

Abstract
Over the past decades, Helicoverpa armigera nucleopolyhedrovirus (HearNPV) has been widely used for biocontrol of cotton bollworm, which is one of the most destructive pest insects in agriculture worldwide. However, the molecular mechanism underlying the interaction between HearNPV and host insects remains poorly understood. In this study, high-throughput RNA-sequencing was integrated with label-free quantitative proteomics analysis to examine the dynamics of gene expression in the fat body of H. armigera larvae in response to challenge with HearNPV. RNA sequencing-based transcriptomic analysis indicated that host gene expression was substantially altered, yielding 3,850 differentially expressed genes (DEGs), whereas no global transcriptional shut-off effects were observed in the fat body. Among the DEGs, 60 immunity-related genes were down-regulated after baculovirus infection, a finding that was consistent with the results of quantitative real-time RT-PCR. Gene ontology and functional classification demonstrated that the majority of down-regulated genes were enriched in gene cohorts involved in energy, carbohydrate, and amino acid metabolic pathways. Proteomics analysis identified differentially expressed proteins in the fat body, among which 76 were up-regulated, whereas 373 were significantly down-regulated upon infection. The down-regulated proteins are involved in metabolic pathways such as energy metabolism, carbohydrate metabolism (CM), and amino acid metabolism, in agreement with the RNA-sequence data. Furthermore, correlation analysis suggested a strong association between the mRNA level and protein abundance in the H. armigera fat body. More importantly, the predicted gene interaction network indicated that a large subset of metabolic networks was significantly negatively regulated by viral infection, including CM-related enzymes such as aldolase, enolase, malate dehydrogenase, and triose-phosphate isomerase. Taken together, transcriptomic data combined with proteomic data elucidated that baculovirus established systemic infection of host larvae and manipulated the host mainly by suppressing the host immune response and down-regulating metabolism to allow viral self-replication and proliferation. Therefore, this study provided important insights into the mechanism of host-baculovirus interaction.
Lepidopteran insect pests cause vast damage to crops worldwide. Baculoviruses, which comprise enveloped viruses with large, circular, and double-stranded DNA genomes, are important biological agents in controlling lepidopteran insects. Several hundred baculovirus species (1) have been found from Lepidoptera. Their genomes range from 80 to 180 kb in size, encoding ∼90 to 180 genes. Baculoviruses usually involve a narrow host range, infecting one or several closely related insect species. During the replication cycle, two morphological forms exist, the budded virus (BV), 1 which causes systemic infection in the host, and the occlusion-derived virus (ODV), which infects per os the host insect and transmits infection horizontally (2). Baculoviruses not only could be used as a biopesticide to regulate insect populations in agriculture with high efficiency and specificity but also can be developed and widely used in various applications, such as baculovirus-insect cell expression systems, transduction vectors for protein expression in mammalian cells, and potential gene therapy vectors.
The cotton bollworm (Helicoverpa armigera), belonging to Noctuidae, is one of the most destructive pest insects in agriculture worldwide (3). It causes severe damage to plant crops, resulting in massive economic losses. Over the past decades, cotton bollworm has gradually developed resistance against almost all chemical pesticides. As an effective biological agent (4, 5) against cotton bollworm, Helicoverpa armigera nucleopolyhedrovirus (HearNPV, a group II Alphabaculovirus) has been exploited as a biopesticide to control pest insects in China. The complete genome sequence of HearNPV, 131 kb in size, has been obtained (6). Previously, 135 putative open reading frames (ORFs) were identified by computer-assisted analysis. The past years have seen great progress in HearNPV research. Many of the genes expressed in HearNPV are involved in viral infection. The functions of ODV-associated protein HA100 (6), F-protein (7), and per os infectivity factors (8, 9) in viral infection were also described in detail. Meanwhile, the structural and functional differences between BV and ODV were revealed by multiple proteomics approaches (10). Altogether, the roles that viral genes play in the infection process have been investigated systematically over the past years.
Unlike vertebrates, insects depend on the innate immune system against invading organisms, including viruses and parasites (11, 12). Various defense reactions occur in hemocytes and the fat body. Cellular reactions involve mainly hemocyte-mediated phagocytosis, nodulation, and encapsulation, whereas humoral reactions include the recognition of pathogens and induction of Toll and Imd signaling pathways in the fat body (11). Immunotranscriptomic studies in tobacco hornworm Manduca sexta and H. armigera indicated that microbial infections activate the Toll and Imd pathways, ultimately leading to the synthesis of antimicrobial peptides (AMPs) (13, 14). Apart from immune signaling reactions, melanization has also been shown to be involved in the response to microbial infections (15, 16). Two distinct melanization pathways were identified in the mosquito Aedes aegypti in response to bacteria and fungi (17). Furthermore, CLSP2 was found to inhibit the melanization cascade and regulate the antifungal response in A. aegypti (18, 19). Recently, antiviral immunity in insects has drawn increasing attention (20–24). After ingestion by insect larvae, baculoviruses reach the midgut, and the polyhedra is dissolved. Next, the ODV infects the midgut epithelial cells, and the BV buds out into the circulating hemolymph, causing systemic infection throughout the entire body. A previous study indicated that melanization and encapsulation might be implicated in the defense against baculovirus (25). Besides melanization, it was demonstrated that the RNA interference (RNAi) pathway is also involved in the defense against baculovirus infection at the cellular level (26). In addition, one AMP, gloverin (27), was shown to participate in the response against baculovirus. Apart from the host immune response, other physiological processes related to interactions have been revealed, including transcription, translation, cell cycle, degradation, signaling, and metabolism (28). However, the latter functions were poorly characterized, thus requiring more research efforts.
The advent of next-generation sequencing (NGS) techniques, such as RNA sequencing (RNA-seq), has enabled gene expression profiling on a genome-wide scale. Large-scale sequencing was used to investigate the differential effects of four types of exogenous pathogens on gene expression in H. armigera larvae, demonstrating significant induction of AMPs (29). Besides, much knowledge related to the infection process of baculovirus has been acquired since the application of NGS. The spatial expression pattern of AcMNPV genes in Trichoplusia ni cells was established using RNA-seq (30). Furthermore, the predicted protein-protein interaction networks demonstrated that 22 baculovirus proteins interact with 2,326 host proteins (31). Although changes at the transcriptional level substantially influence host physiological functions, proteins are the pivotal molecules involved in biochemical processes. Mass spectrometry-based proteomics can be used to identify and accurately quantify proteins in complex samples (32, 33).
In this study, we employed a deep-sequencing technique to determine host defense responses against baculovirus infection in H. armigera larvae. The dynamics at the gene expression level in the host following infection were revealed by time course transcriptomic analysis. Additionally, to gain insight into gene expression changes at the protein level, label-free quantitative proteomics analysis was also performed to identify and quantitate proteins in the fat body. To our knowledge, this is the first study to integrate transcriptomic and proteomic approaches to provide a framework for genome-wide gene expression profiles in H. armigera larvae infected with HearNPV at both the mRNA and protein levels, which might contribute to a better understanding of the antiviral immunity mechanism of H. armigera against baculovirus infection, thus providing insights into the development of novel biological control strategies and facilitating more practical applications for baculovirus-based engineering technology.
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
Mock and infected fat body samples were collected from naive and infected H. armigera larvae at 72 h post-infection (hpi), respectively. The samples include three independent biological replicates for each group (n = 3, five larvae per replicate), and each biological replicate consists of three technical replicates. Protein was extracted from the fat body samples and separated by electrophoresis on SDS-polyacrylamide gel. Proteins in the fat body were identified by searching mass spectrometry data against an in-house sequence database of H. armigera proteins. To confirm the occurrence of proteins in one group, we adopted a stringent criteria that each protein must be identified at least twice in both biological and technical replicates. Protein abundances were quantified by label-free quantification using Peak Area Calculation Quantification method and compared by Student's t test to determine significant differences (p < 0.05 and fold change ≥1.5 or <0.67) between the infected and mock group. Pearson correlation coefficients were calculated to analyze the correlations of protein levels and mRNA levels in the fat body. A similar method was used to examine the possible correlations between mRNA and protein level changes.
Insect Rearing, Viral Infection, and Cell Culture
The H. armigera nucleopolyhedrovirus G4 strain (34) was used to infect H. armigera larvae, which were reared on an artificial diet at 27 ± 1 °C under relative humidity of 80% and a 16:8-h (light/dark) photoperiod. Helicoverpa zea cells were cultured in Grace's insect medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum. Occlusion bodies (OBs) were amplified from infected larvae. Day 2 third instar cotton bollworm larvae with a similar body size and weight were used for per os infection in this study.
Oral Infection of H. armigera Larvae Using the Droplet Method
Day 2 third instar H. armigera larvae were selected and divided into the infected and control groups, which had been starved for 16 h before infection. Each larva in the infected group was fed for 10 min on 2 μl of 10% sucrose solution containing edible blue dye contaminated with HearNPV at a final concentration of ∼6.7 × 107 OBs/μl (LC99). For the control group, each larva was provided only 2 μl of 10% sucrose solution containing edible blue dye. The time at which the edible blue dye was consumed was defined as 0 hpi. Hereafter, the larvae in the two groups were transferred to the normal artificial diet. The larvae were harvested at four time points after infection, 0, 24, 48, and 72 h, respectively, and then were dissected for collection of the fat body. Fat body samples were frozen immediately in liquid nitrogen and stored at −80 °C for later use.
RNA Extraction and cDNA Library Construction
Briefly, fat body samples were homogenized thoroughly in Qiazol lysis buffer (Qiagen, Hilden, Germany) using a tissue grinder. Total RNA was extracted from the fat body of larvae using the Qiagen RNeasy kit according to the manufacturer's instructions. The integrity of total RNA was measured using the Nanodrop 2000 spectrophotometer, and the RNA samples were separated by gel electrophoresis. In a subsequent procedure, mRNA was purified from 100 μg of total RNA using an mRNA purification kit (Ambion, Austin, TX). A total of 10 μg of mRNA was used to construct a cDNA library for RNA-seq following the Illumina library construction protocol. Using a magnetic stand and beads, DNA fragments of the appropriate sizes were isolated from the initial PCR products. Quality assessment of the cDNA libraries was evaluated using the Agilent Bioanalyzer 2100 system.
Transcriptomic Analysis and Identification of DEGs
cDNA libraries were paired-end sequenced on the Hiseq 2000 platform. The obtained raw sequencing reads were preprocessed to remove adaptor sequences and reads of low quality and short size, followed by the filtering of bacterial and viral genomes except for HearNPV. Next, the clean reads were de novo assembled into primary transcripts using Trinity (35). For each library, a set of transcripts was produced from the assembly. To remove redundant transcripts, CAP3 (36) was used to assemble overlapping transcripts into larger contigs. Additionally, the CD-Hit suite (37) was employed to remove redundant transcript sequences to obtain non-redundant unigenes. To predict the functions of these transcripts, the Blast2GO (38) pipeline was used to annotate the unigene datasets.
Bowtie was employed to map reads to the corresponding transcripts; subsequently, RSEM (39) was applied to calculate the normalized gene expression value, expressed in reads per kilobase of exon model per million (RPKM) mapped reads. Based on the gene expression values, the DEGseq (40) package in the R environment was implemented for differential gene expression analysis. The genes were considered differentially expressed if they had a p value <0.05 and a fold change of either >1.5 (up-regulated genes) or <0.67 (down-regulated genes).
Gene Ontology (GO) Enrichment Analysis (GOEA)
To determine the enriched GO terms, the significantly up- and down-regulated genes were subjected to GOEA using DAVID (41). First, the corresponding DEG orthologs in Drosophila melanogaster were identified using InParanoid. Next, the orthologs from D. melanogaster were submitted to the DAVID annotation server for GOEA. The entire gene set of D. melanogaster was used as the reference. The enriched GO terms were depicted as a bar plot, with an adjusted p value (Padj) of <0.05 deemed to indicate statistical significance.
