

Abstract
Trypsin dominates bottom-up proteomics, but there are reasons to consider alternative enzymes. Improving sequence coverage, exposing proteomic “dark matter,” and clustering post-translational modifications in different ways and with higher-order drive the pursuit of reagents complementary to trypsin. Additionally, enzymes that are easy to use and generate larger peptides that capitalize upon newer fragmentation technologies should have a place in proteomics. We expressed and characterized recombinant neprosin, a novel prolyl endoprotease of the DUF239 family, which preferentially cleaves C-terminal to proline residues under highly acidic conditions. Cleavage also occurs C-terminal to alanine with some frequency, but with an intriguingly high “skipping rate.” Digestion proceeds to a stable end point, resulting in an average peptide mass of 2521 units and a higher dependence upon electron-transfer dissociation for peptide-spectrum matches. In contrast to most proline-cleaving enzymes, neprosin effectively degrades proteins of any size. For 1251 HeLa cell proteins identified in common using trypsin, Lys-C, and neprosin, almost 50% of the neprosin sequence contribution is unique. The high average peptide mass coupled with cleavage at residues not usually modified provide new opportunities for profiling clusters of post-translational modifications. We show that neprosin is a useful reagent for reading epigenetic marks on histones. It generates peptide 1–38 of histone H3 and peptide 1–32 of histone H4 in a single digest, permitting the analysis of co-occurring post-translational modifications in these important N-terminal tails.
The most widely used “bottom-up” proteomics approach requires the proteolytic digestion of proteins into peptides. These peptides are detected by MS and identified through database searching (1). Trypsin is the standard protease and cleaves almost exclusively after the basic amino acids lysine (Lys) and arginine (Arg) (2). However, in practice, due to the natural distribution of these amino acids in proteomes, a large segment of the proteome is not identifiable. Most peptides are too small for unambiguous identification (56% ≤6 residues) (3). Improvements in proteome representation and the characterization of post-translational modifications (PTMs) 1 and proteoforms are possible with proteolytic enzymes such as LysC, ArgC, AspN, and GluC (3–5). They all share the property of cleaving either the C or N terminus to charged residues. An enzyme with specificity for neutral amino acids would be a welcome addition. For example, an advantage in membrane protein representation was demonstrated recently using WaLP and MaLP (6). These are lower-specificity enzymes that cleave after Thr, Val, Ala, Ser, and Met (WaLP) and Met, Leu, Phe, Tyr, Thr, and Val (MaLP). It would be useful to have enzymes in this general class with even higher specificity to generate longer sequence “read lengths” and possibly improved representation of heterogeneity in PTMs. Enzymes recognizing dibasic motifs such as Sap9 (7) and OmpT (8) show promise in this regard, but candidates that avoid tryptic residues may offer superior complementarity.
Of course, proteases like elastase, chymotrypsin, and pepsin cleave after hydrophobic residues, but their low specificity complicates database-driven peptide identification and limits their utility in proteomics (9–11). Defined cleavage sites permit higher rates of peptide-spectrum matching and reduce search time substantially (12, 13), plus “losing” a defined C-terminal residue appears to add complexity to fragment ion distributions (6). We considered proline-cleaving enzymes as an attractive alternative for number of reasons. Proline (Pro) is less abundant than Lys + Arg (14). It often delineates structural transitions in proteins and is generally unmodified in cellular proteins; thus digestion products could provide a better grouping of PTMs. Unfortunately, there are very few validated prolyl endoproteases (PEPs) currently available. Numerous prolyl oligopeptidases (POPs) are known to cleave after Pro and alanine (Ala), but their activity is restricted to small substrates (15). The gating action of regulatory domains restricts substrates to lengths of ∼30 residues (16). PEPs that are capable of digesting larger substrates are rare and are usually classified as POPs with extended range (17). AN-PEP, a protease of the S28 family, is the only PEP confirmed to offer broad substrate recognition, but cleavage specificity appears to extend beyond Pro and Ala (18).
An effective PEP would be particularly useful for characterizing histone modifications. Histones are Lys- and Arg-rich proteins that form the core components of nucleosomes, around which the DNA is wound in the chromatin of eukaryotes (19). Histones are tightly packed into octamers, but they maintain highly modified N-terminal tails that are flexible and integrate a variety of post-translational modifications. Many of these modifications are dynamically installed and removed. They play a key role in epigenetically regulating cell fate (20), and their deregulation is often associated with cancer (21). The high frequency of positively charged residues in the histone tails complicates tryptic analysis, as the resulting peptides are small, and the connectivity between modifications is lost (22). GluC and AspN enzymes show more promise, especially for presenting the combinatorial modifications of histones H3 and H4, but they need to be used separately to cluster the most important PTM sites (23, 24). A PEP may provide a more universal approach to mapping the “histone code” and complement the existing methods for analysis.
We recently discovered neprosin from the secretions of the carnivorous pitcher plant (25). It appears to be a proline-cleaving enzyme, based on our initial characterizations (26). In this study, we produced neprosin recombinantly and characterized the enzyme for use in proteomics. We show that neprosin is a legitimate low-molecular-weight PEP, active at low concentrations and low pH. We demonstrate strong complementarity with conventional enzymes for whole-proteome analysis and histone mapping.
EXPERIMENTAL PROCEDURES
Preparation of the Neprosin Expression Vector
The gene corresponding to pro-neprosin (residues 25–380) from Nepenthes ventrata was synthesized by Genscript with codon optimization for expression in Escherichia coli and yeast (26). The optimized neprosin gene was then inserted into pET28a(+) at the EcoRI and SalI restriction sites in-frame and downstream of an N-terminal His tag, yielding the plasmid pDS36. The MBP gene was amplified from e3884 (generous gift from Dr. A. Schryvers, University of Calgary) with forward primer LSO13F (5′GGC TCG CATATG AAA ACT GAA GAA GGT AAA CTG GTA ATC TG3′, NdeI site underlined) and reverse primer LSO14R (5′CAC TCA GCTAGC CCT TCC CTC GAT GTT GTT GTT ATT GTT ATT GTT GTT GTT GTT CG3′, the NheI site is underlined, and the factor Xa cleavage site is italicized) and inserted into pDS36 at the NdeI and NheI restriction sites in-frame and between the His tag and neprosin gene, to form the His-MBP-neprosin expression plasmid termed pDS42. The constructs were verified by DNA sequencing.
