

Abstract
Policy Points:
Randomized trials—the gold standard of evaluating effectiveness—constitute a small minority of existing evidence on agents given accelerated approval. One‐third of randomized trials are in therapeutic areas outside of FDA approval and less than half evaluate the therapeutic benefits of these agents but use them instead as common backbone treatments.
Agents receiving accelerated approval are often tested concurrently in several therapeutic areas.
For most agents, no substantial time lag is apparent between the average start dates of randomized trials evaluating their effectiveness and those using them as part of background therapies.
There appears to be a tendency for therapeutic agents receiving accelerated approval to quickly become an integral component of standard treatment, despite potential shortcomings in their evidence base.
Context
Therapeutic agents treating serious conditions are eligible for Food and Drug Administration (FDA) accelerated approval. The clinical evidence accrued on agents receiving accelerated approval has not been systematically evaluated. Our objective was to assess the timing and characteristics of available studies.
Methods
We first identified clinical studies of novel therapeutic agents receiving accelerated approval. We then (1) categorized those studies as randomized or nonrandomized, (2) explored whether they evaluated the FDA‐approved indications, and (3) documented the available treatment comparisons. We also meta‐analyzed the difference in start times between randomized studies that (1) did or did not evaluate approved indications and (2) were or were not designed to evaluate the agent's effectiveness.
Findings
In total, 37 novel therapeutic agents received accelerated approval between 2000 and 2013. Our search of ClinicalTrials.gov identified 7,757 studies, which included 1,258,315 participants. Only one‐third of identified studies were randomized controlled trials. Of 1,631 randomized trials with advanced recruitment status, 906 were conducted in therapeutic areas for which agents received initial accelerated approval, 202 were in supplemental indications, and 523 were outside approved indications. Only 411 out of 906 (45.4%) trials were designed to test the effectiveness of agents that received accelerated approval (“evaluation” trials); others used these agents as common background treatment in both arms (“background” trials). There was no detectable lag between average start times of trials conducted within and outside initially approved indications. Evaluation trials started on average 1.52 years (95% CI: 0.87 to 2.17) earlier than background trials.
Conclusions
Cumulative evidence on agents with accelerated approvals has major limitations. Most clinical studies including these agents are small and nonrandomized, and about a third are conducted in unapproved areas, typically concurrently with those conducted in approved areas. Most randomized trials including these therapeutic agents are not designed to directly evaluate their clinical benefits but to incorporate them as standard treatment.
Keywords: Food and Drug Administration, pharmaceutical policy, market authorization, accelerated approval
The aim of biopharmaceutical regulation within the US Food and Drug Administration (FDA) is to ensure that only effective and safe treatments reach patients.1 FDA guidance suggests that manufacturers submit at least 2 well‐controlled randomized clinical studies, each providing independent evidence of efficacy for their products.2 The FDA reviews the clinical studies of investigational products and evaluates whether they are safe and effective in well‐defined groups of patients before granting approval.
Over the past 3 decades, the FDA introduced significant flexibility to its evidence standards.3 Regulators created several programs aimed at expediting the approval of novel therapeutic agents that address unmet medical needs in the treatment of serious or life‐threatening conditions.4 For example, the “fast track” designation, created in 1988, permits approval of agents treating severely debilitating diseases after a single study. Such expedited approval programs have become more common in recent years.5 Over half of new agents entering the market benefit from expedited approval programs.
Background
FDA's Accelerated Approval Pathway
In 1992, Congress enacted the “accelerated approval” pathway.1 Drugs and biologics expected to provide a meaningful advantage over available therapies for serious conditions have since been eligible for receiving accelerated FDA approval on the basis of surrogate measures.3 Surrogate measures are proxies for clinically meaningful outcomes and are “reasonably likely” to predict clinical benefit. An oft‐cited example of an established surrogate measure is the magnitude of cholesterol reduction, which has been demonstrated to predict the risk of future fatal and nonfatal heart attacks.6 Such established surrogate measures are frequently used in regular FDA approvals.7 However, many surrogate measures used for accelerated approval decisions are not established and have a weak empirical association with important outcomes such as overall survival.8, 9, 10 Hence, agents granted accelerated approval on the basis of surrogate measures are required to conduct studies to confirm the anticipated clinical benefit.4
The objective of using surrogate measures as a basis for FDA‐accelerated approval is to reduce the evidence requirements and considerably shorten the duration of clinical testing required prior to market entry for agents targeting important conditions. The bar for market entry is therefore substantially lower for agents receiving accelerated versus regular approval. Clinical studies used as the basis of regulatory decisions in the accelerated approval pathway are relatively small, have shorter follow‐up durations, often lack comparators, and are less likely to be randomized.11 Collectively, these study features effectively reduce the research and development timelines for agents in the accelerated approval pathway. According to a recent study, oncology agents receiving accelerated approval entered the market on average 4.7 years earlier than those receiving regular approval.12
Limitations of Available Evidence
Therapeutic agents granted accelerated approval have premature data on their clinical benefits and harms at the time of market entry. Substantial research is thus needed to compensate for the limitations of the evidence base and generate meaningful, definitive data confirming clinical benefit following accelerated approval. However, the FDA's evidence standards for accelerated approval may influence evidence generation patterns not only for clinical studies aimed at regulatory approval, but also for the much larger numbers of studies conducted both before and after market entry.
First, a low bar for initial market entry may have compounding effects on subsequent research activities and may facilitate clinical studies aimed at developing new uses. The industry may fragment and diversify its development activities and pursue research in several therapeutic areas in parallel and seek approval for supplemental indications.13 Previous research has shown that regulatory requirements for supplemental indications are less arduous than those for original indications, potentially presenting an even lower bar for market entry than original accelerated approval. For example, clinical studies submitted for supplemental indications are less likely to have active comparators.14 Regulatory review times for supplemental indication approvals are also shorter than those for original indications.15 Pursuing research in several therapeutic areas in parallel is therefore a financially attractive strategy for the industry. While there may be a scientific rationale for testing the efficacy of a new agent outside of its FDA‐approved indication, such decisions appear to be driven by commercial objectives; approximately 9 out of 10 approvals for supplemental indications occur during the market exclusivity period.16 Supplemental indications ultimately account for a substantial share of drug utilization, often surpassing the levels of use within the original indication.17
Second, FDA approval of a novel therapeutic agent may be misinterpreted by the research community as regulatory backing for its immediate and widespread use as standard therapy.18, 19 The agent granted accelerated approval may thus be used in trials of new drugs either as part of combination therapy or as a background treatment given to all patients. While there may be strong scientific rationale for evaluating the effectiveness of a new drug given in combination with an accelerated approval agent, such studies would not be able to generate evidence on the clinical benefits of the accelerated approval agent. Studies giving the accelerated approval agent to all patients would be warranted only after the clinical benefit of the accelerated approval agent has been established. If research evaluating the effectiveness of the accelerated approval agent is underway concurrently with research using it as background therapy, this may indicate that the research community has accepted it as an effective treatment option before valid evidence is available to confirm benefit. The speed with which accelerated approval drugs are tested in different combinations and as background therapy would thus be indicative of the strength of the regulatory signal sent to the research community with each accelerated approval decision.18
Objectives
To date, there has not been a systematic evaluation of the clinical evidence on agents receiving accelerated approval. Here, we explore the extent to which the objectives, nature, and timing of research activity are aimed at addressing the limitations of the data available on agents receiving accelerated approval between 2000 and 2013. First, we document the total number of clinical studies, their sample size, and design characteristics. Second, we investigate the extent to which clinical evidence on novel therapeutic agents is generated in therapeutic areas for which the FDA granted accelerated approval versus in other indications. Third, we describe the types of available treatment comparisons in identified randomized controlled trials and determine whether these comparisons allow for evaluating the effectiveness of accelerated approval agents. We also examine the relative timing of randomized controlled trials done within versus outside of approved indications and those conducted for assessing effectiveness versus background use of these agents.
