

Abstract
Background
Aspirin Exacerbated Respiratory Disease (AERD) is a chronic medical condition that encompasses asthma, nasal polyposis, and hypersensitivity to aspirin and other non-steroidal anti-inflammatory drugs. Several previous studies have shown that part of the genetic effects of the disease may be induced by the interaction of multiple genetic variants. However, heavy computational cost as well as the complexity of the underlying biological mechanism has prevented a thorough investigation of epistatic interactions and thus most previous studies have typically considered only a small number of genetic variants at a time.
Methods
In this study, we propose a gene network based analysis framework to identify genetic risk factors from a genome-wide association study dataset. We first derive multiple single nucleotide polymorphisms (SNP)-based epistasis networks that consider marginal and epistatic effects by using different information theoretic measures. Each SNP epistasis network is converted into a gene-gene interaction network, and the resulting gene networks are combined as one for downstream analysis. The integrated network is validated on existing knowledgebase of DisGeNET for known gene-disease associations and GeneMANIA for biological function prediction.
Results
We demonstrated our proposed method on a Korean GWAS dataset, which has genotype information of 440,094 SNPs for 188 cases and 247 controls. The topological properties of the generated networks are examined for scale-freeness, and we further performed various statistical analyses in the Allergy and Asthma Portal (AAP) using the selected genes from our integrated network.
Conclusions
Our result reveals that there are several gene modules in the network that are of biological significance and have evidence for controlling susceptibility and being related to the treatment of AERD.
Keywords: Aspirin exacerbated respiratory disease (AERD), Asthma, Mutual information (MI), Information gain (IG), Genome-wide association study (GWAS), Epistasis, Integrated network, Single nucleotide polymorphisms (SNP)
Background
Aspirin Exacerbated Respiratory Disease (AERD) is a chronic medical condition that is also called Aspirin-induced Asthma (AIA). Asthma, nasal polyposis, and hypersensitivity reactions to aspirin and non-steroidal anti-inflammatory drugs (NSAIDs) were referred to as AERD [1]. Many previous studies have been conducted to identify genetic variants that affect AERD and related disease [2–8], which revealed that some genetic effects for the disease may be induced by the interaction of multiple genetic variants. However, heavy computational cost as well as the complexity of the underlying biological mechanism has prevented thorough investigation of epistatic interactions [9]. Thus, most previous studies have typically considered only a small number of genetic variants at a time.
Many previous studies proposed different methods to detect high-order genetic interactions using machine-learning approaches or heuristic algorithms [10–17]. However, most of the methods have a limitation that they can detect multi-order interactions only with a small number of SNPs or genes [18]. Furthermore, considering high-order interaction is not typically feasible for the GWAS dataset [19]. In the network analysis, many studies use information theoretic measures such as mutual information or information gain to obtain the strength of association between a pair of SNPs and a trait and then to construct an epistasis network. An extension of relevance network by Butte and Kohane, which uses mutual information with permutation test [18], was applied to a genome-wide data of Korean population and identified potential gene-gene interaction factors that affect the susceptibility to gastritis [19]. McKinney et al. proposed GAIN (Genetic Association Interaction Network) methods, which constructs an interaction network using information gain with SNP prioritization with Evaporative Cooling [20]. Davis et al. extended the McKinney et al.’s work using a network eigenvector centrality algorithm (SNPRank), which is analogous to Google PageRank algorithm to detect interactions that have the weak effect [21], and GAIN with a generalized linear model (reGAIN) was also proposed [22]. Hu et al. proposed SEN (Statistical Epistasis Networks), which use the difference of the number of largest connected components between random networks for network selection, demonstrated the method on bladder cancer [23]. Also, they extended the work that characterizes genes using dyadicity and heterophilicity analyses [24]. Although different methods have shown to be effective for different purposes, few studies enabled systematic and efficient analysis of interacting gene modules in a gene-based network framework given a GWAS dataset.
In this study, we propose a gene network based analysis framework to identify multiple genetic risk factors associated with the disease from a genome-wide association study dataset. We construct SNP-based epistasis networks using different information theoretic measures of mutual information and information gain. Mutual information and information gain mainly consider marginal and epistatic effects, respectively, and can give complementary information. We convert the SNP epistasis networks into gene-gene interaction networks by using the SNPs mapped to each gene and the association strengths of the corresponding pairs of SNPs. Our method improves the previous study [19] for the conversion and gets rid of the dependency of the resulting edge weight on the gene size. We integrate the resulting two gene networks as one to have a better view on the interaction mechanism. We validate our method using existing knowledge databases of DisGeNET [25] for known gene-disease associations and GeneMANIA [26] for biological interaction between genes.
We demonstrate our proposed method on a Korean GWAS dataset, which has genotype information of 440,094 SNPs for 435 unrelated Korean patients. The genotypic data and clinical information of the patient were previously collected with written informed consent and with the approval of the Ethics Review Board of the Ajou University Hospital (AJIRB-GEN-GEN-11-304) in the genome-wide association study of AERD [27]. The SNP data were anonymized and then used for an epistasis analysis for this study. The topological properties of the generated networks are examined for identifying statistically significant edges. For further validation, pathway and gene ontology enrichment tests are performed in the Allergy and Asthma Portal (AAP). AAP is built upon InnateDB [28] that is a previously developed integrated analysis platform for innate immune responses.