Protein Expression and Purification of Recombinant Neprosin
The His-MBP-pro-neprosin expression plasmid (pDS42) was transformed into E. coli Artic Express (generous gift from Dr. Peter Facchini, University of Calgary) competent cells. The pDS42-transformed cells (DSB90) were grown in 2YT with kanamycin 50 μg ml−1 at 37 °C until an OD of ∼0.8 was reached, and then expression was induced with 0.3 mm isopropyl β-d-thiogalactopyranoside at 16 °C overnight with shaking. Cells were harvested by centrifugation and frozen at −80 °C. The cells were then thawed and resuspended in lysis buffer (50 mm Tris-HCl, pH 8.0, 0.5 m NaCl, 10% glycerol, 1 mm DTT, 1 mm PMSF, 0.5% Triton X-100, 0.025% sodium azide, 1 μg ml−1 lysozyme, RNase A) supplemented with a mixture of protease inhibitors (Roche Applied Science) at a ratio of 10 ml of buffer per 1 g of wet cells. The cells were lysed by sonication (Qsonica Sonicators) on ice at 30% amplitude using six 30-s pulses with a 30-s rest between pulses. Following centrifugation at 15,000 rpm at 4 °C for 30 min, the supernatant was applied to a 5-ml HisTrap HP (GE Healthcare) pre-equilibrated with 50 mm Tris-HCl, pH 8.0, 0.5 m NaCl (buffer A). After washing with 20 ml of buffer A, His-MBP-pro-neprosin was eluted with 50 ml of a linear gradient of 0–100% buffer B (50 mm Tris-HCl, pH 8.0, 0.5 m NaCl, 0.5 m imidazole) at a flow rate of 1 ml min−1. Fractions containing His-MBP-pro-neprosin were pooled and dialyzed against 50 mm Tris-HCl, pH 7.5, 150 mm NaCl. Multiple rounds of dialysis against 100 mm Gly-HCl, pH 2.5, at 37 °C for 7 days allowed for acid auto-activation to the mature, active neprosin. Neprosin was then concentrated in a 10-kDa molecular mass cutoff filter (Millipore) and analyzed by SDS-PAGE. The concentration of active recombinant neprosin was estimated by comparing its activity to native neprosin purified from Nepenthes digestive fluid, using the GFP activity assay (below).
Neprosin Activity Assay via GFP Proteolysis
The green fluorescent protein (GFP) proteolysis assay was performed as reported (27) with minor modifications. Briefly, 0.1 mg ml−1 of GFP S65T (in 10 mm Tris-HCl, pH 8) was denatured with 0.1× volume of 1 m Gly-HCl, pH 2.4 (final pH of solution was 2.5), in the absence or presence of neprosin (1 nm) at 37 °C (or the indicated temperature) for 1 h. Reaction was quenched, and the GFP was renatured by addition of 0.25× volume of 1 m Tris-HCl, pH 8.4, and 0.1× volume of 1 m DTT (final pH of solution was 8). Fluorescence of renatured GFP S65T was determined using a Molecular Devices Filter Max F5 microplate spectrophotometer with excitation at 485 nm, and emission was monitored at 535 nm. For the analysis of the effect of protease inhibitors on proteolytic activity, neprosin was pre-incubated with the protease inhibitor in the reaction volume at room temperature for 30 min prior to the addition of GFP S65T and acid denaturation. For the analysis of the effect of pH on neprosin activity, GFP S65T and neprosin were incubated in 100 mm solution of various buffers (pH 2–9: Gly-HCl, ammonium formate, sodium phosphate, Tris-HCl). Neprosin activity was expressed as percentage of fluorescence lost (100% − % fluorescence recovery) relative to the untreated GFP S65T under the same reaction conditions.
Neprosin Activity Assay of Standard Proteins
For gel-based analysis using recombinant neprosin, 1 mg ml−1 of BSA was reduced with 10 mm DTT at 50 °C for 30 min. The reduced protein substrate (0.05 mg ml−1) was incubated with recombinant Npr1 (∼1 nm) and different concentrations of urea in 100 mm Gly-HCl, pH 2.5, at 37 °C for 1 h and analyzed by SDS-PAGE.
HeLa Cell Lysate Preparation
HeLa S3 cells were grown at the National Cell Culture Centre in Joklik-modified minimum Eagle's medium supplemented with 5% newborn calf serum to high cell density (1 × 106 cells/ml). Cells were collected by centrifugation at 2500 × g, followed by two washes in warm 37 °C PBS (Ca/Mg free) and stored at −80 °C. After thawing, the cells were resuspended in 50 mm HEPES buffer (pH 8, 150 mm KCl, 1 mm MgCl2), 10% glycerol, 0.5% Nonidet P-40, 5 units/ml benzonase supplemented with a mixture of protease inhibitors (Roche Applied Science). The cells were lysed by sonication on ice at 30% amplitude for six rounds of 30-s pulses with a 30-s rest between pulses. Following centrifugation at 37,000 rpm at 4 °C for 30 min, the supernatant lysate was aliquoted, frozen in liquid nitrogen, and stored at −80 °C. Protein concentration was determined by Bradford assay, using BSA as standards.
HeLa Cell Lysate and Histone Digestion
HeLa whole-cell lysate was digested with recombinant and endogenous neprosin, AN-PEP, LysC, and trypsin using the FASP protocol (28). Briefly, 100 μg of precipitated lysate was loaded onto a 10-kDa filter device and subsequently denatured, reduced, and alkylated at pH 8.5. Depending on the protease, buffer exchange was conducted using a volume of 120 μl of 100 mm Gly-HCl, pH 2.5, for both neprosin preparations and AN-PEP or 50 mm ammonium bicarbonate, pH 8.5, for LysC and trypsin. Addition of buffer was followed by a centrifugation step for 30 min at 14,000 × g. This was repeated three times for a complete buffer exchange. The enzyme was then added to an estimated enzyme-to-substrate ratio of 1:50 (w/w) in case of trypsin and LysC, 1:100 for AN-PEP, and 1:500 for both neprosin preparations. Samples were subsequently incubated overnight at 37 °C. Released peptides were eluted from the filter device in three steps. First, the filter unit was centrifuged for 30 min at 14,000 rpm. Second, 50 μl of the corresponding buffer solution was added to the filter unit, followed by a second centrifugation step for 30 min at 14,000 rpm. Final elution was performed by adding 50 μl of 0.5 m NaCl to the filter device and subsequent centrifugation for 15 min at 14,000 rpm, and the eluates were combined. Prior to mass spectrometric data acquisition, all samples were desalted and concentrated using Stage tips (29). Peptides were eluted with 50% ACN in 0.1% TFA and evaporated to dryness using a SpeedVac. Finally, peptides were reconstituted in 0.1% FA for mass spectrometric analysis. Cleavage specificity experiments using recombinant and endogenous neprosin, AN-PEP, and trypsin were conducted in biological duplicates. Experiments for the evaluation of proteome coverage with recombinant neprosin, trypsin, and LysC were done in single experiments. To evaluate whether acidic solution induced unspecific cleavages under long incubation times at pH 2.5, tryptic digests were reconstituted in 20 μl of H2O and split in 2 aliquots of 10 μl. One aliquot was kept at −20 °C until analysis. The other aliquot was adjusted to pH 2.5 with 100 mm Gly-HCl buffer and incubated at 37 °C overnight.
For whole histone analysis, unfractionated whole histone from calf thymus (Sigma-Aldrich) was dissolved in Gly-HCl, pH 2.5, to a final concentration of 1 μg μl−1. Recombinant neprosin was added to an estimated enzyme-to-substrate ratio of 1:500. Samples were subsequently incubated overnight at 37 °C.
