers have focused mainly on understanding the deregulation of CD4+ T cells in SLE, it is still largely unknown. Previous studies have shown that abnormal DNA hypomethylation in T cells is generally considered as an important player in the pathogenesis of SLE.27, 28, 29 The impaired DNA methylation contributes to the activation of CD4+ T cells, which might be regulated by microRNAs.30, 31, 32 Besides, the increased expression of co‐stimulatory molecules, such as CD80, CD134 and CD154, is associated with the activation of CD4+ T cells in SLE.33 However, on the other hand, to achieve an appropriate immune response, our immune system has some negative regulation mechanisms to avoid this over‐amplified inflammatory response. PD‐1 and Tim‐3, known as important inhibitory receptors, are associated with the programmed cell death or apoptosis of CD4+ T cells.34, 35 Recent studies have shown that the expression of PD‐1 or Tim‐3 is elevated on T lymphocytes and associated with SLE activity.36, 37 The limitation of these studies, however, is that they did not analyse the role of these receptors in the pathogenesis of SLE. In this study, we first reported that TIGIT is an inhibitory receptor involved in down‐regulation of the function of CD4+ T cells in SLE. This finding provides a mechanism by which our immune system regulates the over‐amplified CD4+ T‐cell responses in this disease.
Another question is why we focused on the association between TIGIT pathway and CD4+ T‐cell function, while TIGIT can be expressed on other cell types, including CD8+ T cells and NK cells. For one thing, SLE is a T‐cell‐dependent autoimmune disease where CD4+ T cells have an important pathogenic role. Depletion of CD4+ cells blocks disease onset in mice,38 and in humans the effects of HIV infection on CD4+ lymphocytes can ameliorate the clinical activity of SLE.39 Autoimmune T helper cells have been cloned in mouse and human SLE and consist of CD4+ T cells, which provide the necessary help for IgM‐producing B cells to secrete pathogenic anti‐DNA antibodies.40 These data suggest that CD4+ T cells are the predominant helper subset in SLE. For another, in patients with SLE, the expression of TIGIT was increased on CD4+ T cells, but not on CD8+ T cells and NK cells. It seems that the TIGIT pathway may play an important role in the pathogenesis of SLE through regulation of CD4+ T‐cell function. For the above reasons, we did not further analyse the effect of TIGIT on the function of CD8+ T cells and NK cells in patients with SLE. Furthermore, our results also showed that TIGIT was highly expressed on other CD4+ T‐cell subsets, including Treg cells and Tfh cells. Previous study has shown that ligation of TIGIT on Treg cells promotes Treg‐cell‐mediated suppression of T effector cell proliferation.10 Regarding the effect of regulation of TIGIT pathway on Tfh cell function, our results showed that recombinant CD155 protein had no significant effect on the activation of Tfh cells from both healthy individuals and patients with SLE (see Supplementary material, Fig. S2). Hence, these data suggest that the effect of regulation of the TIGIT pathway on the delayed development of SLE may be not only caused by down‐regulation of CD4+ IFN‐γ + T helper type 1 cells but also by promotion of Treg cell‐mediated suppression function.
Recently, more and more published data indicate that TIGIT acts as a checkpoint inhibitor of the immune system. For instance, the increased expression of TIGIT can inhibit the function of CD4+ T cells, which contributes to ameliorate the immune‐mediated bone marrow failure of aplastic anaemia.41 Furthermore, TIGIT overexpression down‐regulates the function of CD4+ T cells and reduces the severity of rheumatoid arthritis in mice.16 These studies and our data suggest that activation of the TIGIT pathway may offer a new therapeutic strategy for the treatment of autoimmune diseases. On the contrary, blockade of TIGIT specifically enhanced CD8+ T‐cell effector function, resulting in significant viral clearance and tumour rejection, respectively.17, 42 Further study showed that blockade of TIGIT restored HIV‐ or SIV‐specific CD8+ T‐cell effector responses, which indicated that TIGIT may be a novel therapeutic target to reverse T‐cell exhaustion.43 These studies have suggested that regulation of the TIGIT pathway may be a potential therapeutic approach for autoimmune disease, malignancy and infectious disease.
Conclusions
Taken together, we found that TIGIT expression was significantly elevated on CD4+ T cells in patients with SLE and highly correlated with the activity of the disease. TIGIT+ CD4+ T cells displayed a more activated phenotype than TIGIT− CD4+ T cells in patients with SLE. In contrast, the functional potential of TIGIT+ CD4+ T cells was significantly decreased compared with TIGIT− CD4+ T cells. Intriguingly, activation of the TIGIT pathway by using CD155 could down‐regulate the activation, proliferation and cytokine production of CD4+ T cells from patients with SLE in vitro. In vivo administration of CD155 resulted in a delayed development of SLE in MRL/lpr mice. This study reveals another role for TIGIT in the self‐regulation of CD4+ T cells in SLE, which may offer a new approach to the treatment of the disease.