Methods
Data pre-processing
Our raw dataset consists of 440,094 SNPs from 188 Aspirin Exacerbated Respiratory Disease (AERD) samples and 247 Aspirin Tolerant Asthma (ATA) samples. We use AERD samples as cases and ATA as controls. We filter out SNPs with missing values in more than one-third of the samples. A linkage based imputation method [29] is then applied to remaining missing values. We also remove SNPs with minor allele frequency < 0.05. The resulting dataset consists of 320,815 SNPs.
Overview
We first give a brief introduction to the overall process of our proposed analysis framework. Figure 1 illustrates each step of the whole analysis process. First, single nucleotide polymorphisms (SNP)-based epistasis networks are constructed by using information-theoretic measures of mutual information (MI) and information gain (IG), respectively, as association measures between a pair of SNPs and the disease. Second, each SNP epistasis network is converted into a gene-gene interaction network. We experiment with different conversion methods and choose the one that is robust to gene size variation. Each converted network is further cut off with an appropriate threshold for edge weights obtained from permutation strategy. The resulting two gene networks are combined as one for downstream analysis. Details of each step are described in the following sections.
Fig. 1.

Illustration of the overall process of the proposed gene network based framework
Construction of SNP epistasis networks using information-theoretic measures
We construct SNP epistasis networks in which nodes represent the SNPs, and the edge weight is defined as the association strength between a pair of SNPs and the disease. We consider two information-theoretic measures of mutual information and information gain as for defining the edge weights. Mutual information is a non-parametric measure that represents the amount of information obtained about one random variable through the other [30]. It has been used to detect an association between two random variables [12, 19]. Mutual information of two random variables X and Y is defined as:
where H(X) and H(Y) denote the entropy of X and Y, respectively, and H(X,Y) is the joint entropy of X and Y. Mutual information to measure the strength of association between a pair of variables X 1, X 2 and Y can be written as follows:
In this study, X 1 and X 2 are discrete random variables for representing two SNPs and Y denotes the discrete random variable for the disease label.
While mutual information is largely affected by the marginal effect of either SNP, the information gain [31] mainly reflects the synergistic effect by subtracting each marginal effect of X 1 and X 2 from the mutual information [32] as follows.
Therefore, mutual information and information gain can capture different types of interaction mechanisms. Since the two measures can give complementary information, we construct two different networks, compare the major characteristics, and integrate the two for the final downstream analysis.
Gene-gene interaction network construction from SNP epistasis network
To expand the analysis scope from SNPs to genes and enable better interpretation and functional validation in a network framework, we convert the constructed SNP epistasis networks into gene-gene interaction networks. Edge weights of the gene-gene interaction network are computed using the edge weights of SNP epistasis network. As multiple SNPs can be mapped to the same gene, we need an algorithm to determine the weight between two genes given the mapped SNPs and the association strengths between them. Given multiple edge weights between SNPs belonging to two different genes, one may choose different summary statistics as the weight in a gene network such as the sum, average, minimum, or the maximum. Figure 2(a) shows an example of assigning the edge weight of a gene network given SNP epistasis network using different statistics. The summation method suffers from the bias for a longer gene accumulating higher edge weights because more SNPs tend to be mapped to the gene. In contrast, the average method is found to be limited in that the genes having only a couple SNPs tend to have higher degree: if a certain gene has many SNPs in it, it is more likely to contain some SNPs with very low edge weights, and this can substantially affect the average that is sensitive to outliers. The same problem arises in the case of taking the minimum. The maximum method does not suffer from these problems, and the maximum weight can represent the most meaningful interaction between SNPs. So we choose to take the maximum value in the conversion process.
Fig. 2.
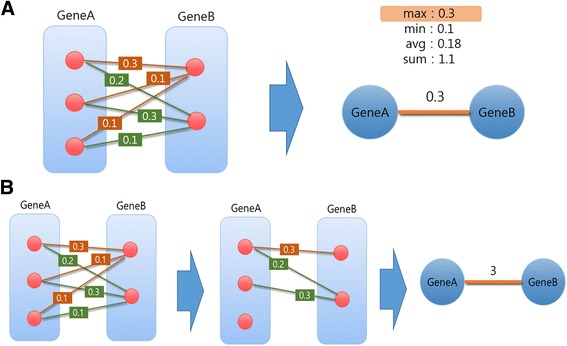
Illustration of the conversion process from a SNP epistasis network to a gene-gene interaction network of our method (a) and the one in a previous study [19] (b). In this figure, red circles represent the SNP and edge weight is the association strength of two SNPs
In a previous work [19] that performs similar network analysis, the SNP epistasis network is first cut off by a threshold obtained from a permutation strategy, and then the number of remaining edges in the SNP epistasis network was used to construct a gene-gene network as illustrated in Fig. 2(b). Finally, the top 5% edges with largest weights are chosen for further analysis. In this scheme, the network thresholding is performed twice, one for the SNP network and the other for the converted gene network. Therefore, one needs to define the cut-off each time. Moreover, as it counts the number of SNP pairs mapped to the corresponding genes, it also has the bias with respect to the gene size. That is, long genes that have many SNPs may become hub genes with a high degree even if they are not high risk factors for the disease. In our method, we directly utilize the edge weight in the SNP network instead of counting the number of edges, and also the thresholding is performed only once for the converted gene network as described in Fig. 2.
We also measured the correlation between gene size and node degree to see if our method and previous method were biased by the gene size. The results are described in Results and Discussion section.
