

Abstract
Fibroblast growth factor (FGF) signaling is vital for many biological processes, beginning with development. The importance of FGF signaling for skeleton formation was first discovered by the analysis of genetic FGFR mutations which cause several bone morphogenetic disorders, including achondroplasia, the most common form of human dwarfism. The formation of the long bones is mediated through proliferation and differentiation of highly specialized cells - chondrocytes.
Chondrocytes respond to FGF with growth inhibition, a unique response which differs from the proliferative response of the majority of cell types; however, its molecular determinants are still unclear. Quantitative phosphoproteomic analysis was utilized to catalogue the proteins whose phosphorylation status is changed upon FGF1 treatment. The generated dataset consists of 756 proteins. We could localize the divergence between proliferative (canonical) and inhibitory (chondrocyte specific) FGF transduction pathways immediately upstream of AKT kinase. Gene Ontology (GO) analysis of the FGF1 regulated peptides revealed that many of the identified phosphorylated proteins are assigned to negative regulation clusters, in accordance with the observed inhibitory growth response. This is the first time a comprehensive subset of proteins involved in FGF inhibitory response is defined. We were able to identify a number of targets and specifically discover glycogen synthase kinase3β (GSK3β) as a novel key mediator of FGF inhibitory response in chondrocytes.
The family of fibroblast growth factors (FGFs) 1 is represented by 23 members. Most of them (excluding FGF11-FGF14) activate FGF receptors (FGFRs) that trigger multiple signaling cascades (1, 2). FGF signaling is vital for numerous biological functions, including skeletal development (3). The formation and growth of long bones and vertebrae is achieved through endochondral ossification, a strictly regulated process that is mediated by chondrocyte proliferation and differentiation. Unlike most cell types, where FGF signaling induces proliferation, chondrocytes undergo growth arrest when exposed to FGFs (4–7). It has been suggested that tissue specific targets may be responsible for this distinct response (8). FGFs 2,9 and, in particular, 18 together with FGFR1 and 3 have been implicated in growth plate development (3, 9, 10), however, the underlying mechanism remains poorly characterized.
Phosphoproteomics has been used to delineate the FGF signaling pathways in different cell types (11, 12). Current advances in multiplexed quantitative proteomics, including stable isotope labeling of amino acids in cell culture (SILAC) (13) or isobaric labeling (14, 15) allow for comprehensive profiling of proteomes. Presently, isobaric labeling strategies allow for simultaneous analysis of up to 10 samples (16), thereby reducing both analysis time and run-to-run variability.
Here we utilized tandem mass tag (TMT) labeling to catalogue FGF1-induced changes in the chondrocyte phosphoproteome. This data set provides an array of proteins whose phosphorylation status is changed upon FGF1 treatment. Immediate FGF1 response was previously characterized by immunoblotting and some differences between signaling pathways induced by FGF1 in chondrocytes compared with cells with proliferative response were reported (4, 17). We chose to investigate FGF1 response at intermediate time points to identify proteins which are targeted by FGF signaling in chondrocytes and therefore might play a role in FGF inhibitory response.
Seven hundred fifty-six FGF regulated peptides were identified in our study, many of which are not known to be FGF targets or to be directly involved in chondrocyte biology. Importantly, similar targets were regulated by FGF2 and FGF18 as assayed by immunoblotting. Gene Ontology (GO) analysis revealed that many of the identified phosphorylated proteins are assigned to negative regulation clusters, thus corroborating the functionality of the obtained targets. Collectively, these analyses, for the first time, allowed for a subset of proteins that are directly involved in FGF inhibitory response to be established. Using this subset, we could identify glycogen synthase kinase 3β (GSK3β) as a novel key mediator of FGF inhibitory response in chondrocytes.
EXPERIMENTAL PROCEDURES
Cell Culture
Rat chondrosarcoma (RCS) cells (18) were maintained in DMEM supplemented with 10% fetal calf serum at 37 °C and 9% CO2. Cells were treated with human recombinant FGF1 (5 ng/ml) (a kind gift from Dr. Mohammadi, NYU School of Medicine) or human recombinant FGF2 and FGF18 (Peprotech) and heparin (5 μg/ml) for the indicated period. We routinely use FGF1 in large scale experiments as FGF1 induced growth arrest is relevant to proliferative chondrocytes systems including primary chondrocytes and we have the same batch of the factor (19). Micromass and organ cultures are described in supplemental information.
Phosphoproteomics
Sample Preparation for Mass Spectrometry
Cells were lysed in a lysis buffer composed of 50 mm Tris-HCl pH 7.4, 150 mm NaCl, 10 mm KCl, 1% Nonidet P-40, 1 mm EDTA in the presence of phosphatase and protease inhibitor mixture (Thermo Scientific). We prepared biological triplicates of untreated cells, 30 min post treatment cells, and duplicates of 2 h and 8 h post treatment cells. Cell lysates were prepared using the filter-aided sample preparation (FASP) method as previously described (20) with modifications to accommodate the milligram levels of protein used for this study. Briefly, 6 mg per sample at ∼1 mg/ml was reduced with DTT (final concentration 1 mm) at 57 °C for 1 h and loaded onto CentriPrep Ultracel-30 Concentrators (Millipore) pre-equilibrated with 5 ml of FASP buffer A (8 m urea, 0.1 m Tris HCl, pH 7.8). Following three washes with FASP buffer A, lysates were alkylated on filter with 3 mm iodoacetamide in buffer A for 45 min. Filter bound lysates were then washed 3 times with FASP buffer A, 3 times with FASP buffer B (100 mm ammonium bicarbonate, pH 7.8) and digested with 60 μg of trypsin (Promega) overnight. Peptides were eluted twice with 0.5 m NaCl. Tryptic peptides were desalted using a Sep-Pak C18 column (Waters) and concentrated in a SpeedVac concentrator.
TMT Labeling
Half of each FASP prepared lysate sample was re-suspended in 100 mm triethylammonium bicarbonate and TMT (Thermo Scientific) labeled according to the manufacturer's recommended protocol for the 10plex isobaric labeling reagent set. TMT labeled samples were combined and cleaned using first strong cation exchange (SCX) chromatography and subsequently strong anion exchange (SAX) chromatography on the SCX flow through as described below. An Agilent non-porous Bio SCX HPLC Column (4.6 × 50 mm, 3 μm, 1000 Å) was used for SCX clean-up on an Agilent 1260 Bio-inert LC system. Peptides were eluted using a 50-min gradient from 90% solvent A (5 mm ammonium formate) with 10% solvent C (100% acetonitrile) to 90% solvent B (500 mm ammonium formate) with 10% solvent C in 8 min with a 9 min hold and back to 90% solvent A with 10% solvent C in 9 min. Fractions were collected every 30 s. Fractions 8–20 were combined and concentrated in a SpeedVac concentrator. Fractions 1–8 were combined and further cleaned on an Agilent PL-SAX column (4.6 × 50 mm, 5 μm, 1000 Å). Peptides were eluted using the same 50 min gradient except solvent A was 5 mm ammonium bicarbonate, solvent B was 500 mm ammonium bicarbonate. Fractions 8–20 from SAX were combined with the concentrated fractions from SCX and further concentrated in a SpeedVac concentrator. The combined SCX and SAX fractions were desalted using a Sep-Pak C18 column.
Global Proteome Analysis
A portion of the TMT labeled sample was fractionated via hydrophilic interaction liquid chromatography (HILIC) on an Agilent 1260 Bio-inert LC system using a TSK gel Amide-80 LC column (250 × 4.6 mm, 5 μm, Tosoh Biosciences) (21). Peptides were eluted from the column using a two-step gradient from 90% solvent B (98% acetonitrile, 0.1% trifluoroacetic acid) to 70% solvent B in 60 min and to 50% solvent B in an additional 10 min. Solvent A was 2% acetonitrile, 0.1% trifluoroacetic acid. Fractions were collected every minute. Fractions 1–5 were not analyzed as they contained no peptides. The following fractions were combined for LCMS analysis: 6–10, 61–65, 66–70, 71–75, and 75–80. The remaining fractions were analyzed individually (11–60).
Phosphoproteome Analysis
A portion of the TMT labeled sample was fractionated using SCX and SAX as described above. Peptides were eluted from the column using a two-step gradient from 80% solvent A, 20% solvent C to 30% solvent B, 20% solvent C in 20 min and to 80% solvent B, 20% solvent C in an additional 2 min and held at that composition for 8 min. Fractions were collected every 30 s. Fractions 1–10 were combined and further fractionated using SAX as previously described above utilizing the same gradient as the SCX fractionation.