Disclosures
The authors declare no conflict of interest.
Supporting information
Figure S1. Analysis of the apoptosis of CD4+ T cells.
Figure S2. The effect of CD155 on the activation of follicular helper T cells.
Table S1. The demographic characteristics and clinical presentations of the patients with systemic lupus erythematosus.
Acknowledgements
This work was supported by the Infectious Diseases Control Project from Ministry of Health of China (2016ZX10004207‐004); and the National Natural Science Foundation of China (81401639).
Contributor Information
Feng Wang, Email: fengwang@tjh.tjmu.edu.cn.
Ziyong Sun, Email: zysun@tjh.tjmu.edu.cn.
References
- 1. Rahman A, Isenberg DA. Mechanisms of disease: systemic lupus erythematosus. N Engl J Med 2008; 358:929–39. [DOI] [PubMed] [Google Scholar]
- 2. Zubler RH, Miescher PA. Role of T lymphocytes in systemic lupus erythematosus. Ann Med Interne (Paris) 1990; 141:208–12. [PubMed] [Google Scholar]
- 3. Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature 1996; 383:787–93. [DOI] [PubMed] [Google Scholar]
- 4. Shlomchik MJ, Craft JE, Mamula MJ. From T to B and back again: positive feedback in systemic autoimmune disease. Nat Rev Immunol 2001; 1:147–53. [DOI] [PubMed] [Google Scholar]
- 5. Mak A, Kow NY. The pathology of T cells in systemic lupus erythematosus. J Immunol Res 2014; 2014:419029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, Diacovo TG et al A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur J Immunol 2009; 39:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD et al Cutting edge: TIGIT has T cell‐intrinsic inhibitory functions. J Immunol 2011; 186:1338–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B et al The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol 2009; 10:48–57. [DOI] [PubMed] [Google Scholar]
- 9. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM et al Vstm3 is a member of the CD28 family and an important modulator of T‐cell function. Eur J Immunol 2011; 41:902–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C et al Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014; 40:569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW et al TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest 2015; 125:4053–62. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12. Godefroy E, Zhong H, Pham P, Friedman D, Yazdanbakhsh K. TIGIT‐positive circulating follicular helper T cells display robust B‐cell help functions: potential role in sickle cell alloimmunization. Haematologica 2015; 100:1415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A et al The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci USA 2009; 106:17858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A et al Restoring immune function of tumor‐specific CD4+ T cells during recurrence of melanoma. J Immunol 2013; 190:4899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stengel KF, Harden‐Bowles K, Yu X, Rouge L, Yin J, Comps‐Agrar L et al Structure of TIGIT immunoreceptor bound to poliovirus receptor reveals a cell–cell adhesion and signaling mechanism that requires cis‐trans receptor clustering. Proc Natl Acad Sci USA 2012; 109:5399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao W, Dong Y, Wu C, Ma Y, Jin Y, Ji Y. TIGIT overexpression diminishes the function of CD4 T cells and ameliorates the severity of rheumatoid arthritis in mouse models. Exp Cell Res 2016; 340:132–8. [DOI] [PubMed] [Google Scholar]
- 17. Johnston RJ, Comps‐Agrar L, Hackney J, Yu X, Huseni M, Yang Y et al The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell 2014; 26:923–37. [DOI] [PubMed] [Google Scholar]
- 18. Wang F, Hou H, Wu S, Tang Q, Liu W, Huang M et al TIGIT expression levels on human NK cells correlate with functional heterogeneity among healthy individuals. Eur J Immunol 2015; 45:2886–97. [DOI] [PubMed] [Google Scholar]
- 19. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF et al The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25:1271–7. [DOI] [PubMed] [Google Scholar]
- 20. Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum 1992; 35:630–40. [DOI] [PubMed] [Google Scholar]
- 21. Chan CJ, Martinet L, Gilfillan S, Souza‐Fonseca‐Guimaraes F, Chow MT, Town L et al The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol 2014; 15:431–8. [DOI] [PubMed] [Google Scholar]
- 22. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H et al Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000; 192:1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. O'Neill S, Cervera R. Systemic lupus erythematosus. Best Pract Res Clin Rheumatol 2010; 24:841–55. [DOI] [PubMed] [Google Scholar]