KOBAS and Principal Component Analysis (PCA)
The KOBAS 2.0 (42) web server was employed for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis following the procedure from annotation to identification. In the annotation step, KEGG orthology was used to annotate the corresponding input GenInfo Identifier entries, which were obtained by a BLAST search against the non-redundant protein sequence database (NCBI Nr). Subsequently, the enriched metabolic pathways and biological processes were identified from the preprocessed datasets. The entire gene set of D. melanogaster was used as the reference for identification of enriched pathways. For statistical analysis, the binomial test method was employed to determine statistical significance using the Benjamin and Hochberg method for false discovery rate (FDR) correction. PCA of eight RNA-seq libraries was performed using the mdatools package within the R statistical environment. The top three principal components, PC1, PC2, and PC3, were selected to show the differential effects on global gene expression levels in eight libraries.
qRT-PCR Analysis of Gene Expression Levels
Total RNA was extracted from the fat body as described previously (14). The RNA samples were treated with DNase I (Invitrogen) to remove the genomic contaminants. In total, 2 μg of total RNA were used for synthesis of first-strand cDNA using reverse transcriptase Moloney murine leukemia virus (Promega, Fitchburg, WI) and oligo-(dT) primers according to the manufacturer's instructions. qRT-PCR was performed to measure the expression levels of the genes of interest. qRT-PCR was carried out using the Stratagene Mx3005p real-time PCR system and SYBR solution (Tiangen, China) based on the manufacturer's instructions. The specificity of the PCR amplifications was evaluated by melting curve analysis of each data point. The H. armigera ribosomal protein S3 transcript was used as an internal reference for normalization of the cDNA templates. One microliter of diluted cDNA was used as the template to perform qRT-PCR. Each amplification reaction was performed using 20 μl of the mixture under the following conditions: denaturation at 95 °C for 2 min followed by 40 cycles of 95 °C for 20 s, 58 °C for 20 s, and 69 °C for 20 s. The melting curves were constructed during the last step over a temperature range of 58–95 °C. The relative gene expression levels were calculated using the 2−ΔΔCt method. All of the PCR amplifications were conducted in four replicates. In the corresponding figure, the normalized mRNA abundance was presented as the mean value and standard deviation of the four replicates. The primers used for qRT-PCR analysis are listed in supplemental Table S1.
Fat Body Protein In-gel Digestion and Nano-LC-MS/MS Analysis
Fat body samples from the control and infected groups were collected as described previously (14). Total protein was extracted using the Tissue Protein Extraction Kit (Cwbiotech Co. Ltd., Beijing, China) following the manufacturer's instructions. The protein concentrations were determined by the BCA method. Equal amounts of total proteins were separated on SDS-polyacrylamide gels and stained with Coomassie Brilliant Blue G-250 for visualization. Subsequently, the gel lanes were excised into six sections and stored in individual clean Eppendorf tubes at −20 °C, respectively.
Fat body proteins were digested as follows according to an in-gel protein digestion protocol described previously (43). Each gel section was cut into smaller pieces for the convenience of subsequent treatment. The samples were washed in ammonium bicarbonate (50 mm) and then reduced using dithiothreitol (10 mm) at 37 °C for 20 min. Samples were further alkylated using 55 mm iodoacetamide for 20 min in the dark. After washing twice with 50 mm ammonium bicarbonate, the gel pieces were destained with 50 mm ammonium bicarbonate/acetonitrile (1:1, v/v) by incubating at room temperature for 20 min. Finally, the gel pieces were immersed in trypsin solution and incubated at 37 °C overnight for adequate in-gel digestion.
The peptides obtained from in-gel digestion were collected by centrifugation and concentrated by drying in a vacuum rotary evaporator. Next, the dried peptides were solubilized in 0.1% trifluoroacetic acid and 2% acetonitrile and were sonicated in an ultrasonic water bath for 5 min. The supernatants were collected from centrifugation and individually analyzed on a Q Exactive mass spectrometer (Thermo Fisher Scientific) coupled to a New Objective PV-550 nanoelectrospray ion source and an Eksigent NanoLC-2D chromatography system. Briefly, peptides were separated using a 5–80% acetonitrile gradient in 0.1% formic acid performed over 120 min at a flow rate of 300 nl/min. Three biological replicates and three technical replicates were used for each mock and infected group, and each gel was excised into six sections. In total, 108 sections were subject to the LC-MS/MS analysis.
Database Search and Protein Quantification
Raw MS/MS data were uploaded into the Proteome Discoverer software system (version 1.4.0.288; Thermo Fisher Scientific, Waltham, MA) to perform tandem mass spectrometry data analysis using the built-in Sequest-HT search engine. The predicted H. armigera protein sequence database (14), which was deduced from the putative ORFs using an in-house Perl script based on the transcriptomic unigene dataset, was used as the reference database for protein identification and database searches. Meanwhile, the protein sequences encoded by HearNPV were also incorporated into the protein sequence database to identify HearNPV proteins. Totally the number of entries actually searched was 37,791 in the protein sequence database. The enzyme specificity was set to trypsin using acrylamide modification of cysteines as a static modification and oxidation of methionines as a dynamic modification. The mass tolerance was set at 10 ppm for precursor ions and 0.05 Da for fragment ions. The false discovery rate was set to 0.01 for peptides. The minimal peptide length was set to six, and up to two missed cleavage sites were allowed. Reversed protein sequences in the protein database were used as the decoy database for calculation of the FDR. In the filtering of the search results, matches with a high peptide confidence for at least one unique peptide were regarded as significant.
For quantification of the relative expression levels of different proteins within one sample, we employed the Peak Area Calculation Quantification method within the Proteome Discoverer software. The relative abundance of proteins within the group was represented as the peak area of the target protein, which was normalized to the summed peak area of the whole proteome using Equation 1,
| (Eq. 1) |
Herein, Ab(p) denotes the normalized relative abundance of protein p; Pa(p) denotes the peak area of protein p, denotes the total peak area for the whole proteome, and n denotes the total number of proteins identified. The ratios of the relative expression levels between groups were calculated to compare the relative protein abundances across different samples. A protein with a fold change ≥1.5 or <0.67 and p value <0.05 was considered to be differentially regulated. Differential protein expression analysis was performed for each biological replicate individually, and the proteins that were significantly regulated in at least two biological replicates were determined to be differentially expressed proteins (DEPs).
Gene Interaction Network Construction
The genes that were simultaneously up- or down-regulated at the transcriptional and translational levels were used for gene interaction analysis. Because of the absence of gene interaction network information for non-model organisms, InParanoid (44) was first used to determine the orthologs of H. armigera DEGs in D. melanogaster. Next, the orthologous gene sets in D. melanogaster were used to retrieve gene interaction networks using the on-line STRING database (45). Interaction networks for the up-regulated and down-regulated gene sets were predicted separately. The networks generated by STRING were exported into Cytoscape (46) for visualization.
Statistical Analysis
qRT-PCR data were analyzed according to the Agilent Technology guide. The remaining data were analyzed using GraphPad Prism 6. File conversion of mass spectrometry data complied with the instructions in PRIDE (47). For qRT-PCR and proteomic analysis, the pairwise Student's t test was used to determine statistical significance. Survival rate analysis between the control and infected groups was performed using the Wilcoxon signed-rank test. The data are shown as means ± S.E. of measurement (S.E.). Differences between comparisons were regarded as significant when p < 0.05.
RESULTS
Baculovirus Infection Alters the Growth of H. armigera Larvae
Viral inoculation of late third instar larvae was performed via oral administration of HearNPV OBs using a droplet method as described previously (5). After infection, the surviving larvae from the treated and control groups were counted each day. Survival rate analysis indicated that the life span of HearNPV-infected larvae was significantly shorter than that of naive larvae, thus proving the potent virulence and lethality of NPV used in this study (supplemental Fig. S1A).
To examine the effect of viral infection on the growth and development of H. armigera larvae, the weights of the larvae were measured. Following the viral infection, each larva in the two groups was weighed at four time points post-infection. Notably, it was observed that the average weight of the larvae in the infected group was much smaller than that in the control group (Fig. 1A). Moreover, the difference in body size increased during the infection process, suggesting that the growth and development of host larvae were strongly retarded by baculovirus infection (supplemental Fig. S1B).
Fig. 1.

Effect of H. armigera nucleopolyhedrovirus infection on cotton bollworm larvae and transcription pattern of viral genes during the infection process. A, weight changes of larvae in the control and infected groups throughout the infection process. The data were presented as means ± S.E. (mg). **, p ≤ 0.01; ***, p ≤ 0.001. Notably, the average weight of the larvae in the infected group was significantly lower than that in the control group. B, mRNA abundance of HearNPV over the infection time course. Each column represents the reads as a percentage of the total reads (Illumina), and the percentage of mRNA derived from baculovirus accounted for is >15% of the total mRNA content at 72 hpi. C, heatmap analysis showing the baculoviral gene expression patterns during the infection process. Each gene at each of the four time points post-infection was represented as a horizontal short line. Most of the genes showed a similar trend in expression level, continuously increasing over time. D, relative expression levels of two representative viral genes, virus-encoded DNA polymerase and lef-6, were measured at 0, 24, 48, and 72 hpi using qRT-PCR, and the results were consistent with those of RNA-seq.
Transcriptomic Sequencing and RNA-seq Data Analysis
The expressed RNA sequences from normal and infected larvae at four time points were generated from paired-end sequencing on the Illumina RNA-seq platform and were analyzed following the workflow shown in supplemental Fig. S2. Basically, the raw reads obtained were preprocessed for complete removal of the adaptor, low quality and short sequences. In addition, the contaminated reads were filtered out using BWA alignment tools against bacterial and viral sequences except for HearNPV. Subsequently, to cover as many full-length transcripts as possible, all of the clean reads from eight libraries were pooled together. Next, the pooled reads were assembled de novo into transcripts using Trinity software with the default parameters, resulting in 94,813 primary transcripts (supplemental Fig. S3A). The mean sequence length of the primary transcripts was 1,010 bp, whereas N50, a statistical measure of the transcriptome sequence library, was up to 2,047. The length distribution of the assembled transcripts is shown as a pie chart in supplemental Fig. S3B, which indicated that most of the transcripts were longer than 300 bp. Therefore, the assembly results indicated that the Illumina RNA-sequencing method combined with Trinity software can be used to determine the quality of the transcriptome reliably. Subsequently, CD-Hit was performed to remove redundant transcripts in the initial assembly, ultimately resulting in a non-redundant unigene set containing 71,033 transcripts.
To annotate these transcripts, a homology search was performed locally using the BLASTX (48) program against the non-redundant protein sequence database (NCBI Nr) with a cutoff E-value of 1e-5, which usually represents meaningful hits with statistical significance. The BLAST results demonstrated that ∼26,317 transcripts had at least one significant hit. The species distribution for the top BLAST hits is shown in supplemental Fig. S3C. Next, BLAST2GO (38) pipeline was employed to determine further functional annotation of the transcripts, such as GO term assignment and EC number identification. In total, 15,009 unigenes were assigned at least one GO term.
Time Course Expression Profiling of Baculoviral Genes
To explore the transcriptional changes in baculoviral genes during the infection period, the expression profile at different time points was determined by RPKM values of viral transcripts with RSEM software. The ORF sequences of the HearNPV G4 genome sequence (GenBankTM accession no. AF271059) were used as the reference to estimate relative expression values. To obtain an overview of HearNPV genes at the transcription level, we performed statistical analysis of the percentage of baculovirus reads among the total reads in each library, and the results indicated that viral mRNA molecules increased sharply during the infection process, even accounting for 15% of the total reads at 72 hpi (Fig. 1B). The heat map in Fig. 1C, representing the expression dynamics indicated that most genes followed a similar trend in their expression levels, rapidly increasing at 24 hpi and peaking at 72 hpi. According to the dendrogram on the left (Fig. 1C), the baculoviral genes were classified into four groups based on their expression pattern. Notably, groups III and IV comprised most of viral genes; moreover, the average expression level of genes in group III was slightly higher than that in group IV. Additionally, group I was comparable with group II in gene number, whereas these two groups differed substantially from one another at the gene expression level. The expression levels of genes within group I were relatively low across the four time points, and all of the genes were unique with uncharacterized functions, suggesting their smaller contribution to viral infection and replication. By contrast, 19 genes in group II exhibited the highest expression levels among the four groups, with most being well annotated and characterized functionally, including early and transcriptional activators (ie-1, vlf-1), occlusion body-related genes (polyhedrin, calyx/pp), virion-associated proteins (P6.9, Odv-e25, Odv-e27), and enzymes involved in insect disintegration (chitinase, cathepsin). In particular, the polyhedrin gene showed the highest expression level at 72 hpi, suggesting that infection has reached the late stage, during which ODVs started to assemble into OBs. To validate the expression levels of viral genes during infection, two representative genes were selected to perform qRT-PCR analysis, including the late expression gene lef-6 and viral DNA polymerase. qRT-PCR showed increasing expression patterns during the infection process, consistent with the RNA-seq data (Fig. 1D). Taken together, these results suggested that oral inoculation of HearNPV successfully established the systemic infection of host larvae.
Global Overview of the H. armigera Fat Body Transcriptome
To assess global gene expression in H. armigera larvae during the infection process, RSEM software was used to calculate gene expression values (RPKM) for each library in the two groups.
PCA facilitates comparative analysis of multiple samples by reducing data dimensionality while maintaining most of the variations present in the original datasets; therefore, it is especially suitable for gene expression profiling. To reveal the relationships among the eight libraries, PCA analysis was performed to determine variations in gene expression levels between the control and infected groups during infection. The PCA results indicated that the first three principal components, PC1, PC2, and PC3, account for most of the contribution to the total data variation, as shown in the three-dimensional scatter plot (supplemental Fig. S4A). To some extent, the variations between the control and infected groups at different time points differed from each other. As depicted by the 3D scatter plot, during the early stage of infection, only very little variation was observed, whereas the distinction became more and more significant over the course of infection, with the greatest variation seen at 72 hpi. Thus, PCA analysis substantially reflects the variance within time course transcriptomic data, elucidating distinct global gene expression patterns between the normal and infected groups during the infection process.