TiO2 Phosphopeptide Enrichment
SCX and SAX fractions were combined in a concatenated fashion for phosphopeptide enrichment. Phosphopeptides were enriched using 5 μm Titansphere TiO2 beads (Gl Sciences, Torrance, CA) as previously described (22). Briefly, peptides were reconstituted in binding buffer, 1 mm monopotassium phosphate in 65% acetonitrile, 2% trifluoroacetic acid. The TiO2 beads were washed three times with washing buffer, 65% acetonitrile, 0.1% trifluoroacetic acid, added to the peptides at a ratio of 1:4 peptides to beads, and incubated for 20 min. The flow through was collected and incubated with a new aliquot of beads. Both sets of beads were washed twice with washing buffer and then loaded onto a C18 STAGE tip where the beads were washed two additional times using washing buffer prior to elution of the peptides (23). Peptides were eluted with 15% ammonium hydroxide in 40% acetonitrile, and concentrated in a SpeedVac concentrator.
LC-MS Analysis
An aliquot of each fraction for global proteome analysis was loaded onto an Acclaim PepMap 100 (2 cm × 75 μm ID) trap column in line with an EASY-Spray 50 cm × 75 μm ID PepMap C18 analytical HPLC column with 2 μm bead size using the auto sampler of an EASY-nLC 1000 HPLC (Thermo Fisher) and solvent A (2% acetonitrile, 0.5% acetic acid). The peptides were gradient eluted into a Q Exactive (Thermo Scientific) mass spectrometer using a 120-min gradient from 5% to 30% solvent B (90% acetonitrile, 0.5% acetic acid), followed by 15 min from 30% to 40% solvent B. Solvent B was then ramped up to 100% and was held at 100% for 10 min for column wash. High resolution full MS spectra were acquired with a resolution of 70,000, an AGC target of 1e6, a maximum ion time of 120 ms, and scan range of 400 to 1500 m/z. Following each full MS scan ten data-dependent high resolution HCD MS/MS spectra were acquired. All MS/MS spectra were collected using the following instrument parameters: resolution of 35,000, AGC target of 1e5, maximum ion time of 250 ms, one microscan, 1.5 m/z isolation window, fixed first mass of 115 m/z, and Normalized Collision Energy (NCE) of 30.
An aliquot of each TiO2 enriched sample was loaded as described above. The peptides were gradient eluted into a Q Exactive (Thermo Scientific) mass spectrometer using a 40 min gradient from 2% to 30% solvent B (95% acetonitrile, 0.5% acetic acid), followed by 60 min from 30% to 50% solvent B. Solvent B was then ramped up to 100% and was held at 100% for 10 min for column wash. The data acquisition parameters were kept same as mentioned above except the NCE was set to 27.
Data Analysis
Peptide and protein identification, as well as reporter ion quantitation was performed using the MaxQuant software suite version 1.5.2.8 (24) against Uniprot Rattus norvegicus database downloaded on January 27, 2016 which contained 29,885 entries. For the first search the peptide mass tolerance was set to 20ppm and for the main search peptide mass tolerance was set to 4.5 ppm. Trypsin specific cleavage was selected with two missed cleavages. Both peptide spectral match and protein FDR were set to 1% for identification. Carbamidomethylation of cysteine was added as a static modification. Oxidation of methionine, deamidation of glutamine and aspargine, acetylation of N termini and phosphorylation of serine, threonine and tyrosine were allowed as variable modifications. Protein quantitation was performed using unique and razor peptides. The global proteome data set was filtered to include proteins identified with two or more unique and/or razor peptides. Both the global proteome and phosphoproteome data sets were filtered to remove any peptides or proteins that were not detected in at least two replicates of at least one treatment time point. A two-sided student's t test was performed correcting for multiple testing by controlling for FDR at 5%. Proteins and/or phosphopeptides with a q-value <0.05 were considered significant. In addition, z-scores were calculated and used to perform hierarchical clustering. The data were analyzed using DAVID Bioinformatics Resources 6.7 database for gene ontology enrichment (25, 26). The settings were as follows: EASE - 0.05; classification stringency - high. Population background was set using proteins identified by global proteomics in RCS cells.
SCX and SAX Clean-up Test Sample
An aliquot of the mixed TMT sample as prepared above was separated into 50 fractions with SCX. The first ten fractions from SCX were combined and fractionated into 60 fractions with SAX. Fractions 1–5 from SAX were not analyzed as they contained no peptides. The following SCX fractions were combined for LCMS analysis: 26–30, 31–33, 34–36, 37–40, 41–43, 44–46, and 47–50. The remaining fractions were analyzed individually (10–25). The SAX fractions were combined for LCMS analysis as follows: 1–4, 5–8, 9–16, 41–45, 46–50, 51–55, 56–57, and 58–60. Fractions 1740 were analyzed individually. An aliquot of each fraction was loaded as described previously. The peptides were gradient eluted into a Orbitrap Fusion Tribrid (Thermo Scientific) mass spectrometer using a 120 min gradient from 5% to 30% solvent B (95% acetonitrile, 0.5% acetic acid), followed by 20 min from 30% to 40% solvent B. High resolution full MS spectra were acquired with a resolution of 120,000, an AGC target of 2e5, with a maximum ion time of 50 ms, and scan range of 400 to 1500 m/z. Following each full MS scan as many HCD MS/MS spectra were acquired as possible in the ion trap. All MS/MS spectra were collected using the following instrument parameters: AGC target of 1e4, maximum ion time of 35 ms, one microscan, 0.8 m/z isolation window, fixed first mass of 120 m/z, and NCE of 28. All fraction raw files were combined by fractionation type and searched against a Uniprot Rattus norvegicus database containing in-house compiled contaminant protein database utilizing Byonic within the Proteome Discoverer 2.1 software suite. The following search parameters were used: decoys were allowed, trypsin digestion with two missed cleavages, precursor mass tolerance of 10 ppm, MS/MS mass tolerance of 0.4 Da. A total of four common and one rare modifications were allowed per peptide. Carbamidomethylation on cysteine and TMT 6-plex on lysine and N termini were set as static modifications. Common modifications included oxidation on methionine, phosphorylation on serine, threonine, tyrosine, and deamidation on glutamine and aspargine was set as a rare modification. Results were filtered to only include peptide spectral matches (PSMs) with a Byonic score of 300 or greater. Reporter ion quantitation was not performed for this test set as we aimed to determine total number of phosphopeptides identified with additional SAX clean up.
Experimental Design and Statistical Rationale
For the phosphoproteomic analysis of FGF1 treated chondrocytes we prepared three replicates of the following time points: control (untreated), 30 min, 2 and 8 h post FGF1 treatment. We expected the 30-min time point to show significant changes as compared with the untreated and later post treatment time points. Because TMT methodology allows for simultaneous labeling of up to ten samples, we chose to use triplicates for the control and 30 min time point and duplicates for the 2 and 8 h time points. Surprisingly, no significant changes in the phosphoproteome were observed at 30 min time point (supplemental Fig. S1). We therefore omitted these samples from the discussion to simplify data presentation and to concentrate on more informative results. Three replicate experiments were performed to validate some of the identified targets by immunoblotting and representative immunoblots are shown in Figs. 4 and Fig. 5. GSK3β inhibition assays were performed with 2 and 3 replicates (Fig. 5C, 5D–5F respectively). IHC analysis was done using 4 different fields on three slides (Fig. 5B and 5F). Data presented in Figs. 5B and 5E–5F were analyzed by a one-way ANOVA analysis with Bonferroni's multiple comparisons correction using GraphPad Prism version 7.0 (www.graphpad.com).
Fig. 4.

Validation of FGF regulated targets. RCS cells (A–E) and micromass cultures (F) were treated with FGF1, FGF2, and FGF18 as marked, and analyzed by immunoblotting using indicated antibodies. Twenty micrograms of total protein from RCS lysates and 10 μg of total protein from micromass lysates was used for immunodetection. The blots are representative of at least three independent experiments. E, Fragments of silver stained 2D electrophoresis. RCS cells were treated with FGF1 for 2 h and lysates obtained from treated and untreated cells were enriched for phosphoproteins following 2D electrophoresis. An identified protein those phosphorylation status is changed upon FGF1 treatment is circled in green. Two independent experiments were performed with similar results.
Fig. 5.