LC/MS
For the determination of cleavage specificity, HeLa digests were analyzed using an EASY-nLC 1000 nano-LC coupled to an Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, San Jose CA) equipped with a Nanospray Flex Ion Source. Peptides were chromatographically separated using a 15-cm PicoTip fused silica emitter with an inner diameter of 75 μm (New Objective Inc., Woburn MA) packed in-house with reversed-phase Reprosil-Pur C18-AQ 3-μm resin (New Objective). The flow rate was 300 nl min−1, and peptides were eluted using a 140-min gradient running linearly from 5 to 40% B (97% ACN in 0.1% FA). Data were acquired using data-dependent MS/MS mode. Each high-resolution product ion scan in the Orbitrap (m/z 300 to 2000, R = 60,000) was followed by high-resolution product ion scans (isolation window 3 Th) in the Orbitrap after HCD fragmentation at 35% NCE. Resolution was set to 7500. Top 10 most abundant signals with a charge state greater than 1 were selected for fragmentation, followed by dynamic exclusion for 60 s. Data acquisition was controlled with Xcalibur software (version 3.0.63).
For the comparative proteomic analysis of HeLa cells digested with LysC, trypsin, or recombinant neprosin, samples were measured using an EASY-nLC 1000 nano-LC coupled to an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific) equipped with an EASY-spray source. Peptides were separated using a 25-cm Easy-Spray PepMap analytical column (Thermo Fisher Scientific) packed with reversed-phase C18 beads (2-μm particle diameter, 100-Å pores). The injection volume was adjusted to achieve similar TIC levels for all samples. The flow rate was set to 300 nl min−1. Peptides were eluted using a 110-min gradient running linearly from 5 to 25% B (100% ACN in 0.1% FA) at a spray voltage of 2.4 kV. Each high-resolution precursor ion scan in the Orbitrap (m/z 350 to 1500, R = 120,000) was followed by product ion scans (isolation window 3 Th) in the linear ion trap with a fixed scan cycle time of 3 s. Dissociation in MS2 mode was carried out via CID and ETD using a data-dependent decision tree (30). The fragmentation mode was chosen depending on the nature of the selected ions. Doubly charged ions, triply charged ions with m/z >650, quadruply charged ions with m/z >900, and quintuply charged ions with m/z >950 were triggered with CID. Triply charged ions with m/z ≤650, quadruply charged ions with m/z ≤900, quintuply charged ions with m/z ≤950, and ions with a charge state of six and higher were fragmented using ETD. CID was performed at 30% normalized collision energy (NCE), and ETD was carried out with a maximum injection time of 100 ms. A target value of 20,000 was selected for MS2 automatic gain control, and precursor ions were dynamically excluded for 30 s.
Whole histone digests were measured on an Orbitrap Fusion Lumos Tribrid mass spectrometer coupled to an EASY-nLC 1200 nano-LC system. Peptides were separated using a 15-cm Easy-Spray PepMap analytical column (Thermo Fisher Scientific) packed with reversed-phase C18 beads (2-μm particle diameter, 100-Å pores) using first a 30-min gradient running linearly from 5 to 15% B (80% ACN in 0.015% FA), followed by increasing B to 60% within the next 10 min. Each high-resolution precursor ion scan in the Orbitrap (m/z 350 to 1200, R = 120,000) was followed by product ion scans (isolation window 2 Th) in the Orbitrap (R = 15,000) with a fixed scan cycle time of 3 s. Charge states of three and less were excluded. Peptides were fragmented using EThcD (maximum injection time of 70 ms and HCD at 25% NCE) at a spray voltage of 1.9 kV. A target value of 50,000 was selected for MS2 automatic gain control, and precursor ions were dynamically excluded for 15 s. Data acquisition was controlled using Xcalibur software (version 4.0).
Informatics
All unprocessed data files (RAW-format) were directly loaded into PEAKS studio (31) (version 7.5; Bioinformatics Solutions), and precursor masses were subsequently corrected in the software. Peptides were identified by de novo sequencing and database search, with matching to the human Swiss-Prot database (downloaded from uniprot.org in May 2016; containing 20,201 entries). For whole proteome analysis of HeLa digests, cleavage sites were restricted in a number of ways. For trypsin (P1: Lys and Arg, with P1′: any amino acid, according to Rodriguez et al. (32)), we allowed up to two missed cleavages for a conventional search. We also performed a semi-tryptic search (unspecific cleavage at one end) and a fully nonspecific search. For LysC (P1: Lys, with P1′: any amino acid), we also allowed up to two missed cleavages for a conventional search, and both semi-LysC and fully nonspecific searches. For neprosin (P1: Pro and Ala, with P1′: any amino acid except Pro; and P1: Asp with P1′: Pro), we allowed a maximum number of seven missed cleavages, and we also allowed for both semi-neprosin and fully nonspecific cleavage in separate searches. Met oxidation was selected as a variable modification with a maximum number of two variable modifications per peptide. Carbamidomethylation of cysteine residues was set as a fixed modification. To decrease FDR while searching for multiple post-translationally modified peptides, cleavages were restricted to the same rules as stated above for the three proteases, but applying strict cleavage rules for both ends. Acetylation of the protein's N terminus and methylation and dimethylation of Lys and Arg were chosen as variable modifications with a maximum of three variable modifications per peptide.
For a minimally biased cleavage site analysis in the proteomics experiments, the enzyme was set to “none,” and a modified database file was used, in which initiator methionine residues and signal peptides (35) were removed, to exclude false-positive N-terminal cleavage sites. The normalized cleavage specificity, taking the natural abundance of corresponding amino acids and dipeptide motifs in Homo sapiens into account, was calculated after Keil (41) as shown previously (36). Cleavage specificity was visualized using heat maps and iceLogo (37) representations. Peptide sequences stored in ProteomicsDB (38), which are associated with the proteins commonly identified in the LysC, trypsin, and neprosin digests, were exported with a 1% FDR filter and requiring a minimum of two PSMs per peptide.
For the analysis of whole histones from calf thymus, a manually curated database file was used containing all known 21 histone variants from Bos taurus. Cleavage sites were restricted to the same neprosin rules as stated above allowing one unspecific cleavage. The following variable modifications were chosen allowing a maximum of four variable PTMs per peptide: methylation of Lys and Arg, dimethylation of Lys, trimethylation of Lys, and acetylation of Lys and the protein's N terminus. PTMs were chosen based on known frequent PTMs in histones (33). To decrease the search space, only those spectra were taken into consideration in PEAKS PTM showing a de novo score of at least 1% (34). Identified PSMs were manually inspected and only those kept for which the PTM site was unambiguously confirmed based on the identification of adjacent fragment ions in PEAKS.
Mass error tolerance of precursor ions was set to 10 ppm for all samples. Mass error tolerance of fragment ions was set in general to 0.6 Da for CID and ETD spectra acquired in the linear ion trap or to 0.015 Da for HCD and ETD spectra acquired in the Orbitrap analyzer. The peptide score threshold was decreased until a false discovery rate of ≤1% on the peptide level was reached. A minimum score of 20 together with at least one unique peptide was set as the lower threshold for identification of protein groups. This yielded an FDR between 0.1 and 0.6% at the protein level. Raw files for all MS experiments are available from Chorus (ID 1262), and annotated fragment ion spectra for identified histone tails are available in the supplemental materials.
RESULTS
We recently discovered an entire class of protease that appears to possess a novel domain structure (DUF239) (supplemental Fig. S1) (26). We tentatively assigned PEP functionality to this class, based on our preliminary analysis of a 29-kDa enzyme we call neprosin. The DUF239 sequence domain is found in a large family of proteins throughout the plant kingdom. Isolated from carnivorous plants of the Nepenthes genus, neprosin functions under solution conditions that suggest utility in proteomics. Initial screening of protease activity was conducted by following the degradation of simple protein substrates. These digests were carried out at pH 2.5, a level at which other proteases isolated from the Nepenthes fluid also showed high activity (39). We recombinantly produced the protease in E. coli to support further studies (supplemental Fig. S2).