Extraction of a statistically significant interaction network
From the converted gene network, we extract statistically significant edges using a permutation strategy used in a previous study [19] that is similar with the one in [18]. For every network edge, we permute the disease label in the dataset 30 times and calculate the average of 30 permuted edge weights. The maximum of the resulting edge weights is chosen as a network threshold θ. The edges with weights above θ can be regarded as significant interactions.
However, the resulting network can have a huge number of edges such as over one hundred million, which makes the analysis process too slow or even infeasible. To allow systematic adjustment of the number of edges in the network, we incorporate another parameter α and test varying thresholds in the form of θ*(1 + α) as used in MINA [33]. We vary α by 0.1 and choose the most appropriate network by using its topological properties. We also refer to the gene set known to be involved in the disease.
Specifically, we examine the scale-freeness of the constructed networks using an R-square value. Moreover, 1153 genes that are the intersection of 1300 genes related to asthma according to DisGeNET database [25] and the genes in our data are considered as ground truth. With the list of asthma-related genes from the database, we computed a p-value that is based on the cumulative distribution function (CDF) of the hypergeometric distribution using our node genes and the ground truth genes in the list. We also calculate Area Under the Curve (AUC) of the network nodes with consideration of the asthma related genes as a ground-truth. For each of mutual information and information gain networks, we select significant edges as described above and examine the network topologies with the major hub genes. Since these two networks can give complementary information on the interaction, we further integrate the two and perform the downstream analysis.
Validation through prior knowledge databases
We use two external databases to validate our framework. One is DisGeNET [25], a comprehensive discovery platform that is one of the largest repositories currently available of its kind. Another is GeneMANIA [26], a flexible interface used for generating hypotheses about gene functions that provides interactive functional association network. Figure 3 is a graphical illustration of our validation process.
Fig. 3.
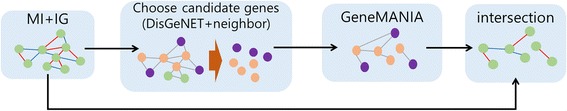
Validation process using DisGeNET and GeneMANIA
First, we extract genes that are related to asthma from DisGeNET database. Neighbors of them in our integrated network are selected as candidate genes. Then we use this candidate gene list as an input to GeneMANIA to obtain gene-gene interactions that are already identified in previous studies. We compare the resulting interaction network with our MI + IG integrated network to check the overlap as shown in the last step of Fig. 3.
Results and Discussion
Network topology
We first examine the network topology of each gene-gene interaction network using mutual information and information gain with a varying threshold of θ*(1 + α) where α is increased by 0.1 and θ is determined through permutation strategy. Tables 1 and 2 summarize the results in the case of mutual information and information gain network, respectively.
Table 1.
Network topologies for gene-gene interaction network measured by mutual information
| Threshold α | Node | Edge | # of component | R2 value | AUC | p-value |
|---|---|---|---|---|---|---|
| 4.7 | 1762 | 5022 | 1 | 0.594 | 0.509 | 3.E-04 |
| 4.8 | 1143 | 3386 | 1 | 0.617 | 0.542 | 1.E-02 |
| 4.9 | 690 | 2328 | 1 | 0.671 | 0.596 | 4.E-02 |
| 5.0 | 430 | 1669 | 2 | 0.708 | 0.587 | 1.E-02 |
| 5.1 | 299 | 1304 | 2 | 0.619 | 0.565 | 5.E-03 |
| 5.2 | 213 | 1047 | 1 | 0.685 | 0.621 | 2.E-02 |
| 5.3 | 171 | 858 | 1 | 0.654 | 0.645 | 2.E-02 |
Table 2.
Network topologies for gene-gene interaction network measured by information gain
| Threshold α | Node | Edge | # of component | R2 value | AUC | p-value |
|---|---|---|---|---|---|---|
| 3.5 | 1204 | 1149 | 293 | 0.885 | 0.508 | 4.E-02 |
| 3.6 | 827 | 763 | 215 | 0.877 | 0.542 | 1.E-01 |
| 3.7 | 526 | 465 | 149 | 0.876 | 0.542 | 9.E-02 |
| 3.8 | 333 | 285 | 100 | 0.769 | 0.504 | 8.E-02 |
| 3.9 | 227 | 188 | 71 | 0.713 | 0.456 | 7.E-02 |
In Table 1, we choose the mutual information network with α = 5.0 because it has the local maxima of R2 value and the p-value from the enrichment test is lower than the conventional significance threshold of 0.05. The chosen network is visualized in Fig. 4. Graphical visualizations of all the networks are produced by using Cytoscape [34]. We find that several hub genes in the network are related to asthma and AERD. For example, in a human study, Transforming Growth Factor Beta Receptor 1 (TGFBR1) has been reported that inhibition of the gene could help treatment of allergy-related conditions, like asthma [35, 36]. The previous study showed that most of Loeys-Dietz syndrome (LDS) patients in the study, who have heterozygous mutations for Transforming Growth Factor Beta Receptor 1 (TGFBR1) or Transforming Growth Factor Beta Receptor 2 (TGFBR2), had been diagnosed with asthma or allergic disease [35, 36]. Moreover, Bradykinin Receptor B1 (BDKRB1), one of the allergic related genes, was reported that the expression of B1 receptor protein for asthma subjects was significantly higher than normal subjects [37]. From the investigation, we presume that the gene-gene interactions of those hub genes and neighbor genes can have an influential role in asthma treatment.
Fig. 4.