GSK3b is essential for mediating FGF inhibitory response in chondrocytes. RCS cells and micromass cultures were treated with FGF1 or GSK3b inhibitors for the times indicated, and analyzed by (A, C, E) immunoblotting. Ten micrograms μg of total protein was used for immunodetection. B, 10 micron sections of PFA-fixed tissue from the growth plate of tibia of newborn mice were analyzed by immunohistochemistry. The proliferating (P), hypertrophic (H) and resting (R) regions are indicated by arrows. Quantification was carried out by counting the number of positively stained cells in six separate fields for each cell type. C–E, RCS cells and micromass cultures were pre-treated with GSK3b inhibitors C–E, CHIR 3uM; SB415286 30 mm as indicated and inhibition was validated by assaying b-catenin stability. C–E, The cell cycle was analyzed either by FACScan™ analysis (C, D) or by BrdU incorporation (E). Numbers on the Y-axis indicate either relative percentage of total cells in the S-phase or percentage of BrdU positive cells. The data are representative of two (C) and three (D, E) independent experiments with the consistent results. F, Metatarsal bone rudiments were isolated from E15.5 embryos and cultivated in vitro for 24 h with or without FGF1 (100 ng/ml). BrdU was added during the last 6 h of FGF1 treatment. After fixation, rudiments were analyzed for immunodetection of BrdU. Representative pictures of immunostaining in the proliferating zone are shown (right panel.) Quantification of BrdU staining in untreated and treated rudiments was carried out by counting the number of positively stained cells in four fields for three different rudiments.
Raw Data Repository
All raw mass spectrometry data and search results have been deposited to the ProteomeXchange Consortium (31) via the MassIVE partner repository with the data set identifierS MassIVE: MSV000080259 and ProteomeXchange: PXD005199.
RESULTS
Analysis of FGF1-stimulated Chondrocytes Reveals a Predominantly Steady Level of the Global Proteome During Intermediate Response
To understand the determinants of chondrocyte FGF inhibitory response we sought to identify the set of proteins whose phosphorylation status is changed upon FGF1 treatment. Rat ChondroSarcoma (RCS) cells were used for our experiments as a well-accepted model of proliferative chondrocytes that respond to FGF signaling with growth arrest (5, 19). Like the proliferating chondrocytes from the growth plate, RCS cells mostly express FGFR3 (supplemental Fig. S2A). Chondrocytes were treated with FGF1 and cell cycle arrest was monitored by FACScan analysis (supplemental Fig. S2B). Phosphorylation of several known targets, was assayed to monitor proper FGF signaling (supplemental Fig. S2C). As expected, ERK1/2 was robustly activated up to 8 h of FGF1 treatment with some residual activation detected even at 24 h. Our primary interest was the intermediate response; therefore, we chose to analyze 2 and 8 h post FGF1 treatment.
The proteomics workflow is summarized in Fig. 1. Three independent experiments were performed to obtain statistically representative datasets. Eight thousand two hundred ninety-nine proteins were quantified in the global proteome analysis using unique and razor peptides. All proteins were identified in at least two replicates of a single treatment set by 2 or more peptides with 1% FDR.
Fig. 1.
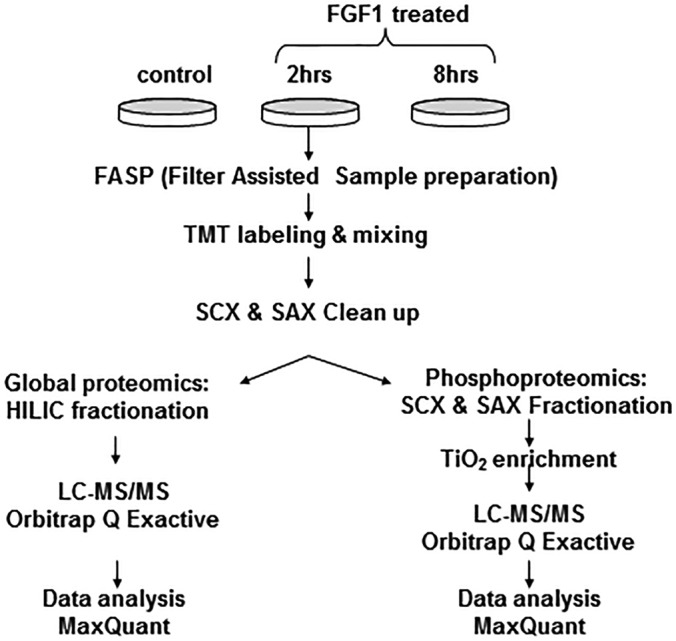
Workflow scheme for the TMT multiplex proteomics analysis.
Very few significant changes occur at the global proteome level in chondrocytes upon FGF1 treatments (Figs. 2A, 2C, and supplemental Fig. S3). The proteins that differ in expression at a statistically significant level were detected only at 8 h of FGF1 treatment and are listed in Table I. Among them S100a6, a member of S100 family of Ca2+-sensor proteins, Syntenin-1 (27), and osteopontin (OPN). OPN has been implicated in chondrocyte differentiation (28), and thus might play a role in defining FGF signaling in this cell type. Accordantly, our previous microarray analysis identified changes in OPN expression upon FGF1 treatment (29). Clearly, the global proteome is predominantly unaffected during intermediate time of FGF1 exposure.
Fig. 2.
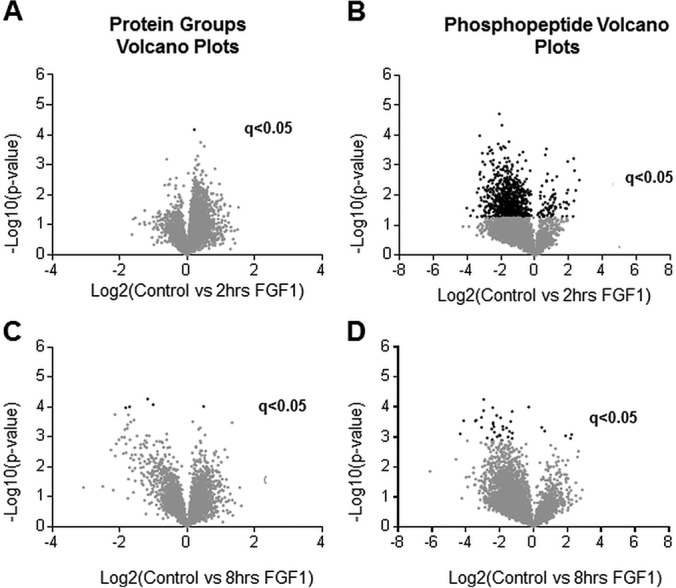
Volcano plots of quantified proteins and phosphopeptides. Volcano plots of global proteome (A, C) and phosphoproteome (B, D). (A) and (B) compare no treatment with 2 h of FGF1 treatment. (C) and (D) compare no treatment with 8 h of FGF1 treatment. A two-sided t test was performed correcting for multiple testing by controlling for FDR at 5%. Proteins or peptides with a q-value of less than 0.05 are considered significant. Data points in black are significant (q < 0.05).
Table I. Proteins that change in intensity upon 8hrs of FGF1 treatment.
| Change in protein intensity | Protein accession number | Genes name | Protein names | |
|---|---|---|---|---|
| 1 | increased | D3ZFS7 | Lyplal1 | Lysophospholipase-Like 1 |
| 2 | decreased | P05964 | S100a6 | S100 Calcium Binding Protein A6 |
| 3 | decreased | P17246 | Tgfb1 | Transforming growth factor beta-1 |
| 4 | decreased | Q9JI92 | Sdcbp | Syntenin-1 |
| 5 | decreased | P08721 | Spp1 | Osteopontin 8 |
Optimization of Phosphopeptide Enrichment
The removal of excess TMT reagent is commonly achieved using SCX; however, this leads to selective losses of acidic peptides including phosphopeptides, which have a neutral or negative charge at pH 2.7. SCX prefractionation was used for enrichment of phosphopeptides as they elute in the flow-through and first few fractions. An SAX cleanup of the SCX flow through was added to the work flow, as SAX preferentially binds phosphopeptides as compared with unmodified peptides (30–32). This allowed for the identification of more than 3000 additional phosphopeptides as compared with SCX alone (supplemental Fig. S4). Sequential SCX and SAX cleanups were used rather than tandem SCX-SAX, as the latter resulted in significant loss of peptides (data not shown). The relative percentage of mono-, di-, tri- and tetra-phosphorylated peptides identified in the SCX and SAX fractions were very similar (supplemental Fig. S4D).
