

Abstract
Prostate cancer is a leading cause of cancer-related deaths of men in the United States. Whereas the localized disease is highly treatable by surgical resection and radiation, cancer that has metastasized remains incurable. Immune cells that primarily scavenge debris and promote prostate cancer angiogenesis and wound repair are M2 macrophages. They are phenotypically similar to M2 tumor-associated macrophages (M2-TAMs) and have been reported to associate with solid tumors and aide in proliferation, metastasis, and resistance to therapy. As an invasive species within the tumor microenvironment, this makes M2-TAMs an ideal therapeutic target in prostate cancer. To identify novel surface glycoproteins expressed on M2 macrophages, we developed a novel method of creating homogeneous populations of human macrophages from human CD14+ monocytes in vitro. These homogeneous M1 macrophages secrete pro-inflammatory cytokines, and our M2 macrophages secrete anti-inflammatory cytokines as well as vascular endothelial growth factor (VEGF). To identify enriched surface glycoproteins, we then performed solid-phase extraction of N-linked glycopeptides followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) on our homogeneous macrophage populations. We discovered five novel peptides that are enriched exclusively on human M2 macrophages relative to human M1 macrophages and human CD14+ monocytes. Finally, we determined whether these surface glycoproteins, found enriched on M2 macrophages, were also expressed in human metastatic castrate-resistant prostate cancer (mCRPC) tissues. Using mCRPC tissues from rapid autopsies, we were able to determine M2 macrophage infiltration by using immunohistochemistry and flow cytometry. These findings highlight the presence of macrophage infiltration in human mCRPC but also surface glycoproteins that could be used for prognosis of localized disease and for targeting strategies.
Prostate cancer is deadly once it metastasizes, and it continues to be an unmet medical need here in the United States (1). Additionally, increasing evidence demonstrates the tumor microenvironment that surrounds cancer cells significantly contributes to cancer progression, circumvention of current therapies (including immune checkpoint therapies), and survival. Metastatic castration-resistant prostate cancer (mCRPC) 1 tumors are often associated with the reactive stroma that is enriched in immune cells known as M2 macrophages that reside in the tumor microenvironment. M2 macrophages that are seen in cancers are known as M2-TAMs due to their phenotypic similarities (2–7). Previous studies from our laboratory partly elucidated how M2-TAMs and reactive stroma associate with primary and metastatic disease and also help promote prostate cancer cell epithelial-to-mesenchymal transition in vitro (7). We believe that M2-TAMs contribute to cancer cell growth by promoting a permissive growth environment through the secretion of cytokines, matrix-degrading enzymes, angiogenic factors, and multiple growth factors (8–14). Growth suppression of M2-TAMs has been shown to also lead to prostate tumor regression in pre-clinical models (15, 16). However, macrophages have two populations (M1 and M2 macrophages) that bear opposing roles of pro- and anti-inflammation, respectively; however, they display surface marker overlap making it difficult to develop targeting strategies solely for surface proteins that are enriched on M2 macrophages.
Glycosylation is one of the most common post-translational modifications that regulates a host of cellular and biological activities (17–19). This is evidenced by glycosylated membrane surface receptor's ability to perform a slew of functional roles (20). Glycoproteomics is a novel sub-proteomic approach that elucidates levels of expression of glycoproteins that are present in a given sample. This technique has been fundamental in the discovery of cancer biomarkers and the diagnosis of human diseases (21–24). Additionally, this approach is promising because glycoproteins are ubiquitous in extracellular secretion, are typically dysregulated in cancer cells, and are likely to enter the bloodstream. Therefore, this makes glycoproteins attractive candidates for cancer biomarkers.
To study the N-linked glycoproteins in a high throughput and reproducible manner, we previously developed a method for the hydrazide chemistry-based solid-phase capture of glycopeptides and coupled it with the use of PNGase F for the specific release of “formerly” N-glycosylated peptides (only the peptides originally attached to glycans) (25, 26). The isolated formerly N-glycosylated peptides are analyzed by liquid chromatography (LC) separation followed by tandem mass spectrometry (MS/MS), which allows the high throughput identification and quantification of a large number of glycoproteins (25, 26). This feature of the technique provides a unique opportunity to detect protein changes in immune cells obtained from peripheral blood. We also recently developed a novel in vitro strategy that yields homogeneous macrophage populations (27). Herein, we used glycoproteomics on human CD14+ monocytes and homogeneous macrophage populations to identify novel surface glycoproteins that may serve as therapeutic targets.
EXPERIMENTAL PROCEDURES
Cell Preparations
Leukopaks from healthy de-identified donors were obtained from the Hemapheresis and Transfusion Center, The Johns Hopkins Hospital, according to established protocols at The Johns Hopkins Hospital (Baltimore, MD). Peripheral blood mononuclear cells (PBMCs) were prepared from buffy coats by Ficoll-Paque (1.077 g/ml density) (GE Healthcare, Little Chalfont, UK; catalogue number 17-1440-02) density gradient centrifugation at 400 × g at 18 °C for 35 min to separate blood constituents. Cells were washed several times using 1× PBS and counted using a Nexcelom Cellometer Auto T4 plus cell counter (Nexcelom Bioscience, Lawrence, MA). Once counted, CD14+ monocytes were isolated from PBMCs by magnetic labeling using mAb CD14-conjugated microbeads (Stem Cell Technologies Cambridge, MA) followed by cells using immunomagnetic cell separation without columns (Stem Cell Technologies catalogue number 18000) according to the manufacturer's instructions.
Cell Culture
Human macrophage phased polarization was performed as described previously (27). Briefly, macrophages differentiated from CD14+ monocytes were seeded at a cell density of 2.0 × 105 to 3.0 × 105 cells/ml in RPMI 1640 medium (Life Technologies, Inc., Invitrogen, Carlsbad, CA) that was supplemented with 2 mm/liter l-glutamine, 10% heat-inactivated FBS (Sigma-Aldrich), 100 units/ml penicillin (Life Technologies, Inc.), and 100 mg/ml streptomycin (Life Technologies, Inc.) at 37 °C in a humidified 5% CO2, 95% atmospheric air. M1 macrophages were cultivated in the presence of 20 ng/ml granulocyte-macrophage colony stimulation factor (GM-CSF) (PeproTech, Rocky Hill, NJ) for 5 days. Once day 5 was reached, media were replaced, and GM-CSF was replenished (20 ng/ml) in concert with interferon-γ (IFN-γ) (20 ng/ml), interleukin 6 (IL-6) (20 ng/ml), and LPS (20 ng/ml) for 4 additional days. Analogous to M1 macrophage groups, M2 macrophages were cultured for 5 days with M-CSF only. On day 5, media were replaced, and cytokines interleukin 4 (IL-4), interleukin 6 (IL-6), and interleukin 13 (IL-13) were added at a concentration of 20 ng/ml on day 5 with M-CSF for an additional 4 days.