Neprosin, a Primary Characterization
Prior to evaluating the enzyme in proteomics experiments, we characterized its basic properties in a modified protease assay using the green fluorescent protein GFP S65T (27). Neprosin was weakly inhibited by the aspartic protease inhibitor pepstatin but was not affected by the POP inhibitor Z-Pro-prolinal inhibitor, and it retained activity under strongly reducing conditions (Fig. 1A). It showed its highest enzymatic activity at pH 2.5, although it retained ∼50% of its activity at pH 4.5, whereas further increases in the pH led to loss of proteolytic activity (Fig. 1B). It also retained activity in the presence of denaturants (i.e. <2 m urea, supplemental Fig. S3). The temperature significantly influenced its activity, and the optimal range was found to be between 37 and 50 °C (Fig. 1C). Based on these results, we conducted all further experiments at pH 2.5 and at a temperature of 37 °C. Although our production yield was modest, the recombinant form retained the high activity we observed with native neprosin, allowing us to use enzyme-to-substrate (E:S) ratios of ∼1:500.
Fig. 1.

GFP S65T proteolysis by neprosin under different conditions. Neprosin activity was expressed as percentage of fluorescence lost (100% − % fluorescence recovery) relative to the untreated GFP S65T (without neprosin) under the same reaction condition. The error bars represent standard deviation from at least three replicates. A, effect of protease inhibitors and reducing agents on neprosin activity. B, effect of pH on neprosin activity. The indicated pH is the final pH of the reaction mixture. C, effect of temperature on neprosin activity. ZPP, Z-pro-prolinal inhibitor.
Neprosin Cleavage Rule
To characterize the cleavage specificity of neprosin, we digested whole-cell lysates followed by single-shot LC-MS/MS experiments. E. coli digests were initially used to evaluate data-dependent acquisition methods that offer high rates of MS2 acquisition, which we then applied to HeLa cell lysate in two biological replicates. Cleavage specificity was calculated according to Keil (41) using sets of 3316 and 3275 unique recombinant neprosin cleavage sites identified from the digests (supplemental Table S1). To validate the method, we determined the cleavage specificities from HeLa digestions using trypsin (supplemental Fig. S4). The observed cleavage specificity was virtually identical with values calculated previously by Huesgen et al. (4). In the neprosin data set, we observed that an average of 61% of all cleavage sites were C-terminal to Pro and 23% were C-terminal to Ala (Fig. 2A). Taking the natural occurrences of Pro and Ala in the human proteome into account, the normalized activity of neprosin toward these residues was 9.8 and 3.1, respectively (Fig. 2B). Endogenous neprosin showed the same cleavage specificity as recombinantly produced neprosin (supplemental Fig. S5), as expected. Neprosin demonstrates substantially greater specificity for C-terminal Pro cleavage than AN-PEP, the only other characterized prolyl endoprotease (supplemental Fig. S6, A and B). For neprosin, charged residues such as Glu, His, Lys, and Arg were disfavored in the P1 position, yielding only 2.6% of all cleavages. The most favored dipeptide motifs were Pro-Xaa, followed by Ala-Xaa (Xaa indicates any amino acid except Pro and to lesser extent Trp) (supplemental Fig. S7A). Cleavage of an Asp-Pro motif also seemed prominent, but it was the only motif for which Pro was found in P1′ position (supplemental Fig. S7, B and C). Because cleavage of Asp-Pro may be chemically induced at pH 2.5, we incubated acidified tryptic HeLa digests overnight at 37 °C to gauge the level of acid-induced cleavages. We observed a significant increase of Asp-Pro cleavage sites, and no increase in cleavages C-terminal to Pro or Ala (supplemental Fig. S8), which supports our assertion that the Asp-Pro cleavage does not arise from the direct action of neprosin.
Fig. 2.

Cleavage specificity of neprosin. A, IceLogo visualization of neprosin preference toward substrate subsites P4–P4′, derived from HeLa lysate digestion (n = 3316 and 3275 unique cleavage sites). Differences are displayed as the amino acid occurrence in a certain position, normalized to the natural occurrence in H. sapiens. B, heat map visualization of neprosin's cleavage specificity toward subsites P4–P4′ normalized to the natural occurrence in H. sapiens.
Proteome Coverage
Next, we analyzed HeLa digests generated using trypsin, LysC, and recombinant neprosin to evaluate its utility in multienzyme proteomics experiments. We chose a precursor-dependent decision tree using CID and ETD for fragmentation to balance acquisition rate with the need to fragment larger peptides, see below (30, 40). Defining a preliminary cleavage rule based on our previous observations increased the number of peptide-spectrum matches (PSMs) over a nonspecific digestion search (supplemental Table S2). The search was established using “semi-PEP,” allowing one site to be nonspecific. With this search output, we calculated missed cleavages throughout the whole dataset to evaluate digestion efficiency. We observed a maximum of two missed Pro cleavages for 90% of all identified peptides, but the number of missing Ala cleavages was considerably higher (supplemental Fig. S9A). Intriguingly, longer incubation times and higher enzyme-to-substrate ratios did not significantly reduce the number of missed cleavages (data not shown). Missed cleavages mostly displayed Pro, Gly, and Ala immediately C-terminal to the Ala or Pro residue (supplemental Fig. S9, B and C). Using neprosin and the semi-PEP rule, we identified 1729 protein groups in a 110-min run, compared with 2826 and 3505 for LysC and trypsin, respectively (Fig. 3A and supplemental Table S3). The sequence coverage contributed by neprosin is strongly complementary to that observed for trypsin and LysC. Of the 1251 proteins identified in common, almost half of the neprosin-generated coverage represents “dark matter” of the proteome sequence and potential PTMs not otherwise found using the two conventional reagents combined (Fig. 3B). When considering a rich data repository like ProteomicsDB (38), which contains a cumulative representation of human proteins identified from numerous projects, the single neprosin data set contributes additional sequence coverage of 6116 amino acids (FDR 1%, supplemental Table S4). We observed no obvious size bias in neprosin protein substrates. Identified proteins showed the same size distribution as proteins identified after digestion with trypsin and LysC (Fig. 4A).
Fig. 3.