Identified FGF1 Regulated Peptides Uncover Novel Targets and Clusters Regulated by FGF1 in Chondrocytes
We identified a substantial increase in the levels of phosphorylated peptides at 2 h of FGF1 treatment (Figs. 2B, supplemental Fig. S5). 957 out of 4191 phosphorylated peptides that were quantified were found to be differentially phosphorylated at a statistically significant level (5% FDR). The difference in phosphorylation of 37 peptides was statistically significant at 8 h (Fig. 2D). A higher level of variance at the 8 h of FGF treatment accounts for the fewer phosphorylated peptides with a q-value less than 0.05, which is not surprising considering that we are looking not at the initial waive of phosphorylation events but at the secondary response that mediates inhibitory FGF signaling. The statistically significant phosphorylated peptides with the greatest change in intensity between treated and untreated cells, and the associated proteins are listed in Tables II and III.
Table II. The top ten phosphorylated peptides that increase the most in intensity and are statistically significant in FGF1 treated chondrocytes as compared to the untreated samples.
| Log2 (Ctrl/treated) | -Log10 p value | q-value | Peptide Sequence | UniProt acc # | Gene name | Protein names | |
|---|---|---|---|---|---|---|---|
| 2 hours | |||||||
| 1 | −4.02 | 1.57 | 0.040 | EPLLSSSENGTGGTEAAPADAR | Q9Z1I6.1 | Arhgef1 | Rho guanine nucleotide exchange factor 1 |
| 2 | −3.80 | 1.31 | 0.049 | LDNTPASPPRSPAEPSDIPIAK | D4A858 | Tacc2 | Transforming, Acidic Coiled-Coil Containing Protein 2 |
| 3 | −3.65 | 1.49 | 0.042 | ENGSDTLPSSPGSGDQTLPDHAPF | Q6PCT3 | Tpd52l2 | Tumor protein D54 |
| 4 | −3.42 | 2.09 | 0.021 | SADNSLENPFSK | F1LMP9 | Dab2 | Disabled homolog 2 |
| 5 | −3.29 | 1.64 | 0.040 | LLSSNEDDASILSSPTDR | Q4V8B3 | Med24 | Mediator of RNA polymerase II transcription subunit 24 |
| 6 | −3.27 | 2.69 | 0.018 | QKSAEPSPTVMSSSLGSNLSELDR | Q1EG89 | Pxn | Paxillin |
| 7 | −3.26 | 3.98 | 0.000* | ALAEEASEDEIPSDVDLNDPYFAEEVKK | Q76MT4 | Esf1 | ESF1 homolog |
| 8 | −3.24 | 1.65 | 0.041 | LTGQESGLGDSPPFEKESEPESPMDVDNSK | G3V8M8 | Parg | Poly(ADP-ribose) glycohydrolase |
| 9 | −3.23 | 2.77 | 0.011 | SSSTSSSTVTSSAGSEQQNQSSSGSESTDK | F1M771 | Rybp | Death Effector Domain-Associated Factor |
| 10 | −3.23 | 1.63 | 0.040 | IGSDPLAYEPK | P26431 | Slc9a1 | Sodium/hydrogen exchanger1 |
| 8 hours | |||||||
| 1 | −4.32 | 3.10 | 0.019 | GESIEPLDPSEK | G3V6N1 | Erbb3 | Receptor tyrosine-protein kinase erbB-3 |
| 2 | −4.13 | 3.54 | 0.000* | TLGLSSPCDNR | Q64346 | Dusp6 | Dual specificity protein phosphatase 6 |
| 3 | −3.43 | 3.53 | 0.000* | LSQVNGSTPVSPVEPESK | O35821 | Mybbp1a | Myb-binding protein 1A |
| 4 | −3.39 | 3.56 | 0.000* | ALAEEASEDEIPSDVDLNDPYFAEEVKK | Q76MT4 | Esf1 | ESF1 homolog |
| 5 | −3.07 | 3.65 | 0.000* | GDSLAYGLR | P08721 | Spp1 | Osteopontin |
| 6 | −2.95 | 3.88 | 0.000* | SWESSSPVDRPELEAASPTTR | B2RYM6 | Zc3hc1 | Zinc Finger, C3HC-Type Containing 1(NIPA) |
| 7 | −2.94 | 4.26 | 0.000* | GSPTGSSPNNASELSLASLTEK | Q4KLM7 | Specc1 | Sperm Antigen With Calponin Homology And Coiled-Coil Domains 1 |
| 8 | −2.73 | 2.97 | 0.047 | EAIEMHENNGSTK | F1M9I4 | Heg1 | Heart Development Protein With EGF-Like Domains 1 |
| 9 | −2.48 | 3.26 | 0.015 | KVMDSDEDDDY | D4ADF5 | Pdcd5 | Programmed Cell Death 5 |
| 10 | −2.43 | 2.91 | 0.048 | KTSASPPLEKSGDEGSEDEAASGED | Q9EPJ0 | Nucks1 | Nuclear ubiquitous casein and cyclin-dependent kinase substrate 1 |
*q-value is less than 0.0009.
Table III. The phosphorylated peptides that decreased the most in intensity and are statistically significant in FGF1 treated chondrocytes as compared to the untreated samples.
| Log2 (Ctrl/treated) | -Log10 p value | q-value | Peptide Sequence | UniProt acc # | Gene name | Protein names | |
|---|---|---|---|---|---|---|---|
| 2 hours | |||||||
| 1 | 2.64 | 2.51 | 0.018 | SVASPVVISIPER | E9PTE1 | Son | SON DNA Binding Protein |
| 2 | 2.43 | 2.16 | 0.022 | NSDLFTVLSR | E9PTY6 | Fry | Furry Homolog |
| 3 | 2.35 | 2.82 | 0.012 | SPFEGAVTESQSLFSDNFR | Q498M7 | Cnot4 | CCR4-NOT Transcription Complex, Subunit 4 |
| 4 | 2.32 | 1.81 | 0.034 | SMSLIPTSPQAPGEWPSPEELGAR | D4A769 | Samd4b | Sterile Alpha Motif Domain Containing 4B |
| 5 | 2.31 | 3.22 | 0.023 | DVFASYLNSNIQSPSVK | D3ZUL8 | Zcchc8 | Zinc Finger, CCHC Domain Containing 8 |
| 6 | 2.27 | 1.29 | 0.048 | TSLMSAESPTSR | O88777 | Psen2 | Presenilin-2 |
| 7 | 1.97 | 2.17 | 0.022 | NIILAPESCEGSPR | G3V6I1 | Llgl1 | Lethal Giant Larvae Homolog 1 |
| 8 | 1.91 | 2.07 | 0.021 | VPSGLFDTNNR | F1LTD7 | Dennd4c | DENN/MADD Domain Containing 4C |
| 9 | 1.90 | 1.3 | 0.048 | LSPEPVAHR | Q5M9G6 | Snip1 | Smad nuclear interacting protein 1 |
| 10 | 1.90 | 1.23 | 0.040 | RRTPSPPPR | B2RYB3 | Srrm1 | Serine/Arginine Repetitive Matrix 1 |
| 8 hours | |||||||
| 1 | 2.24 | 3.08 | 0.025 | NIILAPESCEGSPR | G3V6I1 | Llgl1 | Lethal Giant Larvae Homolog 1 |
| 2 | 2.19 | 2.95 | 0.046 | KGSLGISSR | Q9Z1I6 | Arhgef1 | Rho guanine nucleotide exchange factor 1 |
| 3 | 1.89 | 3.04 | 0.033 | TNATSPGVNSSASPQSTDK | D4A208 | Srgap2 | SLIT-ROBO Rho GTPase-activating protein 2 |
| 4 | 0.66 | 3.20 | 0.014 | SFSKEVEER | D4AD15 | Eif4g1 | eIF4GI |
| 5 | 0.48 | 3.32 | 0.009 | KQQLLDSDEEDTDDER | Q9Z2Y1 | Timeless | Protein timeless homolog |
Canonical FGF Signaling in RCS Cells
We next compared the dataset of FGF regulated proteins with known effectors of proliferative FGF response (Pathway Unification Database (Weizmann Institute of Science, Version 4.1.017)). Proteins from each tier of the MAPK cascade were detected in RCS cells including their upstream activators: FRS2 (fibroblast growth factor receptor substrate 2), GRB2 (growth factor receptor bound 2), SHP2 and SOS (Son of sevenless) (supplemental Table S1 and Fig. 4B). The majority of them, including FRS2, SOS, Ras, Raf, MAP3K3, MAP3K7, MAP2K4, and MAPK14 (p38α), were identified in our data set. We also found that ERK1/2 negative regulator, DUSP6 (33), was robustly phosphorylated on S351 upon FGF1 treatment.