Chemicals
Sodium periodate and hydrazide beads were purchased from Bio-Rad; bicinchoninic acid (BCA) protein assay kit was purchased from Pierce; sequencing-grade trypsin was from Promega (Madison, WI); Sep-Pak C18 1cc Vac Cartridge was from Waters; PNGase F was from New England Biolabs (Ipswich, MA). Other chemicals such as urea, ammonia bicarbonate, acetonitrile (ACN), trifluoroacetic acid (TFA), tris(2-carboxyethyl)phosphine, iodoacetamide, 5 m NaCl, and sodium sulfite were purchased from Sigma-Aldrich.
Protein and Peptide Extraction from Macrophage Cell Types for Proteomic Analysis
Cell pellets from three 10-cm dishes were collected and washed twice with 1× PBS. Thereafter, cells were denatured in 1 ml of 8 m urea and 0.4 m NH4HCO3 and sonicated thoroughly. The protein concentration was measured using a BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL). The proteins were then reduced by incubating in 10 mm tris(2-carboxyethyl)phosphine for 1 h and alkylated in 10 mm iodoacetamide at room temperature for 30 min in the dark. The sample was diluted, and sequence grade trypsin (Sigma-Aldrich) was added at a 1:40 enzyme/protein ratio and incubated overnight at 37 °C. The completion of protein digestion was assessed by SDS-PAGE and silver staining. The peptides were desalted by a C18 cartridge according to the manufacturer's instructions.
Isolation of N-Linked Glycosite-containing Peptides
The solid-phase extraction of glycopeptides (SPEG) method has been described previously (25, 26). Briefly, the glycopeptides were oxidized by incubating with 10 mm sodium periodate in 60% ACN (v/v), 0.1% TFA (v/v) at room temperature for 1 h in the dark. Sodium sulfite was added at a final concentration of 60 mm to quench the oxidation reaction. The samples were then mixed with 50 μl of (50% slurry) hydrazide support prewashed with 1 ml of deionized water. The mixture was incubated with gentle shaking at room temperature overnight for the coupling reaction. The glycopeptide-conjugated hydrazide beads were sequentially washed with 800 μl of 50% ACN (v/v), 0.1% TFA (v/v), then 1.5 m NaCl and water, three times/solution to remove non-coupled peptides. Finally, the beads were washed with 200 μl of 1× G7 buffer twice and incubated with 3 μl of PNGase F in 50 μl of 1× G7 buffer at 37 °C overnight. To specifically release N-linked glycosite-containing peptides, which were desalted by a C18 cartridge, they were dried in a speed-vac and resuspended in 40 μl of 0.1% formic acid prior to mass spectrometric MS analysis.
LC-MS/MS Analysis
The peptides (∼1 μg) were separated through a Dionex Ultimate 3000 RSLC nanosystem (Thermo Fisher Scientific) with a 75-μm × 15-cm Acclaim PepMap100 separating column (Thermo Fisher Scientific) protected by a 2-cm guarding column (Thermo Fisher Scientific). The mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid 95% acetonitrile (B). The flow rate was 300 nl/min. The gradient profile was set as follows. 4–6% B for 9 min, 6–35% B for 83 min, 35–90% B for 5 min, and 90% B for 10 min and equilibrated in 4% B for 12 min. MS analysis was performed using a Q Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific). The spray voltage was set at 2.2 kV. Orbitrap spectra (AGC 1 × 106) were collected from 350 to 1800 m/z at a resolution of 70K followed by data-dependent HCD MS/MS (at a resolution of 17.5K, NCE 28%, AGC target 1 × 105, activation time 100 ms) of the 15 most abundant ions using an isolation width of 2.0 Da. Charge state screening was enabled to reject unassigned, singly, seven, and more than seven charged ions. A dynamic exclusion time of 15 s was used to discriminate against previously selected ions.
Protein Identification and Quantification
Proteins were identified using SEQUEST in Proteome Discoverer software (Thermo Fisher Scientific, version 1.4). The NCBI human database (release date 03/2015) containing 53,918 protein entries was used (28). The precursor mass tolerance was set at 20 ppm, and the MS/MS tolerance was set at 0.06 Da. Parameters of the search were modified as follows: oxidized methionines (add Met with 15,995 Da) and a PNGase F-catalyzed conversion of Asn to Asp (add Asn with 0.984 Da) and Cys modification (add cysteine with 57 Da). A maximum of two missed tryptic cleavage sites were allowed. Percolator node in SEQUEST in Proteome Discoverer was used to filter protein identification at 1% false-discovery rate (FDR). Only peptides with at least two spectral counts were reported and used for quantification. The label-free semi-quantitative differential expression analysis of the identified protein was conducted using SIEVE software (Thermo Fisher Scientific, version 2.0). The raw files were imported into SIEVE and the chromatograms were aligned. Frame parameters were set as follows: frame for all MS2 peaks, retention time from 0 to 120 min, m/z from 350 to 1800, frame time retention time width 2.5 min, and frame m/z width 10 ppm. After framing, the Proteome Discoverer result files were imported with a 1% FDR. The integrated intensity (peak area) of glycosite-containing peptides was calculated and normalized by the total ion chromatogram (TIC) of each sample and used to calculate a p value.
