


Abstract
Pre-tRNA splicing is an essential process in all eukaryotes. It requires the concerted action of an endonuclease to remove the intron and a ligase for joining the resulting tRNA halves as studied best in the yeast Saccharomyces cerevisiae. Here, we report the first characterization of an RNA ligase protein and its gene from a higher eukaryotic organism that is an essential component of the pre-tRNA splicing process. Purification of tRNA ligase from wheat germ by successive column chromatographic steps has identified a protein of 125 kDa by its potentiality to covalently bind AMP, and by its ability to catalyse the ligation of tRNA halves and the circularization of linear introns. Peptide sequences obtained from the purified protein led to the elucidation of the corresponding proteins and their genes in Arabidopsis and Oryza databases. The plant tRNA ligases exhibit no overall sequence homologies to any known RNA ligases, however, they harbour a number of conserved motifs that indicate the presence of three intrinsic enzyme activities: an adenylyltransferase/ligase domain in the N-terminal region, a polynucleotide kinase in the centre and a cyclic phosphodiesterase domain at the C-terminal end. In vitro expression of the recombinant Arabidopsis tRNA ligase and functional analyses revealed all expected individual activities. Plant RNA ligases are active on a variety of substrates in vitro and are capable of inter- and intramolecular RNA joining. Hence, we conclude that their role in vivo might comprise yet unknown essential functions besides their involvement in pre-tRNA splicing.
INTRODUCTION
RNA ligases are engaged in a variety of inter- and intramolecular nucleic acid joining reactions such as the repair of broken tRNAs, ligation of tRNA halves and circularization of linear polyribonucleotides including excised introns. Bacteriophage T4 RNA ligase (Rnl 1) is the type member of this group whose reaction mechanisms and structural features have been intensively studied within the last 20 years (1–3). It catalyses the formation of a 3′,5′-phosphodiester bond between RNA strands containing a 5′ terminal phosphate and a 3′ terminal hydroxyl group. The enzymatic joining reactions involve three reversible steps. First, the enzyme reacts with ATP to form a covalently adenylated enzyme intermediate and pyrophosphate. Second, the AMP is transferred from the ligase–adenylate to the 5′-phosphate end of an RNA molecule to form an adenylated RNA intermediate, and finally, ligase catalyses the formation of a phosphodiester bond by the reaction of the 3′-hydroxyl group with the adenylated 5′ RNA end and simultaneous release of AMP.
The function of T4 RNA ligase 1 in vivo is to repair a break in the anticodon of Escherichia coli tRNALys triggered by phage activation of a host-encoded anticodon nuclease (4,5). The bacterial tRNALys-specific enzyme cleaves its natural substrate 5′ to the wobble base, yielding 2′,3′-cyclic phosphate and 5′-OH ends which are not substrates for T4 RNA ligase as mentioned above. Consequently, the broken ends must be modified before they can be sealed. This mission is performed by the bacteriophage T4-encoded polynucleotide kinase/3′-phosphatase. The bifunctional enzyme converts 2′,3′-cyclic phosphate to a 3′-phosphate and then hydrolyses the phosphate leaving a 3′-OH group. Second, it transfers the γ-phosphate from ATP to the hydroxyl group at the 5′ end of the cleaved tRNALys to generate a 5′-phosphate (4,6,7).
A novel RNA ligation pathway was discovered in wheat germ extract by Konarska et al. (8). A 73-nt long RNA oligonucleotide called Ω fragment (derived from tobacco mosaic virus RNA after RNase T1 cleavage) was circularized upon incubation with S30 wheat germ extract provided that the substrate carried 2′,3′-cyclic phosphate and 5′-hydroxyl or 5′-phosphate termini. Moreover, it was subsequently shown that the plant extract catalysed the formation of an unusual 2′-phosphomonoester, 3′,5′-phosphodiester linkage in which the 2′-phosphate is derived from the RNA substrate (8,9). Later on, it was reported that the structures of the substrates and products in the yeast ligase reaction are identical to those described for the wheat germ ligase and that yeast RNA ligase participates in vivo in the splicing of tRNA precursors (10).
Intron-containing tRNA genes were first discovered in the yeast Saccharomyces cerevisiae (11). Subsequently, the mechanism of pre-tRNA splicing was deduced in this organism. It turned out that it differs fundamentally from the spliceosome-mediated mRNA splicing and from the self-splicing of group I or group II introns. Three enzymes participate in the tRNA splicing process. Intron-containing pre-tRNAs are first cleaved by a tRNA splicing endonuclease at the 5′ and 3′ boundaries of the intervening sequence producing paired tRNA halves with 2′,3′-cyclic phosphate, 5′-OH ends and a linear intron. Second, the halves are joined by a tRNA ligase requiring GTP and ATP leaving a 2′-phosphate and 3′-OH at the splice junction. Finally, this phosphate is removed by a NAD+-dependent phosphotransferase (12).
The tRNA ligase protein of S.cerevisiae was purified by five chromatographic steps including an affinity elution. The amino acid sequence of the N-terminal end of the protein was determined and utilized for the isolation of the corresponding gene from a library of yeast DNA. The purified yeast tRNA ligase is a monomeric 92 kDa protein that contains three intrinsic activities: an N-terminal adenylyltransferase domain that resembles T4 RNA ligase 1, a central domain that resembles T4 polynucleotide kinase (without 3′-phosphatase activity) and a C-terminal cyclic phosphodiesterase (CPDase) domain that is reminiscent of the 2H phosphotransferase enzyme superfamily (13–15). The multifunctional yeast tRNA ligase performs a series of reactions: (i) the 2′,3′-cyclic phosphate at the end of the 5′ tRNA half is hydrolysed by the CPDase activity to generate a 2′-phosphate and 3′-OH; (ii) the 5′-OH group at the end of the 3′ tRNA half is phosphorylated by the GTP-dependent kinase activity and (iii) the ligase protein is adenylylated and the AMP is transferred to the 5′-phosphate of the 3′ tRNA half followed by the formation of the 2′-phosphomonoester–3′,5′-phosphodiester bond and the release of AMP (Figure 1).
Figure 1.

The tRNA splicing pathway of plants and fungi. Intron-containing pre-tRNAs are first cleaved by a tRNA endonuclease at the 5′ and 3′ boundaries of the intervening sequence producing paired tRNA halves with 2′,3′-cyclic phosphate, 5′-OH ends and a linear intron. Second, the halves are ligated by a complex reaction requiring GTP and ATP. Finally, the 2′-phosphate at the splice junction is removed by a NAD-dependent phosphotransferase (12,46).