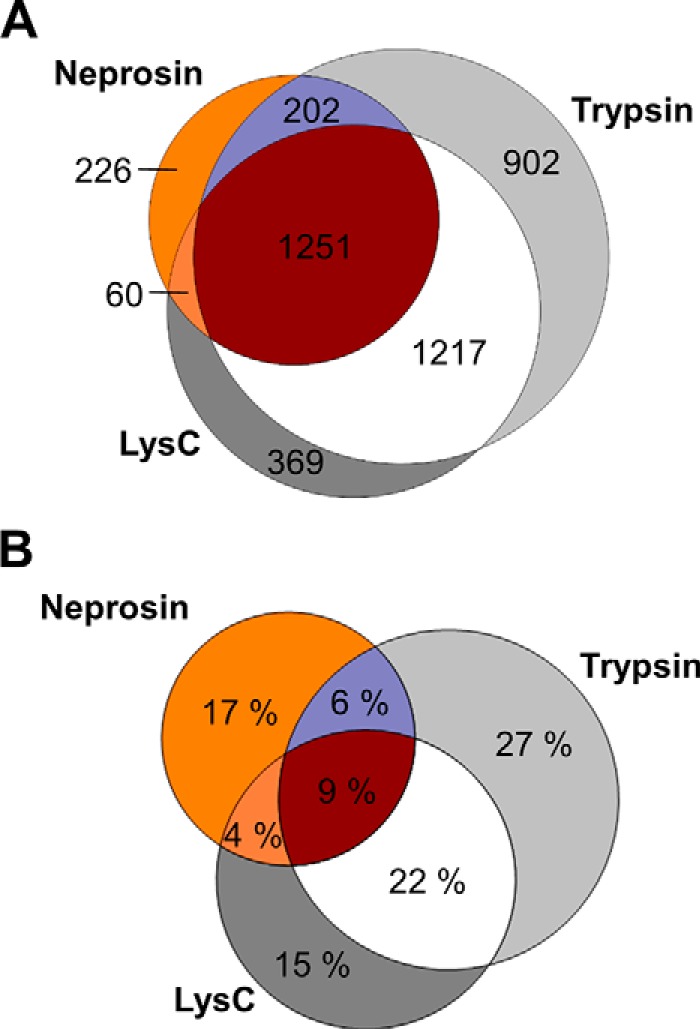
Proteomic analysis of HeLa whole-cell lysate after digestion with neprosin, trypsin, and LysC. A, Venn diagram visualization of identified protein groups for HeLa lysate digested with trypsin, LysC, and neprosin. B, Venn diagram visualization of the observed sequence coverage for the 1251 proteins commonly identified from HeLa digests with trypsin, LysC, and neprosin.
Fig. 4.
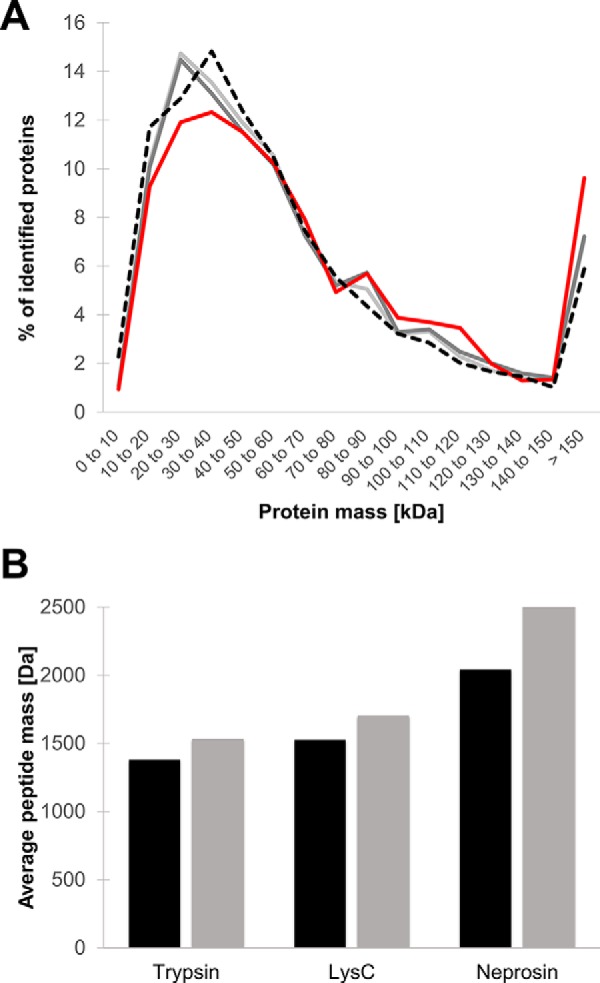
Size distribution of identified proteins and peptides after HeLa whole-cell lysate digestion. A, size distribution of identified protein groups in HeLa lysate digested with neprosin (solid red line), LysC (solid dark gray line), and trypsin (solid light gray line) plotted against the in silico calculated size distribution of protein groups in the human proteome (dotted black line). B, average mass of identified peptides (black bars) and all MS-triggered peptide ions (gray bars) after digestion of HeLa cell lysate with trypsin, LysC, and neprosin as indicated.
MS/MS of Neprosin-generated Peptides
The average mass of a triggered peptide after digestion with neprosin was 2521 units, significantly higher than both trypsin and Lys-C (Fig. 4B). This number represents all MS/MS-triggering events. Only 20% of the MS2 spectra were converted to PSMs, a conversion rate considerably lower than either trypsin (52%) or Lys-C (46%) in our experiments (supplemental Table S2). This was somewhat unexpected as peptide feature sets were equally rich, although lower conversion rates for non-tryptic enzymes have been documented (43). We note that more than half of the PSMs were identified by ETD, whereas LysC and tryptic peptides were mainly identified by CID (supplemental Fig. S10). In separate experiments, we further observed that HCD fragmentation of peptides with a C-terminal Pro residue led to the generation of very abundant y1 ions independent of the position of charged residues within the peptide (supplemental Fig. S11, A and C).
Histone Mapping
The ability to drive digestion to a stable end point while preserving a larger average peptide mass should be useful for PTM mapping exercises. To assess the potential of neprosin for the analysis of complex PTM patterns, we observed improved coverage for histones, the core components of chromatin known for their highly basic and multiply modified N termini (44). With neprosin, even a single untargeted analysis yielded a strong representation of histone tails from H3 and H4, relative to trypsin and LysC (supplemental Tables S5 and S6). Proline positions in these tails in particular appear well suited for presenting the histone code in a minimally biased manner. To explore this further, we digested and analyzed unfractionated histones from calf thymus via LC-MS/MS to expand our initial analysis, and we used EThcD for fragmentation (45). We observed prominent cleavages after Pro-32 for histone H4 and after Pro-38 in histone H3, which led to the release of the N-terminal tails 1–32 and 1–38, respectively (supplemental Fig. S12).
The release of these peptides highlights the vast number of histone modifications present, and we could identify a variety of co-occurring PTMs in what we could describe as an extended bottom-up approach for H3 (Fig. 5A) and H4 (Fig. 5B). In this crude extract, H3 peptide 1–38 showed many co-occurring modifications at Lys-9, -14, -18, -23, -27, and -36, including methylation, dimethylation, trimethylation, and acetylation. Given their hydrophilic character, these peptides elute early in reversed-phase chromatography, mostly independent of the number of modifications (supplemental Fig. S12). H4 peptide 1–32 revealed an acetylated N terminus, together with acetylated Lys-5, -8, -12, and -16 and a methylated or dimethylated Lys-20. Using EThcD, we observed rich fragment ion spectra for these peptides, which allowed us to unambiguously assign the modified amino acids (e.g. Fig. 5C).
Fig. 5.
