

Abstract
Major depressive disorder (MDD) affects around 350 million people worldwide; however, the underlying genetic basis remains largely unknown. In this study, we took into account that MDD is a gene-environment disorder, in which stress is a critical component, and used whole-genome screening of functional variants to investigate the ‘missing heritability’ in MDD. Genome-wide association studies (GWAS) using single- and multi-locus linear mixed-effect models were performed in a Los Angeles Mexican-American cohort (196 controls, 203 MDD) and in a replication European-ancestry cohort (499 controls, 473 MDD). Our analyses took into consideration the stress levels in the control populations. The Mexican-American controls, comprised primarily of recent immigrants, had high levels of stress due to acculturation issues and the European-ancestry controls with high stress levels were given higher weights in our analysis. We identified 44 common and rare functional variants associated with mild to moderate MDD in the Mexican-American cohort (genome-wide false discovery rate, FDR, <0.05), and their pathway analysis revealed that the three top overrepresented Gene Ontology (GO) processes were innate immune response, glutamate receptor signaling and detection of chemical stimulus in smell sensory perception. Rare variant analysis replicated the association of the PHF21B gene in the ethnically unrelated European-ancestry cohort. The TRPM2 gene, previously implicated in mood disorders, may also be considered replicated by our analyses. Whole-genome sequencing analyses of a subset of the cohorts revealed that European-ancestry individuals have a significantly reduced (50%) number of single nucleotide variants compared with Mexican-American individuals, and for this reason the role of rare variants may vary across populations. PHF21b variants contribute significantly to differences in the levels of expression of this gene in several brain areas, including the hippocampus. Furthermore, using an animal model of stress, we found that Phf21b hippocampal gene expression is significantly decreased in animals resilient to chronic restraint stress when compared with non-chronically stressed animals. Together, our results reveal that including stress level data enables the identification of novel rare functional variants associated with MDD.
Introduction
Major depressive disorder (MDD) causes considerable morbidity and mortality,1, 2, 3, 4, 5 and is a leading contributor to the global burden of disease.6 However, we know little about its underlying fundamental biology and the genes conferring susceptibility to this disorder.7
Decades of investigation have revealed little of the genetic basis of MDD. Last year, the first two loci for MDD were identified in Chinese women with severe symptoms, one near the SIRT1 gene and the other in an intron of the LHPP gene. However, neither gene has been replicated in European-ancestry populations,8 and a mega-analysis of several genome-wide association studies (GWAS) in MDD did not find any single nucleotide polymorphism (SNP) with genome-wide significance in European-ancestry populations.9 These results highlight the challenges facing this field despite the substantial inheritance of depression (37–38%),10, 11 which is even higher (~70%) when considering diagnostic unreliability.12 There have been various explanations postulated for this genetic conundrum, including disease heterogeneity and the types of genetic variation studied.13 Recently, the risk of depressive symptoms was associated with a rare missense variant in the LIPG gene; in addition, data from over 75 000 and 230 000 individuals with self-reported clinical diagnosis of depression and no self-reported history of depression, respectively, identified 15 genetic loci in European-ancestry populations.14, 15
In MDD, as in other mental disorders, there is a need for new genomic approaches to identify the ‘missing heritability’, which has arisen from applying the ‘common disease, common variant’ hypothesis. An alternative approach proposes that genetic risk can be better ascertained by considering nonlinear interactions between rare genetic variants that have stronger phenotypic effects.16, 17, 18, 19, 20 Coding and non-coding ‘functional’ SNPs (that is, those resulting in amino acid, splicing, regulatory or epigenetic changes) may have a major impact on phenotype.21 Indeed, important fractions of the current missing heritability of complex traits could be due to genetic interactions.20 Following this lead, we previously analyzed a small set of nonsynonymous SNPs and reported that their interactions with clinical and environment variables were significantly associated with MDD.22 Those findings provided a rationale to focus on functional SNPs.
MDD is a gene-environment disorder;3 however, genetic studies have not accounted for the fact that stressful events are a critical factor when selecting a control cohort. Unique features of this study include: (1) a cohort of cases and controls comprised of Mexican-Americans of the greater Los Angeles area who were mainly recent immigrants born in Mexico23 and who experienced significant levels of hyperactivation of the hypothalamic–pituitary–adrenal axis related to challenges, distress and acculturation issues related to immigration;24, 25 (2) type of genetic variations studied; (3) an ethnically diverse replication cohort with stress level data in controls. We performed whole-genome screening specifically of functional variants to investigate whether our experimental design could contribute to explain the ‘missing heritability’ of MDD. We obtained whole-genome sequencing for a small subset of individuals from both cohorts, and also performed an animal study to investigate the functional relevance of our genetic finding.
Materials and methods
The Los Angeles Mexican-American cohort
We used a Los Angeles Mexican-American cohort of 399 subjects aged 19–65 years: 203 with mild to moderate MDD cases (50.88%) and 196 controls (49.12%). There were no differences in age between MDD cases and controls (Supplementary Table S1). Participants gave written informed consent, and detailed demographic, epidemiological and clinical descriptions of this sample have been presented elsewhere.22, 23, 26 Briefly, our Mexican-American participants had at least three grandparents born in Mexico. They participated in a pharmacogenetic study with antidepressant drugs, and were assessed by the Structured Clinical Interview (SCID) for the Diagnostic and Statistical Manual of Mental Disorders (DSM), using the DSM-IV diagnosis of current, unipolar major depressive episode and a HAM-D21 (21-Item Hamilton Depression Rating Scale) score of 18 or greater with item number 1 (depressed mood) rated 2 or greater. This study was approved by the Institutional Review Boards of the Universities of California Los Angeles and Miami, USA, and by the Human Research Ethics Committees of the Australian National University and Bellbery, Australia, and it was registered in ClinicalTrials.gov (NCT00265291).22, 23, 26
Mexican-American MDD patients had comprehensive psychiatric and medical assessments in their primary language using diagnostic and rating instruments fully validated in English and Spanish. Exclusion criteria included active medical illnesses that could be etiologically related to the ongoing depressive symptoms, active suicidal intent, pregnancy/lactation, illicit drug use and/or alcohol abuse in the last 3 months, other major mental disorders, except for anxiety related disorders.22, 23, 26 Control age- and gender-matched Mexican-American individuals were recruited from the same Mexican-American community in Los Angeles and were in general good health but were not screened for medical or psychiatric illnesses.22, 23, 26
The European-ancestry cohort
We used a European-ancestry cohort of 972 individuals (473 (51%) cases and 499 (49%) controls) for our replication study. The effects of age differences between MDD cases and controls were controlled in the linear mixed-effect models (LMEMs) maximization by introducing age as a covariate (Supplementary Tables S1 and S2). Participants gave written informed consent and were recruited under two protocols: (1) Mood disorder studies Münster (comprised of the Münster neuroimaging and the moodinflame studies, which have been conducted by the Department of Psychiatry and Psychotherapy, University of Münster, Münster, Germany), and (2) the Cognitive function and mood disorders study (conducted by the Discipline of Psychiatry, University of Adelaide, South Australia, Australia). The SCID/MINI was used to ascertain that healthy controls were free from lifetime history of psychiatric disorders; for this cohort, the main diagnostic and mood assessment instruments used were also DSM-IV criteria for MDD and HAM-D21, respectively. Previous traumatic events were assessed using the Childhood Trauma Questionnaire/Family Inventory of Life Events in cases and controls. Exclusion criteria included any neurologic abnormalities, substance-related disorders, psychotic/mania or hypomania symptoms, treatment with benzodiazepine, previous electroconvulsive therapy, and usual magnetic resonance imaging (MRI)-contraindications. European-ancestry cohort samples were studied under approved Human Research Ethics Committees protocols at the University of Münster, Germany, and University of Adelaide and Flinders University, South Australia, Australia.
Power analysis for the European-ancestry cohort study
Power estimation was done using the pwr27 package in R.28 Let n1 and n2 be
the number of cases and controls, p1 and
p2 the allele frequency for cases and controls
respectively, and h the Cohen’s effect size, where 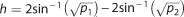 . From this
expression, it follows that
. From this
expression, it follows that 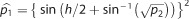 and
and 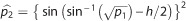 . Provided h and feasible values for
p1 and p2, the difference in allele
frequency between cases and controls can be calculated. Cohen29 suggests that h values of 0.2,
0.5 and 0.8 represent small, medium and large effect sizes, respectively.
. Provided h and feasible values for
p1 and p2, the difference in allele
frequency between cases and controls can be calculated. Cohen29 suggests that h values of 0.2,
0.5 and 0.8 represent small, medium and large effect sizes, respectively.
