

Abstract
Traditional “bottom-up” proteomic approaches use proteolytic digestion, LC-MS/MS, and database searching to elucidate peptide identities and their parent proteins. Protein sequences absent from the database cannot be identified, and even if present in the database, complete sequence coverage is rarely achieved even for the most abundant proteins in the sample. Thus, sequencing of unknown proteins such as antibodies or constituents of metaproteomes remains a challenging problem. To date, there is no available method for full-length protein sequencing, independent of a reference database, in high throughput. Here, we present Database-independent Protein Sequencing, a method for unambiguous, rapid, database-independent, full-length protein sequencing. The method is a novel combination of non-enzymatic, semi-random cleavage of the protein, LC-MS/MS analysis, peptide de novo sequencing, extraction of peptide tags, and their assembly into a consensus sequence using an algorithm named “Peptide Tag Assembler.” As proof-of-concept, the method was applied to samples of three known proteins representing three size classes and to a previously un-sequenced, clinically relevant monoclonal antibody. Excluding leucine/isoleucine and glutamic acid/deamidated glutamine ambiguities, end-to-end full-length de novo sequencing was achieved with 99–100% accuracy for all benchmarking proteins and the antibody light chain. Accuracy of the sequenced antibody heavy chain, including the entire variable region, was also 100%, but there was a 23-residue gap in the constant region sequence.
The main goal of mass spectrometry-based proteomic experiments is typically protein identification. To achieve this goal, current approaches utilize proteolytic digestion of protein samples followed by LC-MS/MS and database searching to identify the peptides, and thereby their parent proteins (1, 2). Although very powerful when analyzing well characterized organisms, the method has several significant drawbacks when analyzing samples that are not well characterized. First, it strictly depends on a protein database that contains the correct sequence of the measured peptides. Unknown protein sequences cannot be identified. Second, it relies on identification of proteolytic peptides, typically tryptic. Trypsin is used for several reasons, including its high efficiency and specificity. However, because trypsin cleaves the protein only after lysine and arginine residues, tryptic digestion of typical proteins results in some peptides that are too short, too long, too hydrophobic, or contain a sequence of residues that is poorly ionized or fragmented. As a result, even for the most abundant proteins in the sample, sequence coverage of a protein (i.e. the percentage of the entire amino acid sequence covered by measured peptides) is almost never 100%, and there are likely to be regions with no overlap between identified peptides. Enzymatic digestion by other proteases is sometimes performed for specific applications, but they too might result in peptides that are not amenable for identification by LC-MS/MS.
Another strategy for proteomic analysis is peptide de novo sequencing, where the peptide sequence is inferred directly from the MS/MS spectrum, without referring to a database (3, 4). This is done by identifying mass differences between peaks in the MS/MS spectrum that correspond exactly to specific amino acids. The advantage of this approach is that no database is required for identification of a peptide. However, the inherent chemical properties of the peptide and inefficiencies of the instrument might lead to gaps in the de novo sequencing, resulting in only partial or imperfect peptide sequences. Thus, obtaining confident and accurate peptide sequences de novo in high throughput is very challenging. Furthermore, even if the peptide was correctly sequenced de novo, inference to its parent protein is, again, strictly dependent on matching the peptide to a known protein in a database, using BLAST 1 search, for example. For an unknown protein, reconstruction of its full amino acid sequence is very challenging using current bottom-up approaches.
Determination of a protein sequence without prior knowledge is an important and rate-limiting step in analysis of poorly characterized protein samples, such as ones derived from unsequenced organisms, environmental samples, and microbiome. Other important cases are antibodies and T-cell receptors for which the variable region sequences are unknown. To infer the amino acid sequence of an unknown monoclonal antibody of interest, typically cDNA from the source hybridoma is produced, sequenced, and translated. However, hybridoma cells are not always available, or the primers used to amplify the cDNA might not match the target antibody DNA sequence. In such cases, the amino acid sequence of the protein has to be determined directly by proteomic techniques.
To date, there are only a few reported methods attempting to perform end-to-end de novo protein sequencing by LC-MS/MS, of which ALPS and meta-SPS (mSPS) are among the most recent ones (5–10). All such bottom-up methods rely on enzymatic digestion by multiple proteases to generate overlapping peptides, followed by de novo peptide sequencing and assembly. Some of these methods use results from searches against a reference protein database for improving the assembly process. If the analyzed proteins or their close homologs are not represented in that database, thus requiring the use of only de novo sequenced peptides for assembly, these methods are expected to have inferior performance. For example, without using results from a database search against an in-house-generated antibody database, ALPS (8) resulted in a fragmented assembly of all light and heavy chains of their analyzed antibodies. Even with the use of the database search results, one of the two heavy chains analyzed resulted in a fragmented assembly by ALPS, specifically at the variable region of the heavy chain (8). A fragmented assembly is detrimental for determination of the full-length sequence of an unknown protein, because without prior knowledge of the protein sequence, it is not possible to determine which among all contigs (the assembled amino acid sequence stretches that cover part of the polypeptide chain) should be used for the assembly. Using de novo data only, the longest contig assembled by mSPS was 194 amino acids long (using analysis of multiple proteolytic digests with three different fragmentation methods) (7), and no protein was reported to be fully assembled (from N to C terminus) by mSPS in a single contig (5). Thus, de novo sequencing of typical full-length proteins is still challenging using current techniques.
Here we present a proof-of-concept for a full-length de novo protein sequencing method that we named Database-independent Protein Sequencing (DiPS). The method is based on cleavage of the protein at semi-random sites by non-enzymatic, microwave-assisted acid hydrolysis (MAAH), enrichment of LC-MS/MS-amenable peptides from the hydrolysate by solid-phase extraction, LC-MS/MS analysis, de novo peptide sequencing of resulting peptides, extraction of peptide tags from the de novo peptide sequences, and their assembly into consensus contigs (Fig. 1). Within minutes of sample processing followed by standard proteomic analysis, full-length de novo protein sequences can be obtained.
Fig. 1.