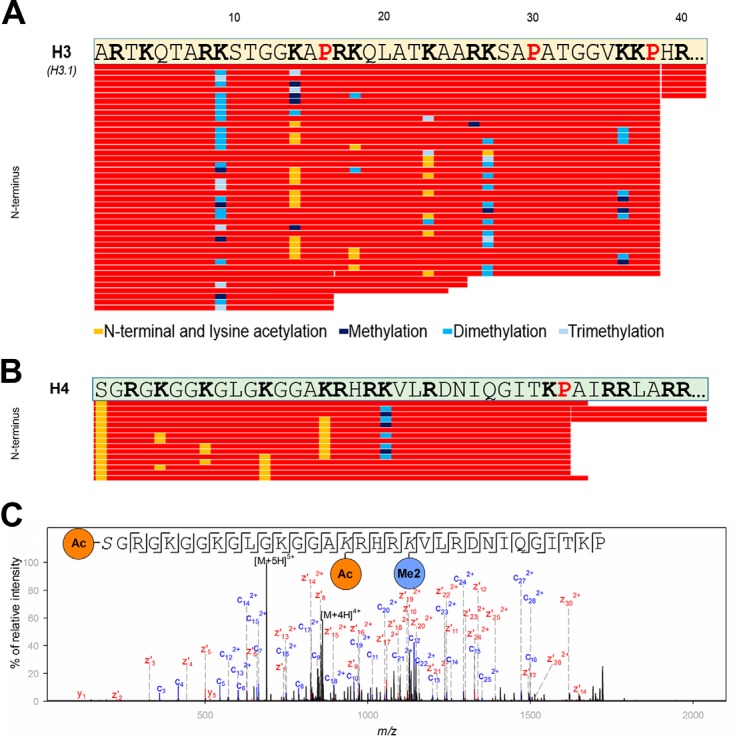
Histone H3 and histone H4 mapping using neprosin as identified within a digest of whole histones from calf thymus. A, sequence coverage plot of the N terminus (amino acids 1–40) of histone H3. Underlying peptides are shown as red bars in a sequence coverage plot. The retention time of the corresponding peptides was between 3 and 8 min. Pro residues are highlighted in red and bold, and Arg and Lys residues are shown in black and bold. Only those sequences are displayed for which the PTM site was confirmed based on manual inspection. Cleavages after Pro-16, Ala-24, and Ala-25 were only observed to minor extent. Legend for identified PTMs is provided in the figure. B, sequence coverage plot of the N terminus (amino acids 1–40) of histone H4. Underlying peptides are shown as red bars. Pro residues are highlighted in red and bold, whereas Arg and Lys residues are shown in black and bold. Only those peptides are displayed for which the PTM site was confirmed based on unambiguous corresponding fragment ions. Legend for identified PTMs is provided in the figure. C, exemplary fragment ion spectrum of an acetylated and dimethylated peptide of histone H4 (residues 1–32; precursor ion was at m/z 688.6046; z = 5) showing almost complete fragment ion series using EThcD.
Histone Mapping, Additional Observations
Even though H3 contains two additional Pro residues at positions 16 and 30, no significant cleavages were observed at these residues (Fig. 5A), which highlights that additional specificity is conferred outside of the P1-P1′ position. For H1 and H2B, we did not observe such a well defined, near-singular N-terminal peptide, likely because of the higher content of Pro residues in their N-terminal tails. We identified several peptides covering the N terminus of H2A (e.g. peptide 1–26), here mostly due to minor cleavages after residues beyond Pro. Nevertheless, the digest still readily demonstrated the presence of an acetylated N terminus and H2AK5Ac (supplemental Fig. S13).
DISCUSSION
Our data show that neprosin cleaves at a combined 85% after Pro and Ala residues, which provides specificity levels approaching trypsin (91%) and its counterpart Lys arginase (92%) (4). This level of specificity permits a limiting of the search space, which reduces the search time compared with other enzymes that cleave at neutral residues. The specificity of neprosin and its ability to digest substrates of any size confirm its characterization as a legitimate PEP. The cleavage of the Asp-Pro motif that we observed is, as we have shown, most likely due to the lability of this peptide bond under acidic conditions (46), rather than a direct result of enzymatic activity. Although we used a “semi-PEP” cleavage rule for our searches, a full PEP cleavage rule was also very effective, and it only reduced the protein group output by 5% (supplemental Table S2).
The number of missed cleavage sites was remarkably high, particularly for Ala, and was not significantly affected by incubation times or E:S ratios. It suggests that we have not yet fully understood the influence of amino acids distal from the P1 and P1′ positions. We have noted that Pro or Ala in the P2 position enhanced catalytic activity, as well as Glu in position P1′ or P2′. There may be other influences that limit activity at the canonical cleavage site, which may only become apparent with much larger data sets and a crystallographic structural model of the enzyme. The “skipping frequency” contributed to an average peptide mass of ∼2500 units, which places neprosin in the category of an enzyme ideal for extended bottom-up proteomics (7). The additional sequence coverage provided by neprosin is considerable, especially considering the lower number of PSMs with respect to trypsin.
The lower conversion rate of MS2 acquisitions into PSMs remains somewhat of a puzzle, especially given the selectivity of neprosin. The phenomenon appears to be the norm for proteases beyond trypsin (43). There can be many reasons for lower performance. Given their smaller average size, tryptic peptides are well matched to the fragmentation methods conventionally used in mass spectrometry, and search algorithms have been exquisitely tuned over many years to reflect fragmentation bias. Our data show an increased reliance upon ETD in the neprosin digests, which at first glance might suggest lower overall spectral quality. However, the intensity and richness of the unidentified MS2 subset was high, which may be due to a higher frequency of internal fragments. Additionally, as the peptide size increases, there is an increased probability of peptides carrying multiple modifications, and these are often missed by search algorithms (47). We anticipate that a combination of different decision-tree algorithms involving fragmentation techniques such as EThcD (45) or UVPD (48), as well as revised search algorithms, could improve PSM rates. In our HCD studies, we observed an abundant y1 ion at m/z 116 in many peptides, arising from the proline effect (49), which may offer a very useful reporter ion and help support alternative search algorithms. Improving the MS2 conversion rate should become a bioinformatics priority, especially for enzymes that exhibit high selectivity and appear to complement trypsin nicely.
The complementarity of neprosin with other enzymes is particularly apparent in the analysis of histone tails. Many disease states in cancer, cognitive dysfunction, and reproductive, respiratory, and cardiovascular illnesses are strongly linked to histone tail-regulated epigenetic mechanisms in the cell, and there is a growing need to understand how these mechanisms drive cellular behavior (50, 51). MS is the method of choice for their investigation (52). Abundant histone tail modifications include the methylation and acetylation of Lys residues, as well as the methylation of Arg, even though the total number of all known histone PTMs is distinctly higher (33). A bottom-up analysis of histones using trypsin requires a derivatization step such as propionylation to partially restrict digestion (53). The peptides generated allow for the quantitation of single PTMs, but information on co-occurring modifications is mainly lost, and new methods are needed that retain such information (54, 55). GluC is generally used to produce peptide 1–50 from histone H3, and AspN is used to produce peptide 1–23 from histone H4. Neprosin provides a single enzyme solution to near-complete profiling of both tails, with additional advantages. Lys-37 is the highest extensively modified amino acid of interest in H3 (56), which means that peptide 1–50 is longer than it needs to be for its analysis. Neprosin reliably generates peptide 1–38, in which all relevant modified amino acids are still present. Its lower mass facilitates efficient fragmentation in ETD mode. Peptide 1–32 from histone H4 is released in the same reliable fashion. The PTM patterns we observed are in good qualitative agreement with previously reported values (23, 56). Our findings lead us to the conclusion that neprosin is a promising alternative to GluC, AspN, and trypsin for the mass spectrometric analysis of histones.