Activation of the phospholipase Cγ (PLCγ) pathway is triggered upon PLCγ binding to FGFR. Six phospholipase isoforms were detected in RCS cells, two of them, PLCδ1 and PLCγ1 were phosphorylated upon FGF1 treatment. Importantly, we determined that PKCα (protein kinase C alpha), a PLCγ downstream target, was phosphorylated on S226 and T228, the residues responsible for its activation.
The PI3K/AKT signaling branch is responsible for survival and anti-apoptotic response. The following PI3K subunits were detected in the RCS global proteome: PIK3CB (p110β), PIK3C3 (p100), PIK3R(p85α), PIK3R4(p150), and PIK3R2(p85β)/PIK3R3(p55γ). No activating tyrosine phosphorylation of p85 was detected, likely because of the transient nature of this phosphorylation. Furthermore, tyrosine phosphorylation is only about 1% of the total phosphoproteome and an additional enrichment step is usually necessary to identify this low-level modification. Nevertheless, PI3K activation was confirmed by detection of S244 (S241 in humans) phosphorylation of PDK1 (3-Phosphoinositide Dependent Protein Kinase 1). The latter is activated by phosphatidylinositols that are in turn phosphorylated by PI3K. This phosphorylation is necessary for PDK1 enzymatic activity and leads to AKT activation by phosphorylation of T308 following S473. It has been shown by our group that AKT phosphorylation is decreased upon FGF1 treatment in chondrocytes (34) (supplemental Fig. S2) and no corresponding AKT phosphopeptides were detected upon FGF1 treatment. Therefore, our data for the first time localize the discrepancy in the PI3K/AKT signaling network in chondrocytes relative to canonical FGF response. Interestingly, we found that FGF1 treatment resulted in S126 phosphorylation of AKT. CK2 (casein kinase 2) was shown to be responsible for this phosphorylation in Jurkat cells where it led to increased AKT activity (35). A possible role of this phosphorylation in mediating FGF response in chondrocytes should be elucidated further. Several signaling molecules and their direct targets detected in our study were confirmed by immunoblotting (Figs. 3 and 4A–4B). To validate our findings, RCS cells were also treated with FGF2 and FGF18, the growth factors whose roles in chondrocyte biology were established (1). All FGFs caused identical changes in the phosphorylation status of assayed targets with FGF18 demonstrating slightly weaker effect likely because of difference in the activity of recombinant proteins.
Fig. 3.
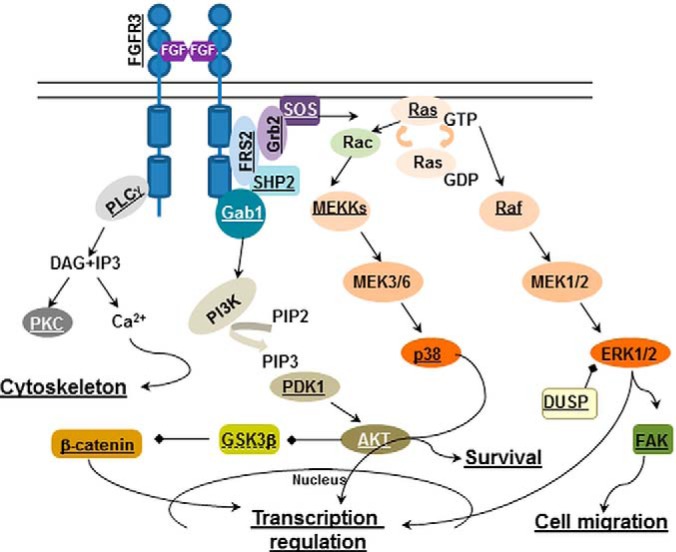
The known FGF signaling pathways in chondrocytes annotated to reflect proteins identified in the inhibitory FGF signaling. Underlined proteins are present in the dataset of targets regulated by FGF1 in RCS cells. GSK3β is underlined with a dotted line, as its role in the inhibitory FGF signaling was determined because of initial identification of its downstream target, b-catenin.
Downstream targets affected by FGF1 signaling in chondrocytes
Most of the identified by phosphoproteomics proteins have not been previously related to the mentioned pathways, and likely represent downstream targets of FGF signaling in chondrocytes. These targets belong to functionally and structurally diverse protein families. We validated some of them by different approaches (Fig. 4A, 4C–4E).
Immunoblotting confirmed phosphorylation of S6K (Ribosomal protein S6 kinase) on T421 and S424. Interestingly, according to the identified phosphopeptides, the p85S6K isoform is more highly phosphorylated as compared with p70. We also identified a novel phosphorylation site at T687 on p85 S6K. CDK1 (cyclin dependent kinase 1) was previously implicated in FGF1 signaling in chondrocytes (36), and accordingly was present in the obtained dataset. CDK1 phosphorylation was validated using phospho-specific antibodies (Fig. 4C). We also confirmed phosphorylation of Stathmin (OP18), a protein that destabilizes microtubules. It was found to be phosphorylated on S25 and S38. Bad (BCL2-associated agonist of cell death), which is involved in programmed cell death, was found to be phosphorylated on S112 at 2 h of FGF1 treatment. All these phosphorylation sites were confirmed by immunoblotting using lysates from the cells treated with FGF1, FGF2, and FGF18 with similar results (Fig. 4C, 4A). Unfortunately, availability of phospho-specific antibodies is a common limitation for validating targets with post-translational modifications. PCDC4 (programmed cell death protein 4) phosphorylation on S76 was detected at 2 and 8 h of FGF1 treatment, however there are no commercially available antibodies recognizing this phosphorylation. It was shown that PDCD4 phosphorylation leads to degradation (37), therefore, observed fluctuations in PDCD4 expression levels might be attributed to the changes in its phosphorylation status/stability (Fig. 5D), which indirectly supports our findings.
We also used 2D electrophoresis to detect changes in the phosphorylation status of proteins in FGF1 treated RCS cells. Protein lysates were enriched for phosphoproteins (supplemental Fig. S6A) and separated using 2D gels (Figs. 4E, (supplemental Fig. S6B and S6C). The silver stained gels were evaluated and several spots with significantly different intensities between treated and untreated samples (circled in green) were analyzed by mass-spectrometry. Two proteins that showed changes were identified as dynein (a motor protein, dynein 1 light intermediate chain 2) and Dynamin-1-like protein. According to our phosphoproteomics data, these proteins were phosphorylated on S194 and S635, respectively. To demonstrate that our targets are likely relative to the situation in vivo we used micromass culture as an ex vivo model of proliferating chondrocytes. The high-density micromass cultures derived from prechondrogenic limb bud mesenchyme from E12.5 embryos and form multiple condensations in which both chondrocyte differentiation and proliferation take place (38). In accordance with our previous results in immortalized cells, CDK1 and S6K were phosphorylated upon FGF1 treatment with kinetics similar to RCS cells (Fig. 4F). Thus, obtained dataset of FGF1 regulated proteins contains potential targets that can serve as a launching point for further studies of FGF signaling in chondrocytes and the relationship of different signaling pathways to the skeletal abnormalities, such as achondroplasia.
GO Analysis Reveals Proteins Assigned to Negative Regulation Clusters
Next, we analyzed our set of FGF regulated peptides for enrichment of biological annotations of gene ontology (GO) terms using DAVID (25). The global proteome identified in RCS cells was used as a background for this analysis. The settings were as follows: EASE - 0.05; classification stringency - high.
One hundred fourteen clusters were identified at 2 h of FGF1 treatment and 4 clusters at 8. Table IV shows the top identified clusters with the enrichment score of 1.5 or higher. We omitted the clusters relative to functional protein domains to simplify the presentation.