Our analysis suggested that 200 cases and 200 controls would be sufficient to detect 80% true positives and a medium size effect (defined by the Cohen’s h parameter; Supplementary Figure S1) when m=100 000 variants are tested for association (a value that overcomes the final number of variants used in the association analysis). On the basis of the effect sizes for the m=19 variants associated with MDD in the Mexican-American sample (Table 1A), the post-hoc power ranges between >60% (exm2249659, h=0.335) and >99% (exm1508600, h=0.643). Overall, these figures indicate that our study had power to detect medium to large effect sizes regardless of the strategy used (that is, GWAS or sequential-based).
Table 1A. Genome-wide variants significantly associated with major depressive disorder.
| SNP | Chr | rs ID | Gene | Major allele | P-value | P-value FDR | Minor allele freq. (cases) | Minor allele freq. (controls) | HGVS coding 1 | HGVS protein |
|---|---|---|---|---|---|---|---|---|---|---|
| exm167893 | 1 | rs41310573 | OR2T12 | T | 4.39E−20 | 3.59E−15 | 0.30 | 0.07 | c.115A>G | p.Ser39Gly |
| exm1508600 | 19 | rs201935337 | TMEM150B | C | 4.77E−11 | 1.95E−6 | 0.10 | 0.00 | c.136G>A | p.Gly46Arg |
| exm1616604 | 22 | rs140395831 | PRR5-ARHGAP8 | A | 3.01E−10 | 6.16E−6 | 0.09 | 0.00 | c.1252A>C | p.Ser418Arg |
| exm875366 | 11 | rs56293203 | MUC5B | G | 1.57E−9 | 2.57E−5 | 0.13 | 0.02 | c.442G>A | p.Val148Ile |
| exm283068 | 2 | rs78562453 | C2orf54 | C | 6.23E−8 | 7.28E−4 | 0.06 | 0.00 | c.36G>C | p.Arg12Ser |
| exm445797 | 5 | rs115054458 | TRIO | C | 3.43E−7 | 3.51E−3 | 0.08 | 0.01 | c.7391C>T | p.Ala2464Val |
| exm1441979 | 19 | rs143696449 | ANO8 | G | 4.55E−7 | 4.13E−3 | 0.06 | 0.00 | c.3206C>T | p.Ala1069Val |
| exm669085 | 7 | rs748441912 | KRBA1 | A | 4.94E−7 | 4.04E−3 | 0.11 | 0.02 | c.2535G>A | Synonymous |
| exm75804 | 1 | rs62001028 | BCAR3 | G | 5.32E−7 | 3.96E−3 | 0.07 | 0.00 | c.820C>T | p.Arg274Trp |
| exm1355772 | 17 | rs150952348 | UNC13D | T | 6.71E−7 | 4.57E−3 | 0.06 | 0.00 | c.3160A>G | p.Ile1054Val |
| exm1044842 | 12 | Rs782472239 | ORAI1 | C | 9.12E−7 | 5.74E−3 | 0.08 | 0.00 | c.638C>A | p.Pro213His |
| exm1435859 | 19 | rs112610420 | EMR2 | T | 1.22E−6 | 7.15E−3 | 0.07 | 0.00 | c.875A>C | p.Tyr292Ser |
| exm1325307 | 17 | rs142029931 | CNTNAP1 | G | 1.31E−6 | 7.16E−3 | 0.06 | 0.00 | c.2501G>A | p.Arg834His |
| exm1505393 | 19 | rs201483250 | LILRA1 | C | 1.48E−6 | 7.55E−3 | 0.17 | 0.06 | c.535C>T | p.Arg179Trp |
| exm1369092 | 17 | rs200897153 | FASN | C | 2.49E−6 | 0.01 | 0.10 | 0.01 | c.5801G>A | p.Arg1934His |
| exm1293569 | 17 | rs3744550 | MYH13 | T | 3.57E−6 | 0.02 | 0.30 | 0.15 | c.5585A>G | p.His1862Arg |
| exm782507 | 9 | rs115668237 | SLC2A8 | C | 5.87E−6 | 0.03 | 0.04 | 0.00 | c.1239C>G | p.Cys413Trp |
| exm2249659 | 11 | rs56344012 | MUC5B | A | 1.04E−5 | 0.04 | 0.07 | 0.01 | c.15861A>G | Synonymous |
| exm2275308 | 19 | rs200520741 | HOMER3 | C | 1.05E−5 | 0.04 | 0.05 | 0.00 | Splicing | Splicing |
Abbreviations: Chr, chromosome; FDR, false discovery rate; Freq., frequency; HGVS, Human Genome Variation Society; rs ID, reference SNP identification number; SNP, single nucleotide polymorphism.
Genotyping and analyses
Whole-exome genotyping
Both cohorts were genotyped by the Australian Genome Research Facility (www.agrf.org.au) using the Illumina HumanExome BeadChip-12v1_A, which exonic content consists of >250 000 markers representing diverse populations and a range of common conditions. Samples with calls below the Illumina (Illumina Australia and New Zealand, Scoresby, VIC, Australia) expected 99% SNP call rates were excluded. An individual was duplicated to test genotyping reliability and quality. The identity by descent (IBD) matrix between all pairs of individuals was estimated after linkage disequilibrium (LD) pruning and used for quality control and for the mixed linear models analyses.
Quality control and filtering for functional and rare variants
GenomeStudio data were analyzed by SNP & Variation Suite (SVS) 7.6.7, Golden Helix’s (Golden Helix, Bozeman, MT, USA (http://www.goldenhelix.com). Parameters for excluding markers from analyses included: (i) deviations from Hardy–Weinberg equilibrium with P-values <0.05/m (where m is the number of markers included for analysis); (ii) a minimum genotype call rate of 90% (iii) the presence of more than two alleles; and (iv) monoallelism. The minor allele frequency (MAF) of 0.01 was the criterion for defining common (⩾0.01) and rare variants (<0.01).
Functional variants filtering and classification
We filtered against functional prediction information available in the dbNSFP_NS_Functional_Predictions annotation track GRCh_37 to define variation with potential functional effect.30 This filter uses SIFT, PolyPhen-2, MutationTaster, Gerp++ and PhyloP.31, 32, 33 We used the SVS 7.6.7, Golden Helix’s Variant Classification module to examine the interactions between variants and gene transcripts to categorize variants based on their potential functional effects. Non-coding variants were tested for being harbored in splicing sequences, enhancers, CTCF (CCCTC-binding factor) binding sites, transcription factor binding sites, open chromatin regions, CpG islands, DNA and histone methylation sites, polymerase binding, and target sites of microRNAs and RNA-binding proteins.
GWAS analysis of common and rare variants
We studied the association of MDD to functional variations using single- and
multi-locus LMEMs34 with up to 10
steps in the backward/forward optimization algorithm. These models include
both fixed (genotype markers, sex and years of education) and random effects
(family or population structure), the latter to account for potential
inbreeding by including the IBD matrix (which was estimated between all pairs
of individuals after LD pruning in our analysis).34 A single-locus LMEM assumes that all loci have a
small effect on the trait, whereas a multi-locus LMEM assume that several loci
have a large effect on the trait.34
Both types of models were implemented in SVS 8.3.0. (Golden Helix). The optimal
model was selected using a comprehensive exploration of multiple criteria
including the Extended Bayes Information Criteria (eBIC), the Modified Bayes
Information Criteria (mBIC) and the Multiple Posterior Probability of
Association (mPPA). After the estimation process using the forward/backward
algorithm was finished, the coefficients 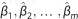 were extracted and a hypothesis test of the
form H0,i: βi=0 vs
H1,i: βi≠0 was
performed for the ith common exonic functional variants to obtain the
corresponding P-value (i=1,2,…,m). Thus,
the collection P1,
P2,…,Pm of
P-values were corrected for multiple testing using the false
discovery rate (FDR)35 and a method
based on extreme-values theory.36
Because hypotheses testing were of the same type, correction was only performed
on the resulting m P-values.35,
36 For the single-locus models, we
estimated the Genomic Control ‘inflation factor’ λ,
to evaluate potential stratification effects.
were extracted and a hypothesis test of the
form H0,i: βi=0 vs
H1,i: βi≠0 was
performed for the ith common exonic functional variants to obtain the
corresponding P-value (i=1,2,…,m). Thus,
the collection P1,
P2,…,Pm of
P-values were corrected for multiple testing using the false
discovery rate (FDR)35 and a method
based on extreme-values theory.36
Because hypotheses testing were of the same type, correction was only performed
on the resulting m P-values.35,
36 For the single-locus models, we
estimated the Genomic Control ‘inflation factor’ λ,
to evaluate potential stratification effects.