Methods
Eligible Agents
We used the Drugs@FDA online database to identify the novel therapeutic agents (ie, new molecular entities or novel biologic drugs) approved by the FDA.20 Drugs@FDA includes drug approval and labeling decisions for all currently marketed prescription drugs and biologics. Using several publicly available FDA documents in combination with Drugs@FDA,20, 21 3 investigators identified agents approved through the accelerated approval pathway between January 1, 2000, and December 31, 2013, and the indication(s) for which they were initially approved. Agents were included if they were categorized by the FDA as “S” (“surrogate”), which designates an approval that was based on a surrogate endpoint or an effect on a clinical endpoint other than survival or irreversible morbidity. We excluded 2 agents with “R” (“restricted”) designation, which indicates that an approval had restrictions to ensure safe use. If an agent received multiple approvals for different indications, doses, or routes of administration during this period, we considered only the initial approval (ie, therapeutic agents that received accelerated approval in a supplemental indication were not eligible for inclusion). We confirmed the consistency of our selected sample with a published report on FDA approvals.22
Identification and Categorization of Clinical Studies
We screened ClinicalTrials.gov to identify the clinical studies of therapeutic agents receiving accelerated approval. ClinicalTrials.gov is a publicly available comprehensive clinical study registry and results database developed and maintained by the US National Library of Medicine.23 It contains 232,506 study records as of December 2016.24
A data set comprising all registered clinical studies of eligible agents was downloaded from ClinicalTrials.gov on June 8, 2016. The data set included the following information:
Disease or condition
Treatment comparator(s)
Recruitment status (whether the study was still recruiting participants)
Enrollment (sample size)
Study design (whether treatments were randomly allocated)
Study start and end dates
Three investigators reviewed the clinical studies. Using predetermined criteria, we categorized each identified study according to its design, recruitment status, therapeutic area, and available treatment comparisons. As detailed below, we reviewed the identified studies in 3 levels. At the first level, we determined the proportion of available evidence that was generated in randomized controlled trials. Randomized controlled trials are considered to be the “gold standard” for establishing whether an investigational agent works or if it works better than another.25 Decades of research have shown that nonrandomized study designs (ie, observational studies) are more likely to produce biased findings about the efficacy of a treatment.26, 27, 28
Second, we identified the subset of randomized trials with advanced recruitment status and examined the proportion of available trials conducted in diseases or conditions with granted accelerated approval (ie, FDA‐approved indications). Finally, we examined the types of treatment comparisons available in randomized controlled trials conducted in initially approved indications.
Level 1: Study Design
Studies identified in ClinicalTrials.gov were considered to be randomized controlled trials if they randomly allocated participants into two or more treatment arms. All other designs were categorized as nonrandomized studies. For each agent, we counted the number of randomized versus nonrandomized studies and the number of participants included.
Among studies with randomized designs, we focused on studies with advanced recruitment status. We therefore excluded studies that were still recruiting participants and withdrawn studies. Consistent with a previous review of ClinicalTrials.gov,29 studies were eligible for inclusion if they had any of the following recruitment designations: “active, not recruiting” (participants no longer enrolled, study may be ongoing); “completed” (trial ended normally); or “terminated” (trial was stopped early for any reason). We included “active, not recruiting” because investigators do not consistently update this designation after completing enrollment; many studies with published results have this listing. We also checked and confirmed that no studies that were still recruiting had results available in ClinicalTrials.gov.
Level 2: Condition
We determined whether randomized controlled trials were conducted within or outside of indications for which agents received accelerated approval, as identified using the Drugs@FDA database. ClinicalTrials.gov specifies for each study the primary disease or condition being evaluated according to the Medical Subject Headings (MeSH) controlled vocabulary used by the National Library of Medicine. We considered randomized controlled trials to be conducted outside of approved indications if done in patient populations with different diseases or conditions (eg, multiple myeloma vs lymphoma; colorectal carcinoma vs head and neck cancer) to those specified in drug labels following initial accelerated approval or subsequent approval in new indications.14 Our categorization was conservative: clinical studies in expanded patient populations (eg, for first‐line vs second‐line treatment) or modified indications (eg, adjunctive treatment vs monotherapy) were still considered to be conducted in approved areas.14 For each agent, we counted the number of studies conducted within and outside of FDA‐approved indications and the total number of participants.
Level 3: Comparison
Among randomized controlled trials with advanced recruitment status conducted within initially approved indications, we separated “evaluation” trials from “background” trials. Evaluation trials included placebo‐controlled or head‐to‐head trials that evaluated the clinical benefits of agents receiving accelerated approval (eg, trials comparing strategies A + B vs B, or A vs B, where A is the agent of interest, and B is either an active or inactive control/agent). Background trials included randomized controlled trials where treatment effects of agents with accelerated approval could not be isolated because they were used as common background (“backbone”) treatment in both arms (eg, trials comparing strategies A + B versus A, or A + B versus A + C, where A is the agent of interest, and B and C are other agents). We also considered trials comparing the effectiveness of different treatment sequences as background trials since the effect of agents receiving accelerated approval could not be isolated in such trials. We counted for each agent the total number of evaluation and background trials and the total number of participants included.
Evaluation of Time Lag
We plotted and visually inspected the start dates and durations of identified randomized controlled trials. For each agent, we then used descriptive statistics to examine the similarity in start dates of trials conducted in therapeutic areas receiving initial accelerated approval and those conducted in other areas. In a similar fashion, we evaluated whether there was any detectable time lag between the evaluation and background trials for each agent. For both sets of comparisons, we first used t tests at the drug level to statistically evaluate the differences in trial start dates between the two sets of studies that (1) did or did not evaluate approved indications and (2) were or were not designed to evaluate the drug's effectiveness. We then inspected the variability in average time lag across different therapeutic agents using the I2 measure. Point estimate of I2 describes the percentage of the observed variability that is due to heterogeneity rather than chance.30 I2 over 50% is conventionally considered to indicate large heterogeneity.