Gene-gene interaction network based on mutual information
Table 2 summarizes the network properties of the information gain network, from which we choose the network with α = 3.7 that has high R2 value, p-value from the enrichment test lower than 0.1, and the local maximum of AUC. We observe that the general topology of the information gain network is very different from the one using mutual information. In the case of the mutual information network, there are only one or two connected components across different values of α as shown in Table 1 and illustrated in Fig. 4. In contrast, the information gain network consists of many smaller components as presented in Table 2. We show the top-10 largest components of the chosen network in Fig. 5 as an illustration.
Fig. 5.

Top-10 largest components of gene-gene interaction network based on information gain
Several hub genes in the network were reported as disease-related in previous studies. Roundabout Guidance Receptor 1 (ROBO1), for example, has been reported as an asthma gene and was differentially expressed during human lung development [38]. Moreover, genetic variants near intragenic region of the gene were reported to have suggestive associations with asthma [39]. Regarding Cadherin 13 (CDH13), lack of its protein (T-cad) has been known to cause reduction of allergic airways disease in the mice study [40]. Existence of strong association between Cadherin 13(CDH13) gene promoter methylation and lung carcinoma risk is also reported [41]. Additionally, we could find that a SNP of Protocadherin 7 (PCDH7) located in flanking 3′ UTR region of the gene, was reported as having significant association in an association study between SNPs and asthma-related quantitative traits [42].
The intersection of the two networks from mutual information and information gain produced only one network edge. Since these two networks are highly different and convey different information for the disease, we take the union network as a final network for the downstream analysis. Table 3 shows the network topology of the resulting network. Figure 6 shows the largest component of the integrated network.
Table 3.
Network topologies for M.I. and I.G integrated gene-gene interaction network
| Node | Edge | # of component | R2 value | p-value |
|---|---|---|---|---|
| 911 | 2133 | 120 | 0.725 | 7.E-03 |
Fig. 6.

The largest component of MI and IG integrated gene-gene interaction network
An empirical evaluation of the gene-gene interaction networks for gene length bias
We also measured the correlation between gene size and node degree. Figure 7(a) and (b) shows correlation between node degree and gene size of our method and previous method respectively. As we can see previous method is biased by gene size but proposed method is not. The correlation coefficient of previous method is about 0.66, while the correlation coefficient of the proposed method is about 0.04.
Fig. 7.

Scatter plots between node degree and gene size of our method (a) and previous method [19] (b)
Validation
We first choose 71 genes in our network from DisGeNET data and their neighbor nodes in our network. The resulting 279 genes are used as query genes on GeneMANIA that produces known gene-gene interactions among query genes. The output network from GeneMANIA has 304 nodes and 7725 edges. The intersection of this network with our final network has 279 genes and 67 interactions as shown in Fig. 8, which visualizes those genes that have at least one interaction. In our network, the number of edges between the 279 genes is 820, and the one from GeneMANIA has 6412 edges between 279 genes, so 8.17% edges of our network overlap with the known edges.
Fig. 8.

Gene-Gene interaction network constructed using the GeneMANIA Cytoscape plugin. Input genes are intersection of the genes in the network and DisGeNET genes
Functional enrichment analysis
We performed pathway and gene ontology enrichment analyses in the Allergy and Asthma Portal using the genes in our final integrated network. The result of pathway enrichment test is shown in Table 4.
Table 4.
Top 10 pathways having the largest gene count from enrichment analysis (p-value < 0.05)
| Pathway Name | Pathway Id |
Source Name |
Gene count | Genes in InnateDB for this entity |
p-value |
p-value (corrected) |
|---|---|---|---|---|---|---|
| Signaling by GPCR | 17449 | REACTOME | 30 | 1035 | 9.60E-04 | 4.54E-02 |
| GPCR ligand binding | 19266 | REACTOME | 20 | 433 | 1.95E-05 | 4.61E-03 |
| G alpha (q) signalling events | 13217 | REACTOME | 13 | 186 | 8.92E-06 | 4.22E-03 |
| Gastrin-CREB signalling pathway via PKC and MAPK | 13219 | REACTOME | 13 | 212 | 3.60E-05 | 5.67E-03 |
| G alpha (i) signalling events | 13220 | REACTOME | 13 | 231 | 8.71E-05 | 8.24E-03 |
| Neuroactive ligand-receptor interaction | 416 | KEGG | 13 | 275 | 4.84E-04 | 3.27E-02 |
| GPCR signaling | 16218 | INOH | 13 | 293 | 8.76E-04 | 4.60E-02 |
| Class A/1 (Rhodopsin-like receptors) | 13250 | REACTOME | 13 | 307 | 0.001341054 | 4.88E-02 |
| Transport of inorganic cations/anions and amino acids/oligopeptides | 13174 | REACTOME | 7 | 94 | 7.75E-04 | 4.58E-02 |
| LPA receptor mediated events | 15008 | PID NCI | 6 | 46 | 8.69E-05 | 1.03E-02 |
In the enrichment analysis of pathways, we can find many modules in the integrated network enrich to G-protein-coupled receptors (GPCR) associated pathways (GPCR ligand binding, Signaling by GPCR, and GPCR signaling), and these pathways can be direct or indirect targets of treatment of AERD or asthma [43]. Several GPCR antagonists have been showed to improve asthma control and reduce exacerbation in clinical trials, e.g. antagonist of P2Y12 (G-protein-coupled purinergic receptor) in a phase II study for AERD treatment [43], antagonist of CRTh2 (G-protein-coupled chemokine receptor homologous molecule expressed on Th2 lymphocytes) in a phase II study for treatment of uncontrolled allergic asthma [44], and antagonist of cysteinyl leukotriene receptor 1 (cysLTR1) for AERD treatment [45]. Furthermore, several genes related to GPCR pathway, such as PDE4A, PDE4D, and ANXA1, were found to be associated with AERD in our data. Drugs that target PDE4 subtypes could play a role in regulating allergic inflammation by attenuating pulmonary eosinophil recruitment, inhibiting lymphocyte proliferation and TFN-α release [46]. Recently, several PDE4 inhibitors have been developed for the treatment of respiratory diseases [47]. Additionally, anti-inflammatory and anti-allergic effects of Annexin A1 have been suggested. Administration of annexin protein prior to an ovalbumin challenge significantly reduced airway hyperresponsiveness, attenuated the production of inflammatory cytokines, such as IL-4, IL-5, and IL-13, as well as ovalbumin-specific IgE in a mouse model of asthma [48]. Meanwhile, neuroactive ligand-receptor interaction, which is another enriched pathway, was reported as a significantly enriched pathway in a differential gene expression study with AERD and ATA subjects [49]. Therefore, we suppose the major modules in the integrated network can play an important role in the pathways related to the treatment of AERD.