Immunoblotting
Total whole-cell lysates were prepared for immunoblotting as described previously (29, 30). Briefly, cells were lysed on ice with 1× MAPK lysis buffer (50 mm Tris, pH 7.5, 0.5 mm EDTA, 50 mm NaF, 100 mm NaCl, 50 mm β-glycerophosphate, 5 mm sodium pyrophosphate, 1% Triton X-100, 1 mm Na3VO4, 1 mm PMSF, 5 μg/ml leupeptin, 5 μg/ml pepstatin, 10 μg/ml aprotinin, 1 mm benzamide). For immunoblotting, 50 μg of total cell lysates in 2× SDS sample buffer were boiled for 10 min, run on SDS-polyacrylamide gels following standard SDS-PAGE protocols, and then transferred to PVDF membranes using the TransBlot Turbo (Bio-Rad, Hercules, CA). Membranes were blocked in 5% BSA in TBST for 2 h at room temperature and then were probed with primary antibody for 2 h at room temperature. After incubation with horseradish peroxide-conjugated secondary antibodies (Bio-Rad) in 5% BSA in TBST for 1 h at room temperature, a signal was detected by chemiluminescence with a CCD camera in a Chemi-Doc Imaging System using Quantity One software version 4.5.2 (Bio-Rad). Primary antibodies were as follows: anti-CD206 monoclonal antibody (LifeSpan, Biosciences, Inc., Seattle, WA); anti-legumain polyclonal antibody (Abcam, Inc., Cambridge, MA); anti-cathepsin L monoclonal antibody (Abcam, Inc.); anti-integrin α3 (Abcam, Inc.); anti-sodium hydrogen exchanger (LifeSpan, Biosciences, Inc.).
Multiplex Cytokine Array Analysis
Human CD14+ monocytes were obtained from whole blood as described above. Following isolation, the cells were plated into 6-well tissue culture plates (Falcon) at a density of 2.0 × 105 cells/ml in RPMI 1640 medium plus 10% heat-inactivated FBS (Sigma-Aldrich), 100 units/ml penicillin (Life Technologies, Inc.), and 100 mg/ml streptomycin (Life Technologies, Inc.). Cells were then treated with cytokines for polarization toward M1 and M2 macrophage phenotypes. Cell culture supernatants were isolated after 10 days and then centrifuged at 1000 rpm for 3 min followed by aspiration. A Bioplex 200 platform (Bio-Rad) was then used to efficiently determine the concentration in picograms/ml of multiple target proteins in cellular supernatants. Luminex bead-based immunoassays were performed following the manufacturer's protocols using the supplied cytokine standards, and the concentrations were determined using a 5-parameter log curve fits. IL-10, IFN-γ, IL-6, IP-10, and VEGF were multiplexed using HCTOMAG-60K (Millipore, Billerica, MA). Experiments were performed in triplicate.
Procurement of Metastatic Castrate-resistant Prostate Cancer Tissues
The rapid autopsy program was approved by the Institutional Review Board (IRB number NA_00036610) at The Johns Hopkins University School of Medicine. The patients whose disease had progressed despite conventional therapies included listing as posthumous tissue donors. The objectives and procedures for tissue donation were explained to the patient. Having agreed to participate in this Institutional Review Board-approved tumor donor program, permission for autopsy was obtained before death, with consent provided by the patient or by next of kin. After death, consent was obtained by monitored telephone call to the next of kin. Four human mCRPC samples were obtained from different patients. Tissues were isolated 0–24 h post mortem from men with mCRPC.
Immunohistochemistry
Sections of excised mCRPC tissues, acquired from rapid autopsy, were formalin-fixed in 10% neutral buffered formalin (Sigma-Aldrich) for 48 h and then paraffin-embedded (FFPE). Serial sections from each of the tissues were cut (5 mm) onto Colormark Plus microscope slides (Cardinal Health, Waukegan, IL) by The Johns Hopkins Histology Core Laboratory. Slides were then de-paraffinized using xylene (Fisher) twice for 10 min, rehydrated through graded alcohol, and then subjected to antigen retrieval using 1× EDTA, pH 8.0 (Thermo Fisher Scientific), for 45 min. Slides were cooled, then washed three times in 1× phosphate-buffered saline/Tween (PBST), and blocked for 5 min in Dual Endogenous Enzyme block (DAKO, Carpentaria, CA) at room temperature. Slides were then incubated with primary antibody CD206 (1:400, LifeSpan Biosciences, Inc.) at room temperature for 45 min followed by three washes in PBST. Slides were then incubated with PowerVision+ poly-HRP (Leica Biosystems, Buffalo Grove, IL) for 1 h at room temperature and then washed three times in TBST. 3,3′-Diaminobenzidine tablets (Sigma-Aldrich) were dissolved in distilled H2O and filtered, and tissues were incubated for 20 min. Thereafter, slides were washed three times with 1× Tris-buffered saline/Tween (1× TBST) and stained with Mayer's hematoxylin stain (Dako, Carpentaria, CA). Images were taken using the Zeiss AxioImager microscope (Zeiss, Thornwood, NY).
Flow Cytometry
Four tissue samples obtained from rapid autopsy were subjected to single-cell homogenization and then added to collected media/adherent cells to be washed. Suspended cells were centrifuged and washed (1× PBS, 0.5% BSA, 2 mm EDTA) twice, counted, and then incubated with fluorophore-conjugated primary antibodies against CD206(FITC), CD86(PE), CD163(PE-Cy7), in the dark for 45 min at 4 °C. Fluorescence was detected by a S3TM Cell Sorter (Bio-Rad).
Experimental Design and Statistical Rationale
The mass spectrometry experimental design was composed of two biological replicates using freshly isolated human CD14+ monocytes and human macrophages. SEQUEST version 1.4 was used to identify unique glycosite-containing peptides from 114 proteins with 1% false discovery rate (FDR). Raw files imported into SIEVE 2.0 were used to quantify all the peptides identified on CD14+ monocytes, M1 and M2 macrophages. Only peptides with at least two spectral counts were reported and used for quantification. The label-free, semi-quantitative differential expression analysis of the identified protein was conducted using SIEVE 2.0 software. The chromatograms were also aligned using SIEVE 2.0. Frame parameters were set as follows: frame for all MS2 peaks, retention time from 0 to 120 min, m/z from 350 to 1800; frame time retention time width 2.5 min and frame m/z width 10 ppm. After framing, the Proteome Discoverer result files were imported with a 1% FDR. The integrated intensity (peak area) of glycosite-containing peptides was calculated and normalized by the TIC of each sample and used to calculate a p value. Furthermore, all data from mass spectrometry was uploaded into PRIDE (URL https://www.ebi.ac.uk/pride/archive/project accession number PXD005080). Three technical replicates were performed using multiplex cytokine analysis using media after 9 days of differentiation and polarization of macrophages. For the rapid autopsy studies, a total of four mCRPC tissues isolated post mortem were used in this study to assess macrophage infiltration.