We performed meta‐analyses to quantify the magnitude of mean time lags across all agents. We adopted the Hartung‐Knapp‐Sidik‐Jonkman method to perform random‐effects meta‐analyses,31, 32 since it outperforms other common random‐effects methods.33 We report mean differences and 95% confidence intervals (CIs). Statistical analyses were performed in STATA version 14 (StataCorp). Two‐tailed P values less than 0.05 were considered statistically significant.
Results
Overview of Approved Agents and Their Clinical Studies
The FDA granted accelerated approval to 37 novel therapeutic agents between 2000 and 2013. During this period, 2 to 4 new therapeutic agents per year entered the market in the accelerated approval pathway. Oncological and antiviral agents accounted for four‐fifths of accelerated approvals during this period (Figure 1).
Figure 1.
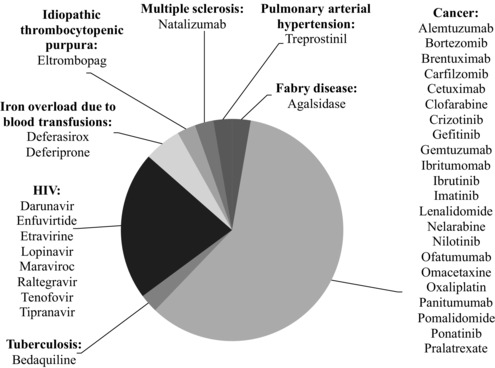
Novel Therapeutic Agents Granted Accelerated Approval From 2000 to 2013 According to Therapeutic Area
Figure 2 shows the 3 levels of our review and the flow of identified records in the study. Our search on ClinicalTrials.gov yielded 8,951 records, corresponding to 7,757 individual studies (excluding duplicates) of agents granted accelerated approval. In total, there were 1,258,315 participants included in these studies. We observed significant variation in the number of studies per accelerated approval agent, ranging from 18 (for omacetaxine mepesuccinate) to 1,417 (for oxaliplatin). Most agents (23 agents; 62.2%) had 100 or more study records and 6 agents (16.2%) had 500 or more.
Figure 2.
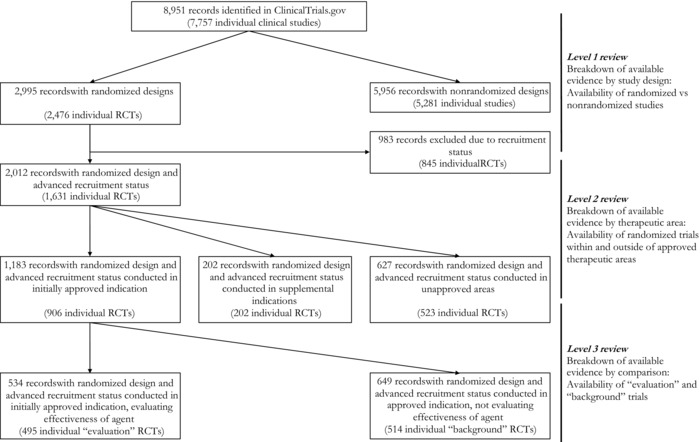
Flow of Identified ClinicalTrials.gov Records in the Study According to 3 Levels of Reviewa
Abbreviations: RCT, randomized controlled trial.
aTotal number of records at each level of the review may include duplicates; trials including more than one accelerated approval agent are counted separately for each therapeutic agent. Numbers in parentheses refer to the total number of studies without any duplicates.
Most identified studies were very small, with 5,469 studies (70.5% of total) including 100 or fewer participants. For 9 agents, studies with fewer than 100 participants accounted for more than 80% of all available studies. Only 502 studies (6.47% of total) included 500 or more participants.
Level 1: Study Design
We categorized approximately one‐third (n = 2,995) of identified records as randomized controlled trials, corresponding to 2,476 individual studies after removing duplicates (see Figure 2). The number of randomized controlled trials ranged from 1 for omacetaxine mepesuccinate to 536 for oxaliplatin; 5 agents had fewer than 10 randomized studies while 10 agents had 100 trials or more (Table 1). Overall, 681,834 participants (45.5%) were included in randomized controlled trials; the number of participants in randomized trials ranged from 680 for pralatrexate to 200,763 for oxaliplatin. Proportions of participants included in randomized controlled trials ranged from 22.0% for oxaliplatin to 91.2% for natalizumab (see Table 1).
Table 1.
Total Number and Distribution of Clinical Studies and Study Participants for Each Agent According to 3 Levels of Review
| Identified studies | Level 1: Availability of randomized controlled trials | Level 2: Availability of randomized controlled trial in FDA‐approved indication | Level 3: Availability of randomized controlled trial evaluating agent's effectiveness | |||||
|---|---|---|---|---|---|---|---|---|
| Accelerated approval agents | Total number of clinical studies identified | Number of participants included | Number of randomized trials (%) | Number of participants (%) | Number of trials (%) | Number of participants (%) | Number of “evaluation” trials (%) | Number of participants (%) |
| Agalsidase | 36 | 11,437 | 7 (19.4) | 10,541 (92.2) | 4 (100.0) | 193 (100.0) | 4 (100.0) | 193 (100.0) |
| Alemtuzumab | 326 | 26,962 | 59 (18.1) | 16,136 (59.8) | 8 (20.0) | 969 (11.9) | 7 (87.5) | 923 (95.3) |
| Bedaquiline | 28 | 6,594 | 18 (64.3) | 2,661 (40.4) | 6 (50.0) | 781 (77.0) | 4 (66.7) | 436 (55.8) |
| Bortezomib | 804 | 79,503 | 168 (20.9) | 35,936 (45.2) | 67 (59.3) | 22,388 (78.8) | 28 (41.8) | 7,462 (33.3) |
| Brentuximab | 107 | 9,256 | 16 (14.9) | 4,096 (44.3) | 6 (100.0) | 685 (100.0) | 3 (50.0) | 491 (71.7) |
| Carfilzomib | 121 | 11,882 | 17 (14.0) | 6,076 (51.1) | 6 (87.5) | 2,996 (98.9) | 4 (66.7) | 2,918 (97.4) |
| Cetuximab | 750 | 102,336 | 257 (34.3) | 31,333 (30.6) | 75 (42.4) | 29,870 (57.3) | 41 (54.7) | 24,700 (82.7) |