Table IV. Clustered enrichment analysis of GO terms describing biological processes (control vs treated) with an enrichment score cutoff of 1.5.
| Cluster | Count of terms | Enrichment score | p value | |
|---|---|---|---|---|
| 2 hours | ||||
| 1 | Transcription regulation | 40 | 3.11 | 2.50E-04 |
| 2 | Regulation of cellular response to stress | 10 | 2.85 | 2.30E-03 |
| 3 | Organelle lumen | 59 | 2.63 | 3.10E-02 |
| 4 | Negative regulation of cellular biosynthetic process | 25 | 2.43 | 2.30E-03 |
| 5 | mRNA processing | 20 | 2.22 | 3.30E-03 |
| 6 | Regulation of microtubule cytoskeleton organization | 7 | 2.09 | 1.80E-03 |
| 7 | Protein serine/threonine kinase activity | 22 | 2.08 | 2.70E-0.3 |
| 8 | Negative regulation of protein kinase activity | 8 | 2.05 | 6.50E-03 |
| 9 | Regulation of phosphorylation | 20 | 2.02 | 1.00E-02 |
| 10 | Negative regulation of transcription | 20 | 1.91 | 1.10E-02 |
| 11 | Positive regulation of cellular biosynthetic process | 26 | 1.83 | 8.40E-03 |
| 12 | Cell projection morphogenesis | 12 | 1.74 | 2.10E-02 |
| 13 | Nuclear mRNA splicing, via spliceosome | 11 | 1.63 | 2.30E-02 |
| 14 | Regulation of nervous system development | 12 | 1.6 | 2.30E-02 |
| 15 | Cytoplasmic dynein complex | 4 | 1.51 | 8.80E-03 |
| 8 hours | ||||
| 1 | Transcription | 5 | 1.5 | 6.20E-02 |
Remarkably, several top clusters were assigned to negative regulation of cellular biosynthetic processes, protein kinase activity, and transcription, with the enrichment scores higher than positive regulation of cellular biosynthetic process (Tables IV, (supplemental Table S2). To compare two time points, we determined clustering of the peptides which are significantly different between 2 and 8 h post FGF1 treatment (supplemental Table S3). One of the top clusters was “Negative regulation of cellular protein metabolic process,” and no targets were assigned to cell proliferation with the enrichment score higher than 1.5. This finding suggests that whereas initial response to FGF signaling harbors signatures of both proliferative and inhibitory responses, later response solely directs inhibition of cell proliferation.
Pathway analysis performed by DAVID and Panther identified FGF, VEGF, EGF and PDGF, focal adhesion and ERBB pathways involved in FGF response of chondrocytes. The similarity between signaling by different growth factors is likely responsible for identification of several closely related growth factor pathways.
GSK3β is Crucial for Mediating FGF Inhibitory Response in Chondrocytes
Next, we thought to investigate the functional importance of targets comprising our newly identified set of FGF1 regulated peptides. One of the proteins assigned to the “negative regulation of cellular biosynthetic processes” cluster was β-catenin, a known player in bone development (39). In line with our phosphoproteomics data, β-catenin phosphorylation was increased following FGF1 treatment while total protein expression was decreased as phosphorylated β-catenin is targeted for degradation (Fig. 5A). Importantly, these changes were mirrored in the growth plate of newborn mice. Endochondral bone formation is driven by changes in the growth plate, where proliferative (P) chondrocytes differentiate into hypertrophic (H) (Fig. 5B). FGFR3 is primarily expressed in proliferating chondrocytes, where it regulates proliferation and the transition to hypertrophic differentiation (3, 40). This implies that in vitro FGF-treated cells would represent more differentiated chondrocytes compared with untreated ones. As shown in Fig. 5B, the percentage of β-catenin positive cells is significantly less in hypertrophic chondrocytes compared with proliferating and resting zones, supporting our hypothesis that β-catenin expression is likely modulated by FGF signaling in vivo. We therefore investigated the activity of GSK3β, an upstream kinase of β-catenin.
GSK3β activity is negatively regulated by AKT-mediated phosphorylation on S9 (41). Thus, canonical FGF signaling is expected to inactivate GSK3β through AKT activation. Clearly, this is an unlikely scenario in our case, as we observed both increased β-catenin phosphorylation and decreased AKT phosphorylation/activity (supplemental Fig. S2C). We detected an increase in GSK3β activating S215 phosphorylation, indicating that the kinase is activated upon FGF1 treatment. In line with these data, immunoblot analysis confirmed a decrease in S9 inhibitory phosphorylation following FGF1 treatment (Fig. 5A).
Next, we used a panel of GSK3β inhibitors to validate its role in the FGF1 response of chondrocytes. CHIR and SB415286 treatment resulted in increased β-catenin stability (Fig. 5C). Importantly, GSK3β inhibition by any compound completely prevented FGF induced growth inhibition as assayed by FACScan analysis (Fig. 5C). This effect was dose-dependent, validating the specificity of the used compounds (Fig. 5D). To validate the importance of GSK3β activity in vivo we used two ex vivo models (micromass cultures, described above, and organ culture of metatarsal bone rudiments with normal cartilage architecture (42)). The cells and rudiments were pretreated with GSK3β inhibitors following FGF1 treatment (Fig. 5E, 5F). The inhibition of GSK3β was confirmed in micromass culture by increased β-catenin expression (Fig. 5E). In line with our data in RCS cells, in both system inhibition of GSK3β activity prevented FGF1 induced growth arrest (Fig. 5E, 5F) supporting our hypothesis that GSK3β activity is crucial for mediating FGF response in chondrocytes.
Taken together, our findings demonstrate that the obtained dataset of FGF regulated peptides can be successfully used to discover novel important modulators and targets of FGF inhibitory response in chondrocytes. Moreover, these data can be used to further delineate the mechanism of this unique response and find novel therapeutic approaches in cases when this axis is dysregulated.
DISCUSSION
For the first time phosphoproteomics was used to study the FGF inhibitory response. We could comprehensively characterize the chondrocyte global proteome (8299 proteins) and to catalogue proteins whose phosphorylation status is modulated by FGF1 signaling. One constraint of our study which should be considered is an identification of low abundance molecules. For example, ERK1/2 was not identified in the global chondrocyte proteome, but was detected by immunoblotting. This concern might be addressed in the future by either depletion of high abundance proteins or by enrichment of low abundance polypeptides. Despite this shortcoming, we built up an impressive set of FGF1 regulated peptides that consists of 756 proteins, cataloguing many novel FGF targets.
Although our major goal was to identify novel FGF targets, the phosphoproteomic approach also allowed us to extensively characterize the FGF signaling pathway itself. We documented isoform specific expression and activation for signaling molecules comprising MAPK, PI3K/AKT and PLCγ pathways and localized the disruption of the PI3K/AKT pathway. FGF1 induced PDK1 activation did not result in AKT phosphorylation, implying that a phosphatase responsible for dephosphorylation of S473 and/or T308 might overpower kinase activity of PDK1. PH domain leucine-rich repeat protein phosphatase 1 (PHLPP) specifically dephosphorylates S473 (43). Phlpp1-deficient mice were previously reported to have slightly decreased snout-to-tail lengths (44) and, as was confirmed later, shorter femurs (45). It was suggested that Phlpp1 effects chondrocytes proliferation through its direct target AKT2 that manipulates FoxO1 levels and consequently FGF18 expression (45). Our data indicate that Phlpp1 activity is, at least partially, modulated by FGF signaling and it will be important to digest this axis in more details in the future.
Another interesting insight into FGF signaling in chondrocytes involves DUSP6. Although we detected DUSP6 phosphorylation (S351), we did not identify phosphorylation of S159 and S197, the residues that are responsible for FGF-induced DUSP6 degradation in other instances of FGF signaling (46). It would be noteworthy to see if there is any preference for this phosphorylation upon inhibitory signaling as compared with proliferative FGF response and to determine the functional importance of S351 phosphorylation. Taking into account that DUSP6−/− mice have many abnormalities similar to those found in animals expressing the mutant (activating) form of FGFR3 (47, 48). DUSP6 might be an important regulator of FGF signaling in the growth plate.
Some of the identified proteins, such as Snail (3, 49), Sox9, CDK1, and 4E-BP1 were implicated in chondrocyte specific FGF response (36, 50, 51). Sox9, for example, becomes phosphorylated on S109 following FGF1 treatment but importance of this modification has not been addressed yet. Surprisingly, we did not detect any phosphorylation of STAT1 (Signal transducer and activator of transcription 1) (6, 52) which might be attributed to limitations of our approach. Yet, these data are in line with the recent work of Kreci et al. (53) suggesting that the role of STAT1 in chondrocytes might be reevaluated, and phosphorylation independent STAT1 functions should be considered.
GO analysis of the FGF regulated peptides revealed that numerous proteins are assigned to the clusters of “negative regulation.” This result suggests that FGF inhibitory response is a team effort with many important players and would explain why we can counteract FGF-induced inhibition by modulating activity of functionally different targets: p107, CDK2, PP2A (protein phosphatase 2A) and 4E-BP1 (19, 36, 51, 54).