Host Responses against Baculovirus Infection
According to the above PCA analysis, there was a clear difference in transcriptomes between the control group and infected groups. In response to infection, the expression levels of various host genes were altered remarkably. To identify DEGs between the groups over the course of infection, the DEGseq package was employed to perform differential gene expression analysis using the MARS method. The differential analyses were performed for any combination of samples at each time point. The number of DEGs was depicted in several representative plots (supplemental Fig. S4B). The scatter plots in supplemental Fig. S4B represent the trend in DEGs between the control and infected groups during the infection process. The number of up-regulated genes was 76 at 24 hpi, increasing to 224 genes at 48 hpi, and peaking to 394 genes at 72 hpi; meanwhile, the number of genes that were down-regulated appeared to show a similar trend, namely increasing (from 155 to 1,322) during the infection process. However, the number of down-regulated genes was substantially more than that of up-regulated ones, suggesting that more genes were repressed at the transcriptional level upon infection.
Furthermore, the distribution of DEG fold changes is presented in Table I. As shown in Table I, the fold changes in DEGs were differentially distributed between the up- and down-regulated gene cohorts during the infection process. During the early stage of infection (24 hpi), ∼71.1% of DEGs were altered by 1.5–5-fold at the mRNA level, whereas fewer DEGs (17.1%) were altered more than 5–10-fold, and only 11.8% of DEGs were altered >10-fold. As the infection processes, more genes were up-regulated to a greater degree (32.1% ≥5-fold at 48 h, 55.3% ≥5-fold at 72 h). However, the distribution of the down-regulated genes was distinct from that of the up-regulated genes, as reflected by the percentages of highly down-regulated (≥5-fold) genes (35.5% at 24 h, 68.5% at 48 h, and 88.1% at 72 h). Selected DEGs are presented in Table II. Among these DEGs, many were related to metabolic function in the host insect, such as alcohol dehydrogenase (∼25.89-fold), acyl-CoA desaturase (∼13.54-fold), and carboxyl/choline esterase (>11.90-fold). In addition, a considerable proportion of immunity-related genes were differentially expressed. Interestingly, most of the differentially regulated immune genes were down-regulated, such as PPO2 (approximately −18.16-fold), βGRP1 (approximately −4.89-fold), scavenger receptor class B (approximately −24.16-fold), and CTL5 (−2.7-fold). Generally, the results implied that a considerable portion of genes involved in various cellular and physiological processes were strongly affected at the transcriptional level following viral infection.
Table I. Numbers of up- and down-regulated unigenes at each time point post-infection.
| Category | Fold change | Time points |
|||
|---|---|---|---|---|---|
| 0 h | 24 h | 48 h | 72 h | ||
| Up-regulated | ≥10 | 4 | 9 | 42 | 132 |
| 5–10 | 9 | 13 | 30 | 86 | |
| 1.5–5 | 42 | 54 | 152 | 176 | |
| Down-regulated | 1.5–5 | 68 | 100 | 345 | 157 |
| 5–10 | 3 | 17 | 205 | 536 | |
| ≥10 | 6 | 38 | 545 | 629 | |
| Total number | 132 | 231 | 1319 | 1716 | |
Table II. Selected unigenes that were differentially expressed upon baculovirus infection.
| Time | Unigene | FCa | p value | Accession no. | E-value | Descriptionb |
|---|---|---|---|---|---|---|
| 24 h | UN009540 | 13.22 | 8.47E-05 | EHJ73474.1 | 2.49E-137 | Hypothetical protein KGM_09706 (Danaus plexippus) |
| 24 h | UN017435 | 7.22 | 7.42E-09 | BAM17901.1 | 2.94E-06 | Serine protease (Papilio xuthus) |
| 24 h | UN024701 | 4.74 | 0.038 | AAX51193.1 | 1.97E-24 | HacD (H. armigera) |
| 24 h | UN022186 | 3.52 | 2.34E-03 | BAM17783.1 | 8.22E-65 | Glutathione S-transferase S1 (P. xuthus) |
| 48 h | UN008880 | 83.55 | 4.95E-34 | ACV32640.1 | 0 | Heat shock protein 70 (H. zea) |
| 48 h | UN018885 | 34.25 | 7.13E-20 | XP_004927349.1 | 2.75E-88 | PREDICTED: heat shock protein Hsp-16.1/Hsp-16.11-like (B. mori) |
| 48 h | UN031693 | 25.89 | 0.010 | AGQ45607.1 | 1.25E-55 | Alcohol dehydrogenase, partial (Agrotis ipsilon) |
| 48 h | UN039097 | 13.83 | 1.92E-18 | |||
| 48 h | UN012238 | 13.54 | 3.89E-07 | AGO45844.1 | 8.37E-129 | Acyl-CoA desaturase HvirIPAE (Heliothis virescens) |
| 48 h | UN008963 | 4.08 | 1.53E-04 | AFP20600.1 | 3.52E-164 | Cytochrome CYP4S8v1 (Spodoptera littoralis) |
| 72 h | UN007340 | 11.90 | 1.06E-80 | ADF43482.1 | 0 | Carboxyl/choline esterase CCE016d (H. armigera) |
| 72 h | UN001606 | 3.62 | 0.011 | ADI61831.1 | 1.18E-34 | Endonuclease-reverse transcriptase (B. mori) |
| 72 h | UN007393 | 5.07 | 1.53E-07 | XP_004923198.1 | 1.01E-136 | PREDICTED: glucose dehydrogenase (acceptor)-like (B. mori) |
| 72 h | UN002465 | 6.11 | 8.70E-05 | EHJ75641.1 | 2.05E-47 | Histone H2B-like protein (Danaus plexippus) |
| 24 h | UN012361 | −2.70 | 2.16E-13 | AFI47450.1 | 0 | C-type lectin 5 (H. armigera) |
| 48 h | UN013510 | −2.38 | 1.03E-04 | AEB26313.1 | 0 | Aldo-keto reductase (H. armigera) |
| 48 h | UN023917 | −2.43 | 0.042 | XP_004922580.1 | 5.11E-73 | PREDICTED: NADH dehydrogenase (ubiquinone) 1β subcomplex subunit 9-like (B. mori) |
| 48 h | UN012650 | −2.69 | 0.034 | AFI47451.1 | 0 | C-type lectin 6 (H. armigera) |
| 48 h | UN008157 | −3.10 | 6.34E-04 | ADD97142.1 | 0 | Carboxylesterase (H. armigera) |
| 48 h | UN001524 | −3.31 | 0.016 | ACH88521.1 | 0 | Trehalose 6-phosphate synthase isoform I (H. armigera) |
| 48 h | UN006294 | −3.50 | 0.034 | AAW24481.1 | 0 | Prophenol oxidase activating enzyme 3 (Spodoptera litura) |
| 48 h | UN006444 | −10.49 | 2.70E-05 | NP_001040411.1 | 0 | Carboxylesterase clade H, member 1 precursor (B. mori) |
| 48 h | UN005429 | −18.16 | 1.18E-07 | AAZ52554.1 | 0 | Prophenoloxidase subunit 2 (H. armigera) |
| 48 h | UN005425 | −24.16 | 2.06E-09 | EHJ75193.1 | 1.79E-145 | Scavenger receptor class B, member 1-like protein (Danaus plexippus) |
| 72 h | UN006178 | −3.08 | 0.029 | BAM18144.1 | 0 | Pyruvate kinase (Papilio xuthus) |
| 72 h | UN008785 | −4.89 | 0.036 | ACI32825.1 | 0 | β-1,3-Glucan recognition protein 1 (H. armigera) |
| 72 h | UN010617 | −6.74 | 5.45E-29 | AEB26314.1 | 0 | Glyceraldehyde-3-phosphate dehydrogenase (H. armigera) |
| 72 h | UN006836 | −7.85 | 4.02E-12 | NP_001093304.1 | 0 | Pyruvate dehydrogenase (B. mori) |
| 72 h | UN012555 | −7.89 | 2.98E-16 | BAM17938.1 | 8.62E-172 | phosphoglyceromutase (P. xuthus) |
a Fold change of DEGs, positive value indicates up-regulation while negative value denotes down-regulation.
b Immunity-related genes were shown in bold.
Analysis of DEGs
To explore the trends in global expression intuitively, hierarchical clustering of DEG expression levels at the four time points was performed using the R package, as shown in Fig. 2A. Noticeably, the DEGs showed distinct expression patterns across the four time points post-infection. At the beginning of infection (0 hpi), the gene expression patterns were highly similar between the control and infected groups as expected. Therefore, the difference at 0 hpi was not considered in the subsequent analysis. As infection progressed, the difference in gene expression increased gradually between the uninfected and infected larvae. A small proportion of genes was up-regulated at 24 hpi, whereas other genes were down-regulated at 24 hpi. Notably, a substantial difference was found at 48 hpi, when a large proportion of genes was significantly down-regulated, whereas fewer genes were up-regulated. During the late stage of infection (72 hpi), most of the host genes showed dramatic down-regulation, whereas a minor proportion remained up-regulated. Interestingly, a large proportion of genes that were induced at 24 hpi appeared to be down-regulated at a later stage (48 or 72 hpi), suggesting that the expression of host genes was strongly repressed during the late stage of viral infection. To analyze the gene cohorts commonly regulated at different time points, a Venn diagram (Fig. 2B) was used to show the overlap among cohorts. According to the Venn diagram, we found 30 DEGs were common among three time points, of which only one (cytochrome CYP4S8v1, UN056945) was commonly up-regulated throughout the infection process, and 13 were simultaneously down-regulated over the time course, including ribosomal protein S15 (UN022113), UDP-glucose pyrophosphorylase (UN005270), topoisomerase 1B (UN001954), PPO2 (UN005429), and CTL-5 (UN012361). Additionally, some genes were uniquely regulated at different time points, most of which were repressed, especially during the late stage of infection.
Fig. 2.

Comparison of fat body transcriptomes between control and infected larvae. A, hierarchical clustering analysis of fat body transcripts that were differentially expressed for any comparison between control and infected samples at the four time points. RPKM values for H. armigera transcripts were calculated using RSEM software. Each gene at each time point is shown as a line color-coded according to expression level. Noticeably, most of the genes were significantly down-regulated in the infected group compared with the mock-infected group. B, Venn diagram analysis of commonly and uniquely regulated genes at three time points post-infection (24, 48, and 72 hpi, respectively). The on-line interactive tool Draw Venn Diagram was used to generate a Venn diagram. C, gene ontology enrichment analysis revealed the biological processes and molecular functions most associated with the DEGs. The plots of second-level GO terms corresponding to the up- and down-regulated enriched genes are shown; herein, enrichment analysis was conducted using the functional annotation tool DAVID. Statistical significance was determined at a p value ≤0.05. *, Padj ≤0.05; **, Padj ≤0.01; ***, Padj ≤0.001. The symbol n.s. indicates no significance for the corresponding GO term. D, KEGG functional classification of baculovirus-regulated transcripts. To explore the effect of viral infection on the mRNA abundance of fat body genes, the DEGs induced and repressed upon viral infection were classified functionally. Functional group abbreviations: OS, organismal systems; ST, signal transduction; R/R, replication and repair; F/S/D, folding, sorting and degradation; Tlat, translation; Tran, transcription; IMM, immunity-related; XBM, xenobiotics biodegradation and metabolism; EM, energy metabolism; AAM, amino acid metabolism; NM, nucleotide metabolism; LM, lipid metabolism; CM, carbohydrate metabolism.
GO Analysis and Functional Classification of DEGs
To better understand the function of genes responsive to baculovirus infection at different time points, the DEGs were subjected to GOEA to determine the enriched GO terms. The up- and down-regulated genes were allocated into three categories according to GO term: molecular function, cellular component, and biological process. Here, the functional annotation tool DAVID was employed to determine the significantly enriched GO terms. The results indicated that the distribution of DEGs differed during different infection stages (Fig. 2C). First, no enrichment results were obtained for the up-regulated DEGs at 24 hpi (data not shown). The enrichment of down-regulated DEGs at 24 hpi was non-significant. In contrast, for the up-regulated genes at 48 hpi, only the DNA metabolic process was significantly enriched. However, for the down-regulated gene cohort, a variety of GO terms were significantly enriched, including amine biosynthetic process, RNA helicase activity, biogenic amine metabolic process, ribosome biogenesis, and carbohydrate binding. Interestingly, more enriched GO terms were found among the down-regulated genes at 72 hpi, including amine biosynthetic process, mitotic cell cycle, and translation, which were also affected at 48 hpi, although antioxidant activity, coenzyme metabolic process, translational elongation, carboxylic acid catabolic process, glucose metabolic process, and transferase activity were uniquely affected at 72 hpi. Collectively, the GOEA results revealed no significant GO enrichment for DEGs during the early infection stage (24 hpi), whereas most of the identified enriched GO terms were associated with the down-regulated gene sets at 48 and 72 hpi. The enrichment results demonstrated that the cell cycle as well as translational machinery were substantially impaired at 48 and 72 hpi. Moreover, glucose metabolism and ATP synthesis were negatively regulated at 72 hpi, implying that energy production was disrupted during the late stage of infection.