| Clofarabine | 156 | 18,312 | 21 (13.5) | 5,728 (31.3) | 1 (10.0) | 405 (11.5) | 0 (0.0) | 0 (0.0) |
| Crizotinib | 102 | 23,216 | 24 (23.5) | 17,116 (73.7) | 9 (64.3) | 1,997 (94.7) | 5 (55.6) | 1,227 (61.4) |
| Darunavir | 206 | 65,040 | 112 (54.4) | 40,580 (62.4) | 64 (73.6) | 17,350 (91.3) | 24 (37.5) | 6,931 (39.9) |
| Deferasirox | 90 | 11,339 | 25 (27.8) | 8,391 (74.0) | 10 (35.7) | 1,000 (48.9) | 8 (80.0) | 1,746 (95.6) |
| Deferiprone | 57 | 7,345 | 35 (61.4) | 2,082 (28.3) | 7 (29.2) | 526 (18.9) | 4 (57.1) | 233 (44.3) |
| Eltrombopag | 102 | 8,614 | 33 (32.4) | 3,922 (45.5) | 8 (30.8) | 736 (20.5) | 8 (100.0) | 736 (100.0) |
| Enfuvirtide | 67 | 7,294 | 30 (44.8) | 3,718 (50.9) | 21 (77.8) | 2,956 (91.6) | 11 (52.4) | 863 (30.0) |
| Etravirine | 79 | 18,824 | 32 (40.5) | 14,142 (75.1) | 19 (67.9) | 3,680 (92.4) | 14 (73.7) | 2,777 (75.5) |
| Gefitinib | 366 | 55,600 | 118 (32.2) | 28,174 (50.7) | 47 (58.8) | 16,934 (80.3) | 35 (74.5) | 12,793 (77.9) |
| Gemtuzumab | 73 | 15,908 | 19 (26.0) | 5,757 (36.2) | 13 (100.0) | 6,759 (100.0) | 11 (84.6) | 6,490 (96.0) |
| Ibritumomab | 105 | 10,615 | 17 (16.2) | 7,526 (70.9) | 10 (100.0) | 1,446 (100.0) | 8 (80.0) | 1,432 (99.0) |
| Ibrutinib | 165 | 25,195 | 27 (16.4) | 18,019 (71.5) | 2 (14.3) | 804 (17.9) | 2 (100.0) | 804 (100.0) |
| Imatinib | 631 | 82,090 | 132 (20.9) | 50,049 (60.9) | 36 (37.9) | 9,776 (47.5) | 24 (66.7) | 7,433 (76.0) |
| Lenalidomide | 696 | 82,986 | 161 (23.1) | 35,899 (43.3) | 7 (6.9) | 1,163 (3.5) | 5 (71.4) | 872 (75.0) |
| Lopinavir | 365 | 109,939 | 210 (57.5) | 52,313 (47.6) | 133 (75.1) | 43,016 (84.0) | 64 (48.1) | 16,615 (40.2) |
| Maraviroc | 140 | 20,631 | 69 (49.3) | 9,708 (47.1) | 42 (73.7) | 7,584 (86.2) | 28 (66.7) | 5,657 (74.6) |
| Natalizumab | 95 | 67,003 | 27 (28.4) | 61,079 (91.2) | 14 (73.7) | 4,230 (81.1) | 13 (92.9) | 4,088 (96.6) |
| Nelarabine | 23 | 4,154 | 2 (8.7) | 1,534 (36.9) | 1 (100.0) | 1,900 (100.0) | 1 (100.0) | 1,900 (100.0) |
| Nilotinib | 167 | 26,758 | 33 (19.8) | 19,316 (72.2) | 10 (47.6) | 2,258 (64.1) | 6 (60.0) | 2,136 (94.6) |
| Ofatumumab | 114 | 12,450 | 28 (24.6) | 4,145 (33.3) | 8 (44.4) | 2,641 (61.2) | 7 (87.5) | 2,371 (89.8) |
| Omacetaxine | 18 | 903 | 1 (5.6) | 898 (99.4) | 1 (100.0) | 5 (100.0) | 0 (0.0) | 0 (0.0) |
| Oxaliplatin | 1,417 | 257,595 | 536 (37.8) | 56,832 (22.1) | 183 (69.3) | 99,762 (87.0) | 36 (19.7) | 23,378 (23.4) |
| Panitumumab | 198 | 28,500 | 64 (32.3) | 11,010 (38.6) | 21 (50.0) | 7,695 (68.2) | 12 (57.1) | 6,994 (90.9) |
| Pomalidomide | 110 | 10,977 | 27 (24.5) | 6,373 (58.1) | 8 (53.3) | 1,414 (65.5) | 5 (62.5) | 920 (65.1) |
| Ponatinib | 30 | 3,177 | 4 (13.3) | 1,720 (54.1) | 1 (100.0) | 307 (100.0) | 1 (100.0) | 307 (100.0) |
| Pralatrexate | 29 | 1,388 | 5 (17.2) | 708 (51.0) | 4 (80.0) | 786 (79.6) | 2 (50.0) | 137 (28.6) |
| Raltegravir | 297 | 49,113 | 153 (51.5) | 24,253 (49.4) | 89 (78.1) | 18,701 (94.0) | 55 (61.8) | 15,439 (82.6) |
| Tenofovir | 716 | 234,714 | 436 (60.9) | 82,261 (35.0) | 212 (66.0) | 71,525 (67.5) | 38 (17.9) | 12,098 (17.2) |
| Tipranavir | 82 | 13,849 | 32 (39.0) | 8,921 (64.4) | 17 (56.7) | 3,455 (83.5) | 7 (41.2) | 1,185 (34.3) |
| Treprostinil | 83 | 13,997 | 40 (48.2) | 10,402 (74.3) | 13 (56.5) | 1,476 (77.0) | 9 (69.2) | 1,448 (98.1) |
Level 2: Condition
Of 1,631 randomized controlled trials with advanced recruitment status, 906 (55.5%) were conducted in therapeutic areas for which agents received initial FDA‐accelerated approval, 202 trials (12.4%) were in supplemental indications, and 523 (32.1%) were in unapproved indications (see Figure 2). The number of available trials in initially approved areas varied considerably across agents, ranging from 1 for omacetaxine mepesuccinate and ponatinib to 212 for tenofovir. As shown in Table 1, trials in initial indications accounted for 50% or less of all randomized controlled trials for 8 agents receiving accelerated approval. For 7 agents, all available randomized controlled trials were in therapeutic areas for which the agents received initial accelerated approval.
In total, trials conducted in initially approved areas included 390,169 participants (70.0% of total); trials in supplemental indications included 52,761 participants (9.5%). Taken together, these participants accounted for less than 20.0% of total trial populations for 3 accelerated approval agents: alemtuzumab (969; 11.9%), clofarabine (405; 11.5%), and deferiprone (526; 18.9%) (see Table 1).