Here we could identify an additional key mediator of FGF signaling-GSK3β kinase. We demonstrated that FGF1 signaling activates GSK3β and its inhibition overturns the FGF inhibitory response of chondrocytes in cell culture and in ex vivo models. Our experiments are in line with ex vivo data published by Dr. Beier's group (55) where prolong GSK3β inhibition in a metatarsal organ culture system caused increased longitudinal growth of endochondral bones mimicking the phenotype of FGFR3 knockout mice (56, 57). Similar effect on the tibia length was reported in cartilage-specific GSK3β KO mice (58). At this point it is not clear why Kapadia et al. (59) had an opposite result showing that a different pharmacological inhibitor reduced chondrocyte proliferation in a metatarsal organ culture model. This discrepancy might be because of the nature of the inhibitors, treatment conditions, or the identity of the monitored skeletal elements. Therefore, the GSK3β role in chondrocytes biology should be investigated further in different contexts, including FGF signaling. It will be also interesting to examine specific roles of different GSK3β targets. Though we identified β-catenin as one of the FGF1 related GSK3β targets, other downstream targets might be important for proper execution of FGF signaling as well, for example, RelA, that was identified as a substrate of GSK3β activity in chondrocytes (60).
In summary, the presented pool of FGF regulated peptides is a powerful resource for future identification of novel targets of FGF signaling in chondrocytes and the characterization of novel phosphorylation sites. This will ultimately broaden our understanding of the pathways involved in both negative and positive FGF regulation of cell homeostasis and will lead to the discovery of novel potential pharmacological targets when the proper signaling is disrupted under pathological conditions.
DATA AVAILABILITY
All raw mass spectrometry data and search results have been deposited to the ProteomeXchange Consortium “http://www.mcponline.org/content/15/10/3090.full” via the MassIVE partner repository with the data set identifier MassIVE: MSV000080259 and ProteomeXchange: PXD005199.
Supplementary Material
Footnotes
Author contributions: J.R.C., B.U., and V.K. designed research; J.R.C., O.K., R.R., S.N., D.M., and V.K. performed research; J.R.C., O.K., D.M., S.N., C.B., B.U., and V.K. analyzed data; J.R.C., S.N., C.B., B.U., and V.K. wrote the paper.
* This work was supported by NIH grant AR063128 (to V.K.) and with a Shared Instrumentation Grant from the NIH/ORIP S10OD010582 for the purchase of an Orbitrap Fusion mass spectrometer. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- FGF
- Fibroblast Growth Factor
- GO
- Gene Ontology
- FGFR3
- Fibroblast Growth Factor Receptor 3
- PLCγ
- Phospholipase C Gamma
- FRS2
- Fibroblast Growth Factor Receptor Substrate 2
- Grb2
- Growth Factor Receptor-Bound Protein 2
- SHP2
- Src Homology Phosphatase 2
- Gab1
- Grb2-Associated Binder-1
- SOS
- Son Of Sevenless Guanine Nucleotide Exchange Factor
- DAG
- Diaglycerol
- IP3
- Inositol Triphosphate
- PKC
- Protein Kinase C
- PI3K
- Phospho Inositide 3-Kinase
- PIP2
- Phosphatidylinositol 4,5-Bisphosphate
- PIP3
- Phosphatidylinositol 4,5-Triphosphate
- PDK1
- Phosphoinositide –Dependent Kinase-2
- GSK3β
- Glycogen Synthase Kinase 3-Beta
- AKT
- Protein Kinase B
- MEKK
- Mitogen Activated Protein Kinase
- GTP
- Guanosine 5′ Triphosphate
- GDP
- Guanosine 5′ Diphosphate
- MEK
- Mitogen-Activated Protein Kinase Kinase
- IHC
- Immunohistochemistry
- TMT
- Tandem Mass Tags
- NCE
- Normalized Collision Energy
- FDR
- False Discovery Rate
- DAVID
- Database for Annotation, Visualization, and Integrated Discovery
- SCX
- Strong Cation Exchange
- SAX
- Strong Anion Exchange.
REFERENCES
- 1. Ornitz D. M., and Itoh N. (2015) The Fibroblast Growth Factor signaling pathway. Rev. Dev. Biol. 4, 215–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goetz R., and Mohammadi M. (2013) Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 14, 166–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ornitz D. M., and Marie P. J. (2015) Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 29, 1463–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aikawa T., Segre G. V., and Lee K. (2001) Fibroblast growth factor inhibits chondrocytic growth through induction of p21 and subsequent inactivation of cyclin E-Cdk2. J. Biol. Chem. 276, 29347–29352 [DOI] [PubMed] [Google Scholar]
- 5. Krejci P., Bryja V., Pachernik J., Hampl A., Pogue R., Mekikian P., and Wilcox W. R. (2004) FGF2 inhibits proliferation and alters the cartilage-like phenotype of RCS cells. Exp. Cell Res. 297, 152–164 [DOI] [PubMed] [Google Scholar]
- 6. Sahni M., Ambrosetti D. C., Mansukhani A., Gertner R., Levy D., and Basilico C. (1999) FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. 13, 1361–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Colvin J. S., Bohne B. A., Harding G. W., McEwen D. G., and Ornitz D. M. (1996) Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat. Genet. 12, 390–7 [DOI] [PubMed] [Google Scholar]
- 8. Dailey L., Ambrosetti D., Mansukhani A., and Basilico C. (2005) Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 16, 233–247 [DOI] [PubMed] [Google Scholar]
- 9. Lazarus J. E., Hegde A., Andrade A. C., Nilsson O., and Baron J. (2007) Fibroblast growth factor expression in the postnatal growth plate. Bone 40, 577–586 [DOI] [PubMed] [Google Scholar]
- 10. Ornitz D. M. (2005) FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 16, 205–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cunningham D. L., Sweet S. M., Cooper H. J., and Heath J. K. (2010) Differential phosphoproteomics of fibroblast growth factor signaling: identification of Src family kinase-mediated phosphorylation events. J. Proteome Res. 9, 2317–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zoumaro-Djayoon A. D., Ding V., Foong L. Y., Choo A., Heck A. J., and Munoz J. (2011) Investigating the role of FGF-2 in stem cell maintenance by global phosphoproteomics profiling. Proteomics 11, 3962–3971 [DOI] [PubMed] [Google Scholar]
- 13. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., and Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 14. Thompson A., Schafer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., Neumann T., Johnstone R., Mohammed A.K., and Hamon C. (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904 [DOI] [PubMed] [Google Scholar]
- 15. Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., and Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 16. McAlister G. C., Russell J. D., Rumachik N. G., Hebert A. S., Syka J. E., Geer L. Y., Westphall M. S., Pagliarini D. J., and Coon J. J. (2012) Analysis of the acidic proteome with negative electron-transfer dissociation mass spectrometry. Anal. Chem. 84, 2875–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Raucci A., Laplantine E., Mansukhani A., and Basilico C. (2004) Activation of the ERK1/2 and p38 mitogen-activated protein kinase pathways mediates fibroblast growth factor-induced growth arrest of chondrocytes. J. Biol. Chem. 279, 1747–1756 [DOI] [PubMed] [Google Scholar]
- 18. Mukhopadhyay K., Lefebvre V., Zhou G., Garofalo S., Kimura J.H., and de Crombrugghe B. (1995) Use of a new rat chondrosarcoma cell line to delineate a 119-base pair chondrocyte-specific enhancer element and to define active promoter segments in the mouse pro-alpha 1(II) collagen gene. J. Biol. Chem. 270, 27711–27719 [DOI] [PubMed] [Google Scholar]
- 19. Kolupaeva V., Laplantine E., and Basilico C. (2008) PP2A-mediated dephosphorylation of p107 plays a critical role in chondrocyte cell cycle arrest by FGF. PLoS One 3, e3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wisniewski J. R., Zougman A., Nagaraj N., and Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 21. McNulty D.E., and Annan R.S. (2008) Hydrophilic interaction chromatography reduces the complexity of the phosphoproteome and improves global phosphopeptide isolation and detection. Mol. Cell. Proteomics 7, 971–980 [DOI] [PubMed] [Google Scholar]