We used the regression- and the permutation-based kernel-based adaptive cluster (KBAC) methods to analyse rare exonic functional variants.18, 37 KBAC, implemented in the Golden Helix’s SVS 8.3.0, catalogs rare variant data within each of a number of regions/transcripts into multi-marker genotypes and determines their association with the phenotype, weighting each multi-marker genotype by how often that genotype was expected to occur according to control and MDD cases data and the null hypothesis that there is no association between that genotype and the case/control status.18, 37 Thus, genotypes with high sample risks are given higher weights that potentially separate causal from non-causal genotypes. A one-sided test was applied due to the weighting procedure and the P-values were estimated using 10 000 permutations.
Sequential test in the Mexican-American cohort
As an instrument of cross-validation we applied a sequential strategy of analysis by examining 50% of the Mexican-American individuals randomly chosen, and using the remaining set of individuals to replicate only those markers reaching genome-wide FDR <0.05 during the first round.
Replication study in the European-ancestry cohort
We used a replication European-ancestry cohort; functional variations harbored in genes, or nearby 30 kb, significantly associated with MDD in the Mexican-American sample were filtered in and tested for GWAS analysis of common and rare variations. For rare variants we performed KBAC with permutation testing on CCDS (consensus coding sequence) genes 15, UCSC (rfs://data.goldenhelix.com:80/rfs/CCDSGenes15-UCSC_2014-02-16_GRCh_37_Homo_sapiens.tsf:1). A matrix of IBD for the LMEMs analyses was generated with common variation filtered out after the targeting of genes associated in the first tier. The criterion for replication was a P-value of 0.10 after correction for multiple comparisons using FDR.
Haplotype analyses
Haplotype analyses were performed using Golden Helix. In the Mexican-American and European-ancestry cohorts, we interrogated the chromosomal region with the strongest replication signals to characterize LD blocks and transmission. Lewontin’s D statistics was applied to those analyses because it is unaffected by rare allele frequencies.38
Whole-genome sequencing and analysis
A small subset of both cohorts (15 Mexican-American samples and 10 Australians of European-ancestry samples) was sequenced using Illumina HiSeq 2000 (BGI, Shenzhen, Guangdong, China) or HiSeq X (Garvan Institute, Sydney, NSW, Australia). Whole-genome sequencing paired-end reads were aligned to the human reference genome (hg19, Genome Reference Consortium GRCh37) using Burrows-Wheeler Aligner (BWA)39 to get SAM (sequence alignment/map) format files. Then SAMtools40 was used to convert the SAM to the BAM (Binary version of a SAM file) format files. After internal sorting, BAM files were merged into one BAM file (where one sample may include several lanes of sequencing reads). We used the mpileup command in SAMtools to collect summary information from the BAM file, calculate the likelihood of data given each possible genotype, and store the likelihoods in a binary file. We then piped the output to SAMtools/BCFtools,41 which makes the SNV (single nucleotide variant)/INDEL (small insertions and deletions) calling to generate the VCF (variant call format) files. After that, we used ANNOVAR42 to annotate the SNP/INDEL information and their classification details in those VCF files.
Pathway and network analyses
We evaluated potential common ontogenetic and cellular function processes of the genes disclosed by the GWAS analysis on rare and common variants using Metacore 6.8 software build 29806 (GeneGo, St. Joseph, MI, USA).
Brain eQTL analyses
We used the UK Brain Expression Consortium (UKBEC) web based server Braineac (Brain eQTL Almanac, http://www.braineac.org/) to understand whether some genetic polymorphisms have significant statistical association with transcription level (expression quantitative trait loci, eQTL) in ten brain regions (cerebellum, frontal cortex, hippocampus, medulla, occipital cortex, putamen, substantia nigra, thalamus, temporal cortex and central white matter).43
Animals
Procedures were approved by the Animal Ethics Committees of the South Australian Health and Medical Research and Flinders University and are in accordance with the Australian Code for the Care and Use of Animals for Scientific Purposes (8th edition, 2013). Virus- and antibody-free young adult male Sprague–Dawley rats (150–200g) obtained from Charles River (Margate, Kent, UK) were individually housed in Green Line IVC Sealsafe PLUS cages (Tecniplast, Varese, Italy) in a temperature- (22±1°C) and light- (12h cycles, lights on at 07:00 hours) controlled, stress-free and specific pathogen-free environment with water and standard regular chow ad libitum. They were allowed to habituate at least 5 days before the initiation of experimental procedures. After the baseline behavioral testing, rats were assigned to one of two groups so that the range of floating time was similarly distributed and there was no floating time differences between groups before treatment: (i) chronic restraint stress (CRS, n=27) and (ii) non-CRS (n=10) groups. Moreover, the CRS group was further classified into CRS resilient and CRS non-resilient subgroups (see ‘Statistical analyses’ section below). The investigator was not blinded to group allocation; however, most of the behavioral data were collected by a camera coupled to a software (EthoVision, Noldus Information Technology, Wageningen, The Netherlands). The number of animals was decided based on pilot studies carried out in the lab.
Chronic restraint stress
Flat-bottom clear acrylic restraint containers (20.3 × 8.3 cm; cat no. 544-RR Plas Labs, Lansing, MI, USA) were used as previously described.44 For 14 consecutive days during daytime (09:00–16:00 hours) CRS rats were submitted to six consecutive hours of restraint, after which they were unrestrained and returned to their home cages. Non-CRS animals were submitted to behavioral testing but not restrained.
Forced swim test
We used the automatic video-tracking EthoVision XT video tracking software (EthoVision) to record and analyze the behavior and activity of our animals during the forced swim test (FST). Tests were performed between 09:00 and 12:00 hours after a 60 min habituation to the testing room. At baseline and after the CRS paradigm, the pre-test (training) was carried out for 10 min and was followed 24 h later by the test. Rats were individually tested in a glass cylinder (45cm height and 30cm diameter), which contained 30 cm of water (the rat’s hindlimbs did not reach the cylinder’s floor) at 23°C for 5min. Activity was recorded by one perpendicular camera located in the front; automated measurements of floating/immobility (<12% of distance moved) and struggling/highly mobile activity (>18.5% of distance moved) were obtained twice per second using the EthoVision XT software.45 After the test, rats were dried and placed in a 30 °C drying environment for ~10–15 min. FST was performed at baseline and four weeks later, at which time the CRS paradigm had been completed.
RNA extraction
After the last behavioral testing, animals were allowed to rest at least 2 days before being killed. To avoid the confounding effects of circadian rhythms, animals were killed between 10:00 and 12:00 hours. Brains were remove from the skull, immediately submerged in RNAlater solution and stored overnight at 4 °C; supernatant was then removed and brains were dissected and stored at −80°C until use. Total RNA was isolated from hippocampi using the Purelink RNA Mini Kit (Life Technologies, Mulgrave, VIC, Australia), and DNase digestion was performed using the Purelink DNase set (Life Technologies). RNA was quantified using a spectrophotometer (NanoDrop 2000, Thermo-Fisher Scientific, Waltham, MA, USA) and 500 ng total RNA was reversed transcribed to cDNA using iScript RT supermix (BioRad, Hercules, CA, USA) and random hexamer primers.
Quantitative real-time reverse transcriptase
Primers for each gene of interest were exon spanning, designed using the IDT primer quest tool (Integrated DNA Technologies, Baulkham Hills, NSW, Australia) or the primer premier 5 software (Premier Biosoft International, Palo Alto, CA, USA). Amplicon lengths were between 76 and 135 bp. We employed the Bestkeeper46 and the geNorm47 softwares to select 2 out of 5 reference genes we tested. The following primer pairs were used:Phf21bF: 5′-CAGCGGAAGGCCTTAAAGAA-3′ Phf21bR: 5′-CACTGTCTTGTGGGTGACATAG-3′ Rps18F: 5′-TTCAGCACATCCTGCGAGTA-3′ Rps18R: 5′-TTGGTGAGGTCAATGTCTGC-3′ GapdhF: 5′-CCATTCTTCCACCTTTGATGCT-3′ GapdhR: 5′-TGTCATACCAGGAAATGAGCTTCA-3′.
A standard curve of pooled, serially diluted cDNA was run for the Phf21b gene and for housekeeping genes (Gapdh and Rps18) using the QuantStudio 7 Flex Real-Time PCR system (Thermo-Fisher Scientific). cDNA samples were diluted 1:4 and run in triplicates. Primer sets were tested for optimal dissociation curves with amplification efficiencies between 94 and 107%. To check for genomic contamination, a minus-reverse transcriptase control of each sample was run in an quantitative real-time reverse transcriptase (RT-qPCR) experiment. The geometric mean of both housekeeping genes was used to calculate the results. Only reactions with threshold cycle (CT) standard deviation values ⩽0.3 were accepted.