Furthermore, the functional classification of DEGs was performed by mapping to the KEGG pathway using KOBAS 2.0. As shown in Fig. 2D, the DEGs were related to a diversity of KEGG pathways, involving immunity and various metabolism-related pathways. During the early infection stage of infection, the molecules participating in several pathways, such as translation, AAM, and CM, were weakly down-regulated. At 48 hpi, AAM and replication/repair were highly activated upon baculovirus infection, together with up-regulation of fold, sorting, and degradation (F/S/D), although genes related to signal transduction and translation processes appeared to be repressed. At 72 hpi, the most profound alterations were observed in the KEGG pathways (Fig. 2D). The gene sets involved in F/S/D exhibited drastic down-regulation. Importantly, most immunity-related genes were strongly down-regulated at 72 hpi; meanwhile, several genes related to CM, energy metabolism, and AAM were significantly inhibited in expression, although several other pathways such as replication/repair were not affected during the active state. These results are consistent with GO analysis. It is worth noting that the translation process was negatively regulated during the infection process, implying inhibition of the host translational machinery following baculovirus infection.
Expression Profiling of Immunity-related Genes
Given that most of the immune genes were significantly regulated after baculovirus infection, we focused mainly on the expression dynamics of immunity-related genes. Previously, 233 immunity-related genes were identified in H. armigera (14) based on sequence similarity. Here, we selected immune genes that were differentially expressed during the baculovirus infection process to analyze their expression patterns. The hierarchical clustering of these genes (Fig. 3A) revealed the expression profiles of immune DEGs at the mRNA level, including the molecules involved in pattern recognition, signal modulation, and execution. The Venn diagram (Fig. 3B) indicated that fewer immune genes were regulated during the early infection stage, although more immunity-related genes were modulated as infection progressed, most of which were suppressed at the transcriptional level. In detail, C-type lectin 19 (CTL19), cecropin 2, cecropin 7, attacin, and lebocin were slightly induced at 24 hpi, whereas 57 immune genes belonging to recognition (βGRPs, CTLs, and PGRPs), signaling transduction (cSPs, SPHs, and serpins), and effectors (lysozymes, gloverins, GSTs, and SOD) were significantly repressed during the late stage of infection. Furthermore, the Venn diagram (Fig. 3B) indicated that two immune genes (CTL5 and PPO2) were common among three time points, both of which were suppressed throughout the infection process. Additionally, the number of down-regulated immune genes increased from 3 at 24 hpi to 40 at 72 hpi as shown in Fig. 3B. We chose immunity-related genes to perform qRT-PCR analysis, such as attacin, lysozyme 1, moricin 4, gloverin 1, and prophenoloxidase-2 (PPO2). The results suggested that most of the immune genes displayed the similar expression patterns as shown in the RNA-seq data (Fig. 3C). Antimicrobial effector attacin exhibited clear down-regulation upon baculovirus infection. Meanwhile, lysozyme 1 and moricin 4 were significantly down-regulated. qRT-PCR analysis demonstrated that C-type lectin 7 (CTL7) was dramatically decreased in expression, in accordance with the RNA-seq data. Together, 60 immune genes were substantially influenced at the transcriptional level, suggesting their potential roles in baculovirus infection.
Fig. 3.

Expression profiling of immunity-related genes in the larval fat body after baculovirus infection. A, hierarchical clustering analysis of immunity-related genes that were differentially expressed following viral infection. The immune genes were divided into three major groups according to their respective functions: recognition molecules, signaling modulators, and immune effectors. The RPKM value of each gene is shown as a rectangle coded in blue (lower expression level) or red (higher expression level). B, Venn diagram showing the unique and common immune genes that were significantly regulated at three time points post-infection. Two genes were commonly down-regulated at three time points. The number of down-regulated genes was apparently more than that of up-regulated ones. C, qRT-PCR analysis of selected immunity-related genes. Pairwise Student's t tests were used for statistical analysis. **, p ≤ 0.01; ***, p ≤ 0.001. The results showed that the recognition molecules C-type lectin 7 (CTL7), lysozyme 1, antimicrobial peptides attacin, moricin 4, and gloverin 1, and the key member of melanization cascade reactions, PPO2, was substantially suppressed at the transcript level at 72 hpi.
Time Course Expression Pattern of CM Genes
Baculoviruses rely largely on host metabolism to provide energy for DNA replication, proliferation, and protein synthesis due to the lack of the corresponding machinery in the virus itself. The fat body is involved in immune response as well as energy storage and various metabolic processes in insects. A large gene cohort participating in CM was highly modulated after viral infection in the fat body. CM is one of the key processes producing ATP in insects; therefore, we characterized the expression dynamics of CM-related genes during infection in the fat body of H. armigera larvae. CM included a series of biochemical processes, mainly including glycolysis, the tricarboxylic acid (TCA) cycle, and the branch pathway pentose-phosphate pathway (PPP). Here, we explored the expression pattern of CM-related enzymes based on these three categories. The hierarchical clustering analysis (Fig. 4A) showed dynamic changes in the expression levels of metabolism genes during the infection process. The transcript levels of a few CM genes, such as phosphofructokinase (PFK), phosphoglycerate kinase in glycolysis, and succinate dehydrogenase, isocitrate dehydrogenase, and succinyl-CoA synthetase within the TCA cycle, were slightly reduced during the early infection stage (24 hpi). At 48 hpi, hexokinase, phosphoglycerate mutase, and PFK in glycolysis and aconitase, succinate dehydrogenase in TCA cycle, and ribulose-5-phosphate isomerase (RPI) in PPP tended to follow the same trend, although the expression of pyruvate kinase (PyK) and α-ketoglutarate dehydrogenase was increased. As infection progressed, the mRNA abundances of more CM-related genes were drastically decreased during the late infection stage (72 hpi), with the exception of hexokinase. Within each stage of sugar metabolism, the genes were largely down-regulated at 72 hpi. Importantly, two rate-limiting enzymes in glycolysis, PyK and PFK, were significantly suppressed at the late infection stage. Additionally, glucose-6-phosphate isomerase (GPI), aldolase, PGK, triose-phosphate isomerase (TPI), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and enolase of glycolysis, succinate dehydrogenase, IDH, pyruvate dehydrogenase, fumarase, malate dehydrogenase (MDH), citrate synthase 1, and succinyl-CoA synthetase of the TCA cycle, and RPI, transketolase, transaldolase, 6-phosphogluconate dehydrogenase, and ribulose phosphate-5-epimerase of the PPP were significantly inhibited transcriptionally. Furthermore, to validate the RNA-seq results, the relative expression levels of CM-related genes were determined by qRT-PCR analysis (Fig. 4B), revealing that genes such as aldolase, PFK, enolase, fumarase, and GPI, showed similar trends as seen in the RNA-seq data. Additionally, the relative expression level of fructose-1,6-bisphosphatase (FBP) was dramatically down-regulated (>30-fold) at 72 hpi according to qRT-PCR. Therefore, the expression profiles of CM-related genes suggested that CM was significantly regulated during the infection time course, with the most profound changes seen during the late infection stage.
Fig. 4.
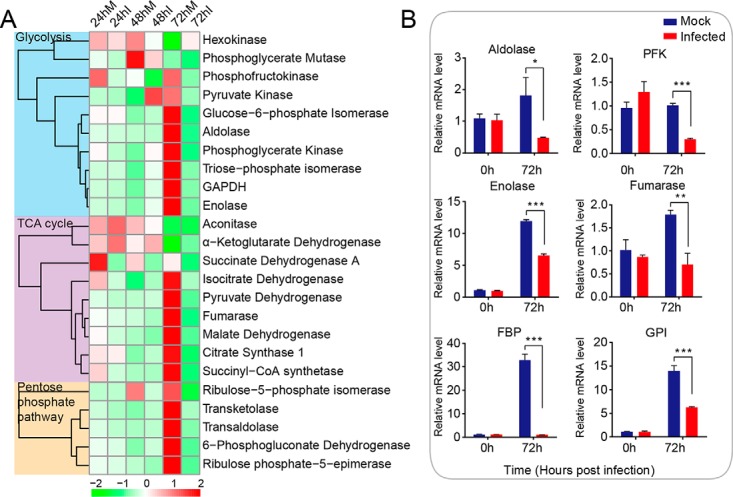
Expression profiling of CM-related genes during the infection process in the normal and infected groups. A, hierarchical clustering of CM-related genes during the infection process. CM genes were divided into three categories: glycolysis, TCA cycle, and pentose-phosphate pathway. B, relative expression levels of selected CM genes at 0 and 72 hpi were validated by qPCR analysis. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. The mRNA abundances of aldolase, phosphofructokinase (PFK), enolase, fumarase, fructose-1,6-bisphosphatase (FBP), and glucose-6-phosphate isomerase (GPI) were decreased at 72 hpi, consistent with the RNA-seq data results.
Overview of Proteins Identified in the H. armigera Fat Body
Based on transcriptomic analysis, we found that several immunity- and metabolism-related genes were significantly down-regulated at 72 hpi, suggesting the importance of understanding transcriptional regulatory mechanism. Here, to understand the changes in protein abundance better, label-free quantitative proteomics research was performed to evaluate the effect of baculovirus infection on host gene expression at the proteome level.
For proteomics studies, proteins were extracted from the fat body of larvae at 72 hpi, designated as mock fat (MFs) body samples and infected fat body samples (IFs). Equal amounts of protein samples were separated by SDS-PAGE, and the gel was stained with Coomassie Brilliant Blue G-250 (supplemental Fig. S5A). There was considerable difference in the protein band patterns between the control and infected groups. Compared with the control group, the intensity of most bands was weaker in the infected group, suggesting substantial changes at the protein level upon baculovirus infection. However, a band of extremely high intensity was observed at ∼30 kDa in the infected group, identified as the putative product of the baculoviral gene encoding polyhedrin, indicating that viral infection had reached the very late stages. Each lane was excised into six sections, in-gel digested with trypsin, and then subjected to analysis by nano-LC-MS/MS; the resulting mass spectrometry data were used for a database search. A flowchart of the LC-MS/MS analysis is shown in supplemental Fig. S5B.
After LC-MS/MS analysis, the obtained raw MS/MS data were uploaded into the Proteome Discoverer software to identify proteins via a search against the in-house H. armigera protein sequence database. For each MF and IF sample evaluated, the numbers of spectra identified by LC-MS/MS were 181,540 (MF1), 170,770 (MF2), 172,037 (MF3), 162,986 (IF1), 157,504 (IF2), and 157,253 (IF3). Pearson correlation coefficients between biological replicates are shown in supplemental Table S2. All of the peptides in the control and infected groups were assembled separately into protein sequences, resulting in 2,273 proteins identified in the fat body of H. armigera larvae. Among these, 1,360 proteins were common between the control and infected groups, as illustrated in the Venn diagram (Fig. 5A). However, 613 proteins were detected exclusively in the control group, although 300 proteins were unique to the infected group. As for the reason why the 913 proteins were not detected in either the mock or infected group, it was mainly caused by the strict filtering criteria used by us. In detail, among the 613 proteins uniquely found in the mock group, 374 (61.01%) were identified in one biological replicate of the infected group. Meanwhile, a similar percentage (60.33%, 181) of the 300 proteins that were exclusively detected in infected group was found in one biological replicate of the mock group. In the subsequent analysis, we mainly focused on the proteins commonly found in the mock and infected group. The commonly identified proteins were categorized based on COG functional class, with 820 proteins assigned to 25 COG clusters (supplemental Fig. S6A). The “J” cluster (translation, ribosomal structure, and biogenesis), “O” cluster (post-translational modification, protein turnover, and chaperones), and “R” cluster (general function prediction) represented the three largest groups, accounting for 10.4, 9.1, and 7.4% of total proteins in the fat body, respectively. Furthermore, GO category assignments (supplemental Fig. S6B) indicated that most of the proteins were related to metabolic processes (primary metabolic, macromolecule metabolic, oxidation/reduction, and regulation of metabolic processes).
Fig. 5.