- 22. Tan H., Wu Z., Wang H., Bai B., Li Y., Wang X., Zhai B., Beach T.G., and Peng J. (2015) Refined phosphopeptide enrichment by phosphate additive and the analysis of human brain phosphoproteome. Proteomics 15, 500–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rappsilber J., Ishihama Y., and Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 24. Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., and Mann M. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang, da W., Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 26. Huang, da W., Sherman B. T., and Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jeon H. Y., Das S. K., Dasgupta S., Emdad L., Sarkar D., Kim S. H., Lee S. G., and Fisher P. B. (2013) Expression patterns of MDA-9/syntenin during development of the mouse embryo. J. Mol. Histol. 44, 159–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barak-Shalom T., Schickler M., Knopov V., Shapira R., Hurwitz S., and Pines M. (1995) Synthesis and phosphorylation of osteopontin by avian epiphyseal growth-plate chondrocytes as affected by differentiation. Comp. Biochem. Physiol 111, 49–59 [DOI] [PubMed] [Google Scholar]
- 29. Dailey L., Laplantine E., Priore R., and Basilico C. (2003) A network of transcriptional and signaling events is activated by FGF to induce chondrocyte growth arrest and differentiation. J. Cell Biol. 161, 1053–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mohammed S., and Heck A. Jr. (2011) Strong cation exchange (SCX) based analytical methods for the targeted analysis of protein post-translational modifications. Curr. Opin. Biotechnol. 22, 9–16 [DOI] [PubMed] [Google Scholar]
- 31. Trinidad J. C., Specht C. G., Thalhammer A., Schoepfer R., and Burlingame A. L. (2006) Comprehensive identification of phosphorylation sites in postsynaptic density preparations. Mol. Cell. Proteomics 5, 914–922 [DOI] [PubMed] [Google Scholar]
- 32. Villen J., and Gygi S.P. (2008) The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc. 3, 1630–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eswarakumar V. P., Lax I., and Schlessinger J. (2005) Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 16, 139–149 [DOI] [PubMed] [Google Scholar]
- 34. Priore R., Dailey L., and Basilico C. (2006) Downregulation of Akt activity contributes to the growth arrest induced by FGF in chondrocytes. J. Cell Physiol. 207, 800–808 [DOI] [PubMed] [Google Scholar]
- 35. Di Maira G., Salvi M., Arrigoni G., Marin O., Sarno S., Brustolon F., Pinna L.A., and Ruzzene M. (2005) Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 12, 668–677 [DOI] [PubMed] [Google Scholar]
- 36. Tran T., Kolupaeva V., and Basilico C. (2010) FGF inhibits the activity of the cyclin B1/CDK1 kinase to induce a transient G(2)arrest in RCS chondrocytes. Cell Cycle 9, 4379–4386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dorrello N.V., Peschiaroli A., Guardavaccaro D., Colburn N.H., Sherman N.E., and Pagano M. (2006) S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314, 467–471 [DOI] [PubMed] [Google Scholar]
- 38. Ahrens P. B., Solursh M., Reiter R. S., and Singley C. T. (1979) Position-related capacity for differentiation of limb mesenchyme in cell culture. Dev. Biol. 69, 436–450 [DOI] [PubMed] [Google Scholar]
- 39. Usami Y., Gunawardena A. T., Iwamoto M., and Enomoto-Iwamoto M. (2016) Wnt signaling in cartilage development and diseases: lessons from animal studies. Lab. Invest. 96, 186–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Laederich M. B., and Horton W. A. (2012) FGFR3 targeting strategies for achondroplasia. Expert Rev. Mol. Med. 14, e11. [DOI] [PubMed] [Google Scholar]
- 41. Katoh M., and Katoh M. (2006) Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol. Ther. 5, 1059–1064 [DOI] [PubMed] [Google Scholar]
- 42. Laplantine E., Rossi F., Sahni M., Basilico C., and Cobrinik D. (2002) FGF signaling targets the pRb-related p107 and p130 proteins to induce chondrocyte growth arrest. J. Cell Biol. 158, 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gao T., Furnari F., and Newton A. C. (2005) PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 18, 13–24 [DOI] [PubMed] [Google Scholar]
- 44. Masubuchi S., Gao T., O'Neill A., Eckel-Mahan K., Newton A. C., and Sassone-Corsi P. (2010) Protein phosphatase PHLPP1 controls the light-induced resetting of the circadian clock. Proc. Natl. Acad. Sci. U S A 107, 1642–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bradley E. W., Carpio L. R., Newton A. C., and Westendorf J. J. (2015) Deletion of the PH-domain and leucine-rich repeat protein phosphatase 1 (Phlpp1) increases fibroblast growth factor (Fgf) 18 expression and promotes chondrocyte proliferation. J. Biol. Chem. 290, 16272–16280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marchetti S., Gimond C., Chambard J.C., Touboul T., Roux D., Pouyssegur J., and Pages G. (2005) Extracellular signal-regulated kinases phosphorylate mitogen-activated protein kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites critical for its proteasomal degradation. Mol. Cell Biol. 25, 854–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brodie S. G., and Deng C. X. (2003) Mouse models orthologous to FGFR3-related skeletal dysplasias. Pediatr. Pathol. Mol. Med. 22, 87–103 [DOI] [PubMed] [Google Scholar]
- 48. Li C., Scott D. A., Hatch E., Tian X., and Mansour S. L. (2007) Dusp6 (Mkp3) is a negative feedback regulator of FGF-stimulated ERK signaling during mouse development. Development 134, 167–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou B. P., Deng J., Xia W., Xu J., Li Y. M., Gunduz M., and Hung M. C. (2004) Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 6, 931–940 [DOI] [PubMed] [Google Scholar]
- 50. Bi W., Deng J. M., Zhang Z., Behringer R. R., and de Crombrugghe B. (1999) Sox9 is required for cartilage formation. Nat. Genet. 22, 85–89 [DOI] [PubMed] [Google Scholar]
- 51. Ruoff R., Katsara O., and Kolupaeva V. (2016) Cell type-specific control of protein synthesis and proliferation by FGF-dependent signaling to the translation repressor 4E-BP. Proc. Natl. Acad. Sci. U S A. 113, 7545–7550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Su W. C., Kitagawa M., Xue N., Xie B., Garofalo S., Cho J., Deng C., Horton W. A., and Fu X. Y. (1997) Activation of Stat1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature 386, 288–292 [DOI] [PubMed] [Google Scholar]
- 53. Krejci P., Salazar L., Goodridge H. S., Kashiwada T. A., Schibler M. J., Jelinkova P., Thompson L. M., and Wilcox W. R. (2008) STAT1 and STAT3 do not participate in FGF-mediated growth arrest in chondrocytes. J. Cell Sci. 121, 272–281 [DOI] [PubMed] [Google Scholar]
- 54. Kolupaeva V., Daempfling L., and Basilico C. (2013) The B55alpha regulatory subunit of protein phosphatase 2A mediates fibroblast growth factor-induced p107 dephosphorylation and growth arrest in chondrocytes. Mol. Cell Biol. 33, 2865–2878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gillespie J. R., Ulici V., Dupuis H., Higgs A., Dimattia A., Patel S., Woodgett J. R., and Beier F. (2011) Deletion of glycogen synthase kinase-3beta in cartilage results in up-regulation of glycogen synthase kinase-3alpha protein expression. Endocrinology 152, 1755–1766 [DOI] [PubMed] [Google Scholar]
- 56. Su N., Xu X., Li C., He Q., Zhao L., Li C., Chen S., Luo F., Yi L., Du X., Huang H., Deng C., and Chen L. (2010) Generation of Fgfr3 conditional knockout mice. Int. J. Biol. Sci. 6, 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eswarakumar V.P., and Schlessinger J. (2007) Skeletal overgrowth is mediated by deficiency in a specific isoform of fibroblast growth factor receptor 3. Proc. Natl. Acad. Sci. U S A 104, 3937–3942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhou J., Chen Y., Cao C., Chen X., Gao W., and Zhang L. (2015) Inactivation of glycogen synthase kinase-3beta up-regulates beta-catenin and promotes chondrogenesis. Cell Tissue Bank 16, 135–142 [DOI] [PubMed] [Google Scholar]
- 59. Kapadia R. M., Guntur A. R., Reinhold M. I., and Naski M. C. (2005) Glycogen synthase kinase 3 controls endochondral bone development: contribution of fibroblast growth factor 18. Dev. Biol. 285, 496–507 [DOI] [PubMed] [Google Scholar]
- 60. Itoh S., Saito T., Hirat a M., Ushita M., Ikeda T., Woodgett J.R., Algul H., Schmid R.M., Chung U.I., and Kawaguchi H. (2012) GSK-3alpha and GSK-3beta proteins are involved in early stages of chondrocyte differentiation with functional redundancy through RelA protein phosphorylation. J. Biol. Chem. 287, 29227–29236 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw mass spectrometry data and search results have been deposited to the ProteomeXchange Consortium “http://www.mcponline.org/content/15/10/3090.full” via the MassIVE partner repository with the data set identifier MassIVE: MSV000080259 and ProteomeXchange: PXD005199.