Statistical analyses
Mirroring our clinical approach, our analyses took into consideration the stress level displayed by animals under non-chronically stressed conditions, as our rats were individually housed and social isolation can increase anxiety- and depressive-like behaviors.48, 49, 50 In both groups, we excluded non-chronically stressed animals that displayed increased floating time at baseline (in the CRS group) or increased averaged floating time (in the non-CRS group), which was defined as floating time higher than the post-CRS FST floating time mean. We classified the CRS group into CRS resilient (below average baseline and post-CRS floating times) and CRS non-resilient. Differences between groups were analyzed by the Student-paired t-test or one-way ANOVA when appropriate. The significance level for each of these effects was set at P<0.05. We used the Pfaffl method to calculate RT-qPCR data.51 Statistical analyses were performed using GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA).
Results
Genotyping analyses
GWAS analysis of common and rare variants in the Mexican-American cohort
We obtained Illumina HumanExome BeadChip-12v1_A genotype data from 399 Mexican-American subjects (203 MDD cases and 196 controls) and 972 European-ancestry subjects (499 MDD cases and 473 controls). After filtering out markers not meeting either quality control criteria or variability requirements 83 898 variants remained for analyses (Figure 1). In the Mexican-American cohort, a total of 19 common SNPs in 18 genes were significantly associated with MDD at the genome-wide FDR <0.05 (Figure 2; Table 1A); two SNPs were found in the MUC5B gene. Sixteen nonsynonymous SNPs out of the nineteen genome-wide associated SNPs predicted functional protein effects and eleven MDD-associated SNPs were rare variants in controls (MAF<0.01). Haplotype analysis reported the clustering of markers with significant association to four conspicuous regions harboring the OR2T12 (Chr 1), TMRM150B (Chr 19), SLX4 (Chr 16) and TRPM2 (Chr 21) genes (Supplementary Figures S2A–D).
Figure 1.

Flow diagram describing the process of filtering out low quality and functionally irrelevant variants. From 247 909 variants, 164 011 variants were discarded because they were monoallelic, had more than two alleles, had a call rate of <90%, or because their genotype proportions deviated from the expected ones as defined by the Hardy-–Weinberg (H–W) equilibrium law in both cases (patients with major depression) and controls at a P-value of 2 × 10−7. The remaining 83 898 variants were used for exome-wide association analysis. After additional functional filtering using several tools (SIFT, PolyPhen-2, MutationTaster, Gerp++ and phyloP) to exclude tolerated and conserved variants, and after the exclusion of non-exonic variants and variants in the control subjects with allelic frequency >0.01, the remaining 47 296 variants were used in rare variant analysis. MDD, major depressive disorder.
Figure 2.

Manhattan plots for a GWAS of major depressive disorder (MDD) in a cohort of Mexican-American patients (n=203) and matched controls (n=196). Results of the GWAS using an additive model after correcting stratification by principal component analysis. In the inset, the first and second principal components that shows absence of genetic stratification (left box), and the Q–Q plot of observed vs expected of the −log10 (P-values, right box). The estimated Genomic Control ‘inflation factor’ λ was 1. GWAS, genome-wide association study.
We next applied the KBAC method to 47 296 rare exonic variants with potential functional effects. Twenty-seven genes were significantly associated with MDD in the Mexican-American cohort (Table 1B). Functional SNPs harbored in five of these genes, ANO8, CNTNAP1, EMR2, HOMER3, UNC13D, were significantly associated with MDD in common and rare variants analyses (Tables 1A and 1B).
Table 1B. Rare variants analysis using the KBAC method after filtering out: tolerated and non-conserved, non-exonic and variants that in controls had a MAF >0.01.
| Chr | Position start | Position stop | Gene name | P-value (one-sided) | KBAC (one-sided) | FDR (one-sided) | Sample size used | # Markers | # Multi-marker genotypes |
|---|---|---|---|---|---|---|---|---|---|
| 19 | 17 434 032 | 17 445 638 | ANO8 | 9.99E−04 | 0.08 | 0.03 | 399 | 2 | 4 |
| 19 | 11 485 383 | 11 487 627 | C19orf39 | 9.99E−04 | 0.06 | 0.03 | 399 | 2 | 4 |
| 19 | 3 136 191 | 3 163 766 | GNA15 | 9.99E−04 | 0.07 | 0.04 | 399 | 3 | 4 |
| 15 | 78 287 327 | 78 369 994 | TBC1D2B | 9.99E−04 | 0.07 | 0.04 | 399 | 3 | 5 |
| 12 | 6 419 602 | 6 437 672 | PLEKHG6 | 9.99E−04 | 0.07 | 0.05 | 399 | 5 | 6 |
| 19 | 19 040 010 | 19 052 041 | HOMER3 | 2.00E−03 | 0.06 | 0.04 | 399 | 3 | 4 |
| 19 | 14 843 205 | 14 889 353 | EMR2 | 2.00E−03 | 0.10 | 0.04 | 399 | 10 | 13 |
| 17 | 73 823 308 | 73 840 798 | UNC13D | 2.00E−03 | 0.09 | 0.04 | 399 | 8 | 14 |
| 12 | 55 945 011 | 55 945 940 | OR6C4 | 2.00E−03 | 0.07 | 0.04 | 399 | 6 | 7 |
| 11 | 67 776 048 | 67 796 743 | ALDH3B1 | 2.00E−03 | 0.07 | 0.05 | 399 | 4 | 5 |
| 10 | 135 051 408 | 135 055 433 | VENTX | 3.00E−03 | 0.06 | 0.04 | 399 | 2 | 4 |
| 9 | 139 607 024 | 139 619 170 | FAM69B | 3.00E−03 | 0.08 | 0.04 | 399 | 1 | 3 |
| 8 | 145 747 761 | 145 752 416 | LRRC24 | 3.00E−03 | 0.07 | 0.05 | 399 | 3 | 5 |
| 7 | 16 639 401 | 16 685 442 | ANKMY2 | 3.00E−03 | 0.09 | 0.05 | 399 | 3 | 4 |
| 4 | 2 965 343 | 3 042 474 | GRK4 | 3.00E−03 | 0.14 | 0.05 | 399 | 7 | 10 |
| 17 | 40 950 854 | 40 963 605 | CNTD1 | 4.00E−03 | 0.07 | 0.04 | 399 | 4 | 5 |
| 17 | 40 834 632 | 40 852 011 | CNTNAP1 | 4.00E−03 | 0.07 | 0.04 | 399 | 4 | 5 |
| 16 | 1 664 641 | 1 727 909 | CRAMP1L | 4.00E−03 | 0.07 | 0.04 | 399 | 6 | 7 |
| 12 | 110 220 892 | 110 271 212 | TRPV4 | 4.00E−03 | 0.05 | 0.04 | 399 | 4 | 6 |
| 11 | 4 615 269 | 4 616 243 | OR52I1 | 4.00E−03 | 0.06 | 0.05 | 399 | 1 | 2 |
| 11 | 1 012 824 | 1 036 706 | MUC6 | 4.00E−03 | 0.11 | 0.05 | 399 | 14 | 18 |
| 9 | 125 486 269 | 125 487 204 | OR1L4 | 4.00E−03 | 0.08 | 0.05 | 399 | 3 | 5 |
| 17 | 48 638 449 | 48 704 542 | CACNA1G | 5.00E−03 | 0.07 | 0.04 | 399 | 5 | 7 |
| 14 | 44 973 354 | 44 976 499 | FSCB | 5.00E−03 | 0.05 | 0.05 | 399 | 5 | 6 |
| 11 | 66 059 373 | 66 064 135 | TMEM151A | 5.00E−03 | 0.05 | 0.05 | 399 | 1 | 2 |
| 10 | 72 530 995 | 72 545 157 | C10orf27 | 5.00E−03 | 0.07 | 0.05 | 399 | 2 | 4 |
| 9 | 130 928 344 | 130 966 662 | CIZ1 | 5.00E−03 | 0.07 | 0.05 | 399 | 6 | 8 |
Abbreviations: Chr, chromosome; FDR, false discovery rate; KBAC, kernel-based adaptive cluster; MAF, minor allele frequency.
Sequential test in the Mexican-American cohort
For cross-validation, we performed a sequential strategy by first analyzing data of 50% randomly chosen Mexican-American individuals, and then using the remainder 50% to replicate only those markers reaching genome-wide FDR <0.05 during the first round. Ten of the thirteen SNPs significantly associated with MDD in the sequential analyses (Table 1C) also achieved significance (FDR<0.05) in the GWAS analysis (Table 1A).