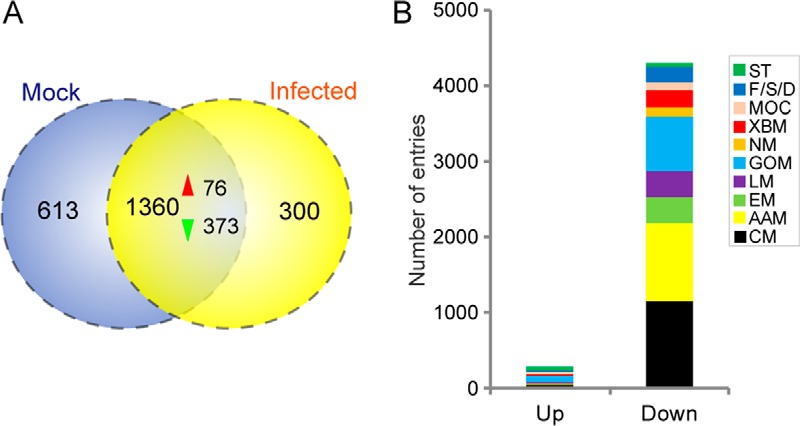
DEPs in the fat body after baculovirus infection. A, proteins identified by LC-MS/MS analysis that were unique and common between the control and infected groups. The red and green arrowheads denote up- and down-regulation at the protein level, respectively. B, KEGG functional classification of proteins that were significantly differentially expressed during proteomics analysis. Functional group abbreviations: ST, signal transduction; F/S/D, folding, sorting, and degradation; MOC, metabolism of cofactors and vitamins; XBM, xenobiotics biodegradation and metabolism; NM, nucleotide metabolism; GOM, global and overview maps; LM, lipid metabolism; EM, energy metabolism; AAM, amino acid metabolism; CM, carbohydrate metabolism.
Apart from the host proteins, proteins encoded by the HearNPV genome were also discovered in the proteomics analysis. As shown in Table III, 87 HearNPV proteins were identified in the infected larval fat body by LC-MS/MS. Polyhedrin, calyx/pep, capsid protein VP39, chitinase, and P10 represented the top five most abundant proteins, further indicating the late stage of infection.
Table III. Identified proteins encoded by baculovirus in fat body proteomics analysis.
Coverage, No. of unique peptides are in whole numbers. Coverage represents the % coverage of all unique peptides covering the entire protein. Area and relative abundance represent the average values among three biological replicates.
| ORF | Description | Coverage | No. of unique peptides | Area | No. of amino acids | Mass | Calculated pI | Relative abundance |
|---|---|---|---|---|---|---|---|---|
| kDa | ||||||||
| ORF1 | Polyhedrin | 77.6% | 45 | 2.17E+11 | 246 | 28.9 | 6.32 | 0.300 |
| ORF120 | calyx/pep | 65.9% | 24 | 5.29E+10 | 340 | 39 | 11.44 | 6.75E-02 |
| ORF78 | vp39 | 73.4% | 19 | 8.75E+09 | 293 | 33.4 | 5.81 | 9.02E-03 |
| ORF41 | Chitinase | 76.3% | 35 | 7.49E+09 | 570 | 63.6 | 5.81 | 8.71E-03 |
| ORF21 | p10 | 49.4% | 4 | 6.54E+09 | 87 | 9.3 | 4.61 | 7.41E-03 |
| ORF68 | Hypothetical protein | 58.6% | 7 | 9.21E+09 | 152 | 17.6 | 5.53 | 7.32E-03 |
| ORF53 | Fp-25k | 77.0% | 24 | 8.85E+09 | 217 | 25.4 | 8.81 | 6.72E-03 |
| ORF82 | odv-e25 | 78.7% | 14 | 4.85E+09 | 230 | 25.9 | 7.83 | 5.19E-03 |
| ORF44 | Hypothetical protein | 56.9% | 17 | 3.55E+09 | 378 | 42.7 | 8.79 | 3.79E-03 |
| ORF118 | p24 | 58.1% | 14 | 3.54E+09 | 248 | 28.4 | 6.52 | 3.71E-03 |
| ORF31 | 39k/pp31 | 75.6% | 24 | 4.49E+09 | 311 | 35.2 | 9.66 | 3.57E-03 |
| ORF9 | p49 | 64.5% | 26 | 2.86E+09 | 468 | 55.2 | 8.78 | 3.13E-03 |
| ORF57 | Hypothetical protein | 75.9% | 11 | 3.46E+09 | 195 | 21.3 | 5.38 | 2.92E-03 |
| ORF94 | Hypothetical protein | 59.8% | 19 | 2.45E+09 | 361 | 41.5 | 6.42 | 2.57E-03 |
| ORF73 | gp41 | 63.4% | 14 | 2.89E+09 | 322 | 36.6 | 7.91 | 2.57E-03 |
| ORF65 | lef-3 | 50.1% | 17 | 2.56E+09 | 379 | 44 | 5.44 | 2.40E-03 |
| ORF10 | odv-e18 | 34.6% | 2 | 2.66E+09 | 81 | 8.8 | 10.67 | 2.05E-03 |
| ORF91 | Hypothetical protein | 61.8% | 22 | 1.96E+09 | 377 | 44 | 6.93 | 2.04E-03 |
| ORF56 | Cathepsin | 41.4% | 14 | 2.30E+09 | 365 | 42 | 5.95 | 1.77E-03 |
| ORF26 | Hypothetical protein | 28.6% | 2 | 2.10E+09 | 133 | 15 | 4.82 | 1.70E-03 |
| ORF25 | dbp | 67.2% | 23 | 1.31E+09 | 323 | 37.5 | 6.14 | 1.57E-03 |
| ORF106 | sod | 27.0% | 4 | 2.20E+09 | 159 | 16.8 | 6.51 | 1.40E-03 |
| ORF96 | odv-e66 | 54.2% | 26 | 1.66E+09 | 672 | 76 | 6.62 | 1.40E-03 |
| ORF130 | pkip-1 | 21.9% | 5 | 1.31E+09 | 169 | 20.3 | 8.69 | 1.34E-03 |
| ORF75 | Hypothetical protein | 40.0% | 8 | 1.15E+09 | 225 | 24.9 | 4.61 | 1.25E-03 |
| ORF24 | lef-6 | 33.7% | 7 | 1.43E+09 | 187 | 22.2 | 9.45 | 1.09E-03 |
| ORF22 | p26 | 59.6% | 17 | 1.11E+09 | 267 | 30.5 | 9.5 | 1.09E-03 |
| ORF58 | gp37 | 64.9% | 8 | 1.44E+09 | 279 | 32.1 | 6.92 | 1.03E-03 |
| ORF100 | Hypothetical protein | 64.7% | 24 | 9.82E+08 | 510 | 58.1 | 4.75 | 9.01E-04 |
| ORF88 | p6.9 | 9.2% | 2 | 6.93E+08 | 109 | 11.5 | 12.82 | 8.59E-04 |
| ORF3 | pk-1 | 46.8% | 11 | 7.10E+08 | 267 | 31.5 | 8.56 | 8.53E-04 |
| ORF72 | Hypothetical protein | 43.6% | 4 | 9.64E+08 | 110 | 12.7 | 8.1 | 8.25E-04 |
| ORF80 | Hypothetical protein | 71.3% | 19 | 6.76E+08 | 254 | 30.8 | 8.12 | 7.15E-04 |
| ORF15 | pif-5 | 41.8% | 9 | 1.10E+09 | 354 | 38.8 | 8.25 | 7.00E-04 |
| ORF123 | 38.7kd | 61.6% | 21 | 6.87E+08 | 385 | 44.4 | 6.54 | 6.84E-04 |
| ORF71 | vlf-1 | 44.9% | 17 | 1.71E+09 | 412 | 47.8 | 7.5 | 6.82E-04 |
| ORF36 | Hypothetical protein | 57.8% | 10 | 8.21E+08 | 223 | 25.8 | 5.16 | 6.64E-04 |
| ORF27 | Hypothetical protein | 29.4% | 8 | 1.01E+11 | 255 | 29.5 | 5.5 | 6.36E-04 |
| ORF63 | Hypothetical protein | 41.6% | 8 | 6.51E+08 | 274 | 31.5 | 8.48 | 6.23E-04 |
| ORF92 | vp80 | 52.9% | 27 | 1.55E+09 | 605 | 69.7 | 7.42 | 5.75E-04 |
| ORF70 | Hypothetical protein | 9.4% | 1 | 6.03E+08 | 85 | 10 | 9.04 | 5.54E-04 |
| ORF66 | Hypothetical protein | 68.8% | 46 | 5.33E+08 | 785 | 88.8 | 5.67 | 5.14E-04 |
| ORF105 | bro | 62.9% | 31 | 6.35E+08 | 501 | 58.2 | 7.97 | 4.85E-04 |
| ORF89 | Hypothetical protein | 62.1% | 17 | 5.23E+08 | 369 | 42.5 | 5.64 | 4.71E-04 |
| ORF69 | hypothetical protein | 63.0% | 7 | 9.84E+08 | 127 | 14.9 | 4.93 | 4.44E-04 |
| ORF76 | vp91 | 52.9% | 38 | 3.96E+08 | 816 | 93.5 | 4.74 | 4.07E-04 |
| ORF11 | odv-e27 | 63.4% | 14 | 3.73E+08 | 284 | 33.3 | 8.35 | 3.92E-04 |
| ORF128 | Hypothetical protein | 48.5% | 11 | 3.11E+08 | 266 | 30.3 | 6.89 | 3.51E-04 |
| ORF14 | ie-1 | 46.3% | 27 | 5.04E+08 | 655 | 75.9 | 9.04 | 3.51E-04 |
| ORF2 | Hypothetical protein | 51.3% | 15 | 3.17E+09 | 413 | 45.9 | 5.68 | 3.45E-04 |
| ORF59 | bro | 65.2% | 13 | 2.15E+09 | 244 | 28.3 | 6.95 | 3.02E-04 |
| ORF60 | bro | 67.7% | 31 | 3.73E+08 | 527 | 59.7 | 7.18 | 2.91E-04 |
| ORF101 | Hypothetical protein | 65.2% | 20 | 2.78E+08 | 253 | 29 | 9.31 | 2.86E-04 |
| ORF133 | Hypothetical protein | 38.0% | 25 | 3.59E+08 | 677 | 78.3 | 6.49 | 2.78E-04 |
| ORF81 | Hypothetical protein | 45.7% | 9 | 3.76E+08 | 162 | 19.1 | 4.6 | 2.62E-04 |
| kDa | ||||||||
| ORF51 | Hypothetical protein | 45.0% | 7 | 4.82E+08 | 160 | 19 | 4.64 | 2.41E-04 |
| ORF126 | egt | 26.2% | 12 | 4.22E+08 | 515 | 58.9 | 7.01 | 2.39E-04 |
| ORF16–17 | me53 | 51.8% | 21 | 2.93E+08 | 359 | 42.7 | 8.72 | 2.21E-04 |
| ORF52 | Hypothetical protein | 45.5% | 3 | 3.17E+08 | 88 | 10.2 | 5.48 | 2.18E-04 |
| ORF114 | alk-exo | 45.3% | 18 | 1.96E+08 | 428 | 49.4 | 8.28 | 1.98E-04 |
| ORF84 | helicase | 39.7% | 43 | 7.94E+08 | 1253 | 145.9 | 7.03 | 1.78E-04 |
| ORF132 | Hypothetical protein | 53.5% | 17 | 6.67E+08 | 383 | 44.5 | 6.15 | 1.64E-04 |
| ORF28 | Ubiquitin | 61.4% | 5 | 1.83E+08 | 83 | 9.2 | 7.25 | 1.37E-04 |
| ORF13 | Hypothetical protein | 46.8% | 7 | 1.83E+08 | 203 | 22.9 | 6.87 | 1.17E-04 |
| ORF67 | DNA pol | 37.0% | 36 | 2.53E+08 | 1020 | 119.2 | 8.29 | 1.12E-04 |
| ORF61 | he65 | 60.2% | 14 | 1.06E+09 | 236 | 27.5 | 8.27 | 1.00E-04 |
| ORF97 | Hypothetical protein | 48.2% | 9 | 9.82E+08 | 276 | 32.4 | 7.96 | 1.00E-04 |
| ORF86 | Hypothetical protein | 49.8% | 14 | 2.10E+08 | 321 | 37.9 | 6.95 | 9.57E-05 |
| ORF64 | Hypothetical protein | 42.1% | 5 | 1.35E+08 | 133 | 15.6 | 5.27 | 7.86E-05 |
| ORF79 | lef-4 | 47.9% | 16 | 2.24E+08 | 461 | 53.9 | 6.3 | 7.57E-05 |
| ORF47 | vp1054 | 34.2% | 11 | 1.94E+08 | 351 | 41.7 | 8.19 | 7.29E-05 |
| ORF38 | lef-8 | 41.2% | 32 | 1.44E+08 | 901 | 104.9 | 8.7 | 7.19E-05 |
| ORF103 | iap-3 | 57.1% | 13 | 2.36E+09 | 268 | 31.5 | 5.99 | 6.68E-05 |
| ORF113 | fgf | 27.6% | 8 | 6.89E+07 | 301 | 34.3 | 8.9 | 6.21E-05 |
| ORF90 | ORF90 | 32.8% | 4 | 3.93E+08 | 122 | 13.8 | 7.43 | 6.18E-05 |
| ORF129 | Hypothetical protein | 29.5% | 21 | 2.10E+10 | 947 | 111.3 | 6.67 | 6.16E-05 |
| ORF111 | Hypothetical protein | 31.1% | 13 | 6.93E+07 | 528 | 60.3 | 6.09 | 5.84E-05 |
| ORF39 | Hypothetical protein | 28.4% | 4 | 2.24E+08 | 194 | 22.5 | 6.34 | 5.54E-05 |
| ORF34 | Hypothetical protein | 51.8% | 14 | 4.12E+08 | 359 | 41.2 | 8.19 | 5.52E-05 |
| ORF35 | p47 | 28.5% | 8 | 6.74E+07 | 333 | 38.9 | 7.49 | 5.46E-05 |
| ORF20 | p74 | 22.5% | 12 | 7.56E+07 | 688 | 78.4 | 5.36 | 4.55E-05 |
| ORF55 | lef-9 | 28.1% | 13 | 3.30E+08 | 519 | 59.5 | 9.11 | 4.35E-05 |
| ORF98 | Hypothetical protein | 22.1% | 3 | 8.05E+07 | 199 | 22.4 | 4.83 | 3.98E-05 |
| ORF32 | lef-11 | 28.3% | 4 | 5.29E+07 | 127 | 14.6 | 6.52 | 3.96E-05 |
| ORF95 | Hypothetical protein | 21.3% | 2 | 5.24E+07 | 94 | 11 | 10.33 | 2.51E-05 |
| ORF74 | Hypothetical protein | 15.4% | 3 | 4.00E+07 | 241 | 27.7 | 8.81 | 2.19E-05 |
| ORF87 | lef-5 | 20.3% | 5 | 1.20E+07 | 315 | 37 | 9.35 | 7.37E-06 |
DEPs after Baculovirus Infection
The Peak Area Calculation Quantification method was employed to determine relative protein abundances within individual samples and across different samples. To determine the DEPs induced by viral infection, protein fold changes between the infected and control groups were calculated and statistically analyzed. For statistical analysis, proteins with a fold change ≥1.5 or <0.67 and a p value <0.05 were considered differentially expressed. Among the identified proteins, 449 were differentially expressed (Fig. 5A), 76 of which were up-regulated and 373 exhibited clear down-regulation. The up-regulated and down-regulated proteins are listed in supplemental Tables S3 and S4, respectively.