Description of DiPS. Proteins are subjected to MAAH by suspension in 3 m HCl and microwaving for 4 min. LC-MS/MS amenable peptides are enriched from the hydrolysates by solid-phase extraction and subjected to standard nLC-MS/MS. Resulting spectra are subjected to de novo sequencing using the PEAKS software. The output from PEAKS is used by pTA to extract and assemble peptide tags into consensus contigs.
EXPERIMENTAL PROCEDURES
Three proteins were subjected to DiPS analysis in three independent replicates. These included bovine serum albumin (BSA), fetuin-A, and myoglobin as benchmarks. Additionally, AR37, a previously un-sequenced monoclonal antibody, was also subjected to DiPS as a test case.
Sample Preparation
All chemicals and proteins were purchased from Sigma-Aldrich, unless stated otherwise. For MAAH, 10 μg of dry protein powder of each BSA (Uniprot accession no. P02769, Sigma-Aldrich catalog no. A2153), fetuin A (Uniprot accession no. P12763, Sigma-Aldrich catalog no. F2379), or equine myoglobin (Uniprot accession no. P68082, Sigma-Aldrich catalog no. M1882) were dissolved in 200 μl of 8 m urea, 0.1 m Tris-HCl, pH 7.9. Dithiothreitol (DTT) was added to final concentration of 5 mm and incubated at 37 °C for 50 min. Iodoacetamide (IAA) was added at a final concentration of 10 mm and incubated 30 min in the dark. Buffer was exchanged to water using an Amicon 3-kDa MWCO filter (Millipore UFC500396) by adding 300 μl of H2O and centrifuging at 14,000 × g until the remaining volume was about 40 μl, and the process was repeated. The remaining volume was collected and transferred to a glass vial with a pre-slit cap (Waters, catalog no. 186000307C). HCl was added to a final concentration of 3 m; the vial was placed on ice in a beaker and microwaved for 4 min (stopping every 1 min to replenish ice) in a standard home microwave (LG Intellowave 1,200 watts) at highest settings. Hydrolysates were then subjected to solid-phase extraction (Oasis HLB, Waters, catalog no. 186001828BA), and peptides were eluted with 80% acetonitrile. Peptide samples were dried using a vacuum centrifuge (Eppendorf Concentrator Plus) and resuspended in 3% acetonitrile, 0.1% formic acid for nanoLC-MS/MS analysis. 1.2 μg of resuspended hydrolyzed protein were loaded onto the chromatography column. AR37 was isolated as described previously (11). Sample processing of AR37 was performed as above with the exception of alkylation with iodoacetate (IAc) (Sigma-Aldrich catalog no. I4386) instead of IAA (identical concentration and conditions to IAA alkylation).
For tryptic digestion, proteins were dissolved in 8 m urea, 0.1 m Tris-HCl, pH 7.9, reduced, and alkylated as described above. Samples were diluted to 2 m urea with 50 mm ammonium bicarbonate. Proteins were then subjected to digestion with trypsin (Promega; Madison, WI) overnight at 37 °C (50:1 protein amount/trypsin), followed by a second trypsin digestion for 4 h. The digestions were stopped by addition of trifluoroacetic acid (1%). Following digestion, peptides were desalted, dried, and resuspended as described above.
Liquid Chromatography
ULC/MS grade solvents were used for all chromatographic steps. Each sample was loaded once (without technical replicates), using split-less nano-ultra performance liquid chromatography (nanoAcquity; Waters). The mobile phase was as follows: A, H2O + 0.1% formic acid; B, acetonitrile + 0.1% formic acid. Desalting of the samples was performed on line using a reversed-phase Symmetry C18 trapping column (180-μm internal diameter, 20-mm length, 5-μm particle size; Waters). The peptides were then separated using an HSS T3 nano-column (75-μm internal diameter, 250-mm length, 1.8-μm particle size; Waters) at 0.35 μl/min. For BSA, fetuin-A, and AR37, peptides were eluted from the column into the mass spectrometer in 3 h using the following gradient: 4–30% B in 140 min and 30–90% B in 25 min, maintained at 95% for 5 min, and then back to initial conditions. For myoglobin, the smallest protein, peptides were eluted from the column into the mass spectrometer in 2 h using the following gradient: 4–30% B in 105 min and 30–90% B in 15 min, maintained at 95% for 5 min, and then back to initial conditions.
Mass Spectrometry
The nano-UPLC was coupled on line through a nano-ESI emitter (10-μm tip; New Objective; Woburn, MA) to a quadrupole orbitrap mass spectrometer (Q Exactive Plus, Thermo Fisher Scientific) using a FlexIon nanospray apparatus (Proxeon).
Data were acquired in data-dependent acquisition (DDA) mode, using a Top20 method. MS1 resolution was set to 70,000 (at 400 m/z), maximum injection time of 20 ms, scan range was 300–1650 m/z, and AGC target of 3e6. MS2 resolution was set to 70,000, maximum injection time of 120 ms, isolation window 1.7 m/z, and AGC target of 1e6. Normalized collision energy was set to 30.
Data Analysis
Raw data were analyzed using the PEAKS 7.0 software (Bioinformatics Solutions Inc, Waterloo, Ontario, Canada) using the de novo module for DiPS or using the database search module for assessment of cleavage efficiency. For BSA, fetuin-A, and myoglobin, analysis parameters included no enzyme specificity, no fixed modifications, and variable modifications as follows: methionine oxidation, cysteine carbamidomethylation, cysteine carboxymethylation, and arginine citrullination. For AR37 (alkylated with IAc), the de novo parameters included no enzyme specificity, fixed modification of cysteine carboxymethylation, and variable modifications as follows: methionine oxidation, arginine citrullination, and glutamine to pyroglutamate conversion. Parent Mass Error Tolerance was 10.0 ppm. Fragment Mass Error Tolerance was 0.02 Da. Maximum variable PTM per peptide was 5. Unfiltered de novo sequenced peptides (minimum “average local confidence score” = 0) were exported as a ‘.csv’ file and used as input for pTA using default parameters: k-mer size = 7, k-mer min overlap = 5, unite overlap size = 5, unite minimum extension = 7, merge minimum quality = 0.7.