Table 5 shows the result of Gene Ontology (GO) enrichment analysis result. From the GO enrichment analysis, we obtain 28 statistical significant terms (after FDR), and most of the statistically significant terms are BP terms (18 terms). We find that several BP terms are related to AERD according to the previous studies. Schäfer and Maune reported that signal transduction (GO:0007165), which showed the second-largest number of associated genes among all BP terms (36 genes), is implicated in AERD and related disease [50]. Several previous studies of the pathogenesis of asthma reported that inflammatory response (GO:0006954) in the airways of asthma patients involves an orchestrated interplay of systems and processes, which drive chronic inflammatory response [51]. Also, another study of AERD pathogenesis reported that the inflammatory disease of AERD is similar to chronic allergic rhinitis and asthma [52].
Table 5.
Top 10 Gene Ontology terms having the largest gene count from enrichment analysis (p-value < 0.05)
| GO Term Name | Term Id |
Source Name |
Gene count | Genes in InnateDB for this entity |
p-value |
p-value (corrected) |
|---|---|---|---|---|---|---|
| plasma membrane | GO:0005886 | cellular component | 85 | 3645 | 1.06E-06 | 1.14E-03 |
| signal transduction | GO:0007165 | biological process | 36 | 1368 | 2.82E-04 | 2.88E-02 |
| integral component of plasma membrane | GO:0005887 | cellular component | 29 | 1063 | 6.50E-04 | 4.97E-02 |
| multicellular organismal development | GO:0007275 | biological process | 20 | 537 | 9.77E-05 | 1.74E-02 |
| signal transducer activity | GO:0004871 | molecular function | 18 | 280 | 1.24E-07 | 2.66E-04 |
| receptor binding | GO:0005102 | molecular function | 16 | 333 | 2.59E-05 | 7.92E-03 |
| synaptic transmission | GO:0007268 | biological process | 15 | 397 | 6.26E-04 | 4.96E-02 |
| dendrite | GO:0030425 | cellular component | 14 | 273 | 4.11E-05 | 9.77E-03 |
| inflammatory response | GO:0006954 | biological process | 14 | 315 | 1.87E-04 | 2.36E-02 |
| positive regulation of neuron differentiation | GO:0045666 | biological process | 8 | 80 | 1.87E-05 | 6.67E-03 |
Conclusion
In this study, we presented a gene network based framework for analyzing a genome-wide association study dataset such as SNPs and identifying important risk factors. Although some genetic effects for the disease are induced by the interaction of multiple genetic variants, most previous studies have typically considered only a small number of genetic variants because of heavy computational cost and the modeling complexity. Our proposed method can identify multiple genetic risk factors associated with diseases from a genome-wide association study dataset in a network-based framework. We generate two SNP epistasis networks using mutual information and information gain that convey complementary information. Two SNP epistasis networks are converted to gene-gene interaction networks and finally integrated as one gene-gene interaction network. In the conversion of a SNP epistasis network to a gene network, we handle the bias for long genes accumulating edge weights over multiple mapped SNPs by taking the maximum weight among candidate weights. This can effectively alleviate the problem of previous approaches that can select long genes regardless of whether they are true risk factors or not. The validation method using existing knowledge databases and functional enrichment analysis shows that our framework could identify essential genes associated with the disease.
One limitation of our framework is that we have to set a threshold to cut the network by checking network topologies manually. We calculate threshold θ for significance level and employ an additional parameter α to control the sparsity, but the selection of appropriate threshold is not fully automatic and rather heuristic. We would explore other methods to address this issue in our future work. We also plan to focus on more biological interpretation of the generated networks to find meaningful interactions between multiple genetic variants. Other future work will include the application of the proposed methods to other diseases.
Acknowledgements
Not applicable.
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning [2014R1A1A3051169 & 2015R1C1A2A01053492]. Publication charge of this article has been funded by NRF of Korea [2014R1A1A3051169] and Ajou University.
Availability of data and materials
All data and material that are not presented in the main paper or additional supporting files are available from the corresponding author on request.