To further characterize the functions of these DEPs, functional classification was performed by mapping these genes to the KEGG pathway The KEGG functional classification results (Fig. 5B) indicated that the down-regulated proteins were involved predominantly in metabolism-related physiological process, including genes involved in AAM, CM, lipid metabolism, and energy metabolism. Furthermore, GOEA was performed for the DEPs (supplemental Fig. S7). The down-regulated proteins were enriched predominantly in catalytic and binding activities among the molecular function category (supplemental Fig. S7A) and metabolic and cellular processes among the biological process category (supplemental Fig. S7B). To characterize the change in CM-related enzymes, we selected several representative proteins, such as GPI, aldolase, TPI, enolase, succinyl-CoA synthetase, fumarase, FBP, trehalose-6-phosphate synthase and transaldolase, whose protein abundances were reduced substantially upon baculovirus infection, consistent with the transcriptional patterns (supplemental Fig. S8).
Taken together, the proteomics analysis of the fat body of larvae indicated that protein expression was substantially influenced by baculovirus infection. A large proportion of proteins were differentially expressed significantly, demonstrating that viral infection strongly regulated the physiological activity of host larvae at the translational level. The manipulation of host larvae by baculoviruses serves to provide more energy and resources required for viral replication and proliferation.
Correlation Analysis between the Proteomic and Transcriptomic Data
Combination of transcriptomic with proteomic analyses provides an opportunity to investigate how reliably the transcriptional profile reflects the translational profile. Therefore, we examined the correlations between mRNA and protein expression profiles at 72 hpi. Proteins identified in both control and infected groups were used to identify correlations between the mRNA and protein levels during baculovirus infection. As illustrated in Fig. 6, for the 1,360 proteins common to the control and infected groups, there was a positive correlation between control protein (CP) and mRNA (CM) levels (Fig. 6A-1, CP-CM: 0.68) and between infected protein (IP) and mRNA (IM) levels (Fig. 6A-2, IP-IM: 0.60). Additionally, we plotted mRNA versus protein levels for the DEPs; the results showed that the correlations observed among the DEPs were comparable with those for the full set of proteins (Fig. 6A-3, CP-CM: 0.67; Fig. 6A-4, IP-IM: 0.54).
Fig. 6.

Correlation analysis between the mRNA and protein abundances and their changes in the fat body. The normalized peak area of proteins to the normalized RPKM of transcripts in the control and infected groups was plotted in the log2 scale, with the y and x axis indicating the relative protein and mRNA levels, respectively. A, correlations between the mRNA and protein levels in fat body samples in the control and infected groups. The upper panel denotes correlations between the mRNA and protein levels for all of the proteins identified in the fat body, and the lower panel represents correlations for the DEPs. B, correlations of the changes between the mRNA and protein levels. The upper panel represents all of the proteins identified in the fat body, and the lower panel represents the genes that show a similar trend at both mRNA and protein levels.
Furthermore, we examined whether a stronger correlation exists between the mRNA and protein level changes after baculovirus infection. For all of the proteins identified in the fat body, the correlation coefficient between the mRNA and protein level changes was only 0.55 (Fig. 6B-1), which is almost the same as that (0.57) reported in the previous study (49), whereas the correlation was stronger for the genes showing similar trends at both the mRNA and protein levels after challenge with baculovirus (Fig. 6B-2, 0.71). A previous study (50) categorized the quantified proteins into seven groups; herein, we grouped the identified proteins into four groups based on the following patterns: group I, the mRNA and protein levels showed the same changes; group II, the directions of mRNA and protein changes were opposite; group III, protein levels were basically unchanged, whereas the mRNA levels were induced or repressed; and group IV, mRNAs were basically unchanged, although protein levels were up- or down-regulated. The 1,360 proteins common to the control and infected groups were distributed among the four groups as follows: group I, 904 (66.47%); group II, 13 (0.96%); group III, 271 (19.93%); and group IV, 172 (12.65%) (supplemental Table S5). This indicated that most proteins showed a positive correlation with their mRNA in expression patterns. The entire list of proteins identified in the larval fat body is presented in supplemental Table S5.
Gene Interaction Network Analysis
To better understand the interaction between HearNPV and host insect, H. armigera, we performed a gene interaction network analysis for the gene sets that were simultaneously up- or down-regulated at the mRNA and protein levels. The interaction network is composed of three types of interaction modes: correlated expression, predicted physical interaction, and known physical interaction. The enriched interaction network for up-regulated genes (Fig. 7A) was related mainly to anatomical structure formation involved in morphogenesis (FDR < 0.036). In contrast, the down-regulated gene set that was much bigger in amount formed a large set of metabolic sub-networks. As shown in Fig. 7B, the down-regulated genes were involved in the interaction networks related to CM (FDR <7.3e-6), fatty acid metabolism (FDR <5.1e-7), and purine metabolism (FDR <9.3e-7). As one of the important steps involved in CM, glycolysis (FDR <8.6e-9) was highlighted among the down-regulated networks. Meanwhile, as an important step in energy production, oxidative phosphorylation (FDR <0.009) was down-regulated significantly. Additionally and of great interest, the cellular translational machinery (FDR <0.0002) in the fat body was also suppressed substantially during the late infection stage, suggesting translational arrest upon viral infection. The components related to ribosome biogenesis were mostly decreased at 72 hpi. Furthermore, elongation factor subunits (e.g. Ef2b and Ef1γ), which are important translation processes, were negatively regulated upon viral infection. Taken together, gene interaction network analysis demonstrated that the networks regulated by baculovirus were associated with diverse aspects of host cellular functions, including the enhancement of anatomical structure formation and down-regulation of a large subset of metabolic pathways, as well as shutdown of the cellular translational machinery.
Fig. 7.

Predicted gene interaction networks regulated by baculovirus infection. The gene interaction networks were retrieved from the STRING protein-protein interaction database. The interaction networks were exported to Cytoscape for visualization. Gray nodes indicate their presence in the corresponding gene sets. Three types of interaction modes were included in the interaction network: the red edge indicates correlated expression; the black edge denotes predicted protein-protein interactions; and the purple edge represents known protein-protein interactions. The false discovery rate for each enriched term is indicated in the figure. A, interaction network for the genes up-regulated by baculovirus infection. The interaction network indicated that anatomical structure formation was up-regulated by baculovirus infection. B, interaction network for the genes down-regulated by immune challenge with baculovirus. Among the down-regulated genes, more interaction networks related to translation and metabolic pathways such as CM, fatty acid metabolism, energy metabolism, and purine metabolism were significantly enriched, suggesting substantial modulation of host physiological functions.
DISCUSSION
The cotton bollworm (H. armigera) is an important pest insect in agriculture throughout the world due to the enormous economic losses caused by its damage. Traditional chemical insecticides have contributed to the accumulation of resistance within insects. The cultivation of transgenic Bacillus thuringiensis cotton has effectively managed the H. armigera population, although resistance is still a barrier to its widespread use. HearNPV is an alternative control strategy for harnessing the H. armigera population that is highly efficient and specific. A better understanding of host-baculovirus interactions would facilitate the research and development of novel biocontrol agents. A global view of host gene expression at the transcript and protein levels was obtained by integrated transcriptomic and proteomic analyses. Meanwhile, the expression dynamics of baculoviral genes was elucidated by transcriptomic analysis. Because the H. armigera genome sequence is not yet available, high-throughput sequencing would provide a wealth of information on host gene expression on a genome-wide scale. Furthermore, differential expression analysis indicated that many genes related to various physiological processes, including translation, signal transduction, cell cycle, and energy metabolism, were differentially regulated at the mRNA level following infection. Importantly, a large proportion of immunity-related genes was significantly regulated after infection, and the majority of these genes was drastically suppressed during the late infection stage.
Rationale for Selecting Four Time Points after Infection
After H. armigera larvae were infected per os, four time points were chosen to analyze changes in host and baculoviral gene expression patterns. Why did we choose the four time points 0, 24, 48, and 72 hpi to characterize the interaction between baculovirus and H. armigera larvae? The 0-hpi time point was representative of the starting point of viral infection. As noted in Fig. 1A, the difference in larval weight between the control and infected groups was statistically significant at 24 hpi; therefore, 24-hpi was used to represent the early stage of infection. During the infection process, the variation between the control and infected groups increased drastically, showing a stronger difference at 48 hpi. However, survival analysis indicated that the larvae started to die at 4 days post-infection (Fig. 1B). This was consistent with the finding that most baculoviral genes were highly elevated during the infection period and that the transcriptional patterns were associated with the potential roles of the genes in viral infection and proliferation. Therefore, viral infection was considered to have reached the late stage by 72 hpi. Additionally, the expression of host genes showed dynamic changes during this infection stage. Previous studies (31, 51) adopted a similar scheme in the selection of time points, further confirming the rationale behind our experimental design. Collectively, this time course transcriptomic analysis may accurately reflect global gene expression in both the insect host and baculovirus.
Expression of Baculoviral Genes at the Transcript and Protein Levels
Previous studies reported time course expression profiling of baculoviral genes during the infection process. The expression phases of different viral genes have been evaluated extensively at the cellular level (31). Particularly, the expression patterns of viral genes expressed at specific stages have been established. The HearNPV genome encodes 135 ORFs, among which 37 were determined to be core genes, primarily comprising virion structure- and viral transcription- and replication-related proteins that play central roles during viral infection. Interestingly, hierarchical clustering analysis showed that most of the core genes (22, 59.5%) belonged to group III (Fig. 1C), such as lef-1, lef-2, helicase, and DNA polymerase involved in viral replication and lef-8 and lef-9 involved in viral transcription. Moreover, six and nine core genes belonged to group II (p74, p6.9, odv-e25, ORF66, vlf-1, and odv-e27) and group IV (lef-4, lef-5, vp91, pif-1–4, ORF64, and ORF86) (Fig. 1C), respectively, most of which encode virion structure-related proteins. In addition to the core genes, 20 unique genes sharing no significant sequence identity with other baculoviruses were found in HearNPV. Among them, eight (40%) and seven (35%) were found in cluster IV and cluster I, respectively, suggesting that most of the unique genes were maintained at a lower transcriptional level, possibly implying their non-essential roles in viral infection. One (ORF29) and two (ORF107 and ORF122) unique genes were found in clusters II and III, respectively, the expression levels of which were several orders of magnitude higher than those of the former ones, potentially implying their importance in viral infection. Notably, eight per os infectivity factors are encoded by HearNPV, and most belong to the core genes except pif-7 (HearNPV ORF95, homologue of Sf58), which is conserved in lepdopteran baculoviruses. Among these eight factors, the mRNA expression levels of six (pif-1–4 and pif-6–7) were low, whereas those of p74 and pif-5 were high, implying their distinct roles in viral infection. Additionally, our proteomic analysis identified 87 baculovirus-encoded proteins. Correlation analysis showed that the transcript and protein levels were poorly correlated (0.122) among baculoviral genes, suggesting that the transcriptional and translational processes are not well coordinated.