An RNA ligase was partially purified from wheat germ and has been shown to be associated with a 2′,3′-cyclic phosphodiesterase and a polynucleotide kinase activity through all chromatographic steps (16,17). Wheat germ and yeast tRNA ligases share many characteristic features. Both are multifunctional enzymes that harbour the same intrinsic activities within one polypeptide and generate an unusual 2′-phosphate at the splice junction. Moreover, yeast tRNA ligase is capable of joining plant tRNA halves derived from intron excision by splicing endonuclease and vice versa (18,19). Despite this apparent overall similarity in structure and function, a search of the fully sequenced genomes of Arabidopsis thaliana and Oryza sativa with the yeast sequence as query did not identify a candidate gene for tRNA ligase in either nuclear genome. In fact, to date specific proteins responsible for tRNA ligation in any archaeal or eukaryotic organism—with the exception of yeast and its fungal relatives—have not yet been identified nor have their genes been cloned.
Here, we report the chromatographic purification of the tRNA ligase protein from wheat germ and the identification of the coding genes in two plant nuclear genomes by means of protein sequencing of the wheat enzyme. Furthermore, we present enzymatic studies with the overexpressed recombinant tRNA ligase from Arabidopsis and S.cerevisiae that demonstrate the functional relationship of plant and yeast ligases.
MATERIALS AND METHODS
Purification of the tRNA ligase from wheat germ
All operations were carried out at 4°C. Buffer A [50 mM Tris–HCl, pH 7.5, 3 mM MgCl2, 0.1% Triton X-100, 4 mM β-mercaptoethanol (β-ME), 0.2 mM PMSF, 0.2 mM benzamidine, 0.4 μg/ml pepstatin A and 0.5 μg/ml leupeptin] was used for all chromatographic steps with the salt concentrations as indicated.
Step 1. Wheat embryos (140 g) were prepared from crude wheat germs (Sigma, W-0125) by immersing the germs successively in 500 ml cyclohexan and the air-dried material was ground in an electric mill. The resulting fine powder was stirred for 10 min with 500 ml of buffer A, containing 200 mM ammonium sulfate, and the homogenate was centrifuged for 15 min at 30 000 g to remove cell debris and organelles. The S30 extract (400 ml; total protein = 60 g) was subsequently centrifuged for 90 min at 100 000 g to remove membranes and ribosomes. The final S100 extract (280 ml; total protein = 28 g) was brought to 0.1% Polymin P (polyethyleneimine; Sigma, München, Germany) by the addition of 2.8 ml of 10% Polymin P, pH 7.5, and stirred for additional 10 min. The precipitated nucleic acids were removed by centrifugation for 10 min at 10 000 g.
Step 2. The supernatant (280 ml) was brought to 50% ammonium sulfate by the addition of 280 ml saturated ammonium sulfate solution in 50 mM Tris–HCl, pH 7.5. After stirring for 1 h, the precipitated proteins were collected by centrifugation for 15 min at 10 000 g. The pellet was dissolved in 1 litre of buffer A and diluted to a conductivity equivalent to that of 180 mM KCl (1600 ml; total protein = 16 g).
Step 3. The diluted (NH4)2SO4 material was loaded onto a 40 ml Heparin Sepharose FF column (Amersham Biosciences, Freiburg, Germany) previously equilibrated with 180 mM KCl in buffer A. The column was eluted with a 400 ml linear gradient of 180–500 mM KCl. The peak fractions containing tRNA ligase activity eluted between 270 and 340 mM KCl. They were pooled and diluted with buffer A to a conductivity equivalent to that of 150 mM KCl (100 ml; total protein = 300 mg).
Step 4. The pooled fractions from the Heparin Sepharose column were applied to a 10 ml Cibacron Blue Trisacryl M column (Serva, Heidelberg, Germany) previously equilibrated with 150 mM KCl in buffer A. The column was eluted with a 100 ml linear gradient of 150–800 mM KCl. The peak fractions eluting between 280 and 540 mM KCl (Figure 2) were pooled and diluted with buffer A to a conductivity equivalent to that of 100 mM KCl (50 ml; total protein = 60 mg).
Figure 2.

Isolation of wheat germ tRNA ligase. (A) Purification scheme. RNA ligase was purified from the soluble protein fraction of wheat embryos (S100 extract) by six consecutive steps. (B) As substrate for assaying tRNA ligase activity, we have used a natural modified Nicotiana pre-tRNATyr (NtY9-T7-M1). The arrows in the two 4 nt bulge loops indicate the 3′ and 5′ splice sites and dots identify the anticodon. (C) Fractionation of wheat germ tRNA ligase by Cibacron Blue Trisacryl M chromatography and ligation activity assay. Partially purified tRNA ligase from the Heparin Sepharose column was applied onto a Blue-Trisacryl M column. Elution of tRNA ligase was performed with a gradient of 150–800 mM KCl. Fractions of 5 ml were collected. Splicing endonuclease co-eluted with tRNA ligase at this purification step, generating 3′ and 5′ tRNA halves. Reaction mixtures (20 μl) contained 20 mM Tris–HCl, pH 7.5, 6 mM Mg(OAc)2, 80 μM spermine, 1 mM ATP, 0.5 mM GTP, 0.1 mM DTT, 0.5% Triton X-100, 40 fmol (4 × 104 c.p.m.) of T7-transcript (NtY9-T7-M1) and 2 μl from eluted fractions. Incubation was for 30 min at 37°C. Products were analysed on a 12.5% polyacrylamide/8 M urea gel. tRNA ligase activity elutes between 280 and 540 mM KCl as revealed by the detection of mature, spliced tRNA.
Step 5. The pooled fractions from the Blue Trisacryl M column were loaded onto a 1 ml Source S15 column (Amersham Biosciences) that had been equilibrated with 100 mM KCl in buffer A. The column was eluted with a 10 ml linear gradient of 100–400 mM KCl. The active fractions eluted between 180 and 240 mM KCl. They were pooled (2.5 ml; total protein = 3 mg) and directly used for the analysis by gel filtration.