Authors’ contributions
SW and HJ designed the study and wrote the first draft of the manuscript. SW performed the experiments. KW and DK helped to develop the study, reviewed and edited the manuscript. HP contributed to the design of the study, recruitment of study subjects and collection of clinical data. SK and KS supervised the entire study and provided guidance on the study design, analysis of the result, the manuscript preparation and the revision. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
About this supplement
This article has been published as part of BMC Medical Genomics Volume 10 Supplement 1, 2017: Selected articles from the 6th Translational Bioinformatics Conference (TBC 2016): medical genomics. The full contents of the supplement are available online at https://bmcmedgenomics.biomedcentral.com/articles/supplements/volume-10-supplement-1.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- AERD
Aspirin exacerbated respiratory disease
- GWAS
Genome-wide association study
- IG
Information gain
- MI
Mutual information
- SNP
Single nucleotide polymorphisms
Contributor Information
Sehee Wang, Email: wsh0509@ajou.ac.kr.
Hyun-hwan Jeong, Email: Hyun-Hwan.Jeong@bcm.edu.
Dokyoon Kim, Email: dkim@geisinger.edu.
Kyubum Wee, Email: kbwee@ajou.ac.kr.
Hae-Sim Park, Email: hspark@ajou.ac.kr.
Seung-Hyun Kim, Email: kimsh@ajou.ac.kr.
Kyung-Ah Sohn, Email: kasohn@ajou.ac.kr.
References
- 1.Kowalski M, Asero R, Bavbek S, Blanca M, Blanca‐Lopez N, Bochenek G, Brockow K, Campo P, Celik G, Cernadas J. Classification and practical approach to the diagnosis and management of hypersensitivity to nonsteroidal anti‐inflammatory drugs. Allergy. 2013;68(10):1219–1232. doi: 10.1111/all.12260. [DOI] [PubMed] [Google Scholar]
- 2.Shin S, Park J, Kim Y, Uh S, Choi BW, Kim M, Choi IS, Park B, Shin H, Park C. A highly sensitive and specific genetic marker to diagnose aspirin-exacerbated respiratory disease using a genome-wide association study. DNA Cell Biol. 2012;31(11):1604–1609. doi: 10.1089/dna.2012.1688. [DOI] [PubMed] [Google Scholar]
- 3.Kim S, Sanak M, Park H. Genetics of hypersensitivity to aspirin and nonsteroidal anti-inflammatory drugs. Immunol Allergy Clin North Am. 2013;33(2):177–194. doi: 10.1016/j.iac.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Kim S, Choi H, Yoon M, Ye Y, Park H. Dipeptidyl-peptidase 10 as a genetic biomarker for the aspirin-exacerbated respiratory disease phenotype. Ann Allergy Asthma Immunol. 2015;114(3):208–213. doi: 10.1016/j.anai.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Park BL, Kim T, Kim J, Bae JS, Pasaje CFA, Cheong HS, Kim LH, Park J, Lee HS, Kim M. Genome-wide association study of aspirin-exacerbated respiratory disease in a Korean population. Hum Genet. 2013;132(3):313–321. doi: 10.1007/s00439-012-1247-2. [DOI] [PubMed] [Google Scholar]
- 6.Palikhe NS, Kim S, Kim J, Losol P, Ye Y, Park H. Role of toll-like receptor 3 variants in aspirin-exacerbated respiratory disease. Allergy, Asthma Immunol. Res. 2011;3(2):123–127. doi: 10.4168/aair.2011.3.2.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shin S, Park BL, Chang H, Park JS, Bae D, Song H, Choi IS, Kim M, Park H, Kim LH. Exonic variants associated with development of aspirin exacerbated respiratory diseases. PLoS One. 2014;9(11):e111887. doi: 10.1371/journal.pone.0111887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S, Jeong H, Cho B, Kim M, Lee H, Lee J, Wee K, Park H. Association of Four-locus Gene Interaction with Aspirin-intolerant Asthma in Korean Asthmatics. J Clin Immunol. 2008;28(4):336–342. doi: 10.1007/s10875-008-9190-7. [DOI] [PubMed] [Google Scholar]
- 9.Hu T, Andrew AS, Karagas MR, Moore JH. Statistical epistasis networks reduce the computational complexity of searching three-locus genetic models. Pac Symp Biocomput. 2013;18:397–408. [PMC free article] [PubMed] [Google Scholar]
- 10.Hahn LW, Ritchie MD, Moore JH. Multifactor dimensionality reduction software for detecting gene–gene and gene–environment interactions. Bioinformatics. 2003;19(3):376–382. doi: 10.1093/bioinformatics/btf869. [DOI] [PubMed] [Google Scholar]
- 11.Wan X, Yang C, Yang Q, Xue H, Fan X, Tang NLS, Yu W. BOOST: A Fast Approach to Detecting Gene-Gene Interactions in Genome-wide Case–control Studies. Am J Hum Genet. 2010;87(3):325–340. doi: 10.1016/j.ajhg.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leem S, Jeong H, Lee J, Wee K, Sohn K. Fast detection of high-order epistatic interactions in genome-wide association studies using information theoretic measure. Comput Biol Chem. 2014;50:19–28. doi: 10.1016/j.compbiolchem.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X, Huang S, Zou F, Wang W. TEAM: efficient two-locus epistasis tests in human genome-wide association study. Bioinformatics. 2010;26(12):i217–i227. doi: 10.1093/bioinformatics/btq186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie M, Li J, Jiang T. Detecting genome-wide epistases based on the clustering of relatively frequent items. Bioinformatics. 2011;28(1):5–12. doi: 10.1093/bioinformatics/btr603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo X, Meng Y, Yu N, Pan Y. Cloud computing for detecting high-order genome-wide epistatic interaction via dynamic clustering. BMC Bioinf. 2014;15(1):1. doi: 10.1186/1471-2105-15-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Liu JS. Bayesian inference of epistatic interactions in case–control studies. Nat Genet. 2007;39(9):1167–1173. doi: 10.1038/ng2110. [DOI] [PubMed] [Google Scholar]