RESULTS
Differential Expression of Glycoproteins on M1 and M2 Macrophages
Our previous report detailed our ability to produce homogeneous human macrophage populations using a phased polarization strategy (27). With the ability to do this in vitro, we wanted to identify novel surface glycoproteins that were exclusively enriched on each macrophage population. To determine the differentially expressed glycoproteins that are expressed on each cell type, cells were subjected to glycoproteomic analysis using SPEG and LC-MS/MS (25, 26). After LC-MS/MS analysis, a database search using SEQUEST identified 176 unique glycosite-containing peptides from 114 proteins with 1% FDR (Fig. 1, A and B). SIEVE software was able to quantify all the peptides identified, and we found that the glycosylation phenotype was significantly different among PBMCs and M1 and M2 cells (Fig. 1). Several proteins that were enriched on M2 macrophages include macrophage mannose receptor 1 (CD206), cathepsin L1, integrin α3, legumain, and sodium/hydrogen exchanger 7 (Fig. 2A). To validate change of the proteins, the cells were again generated and subjected to the same SPEG-based glycoproteomics analysis. In this additional glycoproteomic analysis, cathepsin L1, integrin α3, macrophage mannose receptor (CD206), sodium hydrogen exchanger 7, and legumain were confirmed to have differential expression (Fig. 2 and supplemental Figs. 1–4). Therefore, from these data we were able to discover surface glycoproteins that were enriched on M2 macrophages that were not present on M1 macrophages or their precursor cell, CD14+ monocytes.
Fig. 1.

Differential expression of glycoproteins among the cell types. Volcano plot of glycoproteins among the cell types showed that many glycoproteins were significantly and differentially expressed (A). Unsupervised hierarchy clustering using fold change (log2) showed that the CD14+ monocytes, M1 macrophages (M1), and M2 macrophages (M2) cell types had different profiles of glycoprotein expression with dissimilar patterns (B).
Fig. 2.
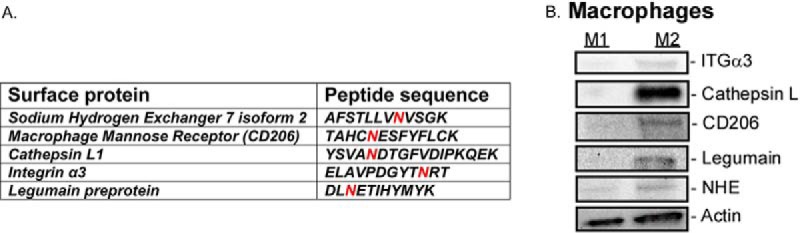
Surface glycoproteins found enriched on human M2 macrophages. SPEG analysis followed by LC-MS/MS identified five novel glycopeptides that are enriched on the surface of M2 macrophages (A). The red N (Asn) represents the site of N-linked glycosylation (A). Expression of enriched glycoproteins found on M2 macrophages was measured using immunoblotting (B). Integrin α3 (ITGα3), macrophage mannose receptor (CD206), cathepsin L, NHE, and legumain have increased expression relative to M1 macrophages (B).
Glycoproteins Found Enriched on Human M1 Macrophages
Human CD14+ monocytes differentiated into M1 macrophages were subject to SPEG analysis followed by LC MS/MS. We found a total of two proteins that were enriched and N-glycosylated that were not present on CD14+ monocytes or human M2 macrophages (supplemental Figs. 6 and 7). Using homogeneous M1 macrophage cell blocks stained with indoleamine 2,3-deoxygenase (IDO1) to assess M1 macrophage presence, we did not find either expressed in human mCRPC tissue from the right tibia (supplemental Fig. 8). However, these glycoproteins that are enriched on macrophages may be useful as diagnostic or prognostic markers in chronic infections and inflammatory diseases.
Homogeneous Populations of Human M1 and M2 Macrophages Secrete Pro- and Anti-Inflammatory Cytokines and Angiogenic Factors
Human CD14+ monocytes obtained from peripheral blood of leukopak donors were differentiated in vitro into M1 or M2 macrophages and then polarized for 9 days thereafter in vitro using our phased polarization method (27). To ensure that the macrophages displayed inflammatory and wound-healing capabilities, we then assessed cytokine and growth factor secretion of both populations. To do this, media were obtained from both M1 and M2 macrophages on day 9. Th1 inflammatory cytokines (IL-6, IFN-γ, and IP-10/CXCL10), Th2 cytokine (IL-10), and pro-angiogenic growth factor vascular endothelial growth factor (VEGF) were quantified in cultured media by multiplex analysis (Fig. 3). We previously demonstrated that these homogeneous populations of M1 macrophages cultured under phased conditions produced anti-angiogenic factor IL-12 (p70) (27). However, they also secrete other pro-inflammatory cytokines IP-10/CXCL10 as well as interferon-γ (Fig. 3, A and B). M1 macrophages also secreted an abundance of IL-6 when compared with the M2 macrophage population (Fig. 3C). Our homogeneous populations of M2 macrophages secreted the anti-inflammatory cytokine IL-10, a potent inhibitor of natural killer cell stimulatory factor/IL-12 (31) as well as an inhibitor of interferon-γ (Fig. 3D) (32). Additionally, we found that our M2 macrophages secreted VEGF-A (Fig. 3E) important for wound healing and angiogenesis. Cells and sections of cells from tissue blocks of M2 macrophages were subject to immunohistochemistry and stained for CD206 (Fig. 4). Altogether, these data demonstrate that the human macrophages used in this study secrete pro- and anti-inflammatory cytokines as well as growth factor VEGF. Additionally, these findings demonstrate that these pro- and anti-inflammatory macrophages bear expression of these glycoproteins.
Fig. 3.

Macrophages secrete known cytokines associated with each cell type. Conditioned media were collected from polarized M1 and M2 macrophages in vitro on day 9. Media were then analyzed for pro- and anti-inflammatory cytokines. M1 macrophages were found to secrete more IFN-γ, IFN-γ-induced protein 10/CXCL10 (IP-10/CXCL10), and interleukin-6 (IL-6) relative to M2 macrophages (A–C). M2 macrophages secreted vascular endothelial growth factor (VEGF-A) and interleukin-10 (IL-10) (D and E).
Fig. 4.