Step 6. The gel filtration was performed with a prepacked HiLoad™ 16/60 Superdex 200 PC column (Amersham Biosciences) equilibrated with 200 mM KCl in buffer A. The pooled fractions from the Source S15 column were applied to the Superdex 200 column and separation was performed with a constant flow rate of 1 ml/min. At this step of the purification procedure, the tRNA ligase protein was identified by its ability to covalently bind AMP upon incubation with [α-32P]ATP. Wheat germ RNA ligase eluted as an enzyme with an apparent native molecular mass of 160 kDa as deduced from comparison with standard proteins. Analysis of the protein pattern by silver staining of appropriate fractions separated by SDS–PAGE revealed the presence of various polypeptides in addition to the tRNA ligase protein (Figure 3A) and a single adenylylated protein of ∼125 kDa in the corresponding autoradiogram (Figure 3B).
Figure 3.

Gel filtration on Superdex™ 200 and adenylyltransferase activity of wheat germ tRNA ligase. (A) Partially purified tRNA ligase from the Source S15 column was subjected to gel filtration on Superdex™ 200. The column (HiLoad™ 16/60) was run with a flow rate of 1 ml/min. Fractions of 2 ml were collected. Aliquots of the elution fraction were analysed on a 7.5% polyacrylamide/0.1% SDS gel. The proteins were visualized by silver staining. (B) Appropriate aliquots of the indicated fractions were incubated in the presence of [α-32P]ATP for 15 min at 37°C. The ligase–[32P]AMP adduct was detected by autoradiography of the dried gel. (C) The peak fractions from the tRNA Sepharose column were concentrated by ultrafiltration and 1/20 of this material was applied onto a 10% polyacrylamide/0.1% SDS gel and stained with Coomassie blue for analytical valuation. The arrows point to the position of the RNA ligase protein with an approximate molecular weight of 125 kDa. Protein size standards in kDa are indicated on the right.
Step 7. The fractions 54–60, indicating the presence of the tRNA ligase protein, were 4-fold diluted with buffer A (8 ml; total protein = 150 μg) to a conductivity equivalent to that of 50 mM KCl and loaded onto an affinity column. Wheat tRNA (Sigma) was coupled to a 1 ml HiTrap NHS-activated Sepharose column (Amersham Biosciences) according to Rösch et al. (20). After equilibration of the column with 50 mM KCl in buffer A, the diluted gel filtration fractions were applied with a flow rate of 0.5 ml/min. Elution was performed with a 10 ml linear gradient of 50–400 mM KCl. Active fractions eluted between 180 and 240 mM KCl (2.5 ml, total protein = 40 μg).
Step 8. The pooled fractions from the tRNA Sepharose column were concentrated by ultrafiltration with Microcon 100 (Millipore, Schwalbach, Germany) and applied to a 10% polyacrylamide/0.1% SDS gel (Figure 3C). After a brief staining with Coomassie brilliant blue, ∼5 μg of the 125 kDa protein was excised from the gel. Protein sequence analysis was performed at the Harvard Microchemistry Facility (Cambridge, MA).
Preparation of RNA substrates
The original clone pNtY9, carrying an intron-containing tRNATyr gene on an EcoRI fragment of 7.5 kb was isolated from a Nicotiana rustica genomic library (21). Subsequent PCR and oligonucleotide-directed mutagenesis yielded a 124 bp long fragment cloned into the SmaI site of pUC19 DNA in which the tRNATyr gene was flanked by a T7 RNA polymerase promoter at the 5′ side and a BstNI restriction site at the 3′ side. The first base pair in the aminoacyl stem of the tRNA was exchanged from C1–G72 to G1–C72 in order to facilitate efficient transcription by T7 RNA polymerase. Furthermore, the intron hairpin loop of original 8 nt was replaced by a stable UUCG loop (22). This clone was named pNtY9-T7-M1. The archeuka tRNA gene is derived from this clone by Ex-site mutagenesis (according to the protocol of Stratagene, Heidelberg, Germany). The tRNATyr-specific anticodon and intron were replaced by the anticodon and intron of tRNATrp from Methanocaldococcus jannaschii with some modifications: the intron hairpin loop of original 33 nt was replaced by the UUCG loop and an additional G–C base pair was inserted into the helical intron region below the 3′ bulge loop.
Intron-containing pre-tRNAs were transcribed by T7 RNA polymerase using the RiboMax T7 transcription kit (Promega, Mannheim, Germany). Reaction mixtures contained 100 μM unlabelled ATP (or UTP) and 10 μM [α32-P]ATP or [α32-P]UTP (4 μCi/μl; 400 Ci/mmol; Amersham Biosciences). The T7-transcripts were purified on an 8% polyacrylamide/8 M urea gel. Purified archeuka pre-tRNA was cleaved with M.jannaschii splicing endonuclease in a 20 μl reaction mixture, containing 10 mM Tris–HCl, pH 7.6, 100 mM KCl, 10 mM MgCl2, 1 mM DTT, 40 μM spermine and 2 μl of 1 μg recombinant enzyme. Incubation was for 15 min at 65°C. After phenol/chloroform extraction, the RNA products were ethanol precipitated and used for ligation assays.
RNA ligase assay
In vitro ligation assays were performed in a total volume of 20 μl containing 10 mM Tris–HCl, pH 7.5, 100 mM KOAc, 0.3 mM spermine, 6 mM Mg(OAc)2, 0.5 mM DTT, 0.5% Triton X-100, 40 fmol (4 × 104 c.p.m.) of T7 transcript and 2 μl of appropriate chromatographic fractions. After incubation for 30 min at 37°C, the reaction mixtures were extracted with phenol/chloroform, ethanol precipitated and dissolved in a urea/dye mixture (8 M urea, 0.02% xylene cyanol and 0.02% bromophenol blue), heated for 3 min at 90°C and applied to a 12.5% polyacrylamide/8 M urea gel. The ligation products were visualized by autoradiography of the gel.
Adenylyltransferase assay
Reaction mixtures (12 μl) contained 50 mM Tris–HCl, pH 7.5, 3 mM MgCl2, 0.1% Triton X-100, 4 mM β-ME, 0.2 μM [α-32P]ATP, 40 fmol T7 transcript (NtY9-T7-M1) and 10 μl of appropriate chromatographic fractions. Incubation was carried out for 15 min at 37°C, followed by the addition of an equal volume of dissociation buffer (0.1 M Tris–HCl, pH 6.8, 4% SDS, 17% glycerol, 0.8 M β-ME and 0.05% BPB) and subsequent gel electrophoretic analysis.
Construction of expression vector
The entire yeast tRNA ligase gene originally contained in the expression vector pDAKC (14) was amplified by PCR and subsequently cloned into the NcoI/XhoI sites of pIVEX WG 1.3 for the expression of a recombinant protein with a C-terminal histidine tag in the RTS 100 wheat germ lysate (Roche Diagnostics, Mannheim, Germany).