- 17.Yang C, He Z, Wan X, Yang Q, Xue H, Yu W. SNPHarvester: a filtering-based approach for detecting epistatic interactions in genome-wide association studies. Bioinformatics. 2009;25(4):504–511. doi: 10.1093/bioinformatics/btn652. [DOI] [PubMed] [Google Scholar]
- 18.Butte AJ, Kohane IS. Mutual information relevance networks: functional genomic clustering using pairwise entropy measurements. Pac Symp Biocomput. 2000;5:418–29. doi: 10.1142/9789814447331_0040. [DOI] [PubMed] [Google Scholar]
- 19.Jeong H, Sohn K. Relevance Epistasis Network of Gastritis for Intra-chromosomes in the Korea Associated Resource (KARE) Cohort Study. Genome Inform. 2014;12(4):216–224. doi: 10.5808/GI.2014.12.4.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKinney BA, Crowe,James E.,,Jr, Guo J, Tian D: Capturing the Spectrum of Interaction Effects in Genetic Association Studies by Simulated Evaporative Cooling Network Analysis. PLoS Genet 2009, 5(3):e1000432 [DOI] [PMC free article] [PubMed]
- 21.Davis NA, Crowe JE J, Pajewski NM, McKinney BA. Surfing a genetic association interaction network to identify modulators of antibody response to smallpox vaccine. Genes Immun. 2010;11(8):630–636. doi: 10.1038/gene.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis NA, Lareau CA, White BC, Pandey A, Wiley G, Montgomery CG, Gaffney PM, McKinney BA. Encore: Genetic Association Interaction Network Centrality Pipeline and Application to SLE Exome Data. Genet Epidemiol. 2013;37(6):614–621. doi: 10.1002/gepi.21739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu T, Sinnott-Armstrong NA, Kiralis JW, Andrew AS, Karagas MR, Moore JH. Characterizing genetic interactions in human disease association studies using statistical epistasis networks. BMC Bioinf. 2011;12:364-2105-12-364. doi: 10.1186/1471-2105-12-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De R, Hu T, Moore JH, Gilbert-Diamond D. Characterizing gene-gene interactions in a statistical epistasis network of twelve candidate genes for obesity. Bio Data mining. 2015;8(1):1. doi: 10.1186/s13040-014-0034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pinero J, Queralt-Rosinach N, Bravo A, Deu-Pons J, Bauer-Mehren A, Baron M, Sanz F, Furlong LI. DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. Database (Oxford) 2015;2015:bav028. doi: 10.1093/database/bav028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, Maitland A, Mostafavi S, Montojo J, Shao Q, Wright G, Bader GD, Morris Q. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(Web Server issue):W214–220. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim S, Cho B, Choi H, Shin E, Ye Y, Lee J, Park H. The SNP rs3128965 of HLA-DPB1 as a genetic marker of the AERD phenotype. PLoS One. 2014;9(12):e111220. doi: 10.1371/journal.pone.0111220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, Winsor GL, Hancock RE, Brinkman FS, Lynn DJ. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013;41(Database issue):D1228–33. doi: 10.1093/nar/gks1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Y, Wu Y, Gonda M, Wu J. A Linkage based Imputation Method for Missing SNP Markers in Association Mapping. J Appl Bioinform Comput Biol. 2015;1:2. [Google Scholar]
- 30.Cover TM, Thomas JA. Elements of information theory 2nd edition. Hoboken: John Wiley & Sons; 2006. [Google Scholar]
- 31.Jakulin A, Bratko I. Analyzing Attribute Dependencies. In: Lavrač N, Gamberger D, Todorovski L, Blockeel H, editors. Knowledge Discovery in Databases: PKDD 2003: 7th European Conference on Principles and Practice of Knowledge Discovery in Databases, Cavtat-Dubrovnik, Croatia, September 22–26, 2003. Proceedings. Berlin, Heidelberg: Springer Berlin Heidelberg; 2003. pp. 229–240. [Google Scholar]
- 32.Moore JH, Gilbert JC, Tsai C, Chiang F, Holden T, Barney N, White BC. A flexible computational framework for detecting, characterizing, and interpreting statistical patterns of epistasis in genetic studies of human disease susceptibility. J Theor Biol. 2006;241(2):252–261. doi: 10.1016/j.jtbi.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 33.Jeong H, Leem S, Wee K, Sohn K. Integrative network analysis for survival-associated gene-gene interactions across multiple genomic profiles in ovarian cancer. J Ovarian Res. 2015;8(1):1. doi: 10.1186/s13048-015-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frischmeyer-Guerrerio PA, Guerrerio AL, Oswald G, Chichester K, Myers L, Halushka MK, Oliva-Hemker M, Wood RA, Dietz HC. TGFβ receptor mutations impose a strong predisposition for human allergic disease. Sci Transl Med. 2013;5(195):195ra94. doi: 10.1126/scitranslmed.3006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bordon Y. Asthma and allergy: TGFβ — too much of a good thing? Nat Rev Immunol. 2013;13(9):618–619. doi: 10.1038/nri3519. [DOI] [PubMed] [Google Scholar]