Neprosin's ability to cleave with high specificity after Pro and Ala, under mildly denaturing conditions and at low E:S ratios, makes it a useful new tool for proteomics. Other opportunities will include protein structure/function applications of mass spectrometry. Neprosin can provide a more selective alternative to aspartic proteases like pepsin in hydrogen/deuterium exchange experiments or the analysis of native disulfide bridges, and as Pro and Ala are inert to most protein chemistries, it should prove useful in processing cross-linked proteins. It remains unclear whether neprosin is the archetype of the entire family of proteins presenting DUF239. Other members may offer PEP-like functionality distinct from neprosin and should be explored for their utility.
DATA AVAILABILITY
Raw files for all MS experiments are available from Chorus (ID 1262), and annotated fragment ion spectra for identified histone tails are available in the supplemental materials.
Supplementary Material
Acknowledgments
Mathias Wilhelm (Technische Universität München, Freising, Germany) is gratefully acknowledged for providing data from ProteomicsDB.
Footnotes
Author contributions: C.U.S. and D.C.S. designed the research; C.U.S., L.L., M.R., and P.M. performed the research; L.L., P.M., S.S., V.Z., and B.L. contributed new reagents or analytic tools; C.U.S., L.L., V.S., B.L., and D.C.S. analyzed the data; and C.U.S. and D.C.S. wrote the paper.
* This work was supported in part by Natural Sciences and Engineering Research Council of Canada Discovery Grant 298351-2010 (to D.C.S.).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- PTM
- post-translational modification
- CID
- collision-induced dissociation
- DUF239
- domain of unknown function 239
- E:S ratio
- enzyme-to-substrate ratio
- ETD
- electron-transfer dissociation
- EThcD
- electron-transfer/higher-energy collision dissociation
- HCD
- higher-energy collisional dissociation
- PEP
- prolyl endoprotease
- POP
- prolyl oligopeptidase
- PSM
- peptide spectrum match
- MBP
- maltose-binding protein
- FA
- folic acid
- ACN
- acetonitrile
- NCE
- normalized collision energy.
REFERENCES
- 1. Aebersold R., and Mann M. (2016) Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355 [DOI] [PubMed] [Google Scholar]
- 2. Olsen J. V., Ong S. E., and Mann M. (2004) Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol. Cell. Proteomics 3, 608–614 [DOI] [PubMed] [Google Scholar]
- 3. Swaney D. L., Wenger C. D., and Coon J. J. (2010) Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J. Proteome Res. 9, 1323–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huesgen P. F., Lange P. F., Rogers L. D., Solis N., Eckhard U., Kleifeld O., Goulas T., Gomis-Rüth F. X., and Overall C. M. (2015) LysargiNase mirrors trypsin for protein C-terminal and methylation-site identification. Nat. Methods 12, 55–58 [DOI] [PubMed] [Google Scholar]
- 5. Tsiatsiani L., and Heck A. J. (2015) Proteomics beyond trypsin. FEBS J 282, 2612–2626 [DOI] [PubMed] [Google Scholar]
- 6. Meyer J. G., Kim S., Maltby D. A., Ghassemian M., Bandeira N., and Komives E. A. (2014) Expanding proteome coverage with orthogonal-specificity α-lytic proteases. Mol. Cell. Proteomics 13, 823–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Laskay Ü. A., Srzentić K., Monod M., and Tsybin Y. O. (2014) Extended bottom-up proteomics with secreted aspartic protease Sap9. J. Proteomics 110, 20–31 [DOI] [PubMed] [Google Scholar]
- 8. Wu C., Tran J. C., Zamdborg L., Durbin K. R., Li M., Ahlf D. R., Early B. P., Thomas P. M., Sweedler J. V., and Kelleher N. L. (2012) A protease for ‘middle-down’ proteomics. Nat. Methods 9, 822–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rietschel B., Arrey T. N., Meyer B., Bornemann S., Schuerken M., Karas M., and Poetsch A. (2009) Elastase digests: new ammunition for shotgun membrane proteomics. Mol. Cell. Proteomics 8, 1029–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vandermarliere E., Mueller M., and Martens L. (2013) Getting intimate with trypsin, the leading protease in proteomics. Mass Spectrom. Rev. 32, 453–465 [DOI] [PubMed] [Google Scholar]
- 11. Ahn J., Cao M. J., Yu Y. Q., and Engen J. R. (2013) Accessing the reproducibility and specificity of pepsin and other aspartic proteases. Biochim. Biophys. Acta 1834, 1222–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pappin D. J., Hojrup P., and Bleasby A. J. (1993) Rapid identification of proteins by peptide-mass fingerprinting. Curr. Biol. 3, 327–332 [DOI] [PubMed] [Google Scholar]
- 13. Kapp E., and Schutz F. (2007) Overview of tandem mass spectrometry (MS/MS) database search algorithms. Curr. Protoc. Protein Sci. Chapter 25, Unit 25.22 [DOI] [PubMed] [Google Scholar]
- 14. Morgan A. A., and Rubenstein E. (2013) Proline: the distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS ONE 8, e53785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gass J., and Khosla C. (2007) Prolyl endopeptidases. Cell. Mol. Life Sci. 64, 345–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moriyama A., Nakanishi M., and Sasaki M. (1988) Porcine muscle prolyl endopeptidase and its endogenous substrates. J. Biochem. 104, 112–117 [DOI] [PubMed] [Google Scholar]
- 17. Shan L., Marti T., Sollid L. M., Gray G. M., and Khosla C. (2004) Comparative biochemical analysis of three bacterial prolyl endopeptidases: implications for coeliac sprue. Biochem. J. 383, 311–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sebela M., Rehulka P., Kábrt J., Rehulková H., Ozdian T., Raus M., Franc V., and Chmelík J. (2009) Identification of N-glycosylation in prolyl endoprotease from Aspergillus niger and evaluation of the enzyme for its possible application in proteomics. J. Mass Spectrom. 44, 1587–1595 [DOI] [PubMed] [Google Scholar]
- 19. Biterge B., and Schneider R. (2014) Histone variants: key players of chromatin. Cell Tissue Res. 356, 457–466 [DOI] [PubMed] [Google Scholar]
- 20. Probst A. V., Dunleavy E., and Almouzni G. (2009) Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 10, 192–206 [DOI] [PubMed] [Google Scholar]
- 21. Audia J. E., and Campbell R. M. (2016) Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 8, a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garcia B. A., Mollah S., Ueberheide B. M., Busby S. A., Muratore T. L., Shabanowitz J., and Hunt D. F. (2007) Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2, 933–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Phanstiel D., Brumbaugh J., Berggren W. T., Conard K., Feng X., Levenstein M. E., McAlister G. C., Thomson J. A., and Coon J. J. (2008) Mass spectrometry identifies and quantifies 74 unique histone H4 isoforms in differentiating human embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 105, 4093–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Young N. L., DiMaggio P. A., Plazas-Mayorca M. D., Baliban R. C., Floudas C. A., and Garcia B. A. (2009) High throughput characterization of combinatorial histone codes. Mol. Cell. Proteomics 8, 2266–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee L., Zhang Y., Ozar B., Sensen C. W., and Schriemer D. C. (2016) Carnivorous nutrition in pitcher plants (Nepenthes spp.) via an unusual complement of endogenous enzymes. J. Proteome Res. 15, 3108–3117 [DOI] [PubMed] [Google Scholar]