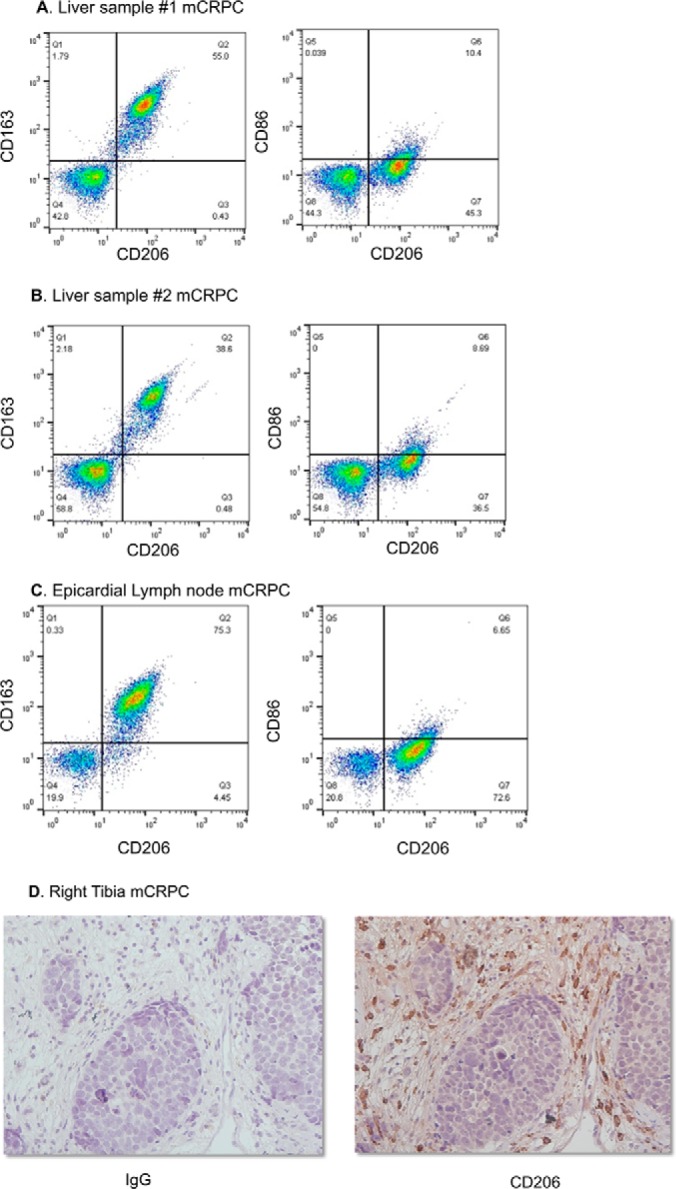
CD206-positive macrophages found in mCRPC samples. Rapid autopsy samples were subject to single cell homogenization followed by flow cytometry (A–C) or FFPE (D). Assessment of CD206 and CD163 in epicardial lymph node and liver metastases was performed using flow cytometry (A–C). Immunohistochemistry of CD206 of mCRPC isolated from right tibia was also performed (D).
Novel Glycoproteins Expression Detected in Metastatic Castrate-resistant Prostate Cancer
To assess whether these findings bore any clinical relevance, mCRPC samples were obtained from The Johns Hopkins rapid autopsy program. Samples were either subject to single cell dissociation or FFPE. We assessed M2 macrophage infiltration into mCRPC by performing flow cytometry using CD206 and M2 macrophage scavenger receptor CD163 (Fig. 4A). Complementary experiments to assess M1 macrophage infiltration in mCRPC tumors were also performed. We used CD86 and CD206 markers by flow cytometry and observed a paucity of M1 macrophage infiltration in mCRPC tumor samples isolated for rapid autopsy. We are able to assess that a bulk of the tumor mass is both CD206- and CD163-positive. Additionally, we assessed macrophage presence in bone metastasis, a common site of mCRPC metastasis, using immunohistochemistry. By staining for CD206 of a right tibia tumor obtained from rapid autopsy, we were able to determine that CD206-positive M2 macrophages were present (Fig. 4B). Altogether, these results demonstrate that CD206, one of the glycoproteins found enriched on M2 macrophages, bears clinical relevance in mCRPC.
DISCUSSION
Myeloid progenitor cells develop into pro-monocytes and are released into the circulation via leukocyte extravasation where they undergo differentiation into monocytes. These cells then migrate into tissues where they differentiate into resident tissue macrophages and help to protect these sites from infection and promote wound healing. When these cells encounter specific environmental cues, such as chemokines and cytokines, monocytes demonstrate functional polarization toward one of two phenotypically different subsets of macrophages: Th1 (also known as M1 macrophages) or Th2 (also known as M2 macrophages). M1 macrophages are known to produce pro-inflammatory cytokines and play an active role in destroying intracellular bacteria and sensitizing cancer cells to FasR-mediated apoptosis (33, 34). In our previous report (27), we demonstrated that differentiating into M1 and M2 macrophages using the phased polarization produced homogeneous populations. Using multiplex cytokine analysis, we assessed cytokine secretion of these homogeneous macrophage cell types. The data from this study demonstrate that homogeneous populations of M1 macrophages secreted IFN-γ, IL-6, and IP-10/CXCL10 (Fig. 3, A–C). These data align with the inflammatory nature of this subset of macrophages. Conversely, homogeneous populations of M2 macrophages produced IL-10 and VEGF-A (Fig. 3, D and E). Together, these results indicate that when polarizing human CD14+ monocytes using the phased polarization method will yield cells that bear the function of either an M1 or M2 macrophage.
To truly identify how different our homogeneous populations of macrophages were from each other, we assessed glycopeptides that were enriched on the cell surfaces of each macrophage population. Using SPEG analysis on CD14+ monocytes, M1 and M2 macrophages, we observed a significant difference of glyco-phenotype. The glyco-phenotypic change was drastic between cell types and indicative of a significant change on cell behavior and function. We, for the first time, have identified two proteins, acid oxidase isoform 1 and lysosome-associated membrane glycoprotein 3, that are N-glycosylated and enriched on the surface of M1 macrophages (supplemental Figs. 6 and 7). Altogether, we were able to identify several enriched glycoproteins that were expressed on M1 and M2 macrophages (Fig. 2) that were not present on other myeloid cell types that we tested. One of these N-linked glycoproteins that was enriched on M2 macrophages was the macrophage mannose receptor (CD206) (Fig. 2B and supplemental Fig. 2). This endocytic receptor binds to mannose, fucose, and N-acetylglucosamines that are found on the cell surface of microorganisms (35). It has also been demonstrated to regulate glycoprotein homeostasis (36). It is not found on expressed tumor cell types making it exclusive to immune cells. Given the important role that M2 macrophages play in eliciting tumor cell epithelial-to-mesenchymal transition (7), tumor cell invasion, and circumvention of therapy as well as being phenotypically similar to M2-tumor-associated macrophages (M2-TAMs), we then measured CD206 expression in mCRPC samples. This lethal form of prostate cancer does not respond to current therapeutics and continues to be an unmet medical need claiming the lives of men annually in the United States and worldwide. Using epicardial lymph node and two liver mCRPC samples obtained from rapid autopsy, we used CD206 as well as the scavenger receptor (CD163) (Fig. 4, A–C) to assess the M2-TAM infiltrate. Although we did not find CD163 to be N-glycosylated or enriched at <0.1 FDR using SPEG, CD163 expression has been long associated with M2 macrophages and M2-TAMs. We found that the mCRPC tumors samples were between 38 and 75% of M2 macrophages that co-expressed CD206 and CD163, although a paucity of CD86- and CD206-positive populations were found (Fig. 4, A–C). The low expression of CD86 may be due to a dearth of M1 macrophage infiltration in these mCRPC samples. Additionally, performing IHC for CD206 (Fig. 4D) further validated the M2 macrophage presence in these rapid autopsy samples.