For AR37 validation, the tryptic digest was searched against a database containing the DiPS determined heavy and light sequences, as well as 123 common laboratory contaminants. The search was performed using the database search module of the PEAKS algorithm with parameters specifying nonspecific digestion, fixed modification of cysteine carboxymethylation, and variable modifications of methionine oxidation, asparagine/glutamine deamidation, and N-terminal asparagine/glutamine to pyroglutamate. Data was filtered at 1% FDR at the peptide level based on a reversed sequence decoy database search.
The pTA executable and example data are provided as a supplemental file. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (12) partner repository with the dataset identifier PXD003804. The pTA tool is available as a Windows executable (supplemental material) and the code is available via https://bitbucket.org/incpm/dips.
RESULTS
Generation of Overlapping Peptides
DiPS is based on assembly of overlapping de novo sequenced peptide tags into a final consensus sequence of the protein. To this aim, a modified MAAH protocol (first described in Ref. 13) was developed as a simple, cost-effective, and rapid method to cleave proteins at semi-random peptide bonds, thus producing peptides overlapping in sequence, which cover the full protein sequence.
Because of the physical and chemical properties of different peptide bonds along the polypeptide chain, the process of generating peptide tags by DiPS is not completely random. To enhance randomization, each step of the process was optimized based on the highest number of unique peptides identified from a BSA hydrolysate subjected to nanoLC-MS/MS and a standard database search as a benchmark. Optimized parameters included hydrolysis time (supplemental Fig. S1), solid-phase extraction elution (supplemental Fig. S2), and normalized collision energy (NCE) for peptide fragmentation (supplemental Fig. S3).
De Novo Peptide Tags Determination and Consensus Sequence Assembly
After MAAH treatment, the resulting hydrolysate was subjected to nanoLC-MS/MS and de novo peptide sequencing using the commercial PEAKS 7.0 software. An algorithm that we named Peptide Tag Assembler (pTA) was developed for extraction of confident peptide tags from the PEAKS de novo output and their assembly into consensus contigs, based on the de Bruijn graph approach. Here, we refer to “peptide tags” as high confidence sections of de novo sequenced peptides. Fig. 2 contains a detailed description of the pTA logic and method of action. Starting with a single seed sequence, the algorithm extends the ends of the growing contig with the most likely residues (in terms of occurrences and confidence scores) at the next positions, as evidenced in the PEAKS de novo output. After initial contig assembly using all unique peptide tags as seeds, pTA performs several refinement steps, including merging of similar contigs into consensus sequences, and uniting these merged contigs into longer contigs if sufficient overlap exists between them. pTA outputs several files summarizing the analysis results at all stages of analysis, including an html report (supplemental Fig. S4).
Fig. 2.

pTA algorithm design and rationale. The input to pTA is the unfiltered PEAKS de novo peptide sequencing output. We define a K-mer as a peptide with K amino acids. pTA has four stages: stage 1, pre-processing, aimed at maintaining a maximal number of peptide tags while removing low confident sequence assignments. Rationale, noise filtering reduces assembly of incorrect tags. Stage 2, contig extension, each K-mer that has not been assembled yet is used as a seed for assembling the longest possible contig, using all original K-mers. Rationale, occasionally the correct K-mer peptide will not have the highest number of occurrences and will not be used for assembly, unless used as a seed. Stage 3, contig merging, contigs are aligned and merged to produce the longest possible sequence. Rationale, premature termination of contig assembly (pTA stage 2) is due to extension of the growing contig by incorrect residues. The correct sequence, overlapping the termination point, can be found in another contig seeded by the correct K-mer or extended in the opposite direction (assembly of the correct sequence will not terminate at that point because there are correct K-mers supporting its further extension). Stage 4, sequence refinement, evaluating whether chemical residue conversions (Gln to Glu or Asn to Asp) occur at given positions or whether mistakes were incorporated in the assembly process. All peptides are aligned onto the assembled sequence and per position evaluation is done. Rationale, remaining unconverted residues at specific positions reveal their identity prior to MAAH. Furthermore, peptide tags generated by enzymatic digestion can additionally be used by pTA for resolving chemical conversion ambiguities.
Certain free amino acids have previously been reported to be modified during acid hydrolysis (14, 15). By examining MS/MS spectra of partially correct de novo assignments of known peptides, we discovered that some of these modifications also result from MAAH in the context of a peptide chain, in addition to unreported modifications (Table I), and they were considered in the data analysis.
Table I. Observed amino acid modifications resulting from microwave-assisted acid hydrolysis sample preparation.
| MAAH-modified residue | Modification | Mass shift | Equivalent mass residue |
|---|---|---|---|
| Da | |||
| Asn | Deamidation | +0.98402 | Asp |
| Gln | Deamidation | +0.98402 | Glu |
| Arg | Citrullination | +0.98402 | |
| Cys | Carboxymethylationa | +58.00548 |
a When IAA is used as the alkylating agent, the majority of cysteines are carboxymethylated and the rest are carbamidomethylated. When IAc is used, only carboxymethylation of cysteines occurs and can thus be considered as a fixed modification for de novo sequencing.
Glutamine and asparagine deamidation into glutamic acid and aspartic acid, respectively, is very common during MAAH. In some cases, little or no evidence for the original Gln or Asn residues remains in the hydrolysate for specific residue positions, especially for poorly covered regions along the protein sequence. In such cases, Glu or Asp residues are selected during the assembly at these specific positions. At positions where the decision is not conclusive regarding the identity of the residue (e.g. potential sequence variants, deamidated Gln/Glu, deamidated Asn/Asp), both options and their coverage are presented at the top panel of the pTA html report (“sequence with potential ambiguities/variants,” supplemental Fig. S4). The final sequence decided upon is presented at the middle panel of the report (“final consensus sequence,” supplemental Fig. S4), where selected residues are color-coded for confidence in assignment. The isobaric leucine and isoleucine cannot be differentiated in the MS/MS spectra, and thus pTA-reported Leu at all relevant positions is regarded as “Leu or Ile.” Finally, pTA reports the sequence coverage at every position along the consensus sequence (i.e. number of peptide tags covering this position) (“coverage graph”, bottom panel, supplemental Fig. S4).