An Arabidopsis size selected (3–6 kb) cDNA library prepared by J. Ecker (23,24) was employed for the amplification of the coding region of the tRNA ligase gene. The cDNA library, ligated into the EcoRI-digested λZAPII DNA and packaged in the Gigapack II Gold packaging extract (Stratagene) was obtained from the ‘Arabidopsis Biological Resource Center, ABRC’, Ohio State University. Amplification was performed via plate lysates utilizing E.coli XL-1 Blue MRF as a host culture. λDNA was purified according to the λDNA purification procedure with tip20 columns (Qiagen, Hilden, Germany). λDNA (100 ng) was used for PCR reactions with gene-specific primers and the Pico Maxx High Fidelity PCR system (Stratagene). A 3.2 kb PCR product was excised from a 1% low-melting agarose gel and the purified PCR product was amplified with primers introducing NotI and SalI restriction sites. This PCR product was cloned into the corresponding restriction sites of pIVEX WG 1.3 vector DNA. Sequence analysis of the inserted cDNA sequence was performed by an Abi Prism 310 Genetic Analyser (Applied Biosystems, Darmstadt, Germany). Two single point mutations were detected and corrected by the QuikChange procedure (Stratagene).
The splicing endonuclease gene of M.jannaschii was amplified by PCR from chromosomal DNA with appropriate primers and cloned into the NcoI and BamHI restriction sites of pIVEX WG 1.4 vector DNA. The expressed recombinant endonuclease carries an N-terminal histidine tag.
Protein expression and purification
The yeast tRNA ligase, the Arabidopsis tRNA ligase and the Methanocaldococcus splicing endonuclease were overexpressed in the RTS 100 WG CECF kit (Roche Diagnostics). Reaction mixtures (50 μl) containing 0.07 mM unlabelled methionine and 0.03 μCi/μl [35S]methionine (SJ1515; Amersham Biosciences) were incubated for 24 h at 24 ± 1°C. The protein mixtures were diluted 5-fold with binding buffer containing 50 mM Tris–HCl, pH 7.5, 200 mM NaCl and 2–5 mM imidazole. The binding to nickel-nitriloacetic acid (Ni-NTA) agarose (Qiagen) was performed in batch for 2 h at 4°C, followed by washing with the same buffer containing 20 mM imidazole and elution of the His-tagged proteins with 500 mM imidazole.
Analysis of ligation products
Digestion of [α-32P]UTP-labelled RNA with RNase T1 (G-specific) was carried out in a total volume of 20 μl containing 50 mM Tris–HCl, pH 8.0, 5 μg carrier tRNA from wheat and 5 U RNase T1 (Calbiochem, Bad Soden, Germany). Incubation was for 2 h at 37°C.
RNase T2 digestion of UTP-labelled RNA was performed in a 10 μl reaction volume, containing 5 mM NH4OAc, pH 4.6, 5 μg carrier tRNA and 2 U of RNase T2 (Invitrogen, Karlsruhe, Germany). Incubation was for 5 h at 37°C.
Complete digestion of UTP-labelled RNA with RNase P1 was carried out in a 20 μl reaction volume containing 50 mM NH4OAc, pH 5.3, 5 μg carrier tRNA and 200 ng P1 nuclease (Roche Diagnostics). Incubation was for 2 h at 50°C. Identification of the labelled nucleotides was by thin-layer chromatography on 20 × 20 cm cellulose plates (Merck, Darmstadt, Germany) using solvent A (isobutyric acid/conc. ammonia/H2O = 57.7:3.8:38.5) or solvent B (isopropanol/32% HCl/H2O = 70:17.5:12.5) according to Nishimura (25). The unlabelled nucleotide 3′ and 5′ monophosphates were visualized under UV light at 254 nm.
RESULTS
Purification of the tRNA ligase protein from wheat germ
Soon after the discovery of a novel ligation mechanism in wheat germ extract (8,9), an attempt to isolate RNA ligase from this tissue was initiated by Furneaux et al. (26). Since wheat germ RNA ligase is active on a variety of synthetic substrates, they used a poly(A) substrate, containing 5′-phosphate and 2′,3′-cyclic phosphate termini in their assays. In this first series, the wheat RNA ligase was partially purified and shown to be associated with 2′,3′-cyclic phosphodiesterase activity. Later on, an improved procedure was developed for the extensive purification of wheat germ RNA ligase and it was shown that RNA ligase, cyclic phosphodiesterase and polynucleotidkinase activity co-purified through all chromatographic steps (16,17).
The putative function of wheat RNA ligase in vivo remained obscure, until the cloning and identification of the first intron-containing tRNA gene in a plant nuclear genome, which codes for a functional Nicotiana tRNATyr (27). Subsequently, a great number of intron-containing tRNA genes coding either for tRNATyr or elongator tRNAMet were characterized in lower and higher plant (including Triticum) nuclear genomes (28–30). Utilizing an intron-containing plant pre-tRNATyr as substrate, we have developed a cell-free processing and splicing system from wheat germ that supports efficient removal of 5′ and 3′ flanking sequences, accurate excision of the intervening sequence and ligation of the resulting tRNA halves (31). Consequently, we have chosen wheat embryos as a source for the isolation of tRNA ligase, because (i) ligase activity has been intensively studied in this tissue (8,31,32) and because (ii) wheat embryos are easy to handle and relatively devoid of proteases and RNases.
The purification procedure (as described in detail in Materials and Methods) yielded ∼5 μg of the 125 kDa protein (Figure 3C) that was sufficient for subsequent protein sequence analyses.
Identification of homologous tRNA ligase proteins and their genes in sequenced plant genomes
The purified wheat ligase protein was subjected to tryptic digestion followed by microcapillary reverse-phase HPLC nano-electrospray tandem mass spectrometry (μLC/MS/MS) on a Finnigan LCQ DECA XP Plus quadrupole ion trap mass spectrometer. About 120 tryptic fragments were evaluated. SEQUEST analysis revealed seven plant expressed sequence tags (ESTs) with a high similarity to an open reading frame (ORF) of the A.thaliana genome [accession no. AC026875; (33)]. These seven plant EST sequences were identified through 91 tryptic fragments that account for 80% of the total ion current. The A.thaliana ORF of ∼120 kDa is incorrectly annotated as translation–elongation factor EF1 alpha and unambiguously codes for a tRNA ligase as discussed below. A partial Arabidopsis cDNA sequence is known (accession no. Z35021) that translates into the C-terminal region of this protein, starting at amino acid position 284 (Figure 4). A full-length cDNA sequence derived from O.sativa (cv. Japonica) expressed mRNA exhibits high amino acid sequence homology to the Arabidopsis protein (accession no. AP005124). The Arabidopsis tRNA ligase gene is located on chromosome I (MATDB–At1g07910) and covers 7674 bp. Its genomic structure consists of 26 exons. The related Oryza gene is located on chromosome VII and spans a region of 12 860 bp, containing 27 exons (accession no. AP005124).