Level 3: Comparison
The majority (495/906; 54.6%) of randomized controlled trials conducted in initially approved indications did not evaluate the effectiveness of accelerated approval agents in these indications (ie, “background” trials). The remaining 411 (45.4%) were “evaluation” trials, testing the effectiveness of accelerated approval agents in FDA‐approved indications. The number of available evaluation trials per agent ranged from 0 to 64, with more than 50 evaluation trials identified for lopinavir‐ritonavir and raltegravir used to treat HIV (see Table 1). While all available randomized controlled trials for 5 accelerated approval agents evaluated their effectiveness, there were no evaluation trials for 2 agents (clofarabine and omacetaxine mepesuccinate).
Overall, 176,133 participants (45.5% of total) were included in randomized trials designed to test the effectiveness of agents granted accelerated approval. The total number of individuals participating in evaluation trials ranged from 0 to 24,700 (see Table 1). The share of participants in evaluation trials accounted for less than half of the total population for 11 agents.
Breakdown of Cumulative Evidence Available on Each Agent
Figure 3 summarizes the findings of the 3 levels of our review for each therapeutic agent. Despite the significant variation in the total number of studies and sample sizes for each agent, a broadly consistent pattern emerged about the relative distribution of available evidence as we restricted our sample from all identified studies to randomized controlled trials evaluating the effectiveness of accelerated approval agents in initially approved indications. Only a small proportion of evidence available on novel therapeutic agents granted accelerated approval had randomized study designs (median: 26.0%; range: 5.6%‐64.3%), while randomized controlled trials in initially approved indications accounted for a median proportion of 10.6% of all available evidence (range: 0.6%‐36.4%), and randomized controlled trials evaluating the effectiveness of accelerated approval agents in initially approved indications accounted for a median proportion of 6.1% (range: 0.0%‐20.0%) of all available evidence.
Figure 3.
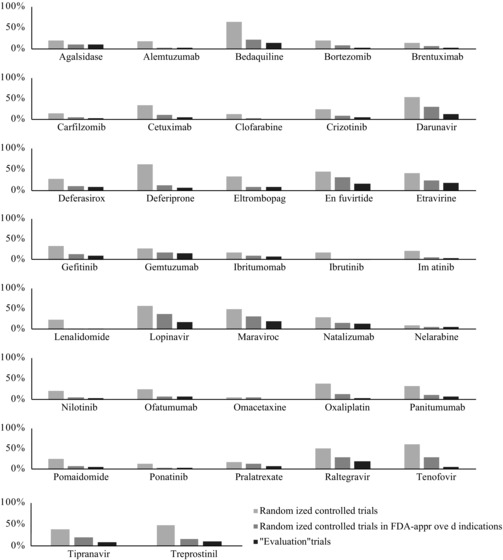
Relative Availability of Evidence in 3 Levels of Review for Each Agenta
a Light gray bars show the relative availability of randomized controlled trials as a subset of all clinical studies. Dark gray bars show the availability of randomized controlled trials in initially approved indications as a subset of all clinical studies. Black bars show the availability randomized controlled “evaluation” trials evaluating the effectiveness of accelerated approval agents relative to all available studies.
Evaluation of Time Lag: Comparing FDA‐Approved and Other Indications
Of 27 agents for which multiple randomized controlled trials conducted both within and outside of initially approved indications were available, we observed similar starting dates for 18 agents. When we meta‐analyzed the difference in start times between randomized trials that did or did not evaluate approved indications, we found no overall difference in average start times across all agents (mean difference = −0.34, 95% CI: −0.95 to 0.27) (Figure 4). There was high between‐agent heterogeneity in mean time lags (I2 = 72.1%). Only 4 agents (deferasirox, deferiprone, etravirine, oxaliplatin) had a time lag of 2 years or more. For gefitinib, randomized controlled trials in initially approved indications started on average 3.54 years later than those in other areas (95% CI: 2.18 to 4.90).
Figure 4.
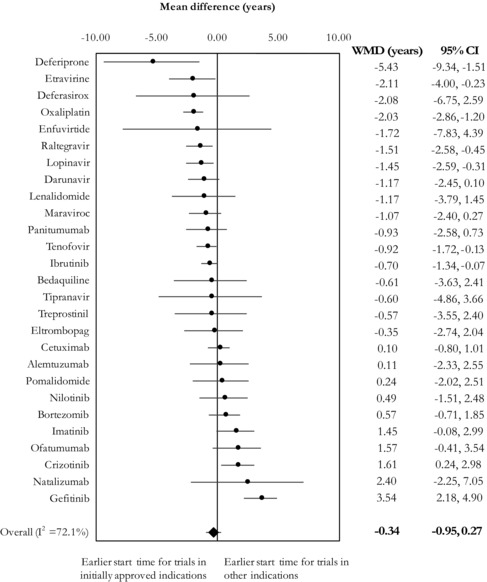
Meta‐Analysis of Mean Time Lags for Each Agent, Comparing the Start Times Between Trials That Were and Were Not Conducted Within Initially Approved Indications
Abbreviations: CI, confidence interval; WMD, weighted mean difference.
I2 refers to the proportion of observed variability that is attributable to heterogeneity rather than chance.
Evaluation of Time Lag: Comparing Evaluation and Background Trials
When we compared the average start times of evaluation and background trials conducted within initially approved indications for the 25 accelerated approval agents for which both sets were available, the summary time lag was −1.52 years (95% CI: −2.17 to −0.87) with substantial between‐agent heterogeneity (I2 = 52.5%) (Figure 5). Of the 25 accelerated approval agents, 22 had on average earlier start dates for evaluation trials than for background trials. However, only 9 agents had a time lag of at least 2 years. Trials evaluating the effectiveness of gemtuzumab started on average 2.18 years (95% CI: 0.34 to 4.02) later than trials using it as background treatment.
Figure 5.
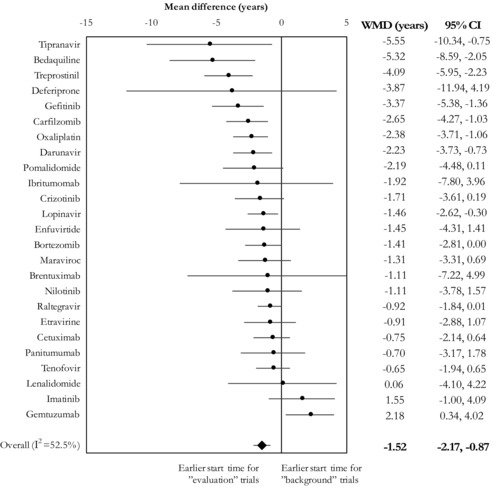
Meta‐Analysis of Mean Time Lags for Each Agent, Comparing the Start Times Between Trials That Were and Were Not Designed to Test the Effectiveness of Therapeutic Agents Receiving Accelerated Approval
Abbreviations: CI, confidence interval; WMD, weighted mean difference.
I2 refers to the proportion of observed variability that is attributable to heterogeneity rather than chance.