Another protein that was enriched on M2 macrophages was the lysosomal protease enzyme, cathepsin L (Fig. 2). The up-regulation of this protein is correlative with poor cancer prognosis and has been identified as a targetable marker in cancer (37). Additionally, studies have shown that phorbol 12-myristate 13-acetate (PMA) treatment can lead to cathepsin L secretion by prostate tumor cells in vitro leading to increased Matrigel invasion (38). Recent data from our group demonstrated that this protein was amplified in prostate cancers and was positively correlated with high Gleason scores (39). However, herein we found that it is also enriched on M2 macrophages, which may allude to the ability of M2 macrophage to aide in tumor invasion and subsequent metastasis. Other proteins that were enriched on M2 macrophages were legumain, an asparaginyl endopeptidase that is overexpressed in solid tumors and abundantly expressed on M2-TAMs (40, 41). Legumain has been implicated in playing a significant role in tumorigenesis and ablation of legumain using prodrug-suppressed angiogenesis and metastasis in pre-clinical models (40). In tumor cells, cathepsin L and legumain both appear to be regulated by tumor suppressor cystatin 6 (CST6); however, this protein does not appear to be secreted by macrophages (42, 43). Additionally, legumain and cathepsin have both been reported to localize to caveolae and are expressed and activated while on the cell surface of tumor cells where they interact with annexin II (44). Legumain was demonstrated in membrane-associated vesicles concentrated at the invadopodia of tumor cells and on cell surfaces where it co-localized with integrins (45). We found both cathepsin and legumain enriched on M2 macrophages and may allude to each of them being in an active state. This result confirms a recent report that demonstrated increased legumain cell-surface expression after monocytes were differentiated into M2 macrophages (46). Together, these proteases suggest the multiple roles that M2 macrophages could play in degrading matrix to aide in tumor escape.
Integrins are heterodimeric transmembrane surface receptors that can mediate adhesion and cell survival (47, 48). We found integrin α3, which forms a heterodimer with integrin β1 to mediate cell adhesion to laminin, enriched on M2 macrophages (Fig. 2). We did not find β integrins enriched on either population. However, this may be relevant to macrophage-tumor interaction during several steps of metastasis. Prostate cancer cells lose integrin β4 and have an increased expression of integrin α6 and β1 (49). Integrin β1 has been reported to be expressed on prostate cancer cells clinically (50), and this increased expression of integrin α3 on M2 macrophages may confer its association with prostate tumor cells. This may allow the formation of the tumor microenvironment of metastasis (TMEM) (51), which is made up of tumor cells, macrophages, and endothelial cells that synergistically allow intravasation in pre-clinical models. More studies are warranted in this regard. Na+/H+ exchangers (NHE) are integral membrane proteins that are responsible for numerous cellular processes, ranging from the fine control of intracellular pH and cell volume to systemic electrolyte, migration, acid-base, and fluid volume homeostasis (52). Additionally, one well studied NHE member, NHE1, has been demonstrated to be pivotal in microglial cell migration and glioma-microglial interactions (53). We find NHE7 enriched on the M2 macrophage cell surface (Fig. 2 and supplemental Fig. 5), which to our knowledge has not been linked to prostate cancer. Additionally, we performed RNA-sequencing analysis on each subset of human macrophages (data not shown), and we did not observe each enriched glycoprotein suggestive that there are differences at the glycoproteomic level that are not observed at the mRNA level. Finally, we found legumain expressed on the cell surface; however, it does not appear to be well studied in M2-TAMs nor in mCRPC and thus also warrants further investigation. Altogether, we believe that using these markers for localized and distant disease could be indicative of how immunosuppressive as well as aggressive a tumor is and what treatment strategies could be used to remedy this malady.
Supplementary Material
Acknowledgments
We thank Christopher Thoburn of the Human Immunology Core Laboratory at The Johns Hopkins University Sidney Kimmel Comprehensive Cancer Center for help with the multiplex cytokine analysis. We also thank The Johns Hopkins Hemapheresis and Transfusion division for leukopaks and the Pathobiology Core for slide processing.
Footnotes
Author contributions: J.C.Z., J.R.H., H.Z., and K.J.P. designed the research; J.C.Z., W.Y., and J.R.H. performed the research; W.Y., H.Z., and K.J.P. contributed new reagents or analytic tools; J.C.Z., W.Y., and J.R.H. analyzed the data; J.C.Z. and W.Y. wrote the paper; H.Z. supervised the performance of research; K.J.P. supervised research and provided funding.
* This work was supported by National Institutes of Health Grants U54CA143803, CA163124, CA093900, and CA143055 from NCI and Grant 1R21AI122382-01 from NIAID, a Write out United Negro College Fund (UNCF)/Merck Postdoctoral Science Research Fellowship (to J. C. Z.), and a Prostate Cancer Foundation Young Investigator Award (to J.C.Z.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- mCRPC
- metastatic castration-resistant prostate cancer
- M2-TAM
- M2-tumor-associated macrophage
- SPEG
- solid-phase extraction of N-linked glycopeptide
- ACN
- acetonitrile
- PNGase
- peptide: N-glycosidase
- FDR
- false-discovery rate
- FFPE
- formalin fixation followed by paraffin-embedding
- NHE
- sodium/hydrogen exchanger
- PBMC
- peripheral mononuclear blood cell.