DEGs after Challenge with Baculovirus
After ingestion of HearNPV ODV, the developmental processes of H. armigera larvae were altered markedly. The normal molting of infected larvae was highly disrupted, resulting in growth retardation. This observation was consistent with findings in silkworm Bombyx mori infected by AcMNPV, influenced by the AcMNPV-encoded ecdysteroid UDP-glucosyltransferase (egt) gene (52). Previous studies have reported that egt inhibits the normal molting and pupation of larvae by inactivating ecdysteroid in baculovirus-infected larvae (53). As an important hormone in the regulation of growth, molting, metamorphosis, reproduction, and immunity, the ecdysone titer could impact multiple aspects of insect physiological functions. Additionally, previous studies have reported that a baculovirus-encoded phosphatase contributed to the enhanced locomotion of the lepidopteran host upon viral infection (2). However, a homologous gene is lacking in the HearNPV genome; an alternative mechanism to achieve the alteration in host behavior is likely employed by HearNPV, which has not been elucidated until now.
The time course expression dynamics of host genes was revealed by comparative transcriptomic analysis, suggesting that more genes were down-regulated upon viral infection than up-regulated. During the infection process, various host genes were differentially regulated, and the majority were repressed at the transcript level. During the early stage of infection (24 hpi), 76 genes were induced in response to viral infection. More genes were significantly inhibited during the late infection stage whereas a proportion of genes was up-regulated, suggesting no global transcriptional repression in our study; this finding was similar to the finding by Jakubowska et al. (54). Recently, digital gene expression analysis was used to compare the whole-body transcriptome between HearNPV-infected and healthy H. armigera larvae (55), identifying many DEGs and showing that more DEGs were down-regulated upon infection, a finding that is highly consistent with ours. However, RNA-seq could yield more comprehensive and detailed information about transcriptomic profiling than can the DGE approach. Furthermore, a single time point experimental design cannot explain the dynamic changes in host-pathogen interactions during the infection process. Regarding functional enrichment analysis of the DEGs, the DAVID analysis results showed that no significant enrichment of GO terms was observed at 24 hpi and that various biological processes such as CM, mitosis, and translation were enriched among the down-regulated genes at 48 and 72 hpi, whereas only the DNA metabolic process was enriched among the up-regulated genes at 48 hpi. Regarding the complicated gene expression patterns during the infection process, multiple factors might contribute to the regulation of gene expression after baculovirus infection, such as hormone levels, the gut microbiota, and other factors. The baculovirus-encoded egt gene inactivates larval 20-hydroxyecdysone (20E), resulting in changes in the expression levels of host genes regulated by 20E. In addition, a previous study reported that the gut microbiota was increased in baculovirus-infected larvae, and in return, the gut microbiota could regulate the expression of host genes. Taken together, the changes caused by baculovirus infection might further enhance the degree of regulation of host gene expression levels.
Innate Immune System versus Baculovirus
After challenge by the foreign stimulus, the immune system of H. armigera is activated. Insect innate immunity is the major defense to protect insects from infectious microorganisms. Humoral and cellular immunity make up the innate immune system, and both share the same immune signaling pathways, such as Toll, Imd, and JAK-STAT pathways. These three signaling pathways have been well investigated in the model insect D. melanogaster (11). Additionally, the genes and pathways related to the immune system in B. mori, M. sexta, A. aegypti, Hepialus xiaojinensis, and Tribolium castaneum have also been evaluated systematically (13, 56–59). Recent studies have suggested that the innate immune response may also play a pivotal role in viral infections (60–63). However, in the interaction between the host and baculovirus, RNA-seq data indicated a lack of significant activation of the immune response during the infection process. During the early stage (24 hpi), there were fewer immunity-related genes induced at the transcript level (Fig. 3A), and their expression levels declined significantly with the infection progress. Moreover, during the late stage of infection, the expression of most of the immune-related DEGs was strongly repressed. Overall, the classical immune signaling pathways Toll, Imd, and JAK-STAT did not show a response against baculovirus infection. Similarly, previous studies reported down-regulation of immune genes in Spodoptera exigua larvae and the larval midgut (54, 64). Despite these significant findings, further functional studies are required to decipher the detailed molecular mechanism.
Relationship between Host Metabolism and Viral Infection
Baculoviruses depend largely on the insect host to provide energy and resources for viral replication and assembly, highlighting the importance of energy metabolism and other metabolic pathways during the infection process. CM is a critical pathway for energy production in insects, which is related to many physiological processes, including development and reproduction. In the mosquito A. aegypti, it was demonstrated that CM was temporally regulated during the reproductive process (65). Interestingly, functional analysis also suggested that the down-regulated genes are enriched mainly in metabolic pathways such as energy metabolism and CM-related pathways. The expression of CM-related enzymes aldolase, enolase, FBP, and PFK was significantly inhibited, as revealed by qRT-PCR analysis. In baculovirus-infected Spodoptera frugiperda Sf9 cells, enzyme activities and cellular factors were analyzed in parallel with changes in metabolites, showing that the metabolic rates were significantly decreased and the enzyme activities of G6PDH, LDH, MDH, AlaAT, AspAT, and GOGAT remarkably decreased upon viral infection (66). Additionally, Tran et al. (67) established a protocol for quantitative intracellular metabolite analysis within baculovirus-infected insect cells, thus facilitating research on insect host metabolism following infection. Collectively, maintenance of host metabolic activity at a lower level might also reflect manipulation of host physiological functions by the baculovirus, implying that host metabolism was modulated to allow viral replication and assembly during the late stage of infection.
Correlation between mRNA and Protein Levels
In this study, consistency between the mRNA and protein levels was explored by correlation analysis, the results of which suggested an acceptable but rather poor association between the transcriptional and translational levels. For the whole proteome, the correlation between the mRNA and protein levels was 0.68 in MF, which was relatively higher than that (0.60) in IF. Likewise, the correlation results for DEPs were comparable with those for the whole proteome (0.67 for MF and 0.54 for IF) (Fig. 6A). A previous work (49) reported the comparison between the fat body mRNA and plasma protein levels in M. sexta larvae after the immune challenge. It was shown that there exist moderately positive correlations (0.41–0.43) between fat body transcript and plasma protein abundances. By contrast, the fat body protein abundances showed a higher correlation (0.60–0.68) to fat body mRNA levels in our study. With regard to the difference in correlation, the plasma proteins came from multiple tissues, including the fat body and hemocytes, although the proteins in the fat body were only derived from itself. This may explain the relatively stronger correlations between the mRNA and protein levels in the fat body. Theoretically, the more mRNA molecules that are transcribed in the cell, the more proteins can be produced. However, several studies have also reported that the mRNA and protein levels were not highly correlated. This result could be due to several reasons as follows: 1) the existence of post-transcriptional modifications (68); 2) the differential half-lives of mRNA and protein molecules (69, 70); 3) different protein stabilities; and 4) other factors contributing to the inconsistency between mRNA and protein levels (71). All of these factors resulted in weak correlation between the mRNA and protein levels.
In summary, the interaction between the host and baculovirus was comprehensively elucidated by the integration of transcriptomic and proteomic analyses in the fat body. Regarding the central role of the fat body in insect immunity and metabolism, we hypothesized that the baculovirus successfully manipulated host larvae mainly by repressing host immune response and decreasing metabolic activity after the establishment of a systemic infection, aimed at benefiting baculovirus self-replication and multiplication. This study advances our understanding of the interplay between the baculovirus and host insect on a genome-wide scale, thus providing insight into the development of novel biological control agents and application of baculovirus in biocontrol fields.
DATA AVAILABILITY
The RNA-seq raw reads in this article have been deposited as project number PRJNA312430 in the Sequence Read Archive of the National Center for Biotechnology Information. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (47) partner repository (http://www.ebi.ac.uk/pride) with the dataset identifier PXD004748.
Supplementary Material
Footnotes
Author contributions: Z.Z. and Z.H. designed the research; L.X., C.Y., M.W., Z.L., B.S., Z.H. and Z.Z. performed the experiment; L.X. and C.Y. analyzed the data; L.X., Z.H. and Z.Z. wrote the paper.
* This work was supported by Strategic Priority Research Program of the CAS Grant XDB11030600, National Key Plan for Scientific Research and Development of China Grant 2016YFD0500300, National Natural Science Foundation of China Grants 31472008 and 31672291, and Open Research Fund Program of State Key Laboratory of Integrated Management of Pest Insects and Rodents Chinese IPM 1515.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- BV
- budded virus
- HearNPV
- H. armigera nucleopolyhedrovirus
- MF
- mock fat body sample
- IF
- infected fat body sample
- DEG
- differentially expressed gene
- qRT-PCR
- quantitative real-time RT-PCR
- FDR
- false discovery rate
- AMP
- antimicrobial peptide
- PRR
- pattern recognition receptors
- CM
- carbohydrate metabolism
- GO
- Gene Ontology
- DEP
- differentially expressed proteins
- NPA
- normalized peak area
- AAM
- amino acid metabolism
- GOEA
- GO Enrichment Analysis
- RPKM
- reads per kilobase of exon model per million
- OB
- occlusion body
- ODV
- occlusion-derived virus
- RNA-seq
- RNA-sequencing
- NGS
- next-generation sequencing
- PCA
- principal component analysis
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- PPP
- pentose-phosphate pathway
- CP
- control protein
- IP
- infected protein
- IM
- infected mRNA
- AcMNPV
- A. californica multiple nucleopolyhedrovirus
- 20E
- 20-hydroxyecdysone.