Trajectory of Evidence for Each Agent
Table 2 summarizes the trajectory of available evidence for each therapeutic agent, combining all 3 elements of the findings: availability, characteristics, and timing of studies. Although most accelerated approval agents had two or more randomized controlled trials in initially approved indications, only 2 agents (oxaliplatin and tenofovir) had at least 2 studies evaluating their effectiveness in approved indications before studies testing them either in other indications or as background therapies were conducted.
Table 2.
Evidence Trajectory for Each Therapeutic Agent
| Accelerated approval agents | (1) Availability of 2 or more randomized controlled trials | (2) Availability of 2 or more randomized controlled trials in FDA‐approved indications | (3) Availability of 2 or more “evaluation” trials | (4) No time lag between trials conducted within and outside of FDA‐approved indications | (5) No time lag between “evaluation” and “background” trials |
|---|---|---|---|---|---|
| Agalsidase | ✓a | ✓ | ✓ | Not enough datab | Not enough data |
| Alemtuzumab | ✓ | ✓ | ✓ | ✗c | Not enough data |
| Bedaquiline | ✓ | ✓ | ✓ | ✗ | ✓ |
| Bortezomib | ✓ | ✓ | ✓ | ✗ | ✓ |
| Brentuximab | ✓ | ✓ | ✓ | Not enough data | ✗ |
| Carfilzomib | ✓ | ✓ | ✓ | Not enough data | ✓ |
| Cetuximab | ✓ | ✓ | ✓ | ✗ | ✗ |
| Clofarabine | ✓ | ✗ | ✗ | Not enough data | Not enough data |
| Crizotinib | ✓ | ✓ | ✓ | ✗✗d | ✗ |
| Darunavir | ✓ | ✓ | ✓ | ✗ | ✓ |
| Deferasirox | ✓ | ✓ | ✓ | ✗ | Not enough data |
| Deferiprone | ✓ | ✓ | ✓ | ✓ | ✗ |
| Eltrombopag | ✓ | ✓ | ✓ | ✗ | Not enough data |
| Enfuvirtide | ✓ | ✓ | ✓ | ✗ | ✗ |
| Etravirine | ✓ | ✓ | ✓ | ✓ | ✗ |
| Gefitinib | ✓ | ✓ | ✓ | ✗✗ | ✓ |
| Gemtuzumab | ✓ | ✓ | ✓ | Not enough data | ✗✗ |
| Ibritumomab | ✓ | ✓ | ✓ | Not enough data | ✗ |
| Ibrutinib | ✓ | ✓ | ✓ | ✓ | Not enough data |
| Imatinib | ✓ | ✓ | ✓ | ✗ | ✗ |
| Lenalidomide | ✓ | ✓ | ✓ | ✗ | ✗ |
| Lopinavir | ✓ | ✓ | ✓ | ✓ | ✓ |
| Maraviroc | ✓ | ✓ | ✓ | ✗ | ✗ |
| Natalizumab | ✓ | ✓ | ✓ | ✗ | Not enough data |
| Nelarabine | ✓ | ✗ | ✗ | Not enough data | Not enough data |
| Nilotinib | ✓ | ✓ | ✓ | ✗ | ✗ |
| Ofatumumab | ✓ | ✓ | ✓ | ✗ | Not enough data |
| Omacetaxine | ✗ | ✗ | ✗ | Not enough data | Not enough data |
| Oxaliplatin | ✓ | ✓ | ✓ | ✓ | ✓ |
| Panitumumab | ✓ | ✓ | ✓ | ✗ | ✗ |
| Pomalidomide | ✓ | ✓ | ✓ | ✗ | ✗ |
| Ponatinib | ✓ | ✗ | ✗ | Not enough data | Not enough data |
| Pralatrexate | ✓ | ✓ | ✓ | Not enough data | Not enough data |
| Raltegravir | ✓ | ✓ | ✓ | ✓ | ✗ |
| Tenofovir | ✓ | ✓ | ✓ | ✓ | ✗ |
| Tipranavir | ✓ | ✓ | ✓ | ✗ | ✓ |
| Treprostinil | ✓ | ✓ | ✓ | ✗ | ✓ |
aIndicates availability of 2 or more randomized controlled trials in columns 1‐3; indicates statistically significant time lag in columns 4‐5.
bThere was not a sufficient number of studies to estimate time lag.
cIndicates a lack of at least 2 randomized controlled trials in columns 1‐3; indicates a lack of statistically significant time lag in columns 4‐5.
dIndicates a statistically significant time lag with trials outside of approved indications (column 4) and “background” trials starting earlier (column 5).
Discussion
In this study, we systematically evaluated the existing clinical studies of novel therapeutic agents given accelerated approval by the FDA between 2000 and 2013. Our review identified a very large number of studies including more than 1 million participants across several therapeutic areas. However, only a small fraction of these studies were randomized controlled trials; a sizeable proportion of randomized evidence was generated in therapeutic areas outside of the FDA approval; and most randomized controlled trials including accelerated approval agents did not evaluate their clinical benefits, but used them instead as common backbone treatments. Randomized controlled trials in approved versus nonapproved indications started on average at the same time, and randomized trials evaluating the effectiveness of agents granted accelerated approval started on average 1.5 years before trials using them as background treatment.
Pharmaceutical and biotechnology companies often cite difficulties in recruiting adequate numbers of patients as the primary reason for failing to conduct large randomized controlled trials to evaluate the clinical effectiveness of accelerated approval agents in a timely manner.34 Patients might be unwilling to participate in clinical studies after an agent is “reasonably likely” to be superior to available therapies. It may also be difficult to achieve large sample sizes due to small patient populations in some rare disease areas.35 Despite these enrollment‐related concerns, our findings show that very large numbers of patients often participate in randomized controlled trials of varying sizes both within and outside of initially approved therapeutic areas. Therefore, large mega‐trials with these agents might have been feasible, if a portion of these participants could have been funneled to such efforts.36, 37, 38 Currently, the thousands of clinical studies that are performed on these agents comprise mostly small investigations with a substantial proportion of nonrandomized studies. These small and nonrandomized studies offer at best questionable evidence on agents with accelerated approval.39 The scientific value of such small and nonrandomized studies compared to their value as marketing tools or seeding trials to propagate their use should be investigated in future studies.40
Perhaps unsurprisingly, accelerated approvals change the calculus of investments on additional research on new agents. Manufacturer sponsors may have a reduced sense of urgency to generate meaningful evidence on clinical effectiveness once products receive accelerated approval.8 They may not wish to risk unfavorable trial findings or even withdrawal from the market due to demonstrated lack of clinical effectiveness. Therefore, accelerated approval—allowing companies to market their products sooner and with less research expenditure—may serve as a disincentive to conduct additional research in therapeutic areas for which the FDA granted initial approval.8, 41
Indeed, our findings suggest that novel therapeutic agents receiving accelerated approval are often tested concurrently in several therapeutic areas, potentially to seek approval in multiple indications and extend market share.17 Overall, there was no major time lag between the start dates of trials testing the effectiveness of accelerated approval agents in initially approved indications and trials testing them in supplemental indications and unapproved conditions, although there was also some diversity across agents.