REFERENCES
- 1. Siegel R. L., Miller K. D., and Jemal A. (2015) Cancer statistics, 2015. CA Cancer J. Clin. 65, 5–29 [DOI] [PubMed] [Google Scholar]
- 2. Lewis C. E., and Pollard J. W. (2006) Distinct role of macrophages in different tumor microenvironments. Cancer Res. 66, 605–612 [DOI] [PubMed] [Google Scholar]
- 3. Mantovani A., Bottazzi B., Colotta F., Sozzani S., and Ruco L. (1992) The origin and function of tumor-associated macrophages. Immunol. Today 13, 265–270 [DOI] [PubMed] [Google Scholar]
- 4. Sica A., Schioppa T., Mantovani A., and Allavena P. (2006) Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur. J. Cancer 42, 717–727 [DOI] [PubMed] [Google Scholar]
- 5. Zhang J., Lu Y., and Pienta K. J. (2010) Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J. Natl. Cancer Inst. 102, 522–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roca H., Varsos Z. S., Sud S., Craig M. J., Ying C., and Pienta K. J. (2009) CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J. Biol. Chem. 284, 34342–34354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roca H., Hernandez J., Weidner S., McEachin R. C., Fuller D., Sud S., Schumann T., Wilkinson J. E., Zaslavsky A., Li H., Maher C. A., Daignault-Newton S., Healy P. N., and Pienta K. J. (2013) Transcription factors OVOL1 and OVOL2 induce the mesenchymal to epithelial transition in human cancer. PLoS ONE 8, e76773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loberg R. D., Day L. L., Harwood J., Ying C., St John L. N., Giles R., Neeley C. K., and Pienta K. J. (2006) CCL2 is a potent regulator of prostate cancer cell migration and proliferation. Neoplasia 8, 578–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murdoch C., Giannoudis A., and Lewis C. E. (2004) Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 104, 2224–2234 [DOI] [PubMed] [Google Scholar]
- 10. Milliken D., Scotton C., Raju S., Balkwill F., and Wilson J. (2002) Analysis of chemokines and chemokine receptor expression in ovarian cancer ascites. Clin. Cancer Res. 8, 1108–1114 [PubMed] [Google Scholar]
- 11. Zarif J. C., Taichman R. S., and Pienta K. J. (2014) TAM macrophages promote growth and metastasis within the cancer ecosystem. Oncoimmunology 3, e941734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lissbrant I. F., Stattin P., Wikstrom P., Damber J. E., Egevad L., and Bergh A. (2000) Tumor associated macrophages in human prostate cancer: relation to clinicopathological variables and survival. Int. J. Oncol. 17, 445–451 [DOI] [PubMed] [Google Scholar]
- 13. Vykhovanets E. V., Maclennan G. T., Vykhovanets O. V., and Gupta S. (2011) IL-17 Expression by macrophages is associated with proliferative inflammatory atrophy lesions in prostate cancer patients. Int. J. Clin. Exp. Pathol. 4, 552–565 [PMC free article] [PubMed] [Google Scholar]
- 14. Sroka I. C., Sandoval C. P., Chopra H., Gard J. M., Pawar S. C., and Cress A. E. (2011) Macrophage-dependent cleavage of the laminin receptor α6β1 in prostate cancer. Mol. Cancer Res. 9, 1319–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Joseph I. B., and Isaacs J. T. (1998) Macrophage role in the anti-prostate cancer response to one class of antiangiogenic agents. J. Natl. Cancer Inst. 90, 1648–1653 [DOI] [PubMed] [Google Scholar]
- 16. Zollo M., Di Dato V., Spano D., De Martino D., Liguori L., Marino N., Vastolo V., Navas L., Garrone B., Mangano G., Biondi G., and Guglielmotti A. (2012) Targeting monocyte chemotactic protein-1 synthesis with bindarit induces tumor regression in prostate and breast cancer animal models. Clin. Exp. Metastasis 29, 585–601 [DOI] [PubMed] [Google Scholar]
- 17. Bielik A. M., and Zaia J. (2010) Historical overview of glycoanalysis. Methods Mol. Biol. 600, 9–30 [DOI] [PubMed] [Google Scholar]
- 18. van Kooyk Y., and Rabinovich G. A. (2008) Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 9, 593–601 [DOI] [PubMed] [Google Scholar]
- 19. Szymanski C. M., and Wren B. W. (2005) Protein glycosylation in bacterial mucosal pathogens. Nat. Rev. Microbiol. 3, 225–237 [DOI] [PubMed] [Google Scholar]
- 20. Grigorian A., Torossian S., and Demetriou M. (2009) T-cell growth, cell surface organization, and the galectin-glycoprotein lattice. Immunol. Rev. 230, 232–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Durand G., and Seta N. (2000) Protein glycosylation and diseases: blood and urinary oligosaccharides as markers for diagnosis and therapeutic monitoring. Clin. Chem. 46, 795–805 [PubMed] [Google Scholar]
- 22. Galonić D. P., and Gin D. Y. (2007) Chemical glycosylation in the synthesis of glycoconjugate antitumour vaccines. Nature 446, 1000–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rek A., Krenn E., and Kungl A. J. (2009) Therapeutically targeting protein-glycan interactions. Br. J. Pharmacol. 157, 686–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Drake P. M., Cho W., Li B., Prakobphol A., Johansen E., Anderson N. L., Regnier F. E., Gibson B. W., and Fisher S. J. (2010) Sweetening the pot: adding glycosylation to the biomarker discovery equation. Clin. Chem. 56, 223–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tian Y., Zhou Y., Elliott S., Aebersold R., and Zhang H. (2007) Solid-phase extraction of N-linked glycopeptides. Nat. Protoc. 2, 334–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang H., Li X. J., Martin D. B., and Aebersold R. (2003) Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 21, 660–666 [DOI] [PubMed] [Google Scholar]
- 27. Zarif J. C., Hernandez J. R., Verdone J. E., Campbell S. P., Drake C. G., and Pienta K. J. (2016) A phased strategy to differentiate human CD14+ monocytes into classically and alternatively activated macrophages and dendritic cells. BioTechniques 61, 33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pruitt K. D., Tatusova T., and Maglott D. R. (2007) NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35, D61–D65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miranti C. K. (2002) Application of cell adhesion to study signaling networks. Methods Cell Biol. 69, 359–383 [DOI] [PubMed] [Google Scholar]