- 37.Bertram CM, Misso NL, Fogel-Petrovic M, Figueroa CD, Foster PS, Thompson PJ, Bhoola KD. Expression of kinin receptors on eosinophils: comparison of asthmatic patients and healthy subjects. J Leukoc Biol. 2009;85(3):544–552. doi: 10.1189/jlb.0508283. [DOI] [PubMed] [Google Scholar]
- 38.Melén E, Himes BE, Brehm JM, Boutaoui N, Klanderman BJ, Sylvia JS, Lasky-Su J. Analyses of shared genetic factors between asthma and obesity in children. J Allergy Clin Immunol. 2010;126(3):631–637.e8. doi: 10.1016/j.jaci.2010.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sleiman PMA, Flory J, Imielinski M, Bradfield JP, Annaiah K, Willis-Owen S, Wang K, Rafaels NM, Michel S, Bonnelykke K, Zhang H, Kim CE, Frackelton EC, Glessner JT, Hou C, Otieno FG, Santa E, Thomas K, Smith RM, Glaberson WR, Garris M, Chiavacci RM, Beaty TH, Ruczinski I, Orange JS, Allen J, Spergel JM, Grundmeier R, Mathias RA, Christie JD, von Mutius E, Cookson WOC, Kabesch M, Moffatt MF, Grunstein MM, Barnes KC, Devoto M, Magnusson M, Li H, Grant SFA, Bisgaard H, Hakonarson H. Variants of DENND1B Associated with Asthma in Children. N Engl J Med. 2010;362(1):36–44. doi: 10.1056/NEJMoa0901867. [DOI] [PubMed] [Google Scholar]
- 40.Williams AS, Kasahara DI, Verbout NG, Fedulov AV, Zhu M, Si H, Wurmbrand AP, Hug C, Ranscht B, Shore SA. Role of the Adiponectin Binding Protein, T-Cadherin (Cdh13), in Allergic Airways Responses in Mice. PLoS One. 2012;7(7):e41088. doi: 10.1371/journal.pone.0041088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhong YH, Peng H, Cheng HZ, Wang P. Quantitative assessment of the diagnostic role of CDH13 promoter methylation in lung cancer. Asian Pac J Cancer Prev. 2015;16(3):1139–1143. doi: 10.7314/APJCP.2015.16.3.1139. [DOI] [PubMed] [Google Scholar]
- 42.Li X, Howard TD, Zheng SL, Haselkorn T, Peters SP, Meyers DA, Bleecker ER. Genome-wide association study of asthma identifies RAD50-IL13 and HLA-DR/DQ regions. J Allergy Clin Immunol. 2010;125(2):328–335.e11. doi: 10.1016/j.jaci.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Erpenbeck VJ, Popov TA, Miller D, Weinstein SF, Spector S, Magnusson B, Osuntokun W, Goldsmith P, Weiss M, Beier J. The oral CRTh2 antagonist QAW039 (fevipiprant): a phase II study in uncontrolled allergic asthma. Pulm Pharmacol Ther. 2016;39:54–63. doi: 10.1016/j.pupt.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 44.Israel E. The protective effects of leukotriene modifiers in aspirin-induced asthma. Postgrad Med. 2000;108(4 Suppl):40–44. doi: 10.3810/pgm.09.15.2000.suppl7.291. [DOI] [PubMed] [Google Scholar]
- 45.Paplinska M, Chazan R, Grubek-Jaworska H. Effect of phosphodiesterase 4 (PDE4) inhibitors on eotaxin expression in human bronchial epithelial cells. J Physiol Pharmacol. 2011;62(3):303. [PubMed] [Google Scholar]
- 46.Abbott‐Banner KH, Page CP. Dual PDE3/4 and PDE4 inhibitors: novel treatments for COPD and other inflammatory airway diseases. Basic Clin Pharmacol Toxicol. 2014;114(5):365–376. doi: 10.1111/bcpt.12209. [DOI] [PubMed] [Google Scholar]
- 47.Murugappa S, Kunapuli SP. The role of ADP receptors in platelet function. Front Biosci. 2006;11:1977–1986. doi: 10.2741/1939. [DOI] [PubMed] [Google Scholar]
- 48.Lee SH, Kim DW, Kim HR, Woo SJ, Kim SM, Jo HS, Jeon SG, Cho S, Park JH, Won MH. Anti-inflammatory effects of Tat-Annexin protein on ovalbumin-induced airway inflammation in a mouse model of asthma. Biochem Biophys Res Commun. 2012;417(3):1024–1029. doi: 10.1016/j.bbrc.2011.12.084. [DOI] [PubMed] [Google Scholar]
- 49.Shin S, Park JS, Kim Y, Oh T, An S, Park C. Differential gene expression profile in PBMCs from subjects with AERD and ATA: a gene marker for AERD. Mol Genet Genomics. 2012;287(5):361–371. doi: 10.1007/s00438-012-0685-9. [DOI] [PubMed] [Google Scholar]
- 50.Schäfer D, Maune S. Pathogenic mechanisms and in vitro diagnosis of AERD. J Allergy. 2012;2012:18. doi: 10.1155/2012/789232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ishmael FT. The inflammatory response in the pathogenesis of asthma. J Am Osteopath Assoc. 2011;111(11_suppl_7):S11–S17. [PubMed] [Google Scholar]
- 52.Stevenson DD, Zuraw BL. Pathogenesis of aspirin-exacerbated respiratory disease. Clin Rev Allergy Immunol. 2003;24(2):169–187. doi: 10.1385/CRIAI:24:2:169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data and material that are not presented in the main paper or additional supporting files are available from the corresponding author on request.