Even when we focused on randomized controlled trials conducted in initially approved indications, most were not designed to evaluate accelerated approval agents versus comparators. Instead, accelerated approval agents were more likely to be included as part of background therapies. Ideally, trials demonstrating the effectiveness of novel agents should be available before they are widely tested and used as part of standard treatment algorithms.28 However, no substantial time lag was apparent for many agents in our sample, and the average time lag was only 1.52 years. There appears to be a tendency for therapeutic agents receiving accelerated approval to quickly become an integral component of standard treatment, despite potential shortcomings in their evidence base.
The speed with which accelerated approval agents are embraced by the research community may reflect the perceived pace of innovation in drug development. However, using accelerated approval agents as common backbone treatment while separate trials are concurrently evaluating their effectiveness raises important questions about the efficiency of the research enterprise.42 The obvious risk of this research strategy is that the latter group of trials may find no demonstrable therapeutic benefit of accelerated approval agents. Indeed, several agents initially granted accelerated approval were later found to be ineffective when tested in large randomized controlled trials. For example, gemtuzumab, originally approved for the treatment of acute myeloid leukemia in May 2000 under the FDA's accelerated approval program, was withdrawn from the market in 2010 when its confirmatory postapproval trial showed no clinical benefit.43 While it was still on the market, gemtuzumab was used as a background therapy in several trials, and these trials generally started earlier than those evaluating its effectiveness.
Limitations
This study has some limitations. Our sample consisted of agents approved as early as 2000. The trials of these agents may have incomplete or inconsistent data elements in ClinicalTrials.gov. Although trial sponsors and investigators often retrospectively enter information on ongoing trials, we may have missed randomized controlled trials if they were not registered in ClinicalTrials.gov or if they had missing enrollment numbers. However, limiting our analysis to 16 agents approved after 2007 yielded largely similar inferences (not shown). In addition, ClinicalTrials.gov may have inadequate detail about the disease, disorder, syndrome, or illness for each clinical study. Our categorization of identified studies according to condition was therefore conservative and we considered all randomized controlled trials conducted in expanded, modified, or new indications as within approved therapeutic areas.
The FDA mandates postapproval confirmatory trials to be completed by industry sponsors to demonstrate clinical benefit. In our study, we did not differentiate between the clinical studies conducted before or after the required postapproval confirmatory trials were completed and published. The FDA's evidence standards are increasingly flexible and the completion of confirmatory trials may not always provide definitive evidence of therapeutic benefit. According to a recent systematic evaluation of accelerated approvals of oncology products, the FDA accepted data from single‐arm (nonrandomized, noncomparative) studies as sufficient evidence to grant regular approval to 2 agents that originally received accelerated approval12—a clear deviation from conventional regulatory requirements for market entry.2 Holding all agents to the same standard, we find that only 2 out of 37 accelerated approval agents had acceptable evidence trajectories—timely availability of randomized controlled trials evaluating their effectiveness in initially approved indications—that would meet the information needs of patients, physicians, researchers, and other decision makers in the health care system.
Conclusion
Taken together, our findings highlight the potential consequences of FDA‐accelerated approval decisions on the patterns and dynamics of evidence generation on novel therapeutic agents. Lack of interested participants cannot be raised as an argument against performing large‐scale pragmatic trials to rigorously test these agents for major, patient‐relevant clinical outcomes. The current research landscape is inefficient and fragmented with thousands of small and nonrandomized studies that provide questionable value. Accelerated approval on the basis of nonestablished surrogate measures may be perceived as full regulatory endorsement for new agents, sending a strong signal to patients, physicians, and researchers regarding a novel therapeutic agent's innovative value, safety, and effectiveness and, in turn, influencing the evolution and nature of scientific evidence on agents.
Funding/Support
None
Conflict of Interest Disclosures: All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. All authors declare that they have no financial, personal, or professional interests that could be construed to have influenced the paper. We declare: no support from any organization for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
Acknowledgments: None.
References
- 1. Carpenter D. Reputation and Power: Organizational Image and Pharmaceutical Regulation at the FDA. Princeton, NJ: Princeton University Press; 2014. [Google Scholar]
- 2. US Food and Drug Administration . Guidance for Industry: Providing Clinical Evidence of Effectiveness for Human Drug and Biological Products. Washington, DC: US Food and Drug Administration; 1998. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatory Information/Guidances/UCM078749.pdf. Accessed March 21, 2017. [Google Scholar]
- 3. Kesselheim AS, Darrow JJ. FDA designations for therapeutics and their impact on drug development and regulatory review outcomes. Clin Pharmacol Ther. 2015;97(1):29‐36. [DOI] [PubMed] [Google Scholar]
- 4. US Food and Drug Administration . Guidance for Industry: Expedited Programs for Serious Conditions—Drugs and Biologics. US Food and Drug Administration: Washington, DC; 2014. www.fda.gov/downloads/drugs/guidancecomplianceregulatory information/guidances/ucm358301.pdf. Accessed March 21, 2017. [Google Scholar]
- 5. Kesselheim AS, Wang B, Franklin JM, Darrow JJ. Trends in utilization of FDA expedited drug development and approval programs, 1987‐2014: cohort study. BMJ. 2015;351:h4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol‐lowering treatment: prospective meta‐analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366(9493):1267‐1278. [DOI] [PubMed] [Google Scholar]
- 7. Yu T, Hsu Y‐J, Fain KM, Boyd CM, Holbrook JT, Puhan MA. Use of surrogate outcomes in US FDA drug approvals, 2003‐2012: a survey. BMJ Open. 2015;5(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fleming TR. Surrogate endpoints and FDA's accelerated approval process. Health Aff (Millwood). 2005;24(1):67‐78. [DOI] [PubMed] [Google Scholar]