- 30. Edick M. J., Tesfay L., Lamb L. E., Knudsen B. S., and Miranti C. K. (2007) Inhibition of integrin-mediated crosstalk with epidermal growth factor receptor/Erk or Src signaling pathways in autophagic prostate epithelial cells induces caspase-independent death. Mol. Biol. Cell 18, 2481–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. D'Andrea A., Aste-Amezaga M., Valiante N. M., Ma X., Kubin M., and Trinchieri G. (1993) Interleukin 10 (IL-10) inhibits human lymphocyte interferon γ-production by suppressing natural killer cell stimulatory factor/IL-12 synthesis in accessory cells. J. Exp. Med. 178, 1041–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ito S., Ansari P., Sakatsume M., Dickensheets H., Vazquez N., Donnelly R. P., Larner A. C., and Finbloom D. S. (1999) Interleukin-10 inhibits expression of both interferon α- and interferon γ-induced genes by suppressing tyrosine phosphorylation of STAT1. Blood 93, 1456–1463 [PubMed] [Google Scholar]
- 33. Jin Y., Lundkvist G., Dons L., Kristensson K., and Rottenberg M. E. (2004) Interferon-γ mediates neuronal killing of intracellular bacteria. Scand. J. Immunol. 60, 437–448 [DOI] [PubMed] [Google Scholar]
- 34. Ruiz-Ruiz C., Muñoz-Pinedo C., and López-Rivas A. (2000) Interferon-γ treatment elevates caspase-8 expression and sensitizes human breast tumor cells to a death receptor-induced mitochondria-operated apoptotic program. Cancer Res. 60, 5673–5680 [PubMed] [Google Scholar]
- 35. Schlesinger P. H., Doebber T. W., Mandell B. F., White R., DeSchryver C., Rodman J. S., Miller M. J., and Stahl P. (1978) Plasma clearance of glycoproteins with terminal mannose and N-acetylglucosamine by liver non-parenchymal cells. Studies with β-glucuronidase, N-acetyl-β-d-glucosaminidase, ribonuclease B and agalacto-orosomucoid. Biochem. J. 176, 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee S. J., Evers S., Roeder D., Parlow A. F., Risteli J., Risteli L., Lee Y. C., Feizi T., Langen H., and Nussenzweig M. C. (2002) Mannose receptor-mediated regulation of serum glycoprotein homeostasis. Science 295, 1898–1901 [DOI] [PubMed] [Google Scholar]
- 37. Sudhan D. R., and Siemann D. W. (2015) Cathepsin L targeting in cancer treatment. Pharmacol. Ther. 155, 105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Colella R., Jackson T., and Goodwyn E. (2004) Matrigel invasion by the prostate cancer cell lines, PC3 and DU145, and cathepsin L+B activity. Biotech. Histochem. 79, 121–127 [DOI] [PubMed] [Google Scholar]
- 39. Tian Y., Bova G. S., and Zhang H. (2011) Quantitative glycoproteomic analysis of optimal cutting temperature-embedded frozen tissues identifying glycoproteins associated with aggressive prostate cancer. Anal. Chem. 83, 7013–7019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lin Y., Wei C., Liu Y., Qiu Y., Liu C., and Guo F. (2013) Selective ablation of tumor-associated macrophages suppresses metastasis and angiogenesis. Cancer Sci. 104, 1217–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gawenda J., Traub F., Lück H. J., Kreipe H., and von Wasielewski R. (2007) Legumain expression as a prognostic factor in breast cancer patients. Breast Cancer Res. Treat. 102, 1–6 [DOI] [PubMed] [Google Scholar]
- 42. Jin L., Zhang Y., Li H., Yao L., Fu D., Yao X., Xu L. X., Hu X., and Hu G. (2012) Differential secretome analysis reveals CST6 as a suppressor of breast cancer bone metastasis. Cell Res. 22, 1356–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. D'Costa Z. C., Higgins C., Ong C. W., Irwin G. W., Boyle D., McArt D. G., McCloskey K., Buckley N. E., Crawford N. T., Thiagarajan L., Murray J. T., Kennedy R. D., Mulligan K. A., Harkin D. P., Waugh D. J., et al. (2014) TBX2 represses CST6 resulting in uncontrolled legumain activity to sustain breast cancer proliferation: a novel cancer-selective target pathway with therapeutic opportunities. Oncotarget 5, 1609–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mai J., Finley R. L. Jr, Waisman D. M., and Sloane B. F. (2000) Human procathepsin B interacts with the annexin II tetramer on the surface of tumor cells. J. Biol. Chem. 275, 12806–12812 [DOI] [PubMed] [Google Scholar]
- 45. Liu C., Sun C., Huang H., Janda K., and Edgington T. (2003) Overexpression of legumain in tumors is significant for invasion/metastasis and a candidate enzymatic target for prodrug therapy. Cancer Res. 63, 2957–2964 [PubMed] [Google Scholar]
- 46. Solberg R., Smith R., Almlöf M., Tewolde E., Nilsen H., and Johansen H. T. (2015) Legumain expression, activity and secretion are increased during monocyte-to-macrophage differentiation and inhibited by atorvastatin. Biol. Chem. 396, 71–80 [DOI] [PubMed] [Google Scholar]
- 47. Hynes R. O. (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 [DOI] [PubMed] [Google Scholar]
- 48. Miranti C. K., and Brugge J. S. (2002) Sensing the environment: a historical perspective on integrin signal transduction. Nat. Cell Biol. 4, E83–E90 [DOI] [PubMed] [Google Scholar]
- 49. Cress A. E., Rabinovitz I., Zhu W., and Nagle R. B. (1995) The α6β1 and α6β4 integrins in human prostate cancer progression. Cancer Metastasis Rev. 14, 219–228 [DOI] [PubMed] [Google Scholar]
- 50. Schmelz M., Cress A. E., Scott K. M., Bürger F., Cui H., Sallam K., McDaniel K. M., Dalkin B. L., and Nagle R. B. (2002) Different phenotypes in human prostate cancer: α6 or α3 integrin in cell-extracellular adhesion sites. Neoplasia 4, 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Robinson B. D., Sica G. L., Liu Y. F., Rohan T. E., Gertler F. B., Condeelis J. S., and Jones J. G. (2009) Tumor microenvironment of metastasis in human breast carcinoma: a potential prognostic marker linked to hematogenous dissemination. Clin. Cancer Res. 15, 2433–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Orlowski J., and Grinstein S. (2004) Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflugers Arch. 447, 549–565 [DOI] [PubMed] [Google Scholar]
- 53. Zhu W., Carney K. E., Pigott V. M., Falgoust L. M., Clark P. A., Kuo J. S., and Sun D. (2016) Glioma-mediated microglial activation promotes glioma proliferation and migration: roles of Na+/H+ exchanger isoform 1. Carcinogenesis 37, 839–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.