Benchmarking DiPS
To benchmark DiPS, it was applied to samples containing BSA (583 amino acids), equine myoglobin (153 amino acids), or bovine fetuin-A (342 amino acids) in triplicate. These proteins were chosen for their diversity in size and structure. The single resulting contig for each experiment matched the respective known protein sequence with 99–100% accuracy, covering 100% of the sequence (after processing of the N-terminal methionine, signal peptide, and propeptide where relevant) (Fig. 3). The only sequencing mistakes were the result of a swap of two residues (e.g. “Asp-Pro” instead of “Pro-Asp”) or the result of Ile:Leu, deamidated Gln:Glu, and deamidated Asn:Asp ambiguities, all of which are identified as potential ambiguities by pTA. We show that incorporating peptide tags from an additional single analysis of a trypsin digest of the benchmarking protein into pTA correctly resolved most ambiguities (supplemental Fig. S5).
Fig. 3.

DiPS results of benchmarking proteins. Equine myoglobin (A), BSA (B), and bovine fetuin-A (C) were subjected to DiPS in triplicates. pTA output sequences were aligned to the known sequence of each protein. Alignment mismatches that are the result of isobaric Ile/Leu, deamidated Gln/Glu (green), or deamidated Asn/Asp (yellow) ambiguities were not counted as sequencing mistakes in the accuracy calculations. A two-residue swap is highlighted in blue.
Sequence Determination of the Antibody AR37
An emerging therapeutic strategy in onco-immunology is the utilization of antibodies to control tumor growth or eradication of cancer altogether using immunotherapy. We sought to demonstrate the utility of DiPS for therapeutic antibody research. Amphiregulin is a member of the epidermal growth factor (EGF) family and has been targeted by inhibitory monoclonal antibodies (11, 16, 17). One of these, AR37, was selected from amphiregulin knock-out mice and shown to retard growth of human tumor cells in vitro and in vivo (data not shown). It was chosen as a test case for DiPS because cDNA amplification and sequencing failed to produce a product when a standard primer mix targeted at the conserved regions flanking the variable regions was used.
For deeper coverage and improved resolution of potential ambiguities, peptide tags from two experiments of a single MAAH preparation (one LC-MS/MS experiment followed by another with an exclusion list containing confidently de novo sequenced peptides from the first experiment) and one experiment of a tryptic digest were included in the analysis. In an attempt to improve de novo sequencing of peptides spanning disulfide bonds (18), which were expected to be crucial in antibody sequencing, IAc was used as the alkylating agent instead of IAA during sample preparation. Cysteine alkylation by IAc results in carboxymethylation (+58.00548 Da) and thus also has the added benefit over IAA alkalization of resolving ambiguities that are the result of the isobaric glycine and carbamidomethyl (+57.02146 Da). In our hands IAA performed better than IAc alkylation in terms of peptide identifications in typical bottom-up workflows, but for DiPS of disulfide-bound proteins, alkylation with IAc should be considered. Cyclization of N-terminal glutamine to pyroglutamate is a common modification of recombinant monoclonal antibodies (19) and was therefore included as a variable modification for the PEAKS de novo peptide-sequencing analysis. When applicable, pTA also reports the number of occurrences of pyroglutamate, glutamine, and glutamic acid at the relevant position. DiPS analysis resulted in three contigs. Because no prior knowledge of the protein in the sample was assumed, an initial characterization of the three contigs was performed by subjecting them to a BLAST search.
Contig1 was 215 residues long. A BLAST search revealed near-perfect identity of contig1 to G0YP42 mouse anti-human langerin 2G3 λ-type light chain (supplemental Fig. S6). The first residue of contig1 aligns to the first G0YP42 residue, after cleavage of the predicted signal peptide. The constant region of G0YP42 is matched perfectly by contig1 (with one deamidated-Q/E ambiguity), and the variable region differs in only five positions. Interestingly, the majority of glutamine residues at position 1 of contig1 were modified to pyroglutamate.
Contig2 was 363 residues long and aligns to the murine IgG heavy chain I6L985, with several differences in the variable region, the majority of which are in the predicted hyper-variable regions (CDR 1–3) (supplemental Fig. S7). The first residue of contig2 aligns to the first residue of I6L985 after cleavage of the predicted signal peptide. Contig2 also covers most of the murine IgG constant region, but weak evidence for adjacent residues results in premature termination of the assembly at this site. As was the case for the light chain, DiPS revealed that a significant portion of N-terminal glutamate residues was modified to pyroglutamate (supplemental Fig. S7). Contig3 covers the rest of the heavy chain constant region, with the exception of the poorly supported 23 residues in-between contigs (supplemental Fig. S7).
Validation of DiPS Determined AR37 Sequence
Initial validation of the antibody sequence included tryptic digestion and database searching against the sequence determined by DiPS using the database search module of the PEAKS algorithm. The database search resulted in 96 or 99% sequence coverage for the light and heavy chains, respectively, with multiple spectra supporting each tryptic peptide. Both variable regions had 100% sequence coverage (supplemental Figs. S6 and S7). The only unsupported sequences are rich in tryptic cleavage sites and thus are expected to produce tryptic peptides that are too short for identification.
Next, based on the close homology of the amino acid sequences determined by DiPS to antibody chains with known DNA sequences, primers flanking the variable regions of the heavy and light chains were custom-designed and used to amplify the variable regions by PCR from cDNA of the AR37 hybridoma. Confident cDNA sequence of the entire heavy chain variable region was determined, whereas DNA sequencing quality of the light chain variable region was suboptimal at a few positions along the sequence. Nonetheless, translation of all confident cDNA sequences confirmed that the antibody amino acid sequence determined by DiPS was 100% accurate (Fig. 4).
Fig. 4.

DiPS results of AR37. AR37 was subjected to DiPS using two nano-UPLC-MS/MS experiments of a single MAAH preparation and one experiment of a tryptic digest. The resulting assembled contigs 1 and 2 are aligned to translated cDNA sequences of the heavy (B) and light chain (A) variable regions, which were PCR-amplified and sequenced based on genes of proteins homologous to determined sequence.