- 9. Fleming TR, Rothmann MD, Lu HL. Issues in using progression‐free survival when evaluating oncology products. J Clin Oncol. 2009;27(17):2874‐2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prasad V, Kim C, Burotto M, Vandross A. The strength of association between surrogate end points and survival in oncology: a systematic review of trial‐level meta‐analyses. JAMA Intern Med. 2015;175(8):1389‐1398. [DOI] [PubMed] [Google Scholar]
- 11. Downing NS, Aminawung JA, Shah ND, Krumholz HM, Ross JS. Clinical trial evidence supporting FDA approval of novel therapeutic agents, 2005‐2012. JAMA. 2014;311(4):368‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson JR, Ning YM, Farrell A, Justice R, Keegan P, Pazdur R. Accelerated approval of oncology products: the Food and Drug Administration experience. J Natl Cancer Inst. 2011;103(8):636‐644. [DOI] [PubMed] [Google Scholar]
- 13. Luijn JC Van, Danz M, Bijlsma JW, Gribnau FW, Leufkens HG. Post‐approval trials of new medicines: widening use or deepening knowledge? Analysis of 10 years of etanercept. Scand J Rheumatol. 2011;40(3):183‐191. [DOI] [PubMed] [Google Scholar]
- 14. Wang B, Kesselheim AS. Characteristics of efficacy evidence supporting approval of supplemental indications for prescription drugs in United States, 2005–14: systematic review. BMJ. 2015;351:h4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DiMasi JA. Innovating by developing new uses of already‐approved drugs: trends in the marketing approval of supplemental indications. Clin Ther. 2013;35(6):808‐818. [DOI] [PubMed] [Google Scholar]
- 16. Langedijk J, Whitehead CJ, Slijkerman DS, Leufkens HG, Schutjens MH, Mantel‐Teeuwisse AK. Extensions of indication throughout the drug product lifecycle: a quantitative analysis. Drug Discov Today. 2016;21(2):348‐355. [DOI] [PubMed] [Google Scholar]
- 17. Berndt ER, Cockburn IM, Grepin KA. The impact of incremental innovation in biopharmaceuticals: drug utilisation in original and supplemental indications. Pharmacoeconomics. 2006;24 Suppl 2:69‐86. [DOI] [PubMed] [Google Scholar]
- 18. Kesselheim AS, Woloshin S, Eddings W, Franklin JM, Ross KM, Schwartz LM. Physicians’ knowledge about FDA approval standards and perceptions of the “breakthrough therapy” designation. JAMA. 2016;315(14):1516‐1518. [DOI] [PubMed] [Google Scholar]
- 19. Krishnamurti T, Woloshin S, Schwartz LM, Fischhoff B. A randomized trial testing US Food and Drug Administration “breakthrough” language. JAMA Intern Med. 2015;175(11):1856‐1858. [DOI] [PubMed] [Google Scholar]
- 20. US Food and Drug Administration . Drugs@FDA: FDA approved drug products. www.accessdata.fda.gov/scripts/cder/drugsatfda. Accessed March 21, 2017.
- 21. US Food and Drug Administration . Accelerated and restricted approvals under Subpart H (drugs) and Subpart E (biologics). www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsare DevelopedandApproved/DrugandBiologicApprovalReports/ucm 121597.htm. Updated June 30, 2008. Accessed March 21, 2017.
- 22. Darrow JJ, Kesselheim AS. Drug development and FDA approval, 1938‐2013. N Engl J Med. 2014;370(26):e39. [DOI] [PubMed] [Google Scholar]
- 23. Zarin DA, Tse T, Williams RJ, Califf RM, Ide NC. The ClinicalTrials.gov results database—update and key issues. N Engl J Med. 2011;364(9):852‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. US National Library of Medicine . ClinicalTrials.gov background. clinicaltrials.gov/ct2/about-site/background. Accessed March 21, 2017. [Google Scholar]
- 25. Higgins JP, Altman DG, Gøtzsche PC, et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ. 2011;343:d5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ioannidis JP, Haidich AB, Pappa M, et al. Comparison of evidence of treatment effects in randomized and nonrandomized studies. JAMA. 2001;286(7):821‐830. [DOI] [PubMed] [Google Scholar]
- 27. Ioannidis JP. Contradicted and initially stronger effects in highly cited clinical research. JAMA. 2005;294(2):218‐228. [DOI] [PubMed] [Google Scholar]
- 28. Naci H, Ioannidis JP. How good is “evidence” from clinical studies of drug effects and why might such evidence fail in the prediction of the clinical utility of drugs? Annu Rev Pharmacol Toxicol. 2015;55:169‐189. [DOI] [PubMed] [Google Scholar]
- 29. Jones CW, Handler L, Crowell KE, Keil LG, Weaver MA, Platts‐Mills TF. Non‐publication of large randomized clinical trials: cross sectional analysis. BMJ. 2013;347:f6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ. 2003;327(7414):557‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hartung J, Knapp G. A refined method for the meta‐analysis of controlled clinical trials with binary outcome. Stat Med. 2001;20(24):3875‐3889. [DOI] [PubMed] [Google Scholar]
- 32. Sidik K, Jonkman JN. A simple confidence interval for meta‐analysis. Stat Med. 2002;21(21):3153‐3159. [DOI] [PubMed] [Google Scholar]
- 33. IntHout J, Ioannidis JP, Borm GF. The Hartung‐Knapp‐Sidik‐Jonkman method for random effects meta‐analysis is straightforward and considerably outperforms the standard DerSimonian‐Laird method. BMC Med Res Methodol. 2014;14(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dagher R, Johnson J, Williams G, Keegan P, Pazdur R. Accelerated approval of oncology products: a decade of experience. J Natl Cancer Inst. 2004;96(20):1500‐1509. [DOI] [PubMed] [Google Scholar]
- 35. Kesselheim AS, Myers JA, Avorn J. Characteristics of clinical trials to support approval of orphan vs nonorphan drugs for cancer. JAMA. 2011;305(22):2320‐2326. [DOI] [PubMed] [Google Scholar]
- 36. Djulbegovic B, Hozo I, Ioannidis JP. Improving the drug development process: more not less randomized trials. JAMA. 2014;311(4):355‐356. [DOI] [PubMed] [Google Scholar]
- 37. Ioannidis JP. Mega‐trials for blockbusters. JAMA. 2013;309(3):239‐240. [DOI] [PubMed] [Google Scholar]
- 38. Yusuf S, Collins R, Peto R. Why do we need some large, simple randomized trials? Stat Med. 1984;3(4):409‐420. [DOI] [PubMed] [Google Scholar]
- 39. Button KS, Ioannidis JPA, Mokrysz C, et al. Power failure: why small sample size undermines the reliability of neuroscience. Nat Rev Neurosci. 2013;14(5):365‐376. [DOI] [PubMed] [Google Scholar]
- 40. Kessler DA, Rose JL, Temple RJ, Schapiro R, Griffin JP. Therapeutic‐class wars—drug promotion in a competitive marketplace. N Engl J Med. 1994;331(20):1350‐1353. [DOI] [PubMed] [Google Scholar]
- 41. Mitka M. Accelerated approval scrutinized. JAMA. 2003;289(24):3227‐3229. [DOI] [PubMed] [Google Scholar]
- 42. Ioannidis JPA, Greenland S, Hlatky MA, et al. Increasing value and reducing waste in research design, conduct, and analysis. Lancet. 2014;383(9912):166‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Food US and Drug Administration. Mylotarg (gemtuzumab ozogamicin): Market Withdrawal 2010. www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedical Products/ucm216458.htm. [Google Scholar]