Figure 4.

Alignment of plant tRNA ligases. The amino acid sequences of the shortest ORFs of A.thaliana, cv. Columbia (accession no. AC026875) and O.sativa, cv. Japonica (accession no. AP005124) are aligned using the Blosum 62 matrix. Identical amino acids are indicated in black and similar amino acids are shaded in grey. Nucleotidyltransferase motifs (Lig I, Lig IIIa and Lig IV), a putative NTP-binding P-loop motif (PNK) and two conserved tetrapeptides (CPD) designating cyclic phosphodiesterase elements are boxed. The lysine residue of the AMP binding site (Lig I) is highlighted by a star. The KFEN sequence element upstream of the Lig I motif resembles an element found at the corresponding position in T4 Rnl1-like ligases (3) and in fungal tRNA ligases (15).
The Arabidopsis and Oryza tRNA ligase proteins of 1104 and 1117, respectively, amino acids express 63% identity and 75% similarity to each other (Figure 4) and no significant overall similarity to the known tRNA ligase protein of S.cerevisiae [(34), accession no. P09880]. However, analysis of the two plant protein sequences reveals a number of conserved motifs as deduced from the signature elements of the yeast enzyme (15) indicating that at least the physical order of the functional domains appear to be identical in plant and fungal tRNA ligases. In the N-terminal adenylyltransferase domain of the plant tRNA ligases, motifs Lig I (KHSG), Lig IIIa (FAAF/YD) and Lig IV (EGLVA) most probably comprise active sequence elements that are also found in DNA ligases and RNA capping enzymes (35), T4 RNA ligases 1 and 2 (3,36) and yeast tRNA ligase (15). The lysine residue in motif I is responsible for the covalent binding of AMP forming a lysyl-AMP intermediate (37). Two clusters of conserved residues upstream of motif I have been identified in T4 Rnl1-like ligases (3), one of which is also present in plant ligases (Figure 4). In the central region of Arabidopsis and Oryza tRNA ligase proteins, a putative NTP-binding P-loop motif (GIPGS/CAKS) defines a polynucleotide kinase domain (38) and in the C-terminal domain two conserved tetrapeptides (HVTL…57…HV/ATL) indicate CPDase signature elements (39).
In vitro expression and purification of recombinant Arabidopsis tRNA ligase
In order to prove that the identified plant tRNA ligase genes encode in fact ligase activity, we have overexpressed the Arabidopsis protein. In parallel, we have produced the recombinant yeast tRNA ligase and used it as a positive control in all assays. The yeast enzyme (∼92 kDa) has been previously overexpressed in E.coli cells and shown to be active, however, the yield of full-length recombinant protein was low due to the generation of many smaller fragments (13,15,37).
Employing the recently available RTS 100 wheat germ lysate for the synthesis of recombinant yeast tRNA ligase, we were able to produce predominantly full-length protein at reasonable amounts that were sufficient for the subsequent purification and activity assays (40). Hence, we have further investigated the in vitro expression of the Arabidopsis tRNA ligase in the same system. The ORF of the Arabidopsis gene (Figure 4) was cloned into the expression vector pIVEX WG 1.3 to generate a recombinant protein with a histidine tag at the C-terminus. Incubation of the vector DNA was carried out in a 50 μl reaction mixture in the presence of [35S]methionine. Analysis of the newly synthesized labelled proteins revealed that a single polypeptide, corresponding in size to the expected full-length recombinant tRNA ligase (∼125 kDa), and virtually no smaller polypeptides had been produced (Figure 5, right panel, lane a). The His-tagged protein was purified by affinity chromatography on a Ni-NTA column under native conditions. The major portion eluted in the fractions with 500 mM imidazole (Figure 5, lanes d and e) and aliquots of this material were subsequently used for functional studies without further treatment.
Figure 5.

In vitro production of Arabidopsis tRNA ligase and purification of the recombinant protein. Overexpression of the Arabidopsis tRNA ligase was accomplished in the RTS 100 wheat germ CECF system. Incubation of the vector DNA, carrying the tRNA ligase cDNA with a C-terminal histidine tag was performed in a 50 μl reaction mixture in the presence of [35S]methionine for 24 h at 24°C. The overexpressed C-tagged tRNA ligase was subsequently purified by Ni-NTA chromatography. Aliquots of 2 μl of the total reaction mixture (lane a), of the flow-through (lane b) and wash fractions (lane c) after binding of the protein mixture to the Ni-NTA agarose as well as of the first fractions eluting with 500 mM imidazole (lanes d and e) were loaded onto a 7.5% polyacrylamide/0.1% SDS gel. After Coomassie brilliant blue staining (left panel), the gel was developed for fluorography (right panel) (61).
Recombinant Arabidopsis tRNA ligase has similar activities as its yeast counterpart
The ultimate function of tRNA ligase is the joining of tRNA halves in the splicing process. We have used a plant intron-containing pre-tRNATyr (NtY9-T7-M1) transcript as a substrate for assaying splicing activity throughout the first chromatographic steps for the purification of wheat tRNA ligase (Figure 2B). The wheat tRNA endonuclease co-purifies together with tRNA ligase until to the Source S15 chromatography and this endogenous activity generates a sufficient amount of tRNA halves to pinpoint ligase activity. At the final purification steps, we have identified the tRNA ligase protein solely by its ability to covalently bind AMP (Figure 3).
For our subsequent functional studies of recombinant Arabidopsis tRNA ligase, we have established the generation of tRNA halves at high quantities in vitro by means of recombinant splicing endonuclease from M.jannaschii and an appropriate substrate. The functional M.jannaschii endonuclease consists of four identical subunits of 21 kDa (41,42). Like all archaeal splicing endonucleases, it recognizes a highly conserved intron structure, the so-called bulge–helix–bulge (BHB) motif, consisting of two bulge loops of 3 nt separated by a helical region of 4 bp (43–45). The intron structure of the plant pre-tRNATyr, a natural substrate, chosen to date for our splicing assays exhibits similarities to the conserved archaeal intron structure, however, the two bulge loops each consist of 4 nt instead of 3 nt (Figure 2B). Hence, we have constructed a chimeric intron-containing pre-tRNA as a substrate for the M.jannaschii endonuclease, containing most of the mature domain from the plant pre-tRNATyr and the intron as well as the anticodon from Methanocaldococcus pre-tRNATrp (Figure 6A). The archaeal splicing endonuclease was efficiently expressed in the RTS 100 wheat germ lysate (not shown) and the recombinant enzyme purified by Ni-NTA chromatography was active, yielding 3′ and 5′ tRNA halves and a linear intron upon incubation with the synthetic archeuka pre-tRNA (Figure 6A). These purified RNA substrates were subsequently used for ligation assays.