REFERENCES
- 1. Jehle J. A., Lange M., Wang H., Hu Z., Wang Y., and Hauschild R. (2006) Molecular identification and phylogenetic analysis of baculoviruses from Lepidoptera. Virology 346, 180–193 [DOI] [PubMed] [Google Scholar]
- 2. Kamita S. G., Nagasaka K., Chua J. W., Shimada T., Mita K., Kobayashi M., Maeda S., and Hammock B. D. (2005) A baculovirus-encoded protein tyrosine phosphatase gene induces enhanced locomotory activity in a lepidopteran host. Proc. Natl. Acad. Sci. U.S.A. 102, 2584–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu K. M., Lu Y. H., Feng H. Q., Jiang Y. Y., and Zhao J. Z. (2008) Suppression of cotton bollworm in multiple crops in China in areas with Bt toxin-containing cotton. Science 321, 1676–1678 [DOI] [PubMed] [Google Scholar]
- 4. Szewczyk B., Hoyos-Carvajal L., Paluszek M., Skrzecz I., and Lobo de Souza M. (2006) Baculoviruses–re-emerging biopesticides. Biotechnol. Adv. 24, 143–160 [DOI] [PubMed] [Google Scholar]
- 5. Sun X., Wang H., Sun X., Chen X., Peng C., Pan D., Jehle J. A., van der Werf W., Vlak J. M., and Hu Z. (2004) Biological activity and field efficacy of a genetically modified Helicoverpa armigera single-nucleocapsid nucleopolyhedrovirus expressing an insect-selective toxin from a chimeric promoter. Biol. Control 29, 124–137 [Google Scholar]
- 6. Luo S., Zhang Y., Xu X., Westenberg M., Vlak J. M., Wang H., Hu Z., and Deng F. (2011) Helicoverpa armigera nucleopolyhedrovirus occlusion-derived virus-associated protein, HA100, affects oral infectivity in vivo but not virus replication in vitro. J. Gen. Virol. 92, 1324–1331 [DOI] [PubMed] [Google Scholar]
- 7. Wang M., Tan Y., Yin F., Deng F., Vlak J. M., Hu Z., and Wang H. (2008) The F protein of Helicoverpa armigera single nucleopolyhedrovirus can be substituted functionally with its homologue from Spodoptera exigua multiple nucleopolyhedrovirus. J. Gen. Virol. 89, 791–798 [DOI] [PubMed] [Google Scholar]
- 8. Fang M., Nie Y., Wang Q., Deng F., Wang R., Wang H., Wang H., Vlak J. M., Chen X., and Hu Z. (2006) Open reading frame 132 of Helicoverpa armigera nucleopolyhedrovirus encodes a functional per os infectivity factor (PIF-2). J. Gen. Virol. 87, 2563–2569 [DOI] [PubMed] [Google Scholar]
- 9. Song J., Wang R., Deng F., Wang H., and Hu Z. (2008) Functional studies of per os infectivity factors of Helicoverpa armigera single nucleocapsid nucleopolyhedrovirus. J. Gen. Virol. 89, 2331–2338 [DOI] [PubMed] [Google Scholar]
- 10. Hou D., Zhang L., Deng F., Fang W., Wang R., Liu X., Guo L., Rayner S., Chen X., Wang H., and Hu Z. (2013) Comparative proteomics reveal fundamental structural and functional differences between the two progeny phenotypes of a baculovirus. J. Virol. 87, 829–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lemaitre B., and Hoffmann J. (2007) The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 25, 697–743 [DOI] [PubMed] [Google Scholar]
- 12. Jiang H., Vilcinskas A., and Kanost M. R. (2010) Immunity in lepidopteran insects. Adv. Exp. Med. Biol. 708, 181–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gunaratna R. T., and Jiang H. (2013) A comprehensive analysis of the Manduca sexta immunotranscriptome. Dev. Comp. Immunol. 39, 388–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xiong G. H., Xing L. S., Lin Z., Saha T. T., Wang C., Jiang H., and Zou Z. (2015) High throughput profiling of the cotton bollworm Helicoverpa armigera immunotranscriptome during the fungal and bacterial infections. BMC Genomics 16, 321–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu A., Zhang Q., Zhang J., Yang B., Wu K., Xie W., Luan Y. X., and Ling E. (2014) Insect prophenoloxidase: the view beyond immunity. Front. Physiol. 5, 252–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang J., Lu A., Kong L., Zhang Q., and Ling E. (2014) Functional analysis of insect molting fluid proteins on the protection and regulation of ecdysis. J. Biol. Chem. 289, 35891–35906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zou Z., Shin S. W., Alvarez K. S., Kokoza V., and Raikhel A. S. (2010) Distinct melanization pathways in the mosquito Aedes aegypti. Immunity 32, 41–53 [DOI] [PubMed] [Google Scholar]
- 18. Shin S. W., Zou Z., and Raikhel A. S. (2011) A new factor in the Aedes aegypti immune response: CLSP2 modulates melanization. EMBO Rep. 12, 938–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Y. H., Hu Y., Xing L. S., Jiang H., Hu S. N., Raikhel A. S., and Zou Z. (2015) A critical role for CLSP2 in the modulation of antifungal immune response in mosquitoes. PLoS Pathog. 11, e1004931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sabin L. R., Hanna S. L., and Cherry S. (2010) Innate antiviral immunity in Drosophila. Curr. Opin. Immunol. 22, 4–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lamiable O., and Imler J. L. (2014) Induced antiviral innate immunity in Drosophila. Curr. Opin. Microbiol. 20, 62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kawai T., and Akira S. (2006) Innate immune recognition of viral infection. Nat. Immunol. 7, 131–137 [DOI] [PubMed] [Google Scholar]
- 23. Richetta C., and Faure M. (2013) Autophagy in antiviral innate immunity. Cell. Microbiol. 15, 368–376 [DOI] [PubMed] [Google Scholar]
- 24. Kingsolver M. B., Huang Z., and Hardy R. W. (2013) Insect antiviral innate immunity: pathways, effectors, and connections. J. Mol. Biol. 425, 4921–4936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Trudeau D., Washburn J. O., and Volkman L. E. (2001) Central role of hemocytes in Autographa californica M nucleopolyhedrovirus pathogenesis in Heliothis virescens and Helicoverpa zea. J. Virol. 75, 996–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jayachandran B., Hussain M., and Asgari S. (2012) RNA interference as a cellular defense mechanism against the DNA virus baculovirus. J. Virol. 86, 13729–13734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moreno-Habel D. A., Biglang-awa I. M., Dulce A., Luu D. D., Garcia P., Weers P. M., and Haas-Stapleton E. J. (2012) Inactivation of the budded virus of Autographa californica M nucleopolyhedrovirus by gloverin. J. Invertebr. Pathol. 110, 92–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nguyen Q., Nielsen L. K., and Reid S. (2013) Genome scale transcriptomics of baculovirus-insect interactions. Viruses 5, 2721–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Q., Liu Y., He H. J., Zhao X. F., and Wang J. X. (2010) Immune responses of Helicoverpa armigera to different kinds of pathogens. BMC Immunol. 11, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang S. S., Chang I. S., Huang L. W., Chen P. C., Wen C. C., Liu S. C., Chien L. C., Lin C. Y., Hsiung C. A., and Juang J. L. (2006) Temporal transcription program of recombinant Autographa californica multiple nucleopolyhedrosis virus. J. Virol. 80, 8989–8999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xue J., Qiao N., Zhang W., Cheng R. L., Zhang X. Q., Bao Y. Y., Xu Y. P., Gu L. Z., Han J. D., and Zhang C. X. (2012) Dynamic interactions between Bombyx mori nucleopolyhedrovirus and its host cells revealed by transcriptome analysis. J. Virol. 86, 7345–7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Old W. M., Meyer-Arendt K., Aveline-Wolf L., Pierce K. G., Mendoza A., Sevinsky J. R., Resing K. A., and Ahn N. G. (2005) Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 4, 1487–1502 [DOI] [PubMed] [Google Scholar]
- 33. Aebersold R., and Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 34. Chen X., IJkel W. F., Tarchini R., Sun X., Sandbrink H., Wang H., Peters S., Zuidema D., Lankhorst R. K., Vlak J. M., and Hu Z. (2001) The sequence of the Helicoverpa armigera single nucleocapsid nucleopolyhedrovirus genome. J. Gen. Virol. 82, 241–257 [DOI] [PubMed] [Google Scholar]
- 35. Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q., Chen Z., Mauceli E., Hacohen N., Gnirke A., Rhind N., et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang X., and Madan A. (1999) CAP3: A DNA sequence assembly program. Genome Res. 9, 868–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang Y., Niu B., Gao Y., Fu L., and Li W. (2010) CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Conesa A., Götz S., García-Gómez J. M., Terol J., Talón M., and Robles M. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 [DOI] [PubMed] [Google Scholar]
- 39. Li B., and Dewey C. N. (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang L., Feng Z., Wang X., Wang X., and Zhang X. (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 [DOI] [PubMed] [Google Scholar]
- 41. Huang da W., Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 42. Xie C., Mao X., Huang J., Ding Y., Wu J., Dong S., Kong L., Gao G., Li C. Y., and Wei L. (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 44. Remm M., Storm C. E., and Sonnhammer E. L. (2001) Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 314, 1041–1052 [DOI] [PubMed] [Google Scholar]
- 45. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., and von Mering C. (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vizcaíno J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Altschul S. F., Gish W., Miller W., Myers E. W., and Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 49. He Y., Cao X., Zhang S., Rogers J., Hartson S., and Jiang H. (2016) Changes in the plasma proteome of Manduca sexta larvae in relation to the transcriptome variations after an immune challenge: evidence for high molecular weight immune complex formation. Mol. Cell. Proteomics 15, 1176–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang J., Mei H., Zheng C., Qian H., Cui C., Fu Y., Su J., Liu Z., Yu Z., and He J. (2013) The metabolic regulation of sporulation and parasporal crystal formation in Bacillus thuringiensis revealed by transcriptomics and proteomics. Mol. Cell. Proteomics 12, 1363–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nguyen Q., Chan L. C., Nielsen L. K., and Reid S. (2013) Genome scale analysis of differential mRNA expression of Helicoverpa zea insect cells infected with a H. armigera baculovirus. Virology 444, 158–170 [DOI] [PubMed] [Google Scholar]
- 52. Shikata M., Shibata H., Sakurai M., Sano Y., Hashimoto Y., and Matsumoto T. (1998) The ecdysteroid UDP-glucosyltransferase gene of Autographa californica nucleopolyhedrovirus alters the moulting and metamorphosis of a non-target insect, the silkworm, Bombyx mori (Lepidoptera, Bombycidae). J. Gen. Virol. 79, 1547–1551 [DOI] [PubMed] [Google Scholar]
- 53. O'Reilly D. R., and Miller L. K. (1989) A baculovirus blocks insect molting by producing ecdysteroid UDP-Glycosyl transferase. Science 245, 1110–1112 [DOI] [PubMed] [Google Scholar]
- 54. Jakubowska A. K., Vogel H., and Herrero S. (2013) Increase in gut microbiota after immune suppression in baculovirus-infected larvae. PLoS Pathog. 9, e1003379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li Z., Lu Z., Wang X., Zhang S., Zhang Q., and Liu X. (2015) Digital gene expression analysis of Helicoverpa armigera in the early stage of infection with Helicoverpa armigera nucleopolyhedrovirus. J. Invertebr. Pathol. 132, 66–76 [DOI] [PubMed] [Google Scholar]
- 56. Tanaka H., Ishibashi J., Fujita K., Nakajima Y., Sagisaka A., Tomimoto K., Suzuki N., Yoshiyama M., Kaneko Y., Iwasaki T., Sunagawa T., Yamaji K., Asaoka A., Mita K., and Yamakawa M. (2008) A genome-wide analysis of genes and gene families involved in innate immunity of Bombyx mori. Insect Biochem. Mol. Biol. 38, 1087–1110 [DOI] [PubMed] [Google Scholar]
- 57. Waterhouse R. M., Kriventseva E. V., Meister S., Xi Z., Alvarez K. S., Bartholomay L. C., Barillas-Mury C., Bian G., Blandin S., Christensen B. M., Dong Y., Jiang H., Kanost M. R., Koutsos A. C., Levashina E. A., et al. (2007) Evolutionary dynamics of immune-related genes and pathways in disease-vector mosquitoes. Science 316, 1738–1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zou Z., Evans J. D., Lu Z., Zhao P., Williams M., Sumathipala N., Hetru C., Hultmark D., and Jiang H. (2007) Comparative genomic analysis of the Tribolium immune system. Genome Biol. 8, R177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Meng Q., Yu H. Y., Zhang H., Zhu W., Wang M. L., Zhang J. H., Zhou G. L., Li X., Qin Q. L., Hu S. N., and Zou Z. (2015) Transcriptomic insight into the immune defenses in the ghost moth, Hepialus xiaojinensis, during an Ophiocordyceps sinensis fungal infection. Insect Biochem. Mol. Biol. 64, 1–15 [DOI] [PubMed] [Google Scholar]
- 60. Dostert C., Jouanguy E., Irving P., Troxler L., Galiana-Arnoux D., Hetru C., Hoffmann J. A., and Imler J. L. (2005) The Jak-STAT signaling pathway is required but not sufficient for the antiviral response of drosophila. Nat. Immunol. 6, 946–953 [DOI] [PubMed] [Google Scholar]
- 61. Sánchez-Vargas I., Scott J. C., Poole-Smith B. K., Franz A. W., Barbosa-Solomieu V., Wilusz J., Olson K. E., and Blair C. D. (2009) Dengue virus type 2 infections of Aedes aegypti are modulated by the mosquito's RNA interference pathway. PLoS Pathog. 5, e1000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xi Z., Ramirez J. L., and Dimopoulos G. (2008) The Aedes aegypti toll pathway controls Dengue virus infection. PLoS Pathog. 4, e1000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kingsolver M. B., and Hardy R. W. (2012) Making connections in insect innate immunity. Proc. Natl. Acad. Sci. U.S.A. 109, 18639–18640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Choi J. Y., Roh J. Y., Wang Y., Zhen Z., Tao X. Y., Lee J. H., Liu Q., Kim J. S., Shin S. W., and Je Y. H. (2012) Analysis of genes expression of Spodoptera exigua larvae upon AcMNPV infection. PLoS ONE 7, e42462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hou Y., Wang X. L., Saha T. T., Roy S., Zhao B., Raikhel A. S., and Zou Z. (2015) Temporal coordination of carbohydrate metabolism during mosquito reproduction. PLoS Genet. 11, e1005309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bernal V., Monteiro F., Carinhas N., Ambrósio R., and Alves P. M. (2010) An integrated analysis of enzyme activities, cofactor pools and metabolic fluxes in baculovirus-infected Spodoptera frugiperda Sf9 cells. J. Biotechnol. 150, 332–342 [DOI] [PubMed] [Google Scholar]
- 67. Tran T. T., Dietmair S., Chan L. C., Huynh H. T., Nielsen L. K., and Reid S. (2012) Development of quenching and washing protocols for quantitative intracellular metabolite analysis of uninfected and baculovirus-infected insect cells. Methods 56, 396–407 [DOI] [PubMed] [Google Scholar]
- 68. Ben-Tabou de-Leon S., and Davidson E. H. (2009) Modeling the dynamics of transcriptional gene regulatory networks for animal development. Dev. Biol. 325, 317–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., and Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 70. Yen H. C., Xu Q., Chou D. M., Zhao Z., and Elledge S. J. (2008) Global protein stability profiling in mammalian cells. Science 322, 918–923 [DOI] [PubMed] [Google Scholar]
- 71. Maier T., Güell M., and Serrano L. (2009) Correlation of mRNA and protein in complex biological samples. FEBS Lett. 583, 3966–3973 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq raw reads in this article have been deposited as project number PRJNA312430 in the Sequence Read Archive of the National Center for Biotechnology Information. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (47) partner repository (http://www.ebi.ac.uk/pride) with the dataset identifier PXD004748.