- 24. Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus 2006; 15:308–18. [DOI] [PubMed] [Google Scholar]
- 25. Fessel WJ. Systemic lupus erythematosus in the community. Incidence, prevalence, outcome, and first symptoms; the high prevalence in black women. Arch Intern Med 1974; 134:1027–35. [PubMed] [Google Scholar]
- 26. Robak E, Robak T. Novel and emerging drugs for systemic lupus erythematosus: mechanism of action and therapeutic activity. Curr Med Chem 2012; 19:438–53. [DOI] [PubMed] [Google Scholar]
- 27. Zhang Y, Zhao M, Sawalha AH, Richardson B, Lu Q. Impaired DNA methylation and its mechanisms in CD4+ T cells of systemic lupus erythematosus. J Autoimmun 2013; 41:92–9. [DOI] [PubMed] [Google Scholar]
- 28. Absher DM, Li X, Waite LL, Gibson A, Roberts K, Edberg J et al Genome‐wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T‐cell populations. PLoS Genet 2013; 9:e1003678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lei W, Luo Y, Lei W, Luo Y, Yan K, Zhao S et al Abnormal DNA methylation in CD4+ T cells from patients with systemic lupus erythematosus, systemic sclerosis, and dermatomyositis. Scand J Rheumatol 2009; 38:369–74. [DOI] [PubMed] [Google Scholar]
- 30. Ding S, Liang Y, Zhao M, Liang G, Long H, Zhao S et al Decreased microRNA‐142‐3p/5p expression causes CD4+ T cell activation and B cell hyperstimulation in systemic lupus erythematosus. Arthritis Rheum 2012; 64:2953–63. [DOI] [PubMed] [Google Scholar]
- 31. Yan S, Yim LY, Lu L, Lau CS, Chan VS. MicroRNA regulation in systemic lupus erythematosus pathogenesis. Immune Netw 2014; 14:138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao S, Wang Y, Liang Y, Zhao M, Long H, Ding S et al MicroRNA‐126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum 2011; 63:1376–86. [DOI] [PubMed] [Google Scholar]
- 33. Yellin MJ, Thienel U. T cells in the pathogenesis of systemic lupus erythematosus: potential roles of CD154‐CD40 interactions and costimulatory molecules. Curr Rheumatol Rep 2000; 2:24–31. [DOI] [PubMed] [Google Scholar]
- 34. Zha Y, Blank C, Gajewski TF. Negative regulation of T‐cell function by PD‐1. Crit Rev Immunol 2004; 24:229–37. [DOI] [PubMed] [Google Scholar]
- 35. Zhu C, Anderson AC, Kuchroo VK. TIM‐3 and its regulatory role in immune responses. Curr Top Microbiol Immunol 2011; 350:1–15. [DOI] [PubMed] [Google Scholar]
- 36. Song LJ, Wang X, Wang XP, Li D, Ding F, Liu HX et al Increased Tim‐3 expression on peripheral T lymphocyte subsets and association with higher disease activity in systemic lupus erythematosus. Diagn Pathol 2015; 10:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dolff S, Quandt D, Feldkamp T, Jun C, Mitchell A, Hua F et al Increased percentages of PD‐1 on CD4+ T cells is associated with higher IFN‐γ production and altered IL‐17 production in patients with systemic lupus erythematosus. Scand J Rheumatol 2014; 43:307–13. [DOI] [PubMed] [Google Scholar]
- 38. Wofsy D, Seaman WE. Reversal of advanced murine lupus in NZB/NZW F1 mice by treatment with monoclonal antibody to L3T4. J Immunol 1987; 138:3247–53. [PubMed] [Google Scholar]
- 39. Molina JF, Citera G, Rosler D, Cuellar ML, Molina J, Felipe O et al Coexistence of human immunodeficiency virus infection and systemic lupus erythematosus. J Rheumatol 1995; 22:347–50. [PubMed] [Google Scholar]
- 40. Desai‐Mehta A, Mao C, Rajagopalan S, Robinson T, Datta SK. Structure and specificity of T cell receptors expressed by potentially pathogenic anti‐DNA autoantibody‐inducing T cells in human lupus. J Clin Invest 1995; 95:531–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang T, Wang J, Zhou X, Liang R, Bai Q, Yang L et al Increased expression of TIGIT on CD4+ T cells ameliorates immune‐mediated bone marrow failure of aplastic anemia. J Cell Biochem 2014; 115:1918–27. [DOI] [PubMed] [Google Scholar]
- 42. Johnston RJ, Yu X, Grogan JL. The checkpoint inhibitor TIGIT limits antitumor and antiviral CD8+ T cell responses. Oncoimmunology 2015; 4:e1036214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chew GM, Fujita T, Webb GM, Burwitz BJ, Wu HL, Reed JS et al TIGIT marks exhausted T cells, correlates with disease progression, and serves as a target for immune restoration in HIV and SIV infection. PLoS Pathog 2016; 12:e1005349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Analysis of the apoptosis of CD4+ T cells.
Figure S2. The effect of CD155 on the activation of follicular helper T cells.
Table S1. The demographic characteristics and clinical presentations of the patients with systemic lupus erythematosus.