Comparing pTA, ALPS, and mSPS
The two previously published methods for protein de novo sequencing, mSPS and ALPS, are based on proteolytic digestions and the De Bruijn graph assembly. We compared pTA assembly performance of these methods, independent of how the peptides were generated or peptide de novo sequences were obtained, by applying pTA to the published data from the ALPS and mSPS and by applying ALPS to our BSA and AR37 antibody data. Despite significant efforts, we were not able to analyze our MAAH-derived hydrolysate data using mSPS.
The key parameters for comparing de novo protein-sequencing methods are as follows: sequencing accuracy, total sequence coverage, and the number of contigs covering the full length of the protein. The latter is crucial for sequencing truly unknown proteins or unknown sequence regions such as the variable regions in antibodies. The more individual fragmented contigs the method produces for a given protein, the more challenging the determination of the full protein sequence becomes.
pTA was specifically designed and optimized for assembly of peptide tags generated by de novo sequencing MAAH-processed proteins. Such data contain deep coverage and high overlap in small increments due to random hydrolysis of peptide bonds, as well as MAAH-derived chemical modifications of certain residues within the context of a peptide. Thus, MAAH data may differ from enzymatic digestion data, which results in lower sequence coverage due to the fact that less unique peptides cover each residue because digestion is not random and occurs at specific residues. Overlap between peptides may occur in large increments (depending on the analyzed protein sequence and the proteases used), and significant chemical modifications resulting from the proteolysis are not expected. Therefore, in addition to de Bruijn graph assembly, pTA performs several pre-processing steps (e.g. replacing all asparagines with aspartic acids and all glutamines with glutamic acids while keeping track of the changed peptide tags) and contig refinement steps (e.g. merging and uniting contigs based on their similarity and overlapping ends) to accommodate these features of the MAAH data (Fig. 2).
pTA Versus ALPS, Hydrolysates
Initially, we compared pTA and ALPS using the hydrolysate data of the BSA and AR37 antibody. Despite the fact that the input data were identical, pTA reached complete coverage of BSA using one contig, whereas ALPS required six (Fig. 5A). For the antibody light and heavy chains, pTA reports three contigs covering both chains, whereas ALPS does not reach comparable coverage even after including 20 contigs, as is seen in Fig. 5B. Furthermore, none of the contigs produced by ALPS covered CDR3 of the light chain, which is the most variable region of the antibody and is key to its characterization. Additionally, as expected, ALPS was not able to identify MAAH-derived deamidation of asparagine and glutamine and reported most of these as aspartate or glutamate, respectively. This emphasizes the superior performance of the combination of MAAH and pTA in de novo sequencing of proteins.
Fig. 5.

Comparison of pTA and ALPS assembly performance on MAAH data. BSA (A) and AR37 (B) antibody were subjected to MAAH and trypsin digestion, followed by LC-MS/MS, peptide de novo sequencing, and analysis by pTA and ALPS. The number of top contigs taken from the output of each algorithm is plotted against the accumulated sequence coverage of the true protein sequence.
pTA Versus ALPS and mSPS, Enzymatic Digestions
Next, we set out to compare the three algorithms in analysis of enzymatic digestions. Table III in the paper by Tran et al. (8) presents a comparison between ALPS and mSPS, where the data acquired from a 6-protein mixture in the paper by Guthals et al. (7) were analyzed by ALPS. Table II is an adaptation of that table. The input to pTA was the same list of peptides (PSM-DDS) used by ALPS' de Bruijn assembler. When applied to this proteolytic digestion data, pTA's pre-processing and refinement steps that are optimized for MAAH data were omitted (i.e. glutamines and asparagine were not replaced, and the results refer to pTA's “contigs.csv” output file, see Fig. 2).
Table II. Assembly performance comparison between mSPS, ALPS, and pTA using proteolytic digest data as input.
A mixture of six proteins was digested with multiple proteases analyzed by LC-MS/MS. A comparison of the assembly performance of mSPS and ALPS on this dataset was presented in Table III of Ref. 8. Here, that table was adapted to contain pTA data, using the same list of peptides used as input by ALPS de Bruijn assembler (PSM-DDS). pTA results refer to assembled contigs in “contigs.csv” file. Green indicates the best result for each category. AA is amino acid.