Table 1C. Genome-wide variants significantly associated with major depressive disorder after using a sequential strategy.
| SNP | Chr | rs ID | Gene | Major allele | P-value | P-value FDR | Minor allele freq. (cases) | Minor allele freq. (controls) | HGVS coding 1 | HGVS protein |
|---|---|---|---|---|---|---|---|---|---|---|
| exm167893 | 1 | rs41310573 | OR2T12 | T | 8.06E−10 | 1.74E−06 | 0.30 | 0.06 | c.115A>G | p.Ser39Gly |
| exm1472172 | 19 | rs201138159 | RABAC1 | 9.87E−08 | 1.07E−04 | 0.17 | 0.00 | c.107A>G | p.Glu36Gly | |
| exm875366 | 11 | rs56293203 | MUC5B | G | 7.48E−07 | 5.38E−04 | 0.15 | 0.00 | c.442G>A | p.Val148Ile |
| exm1508600 | 19 | rs201935337 | TMEM150B | C | 2.39E−06 | 1.29E−03 | 0.13 | 0.00 | c.136G>A | p.Gly46Arg |
| exm1616604 | 22 | rs140395831 | PRR5-ARHGAP8 | A | 1.15E−05 | 4.97E−03 | 0.10 | 0.00 | c.1252A>C | p.Ser418Arg |
| exm445797 | 5 | rs115054458 | TRIO | C | 4.73E−05 | 1.70E−02 | 0.11 | 0.01 | c.7391C>T | p.Ala2464Val |
| exm75804 | 1 | rs62001028 | BCAR3 | G | 5.70E−05 | 1.76E−02 | 0.11 | 0.00 | c.820C>T | p.Arg274Trp |
| exm1577187 | 21 | rs142151549 | TRPM2 | T | 8.95E−05 | 2.42E−02 | 0.12 | 0.00 | c.4364T>G | p.Val1455Gly |
| exm646600 | 7 | rs112002983 | LRWD1 | A | 9.60E−05 | 2.30E−02 | 0.11 | 0.00 | c.794A>C | p.Asp265Ala |
| exm1435859 | 19 | rs112610420 | EMR2 | T | 1.08E−04 | 2.33E−02 | 0.10 | 0.00 | c.875A>C | p.Tyr292Ser |
| exm1355772 | 17 | rs150952348 | UNC13D | T | 1.15E−04 | 2.25E−02 | 0.08 | 0.00 | c.3160A>G | p.Ile1054Val |
| exm1505393 | 19 | rs201483250 | LILRA1 | C | 2.19E−04 | 3.93E−02 | 0.20 | 0.05 | c.535C>T | p.Arg179Trp |
| exm669085 | 7 | rs748441912 | KRBA1 | A | 2.53E−04 | 4.19E−02 | 0.13 | 0.01 | c.2535G>A | Synonymous |
Abbreviations: Chr, chromosome; FDR, false discovery rate; Freq., frequency; HGVS, Human Genome Variation Society; rs ID, reference SNP identification number; SNP, single nucleotide polymorphism.
Fifty percent of the sample was randomly selected for discovery and the remaining 50% for join analyses.
Replication study in the European-ancestry cohort
Functional variations harbored in or 30kb around genes significantly associated with MDD in the Mexican-American sample were next tested by GWAS analysis of common and rare variations in the European-ancestry cohort. The rare variant analysis using the KBAC method identified the PHF21B gene (Table 2), which is adjacent to the PRR5-ARHGAP8 gene within the chromosome 22q.13 region identified in Mexican-Americans. It is noteworthy to mention that the frequencies of genetic variants were different between both cohorts (see ‘WGS analyses’ section below).
Table 2. Rare variants analysis using the KBAC method in the European-ancestry cohort: tolerated and non-conserved, non-exonic and variants that in controls had a MAF >0.01.
| Chr | Position start | Position stop | Gene name | Transcript name(s) | P-value (one-sided) | KBAC (one-sided) | FDR (one-sided) | Sample size used | # Markers | # Multi-marker genotypes |
|---|---|---|---|---|---|---|---|---|---|---|
| 22 | 45248966 | 45435430 | PHF21B | CCDS46727.1 | 0.001 | 0.027 | 0.068 | 972 | 6 | 6 |
| 22 | 45152408 | 45288475 | ARHGAP8 | CCDS33664.1 | 0.003 | 0.027 | 0.102 | 972 | 16 | 8 |
| 22 | 45248966 | 45434692 | PHF21B | CCDS56234.1 | 0.001 | 0.027 | 0.102 | 972 | 6 | 6 |
| 22 | 45248966 | 45435430 | PHF21B | CCDS14061.1 | 0.002 | 0.027 | 0.102 | 972 | 6 | 6 |
| 22 | 45152408 | 45288475 | ARHGAP8 | CCDS14060.2 | 0.003 | 0.027 | 0.123 | 972 | 16 | 8 |
Abbreviations: Chr, chromosome; FDR, false discovery rate; KBAC, kernel-based adaptive cluster; MAF, minor allele frequency.
Haplotype analyses of Chr 22q13.1
Haplotype analyses were performed in Chr 22q13.1 in both cohorts encompassing the region showing the highest replication signals in Table 2, namely the PRR5, ARHGAP8 and PHF21B genes (Supplementary Figure S3). The LD structure of this chromosomal region appears to be more conserved in the European-ancestry cohort than in the Mexican-American cohort. It is evident that this chromosomal region is transmitted as a heterogeneous block. Therefore, diversity of the results in our two cohorts might be explained by the complex evolutionary heterogeneity of this region.
WGS analyses
We performed WGS to better understand variations within and between each of the two cohorts we studied. WGS analyses showed that Mexican-American individuals had significantly more SNVs (50% more) and less INDELs (small insertions and deletions) when compared with Australian individuals of European-ancestry (Table 3; Supplementary Table S3). Our data are compatible with findings from the International HapMap 3 Consortium and the 1000 Genomes Project Consortium, as they respectively showed that individuals with African ancestry carry an increased number of variants and rare variants, and within Europeans the Spanish population carry excess of rare variants.52, 53 As the HapMap Mexican-American samples from Los Angeles and our Mexican-American cohort were recruited from the same location by our bilingual research team, their median ancestry proportions are estimated to be 45% Indigenous American, 49% European and 5% African.54
Table 3. Whole-genome sequencing analysis of 25 human samples.
| Sample | Total SNVs | Total INDELS | Total variations | dbSNP | dbSNP% | Exonic | Synonyms | Non-synonyms | Intronic |
|---|---|---|---|---|---|---|---|---|---|
| AU mean (n=10) | 390 1078 | 546 756.6 | 4 447 834.6 | 3 900 468.9 | 87.7 | 23 942.4 | 12 613.4 | 11 329 | 1 318 630 |
| AU SD (n=10) | 37083.5 | 35237.0 | 67382.1 | 51576.2 | 0.4 | 291.1 | 142.3 | 177.9 | 8851.6 |
| MA mean (n=15) | 772 9021.3 | 517 554.1 | 8 246 575.4 | 3 995 923.3 | 48.6 | 114 430.9 | 31 641.7 | 82 789.1 | 2 927 921 |
| MA SD (n=15) | 413 339 | 5041.5 | 412 190.9 | 43 721.7 | 2.7 | 7998.5 | 1800.7 | 6263 | 179 407.6 |
| T-test, P | 2.09E−15 | 0.028 | 7.05E−16 | 0.00016 | 7.22E−19 | 1.99E−16 | 3.64E−16 | 1.83E−16 | 4.70E−15 |
| Mann–Whitney U, P | 3.59E−05 | 9.65E−04 | 3.59E−05 | 5.27E−04 | 3.59E−05 | 3.59E−05 | 3.59E−05 | 3.59E−05 | 3.59E−05 |
| Sample | Intergenic | Splicing | 3′ UTR3 | 5′ UTR | Down-stream | Up-stream | ncRNA exonic | ncRNA intronic | ncRNA splicing |
|---|---|---|---|---|---|---|---|---|---|
| AU mean (n=10) | 2 246 762 | 111.5 | 25 759.4 | 5859.9 | 25 626.1 | 25 116 | 12 966.1 | 217 112.9 | 71.4 |
| AU SD (n=10) | 25 571.3 | 8.6 | 176.9 | 68.6 | 360.3 | 324.1 | 150.1 | 3070.5 | 5.3 |
| MA mean (n=15) | 4 015 894 | 5630.7 | 66 925.8 | 28 831.7 | 63 537.3 | 82 268.7 | 29 111.9 | 397 645.2 | 338.1 |
| MA SD (n=15) | 198 644.7 | 287.1 | 4324.0 | 5222.3 | 4372.6 | 5407.0 | 1497.9 | 19 516.4 | 22.1 |
| T-test, P | 2.16E−15 | 1.24E−19 | 2.15E−15 | 9.28E−11 | 5.76E−15 | 4.37E−16 | 2.11E−16 | 7.52E−16 | 1.61E−18 |
| Mann–Whitney U, P | 3.59E−05 | 3.58E−05 | 3.59E−05 | 3.59E−05 | 3.59E−05 | 3.59E−05 | 3.59E−05 | 3.59E−05 | 3.53E−05 |
Abbreviations: AU, Australian; dbSNP, the single nucleotide polymorphism database; INDELS, small insertions and deletions; MA, Mexican-American; ncRNA, non-coding RNAs; SD, standard deviation; SNVs, single nucleotide variants; 3′ UTR, three prime untranslated region; 5′ UTR, five prime untranslated region.