Figure 6.

RNA ligase activity of native and recombinant enzymes. (A) A chimeric pre-tRNA, containing most of the mature domain from the plant pre-tRNATyr as shown in Figure 2B, and the intron and anticodon of pre-tRNATrp from M.jannaschii was used as a substrate for the archaeal splicing endonuclease to generate in vitro 3′ and 5′ tRNA halves and a linear intron. It was synthesized by T7 RNA polymerase in the presence of [α-32P]UTP. In an ATP-dependent complex reaction, yeast and plant tRNA ligases convert tRNA halves and the linear intron into spliced tRNA and circular intron molecules, containing a 2′-phosphomonoester, 3′,5′-phosphodiester bond at the splice junction (8–10,46). (B) Archeuka pre-tRNA-derived tRNA halves and linear intron molecules were incubated in 20 μl splicing buffer for 30 min at 37°C in the presence of RNA ligase of different origin. Lane a, undigested archeuka pre-tRNA. In lanes b–g, pre-tRNA after cleavage with splicing endonuclease was used as input RNA; lane b, incubation without protein; lane c, incubation with a protein fraction derived from the expression of a control vector (minus cDNA insert) in the RTS 100 wheat germ extract and subsequent Ni-NTA purification to test whether the observed ligase activities are in fact due to the recombinant tRNA ligases; lane d, incubation with a partially purified tRNA ligase (gel filtration fraction) from wheat germ; lane e, incubation with 50 ng recombinant Arabidopsis tRNA ligase; lane f, incubation with 50 ng recombinant yeast tRNA ligase; and lane g, incubation with a mixture of T4 RNA ligase and T4 polynucleotide kinase/3′-phosphatase. The reaction products were separated on a 12.5% polyacrylamide/8 M urea gel. Stars (*) indicate products that were examined in more detail. (C) Time course of inter- and intramolecular ligation reactions by plant and yeast recombinant tRNA ligases. Archeuka pre-tRNA (400 fmol) was cleaved with splicing endonuclease and the resulting tRNA halves and linear intron molecules were incubated either with 100 ng Arabidopsis (left panel) or with 100 ng yeast (right panel) recombinant tRNA ligase in 100 μl splicing buffer. At the times (min) indicated below, aliquots (10 μl) were removed and the RNA products were analysed on a 12.5% polyacrylamide/8 M urea gel.
Appropriate amounts of tRNA halves together with the linear intron were incubated in splicing buffer under various conditions (Figure 6B). In the two control assays (lanes b and c), the incubated substrates remained unaltered as expected. In all four reactions that contained RNA ligases of different origin including the newly identified recombinant Arabidopsis tRNA ligase, the spliced mature tRNA resulting from ligation of 3′ and 5′ halves was produced efficiently (Figure 6B, lanes d–g). Moreover, the linear intron was converted to its circular form in all assays, albeit in reduced amounts in the presence of yeast tRNA ligase (lane f). In order to emphasize the different substrate specificities of plant and yeast tRNA ligases, we have carried out a time course experiment as shown in Figure 6C. The intermolecular ligation of 3′ and 5′ tRNA halves by plant and yeast RNA ligase proceeds by a similar kinetics whereas the intramolecular joining reaction differs considerably. After 2 min of incubation, the linear intron is converted completely to its circular form in the presence of Arabidopsis tRNA ligase. However, at the same time of incubation ∼95% of the linear intron has not yet been circularized in the presence of equal amounts of yeast tRNA ligase. The amount of synthesized circular intron increases up to 40% upon further incubation for 30 min.
A major feature of plant and yeast tRNA ligases is their ability to generate a 2′-phosphate at the splice junction (8–10,46). We have therefore investigated the spliced tRNA and the circular intron in more detail. For this purpose, we have recovered the [α-32P]UTP-labelled RNA products from preparative gels originating from the incubation with different RNA ligase preparations. The newly formed 2′-phosphomonoester, 3′,5′-phosphodiester linkage is resistant to alkali and a number of ribonucleases, such as RNase T1, RNase P1 and RNase T2 (8). Consequently, cleavage of spliced tRNA—produced by the action of wheat, Arabidopsis and yeast tRNA ligase—yields a long RNase T1-resistant 15mer-oligonucleotide (Figure 7A, lanes a–c) that spans the region around the splice junction (i.e.  ), which does not appear in digests derived from T4-generated spliced tRNA. Instead, an 8mer oligonucleotide occurs that originates from the 5′ end of the 3′ half of archeuka pre-tRNA (see Figure 6A), indicating RNase T1 cleavage at the splice junction (Figure 7A, lane d). The 15mer- and 8mer-oligonucleotides were excised from the gel and subjected to further digestions with either RNase T2 or RNase P1. The occurrence of radioactive products with mobilities expected for the non-cleavable dinucleotides
), which does not appear in digests derived from T4-generated spliced tRNA. Instead, an 8mer oligonucleotide occurs that originates from the 5′ end of the 3′ half of archeuka pre-tRNA (see Figure 6A), indicating RNase T1 cleavage at the splice junction (Figure 7A, lane d). The 15mer- and 8mer-oligonucleotides were excised from the gel and subjected to further digestions with either RNase T2 or RNase P1. The occurrence of radioactive products with mobilities expected for the non-cleavable dinucleotides 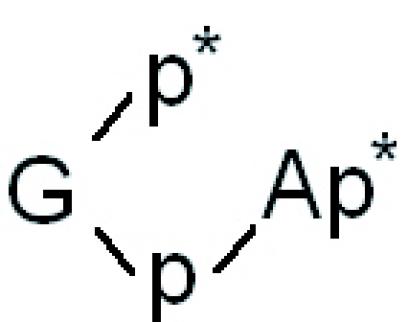 and
and ![]() , respectively (Figure 7B and C, lanes a-c), confirm that all three different eukaryotic tRNA ligases have joined the tRNA halves via a 2′-phosphomonoester, 3′,5′-phosphodiester linkage, whereas the combined action of T4 RNA ligase and T4 polynucleotide kinase/3′-phosphatase has resulted in a normal 3′,5′-phosphodiester bond. A similar picture emerges after cleavage of the circular introns with RNase T2. The non-cleavable dinucleotide
, respectively (Figure 7B and C, lanes a-c), confirm that all three different eukaryotic tRNA ligases have joined the tRNA halves via a 2′-phosphomonoester, 3′,5′-phosphodiester linkage, whereas the combined action of T4 RNA ligase and T4 polynucleotide kinase/3′-phosphatase has resulted in a normal 3′,5′-phosphodiester bond. A similar picture emerges after cleavage of the circular introns with RNase T2. The non-cleavable dinucleotide 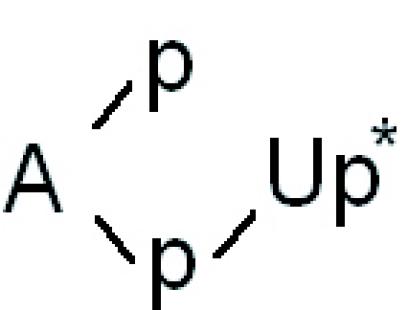 is visible only in molecules produced by the eukaryotic RNA ligases (Figure 7D, lanes a–c).