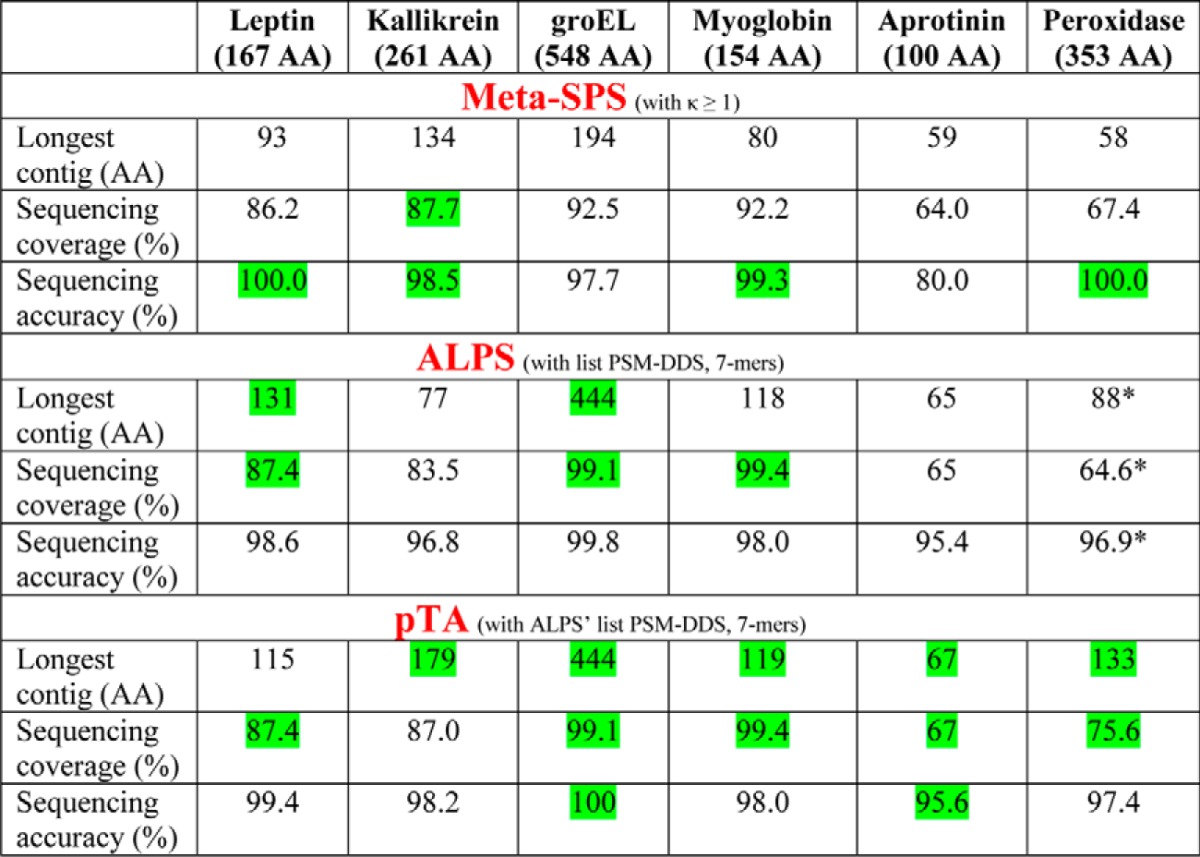
* Slight modification to the original published table (8), which reflects correction of ALPS longest contig (seq5)-assembled sequence, in which the last 4 residues of seq5 are incorrect extensions and thus should not account for the calculations.
Starting from the same input data, pTA performed comparably to or slightly better than ALPS, and much better than mSPS, in almost every parameter (Table II) despite the fact that pTA was not optimized for enzymatic digestions, rather for MAAH-derived data.
DISCUSSION
DiPS is the first method that facilitates rapid, de novo full-length sequencing of proteins. We demonstrate that full-length sequence of proteins of variable sizes can be achieved with nearly perfect accuracy, without the use of a reference database, a reference BLAST homolog, or prior knowledge of the analyzed protein. The other published methods attempting to achieve the same goal, mSPS and ALPS (5–8), require digestion with multiple proteases and separate analysis of each digest. For optimal performance, ALPS requires a database containing homologous sequences of the analyzed protein, and mSPS requires different acquisition modes. These procedures are labor-intensive, costly, resource-demanding, and still result in fragmented assembly and partial sequence coverage of the analyzed protein.
One of the key features in DiPS is the application of MAAH, which is not limited to cleavage of specific residues and is thereby advantageous over enzymatic digestion in its ability to produce multiple overlapping peptides at any position of any sequence. Additionally, during MAAH proteins are assumed to be completely denatured due to high temperature and low pH, thus eliminating the bias against cleavage of peptide bonds that are not solvent-accessible in the three-dimensional structure of proteins under typical digestion conditions. The output from the peptide de novo sequencing of the generated hydrolysate, analyzed while accounting for the MAAH-associated chemical modifications we discovered, provides a greatly improved dataset for the de Bruijn graph assembly compared with enzymatic digestions due to the high coverage and peptide overlap. pTA was specifically designed for and trained on this type of data. Although both mSPS and ALPS also use the de Bruijn graph approach, pTA uses several additional pre-processing and refinement steps to increase the length and accuracy of the assembled contigs (Fig. 2).
For example, due to the high frequency of Asn and Gln deamidation during MAAH, prior to assembly all instances of Asn and Gln are replaced by Asp and Glu, respectively, while noting the changed peptide tags. The determination of the original residue prior to MAAH is made only at the final refinement steps after contig assembly, merging, and uniting, where peptide tags (including the changed ones) are mapped back to the assembled contig, and the modified/unmodified residue ratio is evaluated. At this point, other sequence variants are also evaluated and reported in the final html report if substantial evidence for them exists.
pTA is the only algorithm that addresses MAAH-derived modifications and other sequence variants in the context of protein assembly, and it provides tools for evaluation of the assembled sequences.
Because of the random protein cleavage by MAAH, multiple peptide tags in close proximity on the sequence are used as seeds for the assembly and result in identical or similar contigs. pTA performs a contig merging refinement step for all similar contigs into the most probable consensus sequence. An additional refinement step of uniting overlapping consensus contigs is then performed. Although not optimized for enzymatic digestion as input, we sought to compare pTA's performance to ALPS and mSPS. To this aim, we applied pTA to the published proteolytic digest data of a 6-protein mixture analyzed for ALPS and mSPS benchmarking. Alternatively, we analyzed our BSA and AR37 datasets using ALPS. For the proteolytic digest data, pTA performed comparably to ALPS and superiorly to mSPS in terms of the length of the longest assembled contig, the accuracy, and sequence coverage. For MAAH data, however, pTA performed superiorly to ALPS. Using de novo sequenced peptides only (without additional database search results), ALPS resulted in fragmented assembly of BSA and AR37, with multiple shorter and non-overlapping contigs compared with the near-perfect assembly by pTA. Although pTA correctly identified MAAH-derived Gln and Asn deamidation events at all relevant positions with few exceptions, ALPS incorrectly reported Glu and Asp at most of these positions.
We demonstrated accurate sequencing for samples containing a single protein (or a mixture of two polypeptide chains, heavy and light, for the AR37 antibody). Samples with higher complexity, however, might result in poorer sequence coverage and therefore fragmented assembly. The supplemental Table S1 lists possible ways of increasing coverage and thus improving assembly. Additional de novo peptide tags obtained from partially orthogonal analysis procedures (proteolysis using different proteases, normal-phase LC-MS/MS, different fragmentation techniques, different de novo algorithms, etc.) may also increase coverage at specific poorly covered protein positions and allow assembly of complex samples. As is the case for other de novo methods, the use of a reference database or a BLAST homolog (if available) can assist in assembling fragmented DiPS sequence contigs.