Pathway and network analyses
MDD-associated genes in the Mexican-American cohort were overrepresented in the following Gene Ontology (GO) processes: (1) natural immune response (granuloma formation, natural killer cell degranulation, synaptic vesicle priming, natural killer cell activation, germinal center formation), (2) G-protein-coupled glutamate receptor signaling pathway, (3) detection of chemical stimulus involved in smell sensory perception, (4) cellular response to stimulus, (5) regulation of macromolecule metabolic processes, (6) muscle contraction, (7) cell surface receptor process (G-protein coupled receptor signaling pathway, system process, neurological system process), (8) regulation of ion transmembrane transport (regulation of calcium ion transport and activity) and (9) positive regulation of cellular metabolic process (Supplementary Table S4).
Brain eQTL analyses
Data extracted from the Braineac web server (UK Brain Expression Consortium, UKBEC) revealed that PHF21B gene variants significantly change eQTL in all ten listed brain areas (Supplementary Table S5). Seven listed variants change hippocampus eQTL (Table 4).
Table 4. Hippocampal quantitative gene expression analyses for PHF21B gene variants extracted from the Braineac web server (UK Brain Expression Consortium, UKBEC).
| rs ID | Variant | Expr ID | P-value | Tissue | Genotype_counts | Allele_frequency |
|---|---|---|---|---|---|---|
| rs1003851 | chr22:44782265 | 3963529 | 3.90E−05 | HIPP | CC=5; CT=46; TT=76 | C=20.9% T=79.1% |
| rs117160508 | chr22:45442461 | 3963529 | 1.60E−04 | HIPP | CC=1; CG=12; GG=119 | C=5.2% G=94.8% |
| rs118154683 | chr22:45442465 | 3963529 | 1.60E−04 | HIPP | TT=1; TA=12; AA=119 | T=5.2% A=94.8% |
| rs75806917 | chr22:45442547 | 3963529 | 1.60E−04 | HIPP | GG=1; GT=11; TT=119 | G=4.9% T=95.1% |
| rs8138025 | chr22:45974757 | 3963548 | 1.90E−04 | HIPP | AA=19; AG=63; GG=52 | A=37.7% G=62.3% |
| rs5765490 | chr22:45974913 | 3963548 | 2.10E−04 | HIPP | TT=19; TC=63; CC=52 | T=37.7% C=62.3% |
| rs62228464 | chr22:45113754 | 3963550 | 6.60E−05 | HIPP | CC=18; CA=51; AA=61 | C=32.5% A=67.5% |
Abbreviations: chr, chromosome; Expr ID, expression identification; HIPP, hippocampus; rs ID, reference single nucleotide polymorphism identification number.
Animal study
Our genetic analyses were performed on two cohorts that, uniquely, took into consideration levels of stress data in control groups to minimize the impact of individuals genetically at-risk for MDD in the control population and thereby maximize the likelihood of identifying novel MDD-associated variants. This approach revealed that rare functional variants in the PHF21B gene are associated with MDD in both cohorts and brain eQTL showed PHF21B gene variants could significantly change expression in several brain areas. To further analyze the association between PHF21B, depression and stress, we exposed male Sprague–Dawley rats to CRS (see ‘Materials and methods’ section), and analyzed the effect on floating/immobility time in the FST, which is a measure of behavior despair and depressive-like behavior. CRS resulted in small but significantly increased floating time in the FST in CRS non-resilient animals but not in the CRS resilient ones (Figure 3a; Supplementary Table S6). Importantly, Phf21b mRNA levels were significantly decreased in the CRS-resilient group when compared with the non-CRS group, but not in the CRS non-resilient group (Figure 3b). Therefore, Phf21b gene expression level may reflect a state of resilience to chronic stress.
Figure 3.

Animal studies. (a) Paired t-test showed significantly increased floating time between the first (T1, baseline) and the second (T2, post-CRS) forced swim tests (FST) in the chronic restraint stress (CRS) non-resilient (nonresil, n=8) group but not in the CRS resilient (resil, n=11) and the non-CRS (n=6) groups. (b) One-way ANOVA showed that hippocampal Phf21b gene expression was significantly decreased in the CRS resil (resilient) group (n=11) but not in the CRS nonresil (non-resilient) group (n=8) comparing with the non-CRS group (n=6); columns=mean, bars=standard deviation of the mean. (s), seconds; AU, arbitrary units; **P<0.01; ***P<0.001.
Discussion
We presented results that support the assumption that MDD is a syndrome of considerable genetic heterogeneity, as we identified many common and rare functional variants that confer susceptibility to MDD in the Mexican-American cohort. Those variations withstood the use of stringent correction measures related to genetic stratification and repeated testing. Interestingly, 11 out of 19 SNPs with significant genome-wide association to MDD consisted of rare variants in the control population; the data strongly support a Mendelian type of inheritance for many of the rare variants. Remarkably, five genes were identified in both common and rare variant analyses, and one gene had two SNPs that were significantly associated with MDD in our GWAS analysis.
Our replication strategy in the European-ancestry cohort included testing functional variations harbored in genes, and extended to include nearby 30 kb, significantly associated with MDD, in GWAS analysis of common and rare variations, in the Mexican-American. This strategy replicated the PHF21B (PHD finger protein 21B, also known as BHC80L or PHF4) gene, which encodes the 531 amino acid PHD finger protein 21B. The PHF21B gene and the neighboring ARHGAP8 (Rho GTPase activating protein 8) gene provided the strongest signals in our replication analysis; however, the ARHGAP8 gene did reach the significance threshold (Table 2). Furthermore, as the ARHGAP8 and PRR5 (Proline rich 5) genes were initially thought to be one gene because of readthrough transcripts,55 this chromosomal locus requires further scrutiny.
The PHF21B gene may be a putative tumor suppressor gene whose loss of function results from reduced expression by hypermethylation or gene loss.56 This gene is expressed in the brain and its variants affect its expression in several brain regions, including the frontal cortex and the hippocampus (Brain eQTL Almanac, http://www.braineac.org/); though, its central nervous system functions are unknown. However, its prominent paralog, the PHF21A gene, encodes BHC80 (a component of a BRAF35/histone deacetylase complex), which is a transcriptional repressor during neurodevelopment highly expressed in the brain.57, 58, 59 Both the PHF21A and the PHF21B genes have the PHD zinc finger domain, the DNA-binding domain and the region required for transcriptional repression, and localize in the nucleus.56 The PHF21B gene is located in Chr 22q13.31, which is a genomic region associated with the Phelan-McDermid syndrome (or 22q13.3 deletion syndrome/22q13.3DS), a rare neurodevelopmental disorder that typically presents with generalized developmental delay, intellectual disability, delayed speech and seizures, and involves the SHANK3 gene.60 The SHANK3 gene encodes a protein that has a role in synapse formation and dendritic spine maturation, and mutations in this gene cause autism spectrum disorder and schizophrenia.61, 62 It is noteworthy that the nearby 22q11.2 deletion syndrome [22qDS/velocardiofacial syndrome (VCFS)/DiGeorgio syndrome/CATCH22] is one of the most common multiple anomaly syndromes in humans and it also includes among its manifestations higher rates of psychiatric disorders, including mood and schizophrenia.63, 64 A large percentage (79%) of 22qDS children and adolescents have one psychiatric diagnosis and 12.5–14% meet DSM criteria for MDD.65, 66, 67
Results of our haplotype and sequential analyses support that the TRPM2 gene was significantly associated with MDD; thus, these data may be considered as providing a replication for the association of MDD and the TRPM2 gene, which was previously associated by positional candidate approach with susceptibility to bipolar disorder and unipolar disorder.68 The transient receptor potential cation channel, subfamily M, member 2, TRPM2, is a member of the melastatin-related transient receptor channel family that is involved in oxidative stress-induced cell death and inflammation processes and has a role in endotoxin-provoked cytokine production; TRPM2-mediated calcium influx influences the reactive oxygen species-induced signaling cascade responsible for chemokine production and exacerbates inflammation via the NLRP3 (NLR family, pyrin domain containing 3) inflammasome.69
Pathway analysis offered insight into the complexity of mood regulation and MDD stereotypical behaviors and symptoms. Many of the uncovered enriched processes are novel or underexplored, such as cellular response to stimulus, muscle system process, regulation of metabolic process, sensory perception of chemical stimulus, regulation of ion transmembrane transport and protein–DNA complex disassembly, and others have been active areas of investigation as complementary/alternative hypothesis for the classical monoamine hypothesis of depression, such as neuroimmune mediation/inflammation and glutamate receptor signaling pathway and growth/positive regulation of macromolecule biosynthetic process.70, 71, 72, 73, 74, 75 The olfactory system and its central connections may have a role in affective behavior, and bilateral olfactory bulbectomy has been used as a rodent model for depression since the early 1980’s.76, 77 Our data may help narrow the focus of future investigations toward specific aspects of these broad investigative areas.