is visible only in molecules produced by the eukaryotic RNA ligases (Figure 7D, lanes a–c).
Figure 7.

Analysis of splicing products. (A) The [α-32P]UTP-labelled spliced tRNAs (76 nt) synthesized in the presence of different RNA ligase preparations (see Figure 6), i.e. wheat tRNA ligase (lane a), recombinant Arabidopsis tRNA ligase (lane b), recombinant yeast tRNA ligase (lane c) and a mixture of T4 RNA ligase and T4 polynucleotide kinase/3′-phosphatase (lane d) were recovered from a preparative gel and digested with RNase T1. The labelled T1-oligonucleotides ranging in size from 1 to 15 nt were separated on a 40-cm-long 20% polyacrylamide/8 M urea gel. Oligonucleotides ≤6 nt migrated with the buffer front. (B and C) The 15mer T1-resistant oligonucleotides and the 8mer oligonucleotide were excised from the gel and digested with RNase T2 (B) or with RNase P1 (C). Analysis of labelled Nps or pNs was by thin-layer chromatography (t. l. c.) on cellulose plates in solvent A. (D) The UTP-labelled circular introns (21 nt) synthesized in the presence of different RNA ligase preparations [as described in (A)] were recovered from a preparative gel and digested with RNase T2. Analysis of Nps was by t. l. c. in solvent B. Positions of markers are indicated.
DISCUSSION
After the discovery of a novel RNA ligase activity in wheat germ about 20 years ago (8,9), numerous efforts have been made to isolate the ligase protein. The RNA ligase was eventually purified ∼6000-fold from crude extracts of wheat germ and it was found that it sedimented as a single peak in a glycerol gradient with a coefficient of 6.2S corresponding to a protein doublet of 110 kDa in silver-stained polyacrylamide gels (16,17).
We were able to purify the wheat tRNA ligase to near homogeneity by six consecutive chromatographic steps (Figures 2 and 3) and the amount of 5 μg purified protein was sufficient for subsequent sequence analyses. Moreover, the peptide sequences obtained from the wheat protein identified the corresponding proteins and their genes in Arabidopsis and Oryza databases. The Arabidopsis tRNA ligase protein of 1104 amino acids (Figure 4) has a molecular mass of 123 kDa and a pI of 8.0 and the Oryza protein of 1117 amino acids has a molecular mass of 124 kDa and a pI of 8.5. Although Arabidopsis and Oryza represent plants of dicotyledonous and monocotyledonous origin, they express 63% identity of their amino acid sequences. A number of known ESTs from other higher plants all confirm the remarkable similarity of plant tRNA ligases, whereas the overall concordance of amino acid sequences between plant and fungal tRNA ligases is not significant. This explains why previous database searches with the yeast protein sequence as query failed to identify the corresponding plant proteins. In spite of the deviation in amino acid sequences, the physical order of intrinsic enzymatic activities and the corresponding signature elements have been conserved in plant and fungal tRNA ligases supporting earlier observations about their similar activities in vitro (46).
In the N-terminal adenylyltransferase domain of plant RNA ligases (Figure 4), the sequence KHSG represents the motif Lig I (KxxG) that is conserved in all RNA and DNA ligases (35). The lysine residue forms a covalently ligase (lysyl-N)–AMP intermediate, one of the steps required for the subsequent ligation process. A number of additional motifs are responsible for the formation of an RNA–adenylate intermediate and for phosphodiester synthesis, two of which (i.e. Lig IIIa and Lig IV) are contained in the plant enzymes (Figure 4). In the multifunctional yeast tRNA ligase, the central region harbours a polynucleotide kinase domain (14,15) that is defined by the NTP-binding P-loop motif (GxxGxGKS) identified also in the well-studied coliphage T4 polynucleotide kinase and other phosphotransferases (7,38,47). A putative P-loop element (i.e. GIPGS/CAKS) is present in plant tRNA ligases (Figure 4) that might exert polynucleotide kinase activity. A major difference between the coliphage T4 and the yeast/plant-induced ligation process is the modification at the 3′ end that precedes joining of RNA molecules. The 3′-phosphatase activity contained in the bifunctional T4 polynucleotide kinase enzyme converts the 2′,3′-cyclic phosphate to a 3′-phosphate followed by removal of the phosphate group, whereas the cyclic phosphodiesterase activity (CPDase) in the C-terminal domain of the trifunctional yeast enzyme opens the cyclic terminus at the 3′ end leaving the phosphate in the 2′ position. This 2′-phosphate is retained during the subsequent ligation process generating a 2′-phosphomonoester, 3′,5′-phosphodiester linkage (46). Two conserved tetrapeptides with histidine residues at the first positions are essential for CPDases as well as for CPDase activities contained in multifunctional enzymes (39) and corresponding tetrapeptides can also be detected in the C-terminal region of plant ligases (Figure 4). Evidence for all of the presumed activities in the newly characterized plant tRNA ligases derives from our findings that (i) the purified enzyme is adenylylated in the presence of [α-32P]ATP (Figure 3), that (ii) in vitro synthesized tRNA half molecules carrying 2′,3′-cyclic phosphate and 5′-hydroxyl ends are ligated by recombinant Arabidopsis tRNA ligase (Figure 6, lane e) and that (iii) a free 2′-phosphate is left at the splice junction (Figure 7A–D).