Post-translational Modifications
The goal of DiPS is to resolve the primary sequence of the protein. Initially, DiPS data analysis begins with peptide de novo sequencing using parameters, including only Met oxidation and Cys carbamidomethylation/carboxymethylation as variable modifications, while ignoring all other modifications. To determine PTM presence and position, a standard database search of the same raw data against a database containing the DiPS-determined sequence, with parameters allowing the PTM of interest as variable modification, can be used to identify the modified residues. In fact, the random nature of MAAH peptide bond cleavage can resolve the precise position of MS/MS labile modifications where multiple potential modification sites exist (e.g. determining which residue is phosphorylated in a protein containing the phosphorylated sequence “XXXXSTXXXX” by identifying MAAH-generated peptide “XXXXSp” but not “TpXXXX”). The use of MAAH and database searching has previously been reported for localization of phosphorylation (20, 21) and N-glycosylation, and in theory similar procedures can be followed to localize all other PTMs that are not MAAH-labile (or result in a predictable modified product due to MAAH).
A modified residue will not be sequenced as such if the modification was not specified in the peptide de novo sequencing parameters, and this might lead to adverse effects on contig assembly during DiPS. However, even if a residue at a specific position of the protein is often modified, within a population of millions of molecules of the same protein, the small fraction of the unmodified residues can result in peptide tags that will be used for assembly of the protein. Thus, unless almost all copies of an amino acid at a specific position are modified, resulting in no peptide tags covering the unmodified residue, the full length of the protein can still be sequenced. If specific modifications are suspected to dominate certain residues in a given sample, as is the case for N-terminal pyroglutamate in monoclonal antibodies, the parameters of the peptide de novo sequencing may be tailored for that specific sample, and different variable modifications can be defined.
In this work, we show that DiPS detected a frequent conversion of N-terminal glutamine or glutamic acid to pyroglutamate in both the light and heavy chains of the AR37 antibody (supplemental Figs. S6 and S7). The ability to detect both modified and unmodified N-terminal glutamine or glutamic acid residues by DiPS can provide quality control for the sequenced monoclonal antibody in manufacturing settings.
Signal Peptide, Cleavage Sites, and Determination of Termini
The characteristics of protein termini can have a major impact on protein function, stability, and localization. Even when a gene annotation and a predicted protein product exist, the termini of the mature protein may not be easily inferred due to various factors, including protein truncation, degradation, proteolysis, alternative initiation of transcription/translation, or secretion via the use of a signal peptide. MAAH followed by database searching has been previously used for termini characterization (22). Here, we showed that DiPS was able to precisely and reproducibly determine the N and C termini of all analyzed proteins, because the first and last residues of the DiPS assembled contigs were indeed the N- and C-terminal residues of the respective mature proteins. The only exception was the result of a pre-termination in the assembly of AR37 heavy chain (i.e. the last residue of contig2 was not the C-terminal residue of AR37 heavy chain and the first residue of contig3 was not the first N-terminal residue). However, because this termination occurred in the constant region of the antibody, manual inspection could easily resolve this issue. Moreover, evidence for most of the remaining “missing” sequence can be found in the top assembled contigs prior to the final refinement step.
BSA and fetuin-A, as well as the light and heavy chains of AR37, are all secreted proteins with cleaved signal peptides. BSA also has a 6-residue long propeptide after the signal peptide that is cleaved during maturation of the protein (23). Equine myoglobin is an intracellular protein, whose N-terminal methionine is processed after protein synthesis (24). By determining the exact start residue, DiPS effectively identified the cleavage sites of the respective mature proteins.
In summary, DiPS represents a breakthrough method for database-independent, full-length protein sequencing. We anticipate that DiPS will become instrumental in sequencing proteins of unknown sequences such as antibodies or T-cell receptors. Furthermore, because of its ability to determine protein termini with precision, DiPS represents a new strategy to address challenging questions such as determination of signal peptides or substrate cleavage sites.
DATA AVAILABILITY
The pTA executable and example data is provided as Supplementary File. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE(12) partner repository (https://www.ebi.ac.uk/pride/archive/) with the dataset identifier PXD003804. The pTA tool is available as a Windows executable (Supplemental Information) and the code is available via https://bitbucket.org/incpm/dips.
Supplementary Material
Acknowledgments
We thank Prof. Itai Benhar, Tel Aviv University, for fruitful discussions during the method development. We also thank Dr. Rami Jaschek, Weizmann Institute of Science, for assistance in the early developments of pTA.
Footnotes
Author contributions: A.S., O.A.T., and Y.L. designed the research; A.S., D.E., A.G., O.A.T., and Y.L. performed the research; R.B., Y.Y., and M.L. contributed new reagents or analytic tools; A.S., R.B., O.A.T., and Y.L. analyzed the data; A.S. and Y.L. wrote the paper; R.B. coded the tool.
* The authors declare that they have no conflicts of interest with the contents of this article.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- BLAST
- basic local alignment tool
- pTA
- peptide tag assembler
- DiPS
- database-independent protein sequencing
- PTM
- post-translational modification
- IAA
- iodoacetamide
- IAc
- iodoacetate
- MAAH
- microwave-assisted acid hydrolysis
- mSPS
- meta-SPS.