Our animal study provided evidence that hippocampal Phf21b gene expression modulates the response to chronic stress as CRS-resilient animals had decreased hippocampal Phf21b mRNA levels. However, the lack of non-stressed group housed animals may have restricted our ability to understand the baseline level of this gene in non-socially isolated animals. GO enrichment analysis indicated that MDD-associated genes are involved in regulation of metabolic processes; therefore, food and water restriction during the CRS paradigm could have impacted post-stress gene expression levels. To minimize the effects of food and water restriction in our experiments, CRS was performed during the light phase when rodents do not usually consume a significant amount of food or water, as they are nocturnal animals.
MDD is clearly a gene-environment disorder,3 but most genetic studies have not accounted for stressful life events in the control population. These critical environmental factors have been used in the present work to help minimize the inclusion of genetically susceptible individuals in the control sample. Our Mexican-American cohort is comprised of first-generation individuals (60%)23 who have experienced significant levels of stress and hyperactivation of the hypothalamic–pituitary–adrenal axis related to acculturation issues.24, 25 In contrast to the Mexican-American controls represented a group of individuals who were highly resilient to significant levels of enduring stress, in our replication cohort the stress levels in controls were also included in our analyses. Furthermore, as most of the variations reported here in the Mexican-American cohort were rare, each gene contained low-frequency mutations at many different sites, which could help explain the challenges experienced in this field, as the power of genome-wide association studies in MDD have been greatly reduced because the critical assumption that there were little allelic heterogeneity within loci may not apply;78 this assumption may have been harder to negate in populations of European-ancestry due to their drastically reduced number of SNVs (Table 3; Supplementary Table S3). Our findings suggest that the ‘missing heritability’ in MDD could be at least partly explained by rare variants.
In summary, we identified common and rare variations in a total of 44 genes that may confer susceptibility to MDD in a Mexican-American cohort. Most of these variations were rare and resulted in amino acid (that is, likely functional) changes. Replication of the PHF21B gene in an ethnic diverse population and the finding that Phf21b gene expression modulates the chronic stress response in rats corroborate the strength of our findings. Our findings also provide a set of common and rare genetic variants associated with MDD that require replication. Our clinical cohorts were small; therefore, further replication studies are warranted with larger cohorts recruited under a similar study design to replicate and/or identify additional genes associated with MDD. As PHF21B gene expression could be modulated by methylation,56 future studies should investigate whether the methylation status in this locus is influenced by MDD diagnosis or stress, as epigenetics may account for part of the missing heritability in complex traits.79 No previous studies have looked at Phf21b levels in stress/depressive-like behavior; therefore, additional animal studies are also needed to systematically characterize hippocampal Phf21b gene expression level in non-stressed groups at baseline, and during various types of acute and chronic stress paradigms.
Acknowledgments
We have been supported by grants APP1051931 and APP1070935 (MLW), and APP1060524 (BB) from the National Health and Medical Research Council (Australia), the German Research Foundation (UD, Grant FOR 2107, DA1151/5-1), NIH Grant GM61394 (JL and MLW), the German Australian Institute for Translational Medicine (SRB and JL), and institutional funds from the Australian National University and the South Australian Health and Medical Research Institute. We are grateful for the contributions of Israel Alvarado, Deborah L. Flores, Rita Jepson, Lorraine Garcia-Teague, Patricia Reyes, Isabel Rodriguez, Gabriela Marquez and the University of California, Los Angeles General Clinical Research Center (UCLA GCRC) staff in recruiting and caring of the Mexican-American participants.
Footnotes
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
The authors declare no competing interest and no income from pharmaceutical companies; an intellectual property application has been prepared to include the genetics findings of this work.
Supplementary Material
References
- Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry 2005; 62: 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, McGonagle KA, Zhao S, Nelson CB, Hughes M, Eshleman S et al. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch Gen Psychiatry 1994; 51: 8–19. [DOI] [PubMed] [Google Scholar]
- WHO (ed). WHO depression fact sheet number 369; reviewed April 2016; http://www.who.int/mediacentre/factsheets/fs369/en/.
- Wong ML, Licinio J. Research and treatment approaches to depression. Nat Rev Neurosci 2001; 2: 343–351. [DOI] [PubMed] [Google Scholar]
- Wong ML, Licinio J. From monoamines to genomic targets: a paradigm shift for drug discovery in depression. Nat Rev Drug Discov 2004; 3: 136–151. [DOI] [PubMed] [Google Scholar]
- Lopez AD, Murray CC. The global burden of disease, 1990–2020. Nat Med 1998; 4: 1241–1243. [DOI] [PubMed] [Google Scholar]
- Sullivan PF, Daly MJ, O'Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet 2012; 13: 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONVERGE Consortium. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 2015; 523: 588–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major Depressive Disorder Working Group of the Psychiatric GWAS ConsortiumMajor Depressive Disorder Working Group of the Psychiatric GWAS ConsortiumRipke S Major Depressive Disorder Working Group of the Psychiatric GWAS ConsortiumWray NR Major Depressive Disorder Working Group of the Psychiatric GWAS ConsortiumLewis CM Major Depressive Disorder Working Group of the Psychiatric GWAS ConsortiumHamilton SP Major Depressive Disorder Working Group of the Psychiatric GWAS ConsortiumWeissman MM et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 2013; 18: 497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry 2000; 157: 1552–1562. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Gatz M, Gardner CO, Pedersen NL. A Swedish national twin study of lifetime major depression. Am J Psychiatry 2006; 163: 109–114. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ. The lifetime history of major depression in women. Reliability of diagnosis and heritability. Arch Gen Psychiatry 1993; 50: 863–870. [DOI] [PubMed] [Google Scholar]
- Cohen-Woods S, Craig IW, McGuffin P. The current state of play on the molecular genetics of depression. Psychol Med 2013; 43: 673–687. [DOI] [PubMed] [Google Scholar]
- Amin N, Jovanova O, Adams HH, Dehghan A, Kavousi M, Vernooij MW et al. Exome-sequencing in a large population-based study reveals a rare Asn396Ser variant in the LIPG gene associated with depressive symptoms. Mol Psychiatry 2016; e-pub ahead of print. [DOI] [PubMed]
- Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet 2016; 48: 1031–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnhead NS, Wilding JL, Winney B, Tonks S, Bartlett S, Bicknell DC et al. Multiple rare variants in different genes account for multifactorial inherited susceptibility to colorectal adenomas. Proc Natl Acad Sci U S A 2004; 101: 15992–15997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia G, Bansal V, Harismendy O, Schork NJ, Topol EJ, Frazer K et al. A covering method for detecting genetic associations between rare variants and common phenotypes. PLoS Comput Biol 2010; 6: e1000954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DJ, Leal SM. A novel adaptive method for the analysis of next-generation sequencing data to detect complex trait associations with rare variants due to gene main effects and interactions. PLoS Genet 2010; 6: e1001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet 2008; 40: 695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A 2012; 109: 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009; 461: 272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ML, Dong C, Andreev V, Arcos-Burgos M, Licinio J. Prediction of susceptibility to major depression by a model of interactions of multiple functional genetic variants and environmental factors. Mol Psychiatry 2012; 17: 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ML, Dong C, Flores DL, Ehrhart-Bornstein M, Bornstein S, Arcos-Burgos M et al. Clinical outcomes and genome-wide association for a brain methylation site in an antidepressant pharmacogenetics study in Mexican Americans. Am J Psychiatry 2014; 171: 1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan S, Escobar J, Paris M, Alvidrez J, Dixon JK, Desai MM et al. Cultural influences on causal beliefs about depression among Latino immigrants. J Transcult Nurs 2013; 24: 68–77. [DOI] [PubMed] [Google Scholar]
- Korenblum W, Barthel A, Licinio J, Wong ML, Worlf OT, Kirschbaum C et al. Elevated cortisol levels and increased rates of diabetes and mood symptoms in Soviet Union-born Jewish immigrants to Germany. Mol Psychiatry 2005; 10: 974–975. [DOI] [PubMed] [Google Scholar]
- Dong C, Wong ML, Licinio J. Sequence variations of ABCB1, SLC6A2, SLC6A3, SLC6A4, CREB1, CRHR1 and NTRK2: association with major depression and antidepressant response in Mexican-Americans. Mol Psychiatry 2009; 14: 1105–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champely S.pwr: Basic Functions for Power Analysis. R package version 1.1-2 2015.