- 26. Rey M., Yang M., Lee L., Zhang Y., Sheff J. G., Sensen C. W., Mrazek H., Halada P., Man P., McCarville J. L., Verdu E. F., and Schriemer D. C. (2016) Addressing proteolytic efficiency in enzymatic degradation therapy for celiac disease. Sci. Rep. 6, 30980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malik A., Rudolph R., and Söhling B. (2005) Use of enhanced green fluorescent protein to determine pepsin at high sensitivity. Anal. Biochem. 340, 252–258 [DOI] [PubMed] [Google Scholar]
- 28. Wiśniewski J. R., Zougman A., Nagaraj N., and Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 29. Rappsilber J., Mann M., and Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 30. Swaney D. L., McAlister G. C., and Coon J. J. (2008) Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat. Methods 5, 959–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang J., Xin L., Shan B., Chen W., Xie M., Yuen D., Zhang W., Zhang Z., Lajoie G. A., and Ma B. (2012) PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteomics 11, M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rodriguez J., Gupta N., Smith R. D., and Pevzner P. A. (2008) Does trypsin cut before proline? J. Proteome Res. 7, 300–305 [DOI] [PubMed] [Google Scholar]
- 33. Huang H., Sabari B. R., Garcia B. A., Allis C. D., and Zhao Y. (2014) SnapShot: histone modifications. Cell 159, 458–458 e451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Han X., He L., Xin L., Shan B., and Ma B. (2011) PeaksPTM: mass spectrometry-based identification of peptides with unspecified modifications. J. Proteome Res. 10, 2930–2936 [DOI] [PubMed] [Google Scholar]
- 35. Choo K. H., Tan T. W., and Ranganathan S. (2005) SPdb–a signal peptide database. BMC Bioinformatics 6, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang M., Hoeppner M., Rey M., Kadek A., Man P., and Schriemer D. C. (2015) Recombinant nepenthesin II for hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 87, 6681–6687 [DOI] [PubMed] [Google Scholar]
- 37. Colaert N., Helsens K., Martens L., Vandekerckhove J., and Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 38. Wilhelm M., Schlegl J., Hahne H., Gholami A. M., Lieberenz M., Savitski M. M., Ziegler E., Butzmann L., Gessulat S., Marx H., Mathieson T., Lemeer S., Schnatbaum K., Reimer U., Wenschuh H., et al. (2014) Mass-spectrometry-based draft of the human proteome. Nature 509, 582–587 [DOI] [PubMed] [Google Scholar]
- 39. Rey M., Yang M., Burns K. M., Yu Y., Lees-Miller S. P., and Schriemer D. C. (2013) Nepenthesin from monkey cups for hydrogen/deuterium exchange mass spectrometry. Mol. Cell. Proteomics 12, 464–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frese C. K., Altelaar A. F., Hennrich M. L., Nolting D., Zeller M., Griep-Raming J., Heck A. J., and Mohammed S. (2011) Improved peptide identification by targeted fragmentation using CID, HCD and ETD on an LTQ-Orbitrap Velos. J. Proteome Res. 10, 2377–2388 [DOI] [PubMed] [Google Scholar]
- 41. Keil B. (1992) Specificity of Proteolysis, Springer-Verlag, New York, 8–10 [Google Scholar]
- 42.Delete in proof
- 43. Giansanti P., Tsiatsiani L., Low T. Y., and Heck A. J. (2016) Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat. Protoc. 11, 993–1006 [DOI] [PubMed] [Google Scholar]
- 44. Rodríguez-Paredes M., and Esteller M. (2011) Cancer epigenetics reaches mainstream oncology. Nat. Med. 17, 330–339 [DOI] [PubMed] [Google Scholar]
- 45. Frese C. K., Altelaar A. F., van den Toorn H., Nolting D., Griep-Raming J., Heck A. J., and Mohammed S. (2012) Toward full peptide sequence coverage by dual fragmentation combining electron-transfer and higher-energy collision dissociation tandem mass spectrometry. Anal. Chem. 84, 9668–9673 [DOI] [PubMed] [Google Scholar]
- 46. Piszkiewicz D., Landon M., and Smith E. L. (1970) Anomalous cleavage of aspartyl-proline peptide bonds during amino acid sequence determinations. Biochem. Biophys. Res. Commun. 40, 1173–1178 [DOI] [PubMed] [Google Scholar]
- 47. Chick J. M., Kolippakkam D., Nusinow D. P., Zhai B., Rad R., Huttlin E. L., and Gygi S. P. (2015) A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat. Biotechnol. 33, 743–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Greer S. M., Parker W. R., and Brodbelt J. S. (2015) Impact of protease on ultraviolet photodissociation mass spectrometry for bottom-up proteomics. J. Proteome Res. 14, 2626–2632 [DOI] [PubMed] [Google Scholar]
- 49. Grewal R. N., El Aribi H., Harrison A. G., Siu K. W. M., and Hopkinson A. C. (2004) Fragmentation of protonated tripeptides: the proline effect revisited. J. Phys. Chem. B 108, 4899–4908 [Google Scholar]
- 50. Karch K. R., Denizio J. E., Black B. E., and Garcia B. A. (2013) Identification and interrogation of combinatorial histone modifications. Front. Genet. 4, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weinhold B. (2006) Epigenetics: the science of change. Environ. Health Perspect. 114, A160–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yuan Z. F., Arnaudo A. M., and Garcia B. A. (2014) Mass spectrometric analysis of histone proteoforms. Annu. Rev. Anal. Chem. 7, 113–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sidoli S., Yuan Z. F., Lin S., Karch K., Wang X., Bhanu N., Arnaudo A. M., Britton L. M., Cao X. J., Gonzales-Cope M., Han Y., Liu S., Molden R. C., Wein S., Afjehi-Sadat L., and Garcia B. A. (2015) Drawbacks in the use of unconventional hydrophobic anhydrides for histone derivatization in bottom-up proteomics PTM analysis. Proteomics 15, 1459–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moradian A., Kalli A., Sweredoski M. J., and Hess S. (2014) The top-down, middle-down, and bottom-up mass spectrometry approaches for characterization of histone variants and their post-translational modifications. Proteomics 14, 489–497 [DOI] [PubMed] [Google Scholar]
- 55. Sidoli S., Schwämmle V., Ruminowicz C., Hansen T. A., Wu X., Helin K., and Jensen O. N. (2014) Middle-down hybrid chromatography/tandem mass spectrometry workflow for characterization of combinatorial post-translational modifications in histones. Proteomics 14, 2200–2211 [DOI] [PubMed] [Google Scholar]
- 56. Schwämmle V., Sidoli S., Ruminowicz C., Wu X., Lee C. F., Helin K., and Jensen O. N. (2016) Systems level analysis of histone H3 post-translational modifications (PTMs) reveals features of PTM crosstalk in chromatin regulation. Mol. Cell. Proteomics 15, 2715–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw files for all MS experiments are available from Chorus (ID 1262), and annotated fragment ion spectra for identified histone tails are available in the supplemental materials.