REFERENCES
- 1. Zhang Y., Fonslow B. R., Shan B., Baek M. C., and Yates J. R. 3rd (2013) Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 113, 2343–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yates J. R., Ruse C. I., and Nakorchevsky A. (2009) Proteomics by mass spectrometry: approaches, advances, and applications. Annu. Rev. Biomed. Eng. 11, 49–79 [DOI] [PubMed] [Google Scholar]
- 3. Allmer J. (2011) Algorithms for the de novo sequencing of peptides from tandem mass spectra. Expert Rev. Proteomics 8, 645–657 [DOI] [PubMed] [Google Scholar]
- 4. Seidler J., Zinn N., Boehm M. E., and Lehmann W. D. (2010) De novo sequencing of peptides by MS/MS. Proteomics 10, 634–649 [DOI] [PubMed] [Google Scholar]
- 5. Guthals A., Clauser K. R., and Bandeira N. (2012) Shotgun protein sequencing with meta-contig assembly. Mol. Cell. Proteomics 11, 1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bandeira N., Clauser K. R., and Pevzner P. A. (2007) Shotgun protein sequencing: assembly of peptide tandem mass spectra from mixtures of modified proteins. Mol. Cell. Proteomics 6, 1123–1134 [DOI] [PubMed] [Google Scholar]
- 7. Guthals A., Clauser K. R., Frank A. M., and Bandeira N. (2013) Sequencing-grade de novo analysis of MS/MS triplets (CID/HCD/ETD) from overlapping peptides. J. Proteome Res. 12, 2846–2857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tran N. H., Rahman M. Z., He L., Xin L., Shan B., and Li M. (2016) Complete de novo assembly of monoclonal antibody sequences. Sci. Rep. 6, 31730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vyatkina K., Wu S., Dekker L. J., VanDuijn M. M., Liu X., Tolić N., Dvorkin M., Alexandrova S., Luider T. M., Paša-Tolić L., and Pevzner P. A. (2015) De novo sequencing of peptides from top-down tandem mass spectra. J. Proteome Res. 14, 4450–4462 [DOI] [PubMed] [Google Scholar]
- 10. Bandeira N., Pham V., Pevzner P., Arnott D., and Lill J. R. (2008) Automated de novo protein sequencing of monoclonal antibodies. Nat. Biotechnol. 26, 1336–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carvalho S., Lindzen M., Lauriola M., Shirazi N., Sinha S., Abdul-Hai A., Levanon K., Korach J., Barshack I., Cohen Y., Onn A., Mills G., and Yarden Y. (2016) An antibody to amphiregulin, an abundant growth factor in patients' fluids, inhibits ovarian tumors. Oncogene 35, 438–447 [DOI] [PubMed] [Google Scholar]
- 12. Vizcaíno J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhong H., Zhang Y., Wen Z., and Li L. (2004) Protein sequencing by mass analysis of polypeptide ladders after controlled protein hydrolysis. Nat. Biotechnol. 22, 1291–1296 [DOI] [PubMed] [Google Scholar]
- 14. Fountoulakis M., and Lahm H. W. (1998) Hydrolysis and amino acid composition of proteins. J. Chromatogr. A 826, 109–134 [DOI] [PubMed] [Google Scholar]
- 15. Inglis A. S., Nicholls P. W., and Roxburgh C. M. (1971) Hydrolysis of the peptide bond and amino acid modification with hydriodic acid. Aust. J. Biol. Sci. 24, 1235–1240 [DOI] [PubMed] [Google Scholar]
- 16. Lindzen M., Carvalho S., Starr A., Ben-Chetrit N., Pradeep C. R., Köstler W. J., Rabinkov A., Lavi S., Bacus S. S., and Yarden Y. (2012) A recombinant decoy comprising EGFR and ErbB-4 inhibits tumor growth and metastasis. Oncogene 31, 3505–3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferraro D. A., Gaborit N., Maron R., Cohen-Dvashi H., Porat Z., Pareja F., Lavi S., Lindzen M., Ben-Chetrit N., Sela M., and Yarden Y. (2013) Inhibition of triple-negative breast cancer models by combinations of antibodies to EGFR. Proc. Natl. Acad. Sci. U.S.A. 110, 1815–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Samgina T. Y., Vorontsov E. A., Gorshkov V. A., Artemenko K. A., Nifant'ev I. E., Kanawati B., Schmitt-Kopplin P., Zubarev R. A., and Lebedev A. T. (2011) Novel cysteine tags for the sequencing of non-tryptic disulfide peptides of anurans: ESI-MS study of fragmentation efficiency. J. Am. Soc. Mass Spectrom. 22, 2246–2255 [DOI] [PubMed] [Google Scholar]
- 19. Liu H., Ponniah G., Zhang H. M., Nowak C., Neill A., Gonzalez-Lopez N., Patel R., Cheng G., Kita A. Z., and Andrien B. (2014) In vitro and in vivo modifications of recombinant and human IgG antibodies. MAbs 6, 1145–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang N., and Li L. (2010) Reproducible microwave-assisted acid hydrolysis of proteins using a household microwave oven and its combination with LC-ESI MS/MS for mapping protein sequences and modifications. J. Am. Soc. Mass Spectrom. 21, 1573–1587 [DOI] [PubMed] [Google Scholar]
- 21. Ma C., Qu J., Meisner J., Zhao X., Li X., Wu Z., Zhu H., Yu Z., Li L., Guo Y., Song J., and Wang P. G. (2015) Convenient and precise strategy for mapping N-glycosylation sites using microwave-assisted acid hydrolysis and characteristic ions recognition. Anal. Chem. 87, 7833–7839 [DOI] [PubMed] [Google Scholar]
- 22. Chen L., Wang N., Sun D., and Li L. (2014) Microwave-assisted acid hydrolysis of proteins combined with peptide fractionation and mass spectrometry analysis for characterizing protein terminal sequences. J. Proteomics 100, 68–78 [DOI] [PubMed] [Google Scholar]
- 23. Waldmann T., Rosenoer V., Oratz M., and Rothschild M. (1977) Albumin Structure, Function and Uses. (Rosenoer V. M., Oratz M., and Rothschild M. A., eds) Pergamon Press Inc., Tarrytown, NY [Google Scholar]
- 24. Jahnen W., Ward L. D., Reid G. E., Moritz R. L., and Simpson R. J. (1990) Internal amino acid sequencing of proteins by in situ cyanogen bromide cleavage in polyacrylamide gels. Biochem. Biophys. Res. Commun. 166, 139–145 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The pTA executable and example data is provided as Supplementary File. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE(12) partner repository (https://www.ebi.ac.uk/pride/archive/) with the dataset identifier PXD003804. The pTA tool is available as a Windows executable (Supplemental Information) and the code is available via https://bitbucket.org/incpm/dips.