- Team RDCThe R Project for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Cohen J. Statistical Power Analysis For The Behavioral Sciences, 2nd edn. Lawrence Erlbaum Associates Publishers: Hillsdale, NJ, USA, 1988. [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 2010; 6: e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 575–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 2003; 31: 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7: 575–576. [DOI] [PubMed] [Google Scholar]
- Segura V, Vilhjalmsson BJ, Platt A, Korte A, Seren U, Long Q et al. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat Genet 2012; 44: 825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 1995; 57: 289–300. [Google Scholar]
- Vélez JI, Correa JC, Arcos-Burgos M. A new method for detecting significant p-values with applications to genetic data. Rev Colombiana Estadist 2014; 37: 67–76. [Google Scholar]
- Liu DJ, Leal SM. Replication strategies for rare variant complex trait association studies via next-generation sequencing. Am J Hum Genet 2010; 87: 790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin RC. The interaction of selection and linkage. I. General considerations; heterotic models. Genetics 1964; 49: 49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009; 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011; 27: 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy A, Trabzuni D, Guelfi S, Arghese C, Smith C,, Walker R et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci 2014; 17: 1418–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronardi C, Paz-Filho GJ, Valdez E, Maestre-Mesa J, Licinio J, Wong ML. Long-term body weight outcomes of antidepressant-environment interactions. Mol Psychiatry 2011; 16: 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedou G, Pryce C, Di Iorio L, Heidbreder CA, Feldon J. An automated analysis of rat behavior in the forced swim test. Pharmacol Biochem Behav 2001; 70: 65–76. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—excel-based tool using pair-wise correlations. Biotechnol Lett 2004; 26: 509–515. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002; 3: RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djordjevic A, Djordjevic J, Elakovic I, Adzic M, Matic G, Radojcic MB. Fluoxetine affects hippocampal plasticity, apoptosis and depressive-like behavior of chronically isolated rats. Prog Neuropsychopharmacol Biol Psychiatry 2012; 36: 92–100. [DOI] [PubMed] [Google Scholar]
- Kokare DM, Dandekar MP, Singru PS, Gupta GL, Subhedar NK. Involvement of alpha-MSH in the social isolation induced anxiety- and depression-like behaviors in rat. Neuropharmacology 2010; 58: 1009–1018. [DOI] [PubMed] [Google Scholar]
- Weiss IC, Pryce CR, Jongen-Relo AL, Nanz-Bahr NI, Feldon J. Effect of social isolation on stress-related behavioural and neuroendocrine state in the rat. Behav Brain Res 2004; 152: 279–295. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap ConsortiumInternational HapMap ConsortiumAltshuler DM International HapMap ConsortiumGibbs RA International HapMap ConsortiumPeltonen L International HapMap ConsortiumAltshuler DM International HapMap ConsortiumGibbs RA et al. Integrating common and rare genetic variation in diverse human populations. Nature 2010; 467: 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project ConsortiumGenomes Project ConsortiumAbecasis GR Genomes Project ConsortiumAuton A Genomes Project ConsortiumBrooks LD Genomes Project ConsortiumDePristo MA Genomes Project ConsortiumDurbin RM et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NA, Coram MA, Shriver MD, Romieu I, Barsh GS, London SJ et al. Ancestral components of admixed genomes in a Mexican cohort. PLoS Genet 2011; 7: e1002410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone CN, Castellvi-Bel S, Chang LM, Bessa X, Nakagawa H, Harada H et al. ARHGAP8 is a novel member of the RHOGAP family related to ARHGAP1/CDC42GAP/p50RHOGAP: mutation and expression analyses in colorectal and breast cancers. Gene 2004; 336: 59–71. [DOI] [PubMed] [Google Scholar]
- Bertonha FB, Barros Filho Mde C, Kuasne H, Dos Reis PP, da Costa Prando E, Muñoz JJ et al. PHF21B as a candidate tumor suppressor gene in head and neck squamous cell carcinomas. Mol Oncol 2015; 9: 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi MA, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R. A core–BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc Natl Acad Sci U S A 2002; 99: 7420–7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klajn A, Ferrai C, Stucchi L, Prada I, Podini P, Baba T et al. The rest repression of the neurosecretory phenotype is negatively modulated by BHC80, a protein of the BRAF/HDAC complex. J Neurosci 2009; 29: 6296–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan F, Collins RE, De Cegli R, Alpatov R, Horton JR, Shi X et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature 2007; 448: 718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan K, McDermid HE. The 22q13.3 deletion syndrome (Phelan–McDermid syndrome). Mol Syndromol 2012; 2: 186–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J, Champagne N, Lafreniere RG, Xiong L, Spiegelman D, Brustein E et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc Natl Acad Sci U S A 2010; 107: 7863–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 2007; 39: 25–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegood J, Markx S, Lerch JP, Steadman PE, Genç, Provenzano F et al. Neuroanatomical phenotypes in a mouse model of the 22q11.2 microdeletion. Mol Psychiatry 2014; 19: 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shprintzen RJ. Velo-cardio-facial syndrome: 30 years of study. Dev Disabil Res Rev 2008; 14: 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SX, Yi JJ, Calkins ME, Kohler CG, Souders MC, McDonald-McGinn DM et al. Psychiatric disorders in 22q11.2 deletion syndrome are prevalent but undertreated. Psychol Med 2014; 44: 1267–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolin EM, Weller RA, Jessani NR, Zackai EH, McDonald-McGinn DM, Weller EB. Affective disorders and other psychiatric diagnoses in children and adolescents with 22q11.2 Deletion syndrome. J Affect Disord 2009; 119: 177–180. [DOI] [PubMed] [Google Scholar]
- Gur RE, Yi JJ, McDonald-McGinn DM, TAng SX, Calkins ME, Whinna D et al. Neurocognitive development in 22q11.2 deletion syndrome: comparison with youth having developmental delay and medical comorbidities. Mol Psychiatry 2014; 19: 1205–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuillin A, Bass NJ, Kalsi G, Lawrence J, Puri V, Choudhury K et al. Fine mapping of a susceptibility locus for bipolar and genetically related unipolar affective disorders, to a region containing the C21ORF29 and TRPM2 genes on chromosome 21q22.3. Mol Psychiatry 2006; 11: 134–142. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS et al. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun 2013; 4: 1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ML, Dong C, Maestre-Mesa J, Licinio J. Polymorphisms in inflammation-related genes are associated with susceptibility to major depression and antidepressant response. Mol Psychiatry 2008; 13: 800–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Derecki NC, Lovenberg TW, Drevets WC. Role of neuro-immunological factors in the pathophysiology of mood disorders. Psychopharmacology 2016; 233: 1623–1636. [DOI] [PubMed] [Google Scholar]
- Alt A, Nisenbaum ES, Bleakman D, Witkin JM. A role for AMPA receptors in mood disorders. Biochem Pharmacol 2006; 71: 1273–1288. [DOI] [PubMed] [Google Scholar]
- Miladinovic T, Nashed MG, Singh G. Overview of glutamatergic dysregulation in central pathologies. Biomolecules 2015; 5: 3112–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelucci F, Brene S, Mathe AA. BDNF in schizophrenia, depression and corresponding animal models. Mol Psychiatry 2005; 10: 345–352. [DOI] [PubMed] [Google Scholar]
- Jansen R, Penninx BW, Madar V, Xia K, Milaneschi Y, Hottenga JJ et al. Gene expression in major depressive disorder. Mol Psychiatry 2016; 21: 339–347. [DOI] [PubMed] [Google Scholar]
- Song C, Leonard BE. The olfactory bulbectomised rat as a model of depression. Neurosci Biobehav Rev 2005; 29: 627–647. [DOI] [PubMed] [Google Scholar]
- Yuan TF, Slotnick BM. Roles of olfactory system dysfunction in depression. Prog Neuropsychopharmacol Biol Psychiatry 2014; 54: 26–30. [DOI] [PubMed] [Google Scholar]
- Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet 2001; 69: 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortijo S, Wardenaar R, Colome-Tatche M, Gilly A, Etcheverry M, Labadie K et al. Mapping the epigenetic basis of complex traits. Science 2014; 343: 1145–1148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.