The major ligation reaction mechanism in vertebrates differs from that in fungi or plants. During pre-tRNA splicing in HeLa cell extract, the 3′ terminal phosphate of 5′ half molecules is incorporated into a normal 3′,5′-phosphodiester linkage at the splice junction (48). So far, neither the vertebrate ligase protein nor the exact mechanism of action of this ligation process is known. Recently, it has been demonstrated by Zillmann et al. (49) that a plant-like splicing machinery exists also in HeLa cells. However, a search of the fully sequenced human genome with the plant tRNA ligase as query did not identify a candidate gene for a putative tRNA ligase suggesting that mammalian and plant ligases have diverged as much as have the fungal and plant enzymes.
All known eukaryotic nuclear genomes code for intron-containing tRNA genes and without doubt, tRNA ligases play an essential role in pre-tRNA splicing. The question arises whether tRNA ligases are also involved in other cellular processes. An unexpected link to mRNA metabolism has been the discovery that yeast tRNA ligase is responsible for non-spliceosomal splicing of mRNA in the unfolded protein response pathway (50). A more general task of tRNA ligases might be their participation in the repair of broken tRNAs, as is the major function in vivo of the coliphage T4-induced ligase (5). In this connection, it should be noted that the single-stranded anticodon loop of tRNAs is the most vulnerable part for chemical and/or enzymatic hydrolysis. Interestingly, ligation of endogenous 3′ half molecules to their corresponding 5′ halves of tRNAAla (UGC) and tRNAPro (UGG) via 2′-phosphomonoester, 3′,5′-phosphodiester bonds has been observed in extracts of Chlamydomonas (51). These tRNA halves definitely did not originate from tRNA splicing, because (i) the corresponding tRNA genes in known plant genomes are intronless and (ii) cleavage occurred within the anticodon and not at the highly conserved splice site one nucleotide 3′ of the anticodon.
In animal and plant systems, infectious pathogens are known that rely on yet undefined RNA ligases for their propagation. Hepatitis δ virus (HDV) replicates its circular RNA genome of 1679 nt via a rolling circle mechanism. The long intermediates are cleaved to unit-length RNA molecules by an intrinsic ribozyme. Apparently, a host-specific activity present in mammalian cells is capable of joining HDV molecules in both bimolecular and intramolecular reactions (52). Similarly, a cellular activity might be utilized during the replication of viroids, which are non-coding circular RNAs of about 300 nt that cause severe diseases in plants (53–55). Like HDV, viroids replicate though a rolling-circle mechanism and the oligomeric RNAs are subsequently cleaved by specific RNases or by hammerhead ribozymes. It has been demonstrated that linear viroid RNA molecules were circularized in vitro by incubation with wheat germ or Chlamydomonas extract (56,57). Moreover, monomeric circular forms of a number of viroids were formed in transgenic Arabidopsis plants transformed with dimeric transcripts, indicating the activity of both, RNases and ligases in this host (58).
As mentioned above, wheat germ tRNA ligase and the recombinant Arabidopsis tRNA ligase catalyse not only the joining of tRNA halves but also the circularization of linear introns (Figure 2C; Figure 6B, lanes d and e, and C) and of various natural and synthetic oligo-and polyribonucleotides, provided they carry 2′,3′-cyclic phosphate and 5′-OH ends (8,59). In contrast, yeast tRNA ligase shows a high degree of specificity. Joining of tRNA halves by the yeast enzyme is 104-fold more efficient than joining of artificial oligoribonucleotides (13) and the products of pre-tRNA splicing in yeast nuclear extract are mature tRNAs and linear intron molecules (60). These earlier results are confirmed by our observation that the conversion of linear to circular introns follows different kinetics with plant and yeast RNA ligase, respectively (Figure 6C). The broad substrate range of plant tRNA ligase clearly suggests that the action of this enzyme is not limited to pre-tRNA splicing or tRNA repair but that it participates in other yet undetected cellular processes.
Acknowledgments
We are grateful to Professor H. J. Gross for his stimulating interest during the course of this work and for critical reading of the manuscript. We thank our colleagues Anita Marchfelder (Ulm) and Chris Greer (Irvine, CA) for a gift of genomic DNA from M.jannaschii and the recombinant plasmid pDAKC, respectively. We appreciate the excellent protein sequence analyses done by William S. Lane at the Harvard Microchemistry Facility (Cambridge, MA). We are very much indebted to the Arabidopsis Biological Resource Center (Ohio State University, OH) for providing us with an Arabidopsis cDNA library (CD4-16). This work was mainly financed privately by H.B. The Open Access publication charges for this article were waived by Oxford University Press.
REFERENCES
- 1.Uhlenbeck O.C. T4 RNA ligase. Trends Biochem. Sci. 1983;8:94–96. [Google Scholar]
- 2.Heaphy S., Singh M., Gait M.J. Effect of single amino acid changes in the region of the adenylylation site of T4 RNA ligase. Biochemistry. 1987;26:1688–1696. doi: 10.1021/bi00380a030. [DOI] [PubMed] [Google Scholar]
- 3.Wang L.K., Ho C.K., Pei Y., Shuman S. Mutational analysis of bacteriophage T4 RNA ligase 1. Different functional groups are required for the nucleotidyl transfer and phosphodiester bond formation of the ligation reaction. J. Biol. Chem. 2003;278:29454–29462. doi: 10.1074/jbc.M304320200. [DOI] [PubMed] [Google Scholar]
- 4.Amitsur M., Levitz R., Kaufmann G. Bacteriophage T4 anticodon nuclease, polynucleotide kinase and RNA ligase reprocess the host lysine tRNA. EMBO J. 1987;6:2499–2503. doi: 10.1002/j.1460-2075.1987.tb02532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaufmann G. Anticodon nucleases. Trends Biochem. Sci. 2000;25:70–74. doi: 10.1016/s0968-0004(99)01525-x. [DOI] [PubMed] [Google Scholar]
- 6.Cameron V., Uhlenbeck O.C. 3′-Phosphatase activity in T4 polynucleotide kinase. Biochemistry. 1977;16:5120–5126. doi: 10.1021/bi00642a027. [DOI] [PubMed] [Google Scholar]
- 7.Galburt E.A., Pelletier J., Wilson G., Stoddard B.L. Structure of a tRNA repair enzyme and molecular biology workhorse: T4 polynucleotide kinase. Structure (Cambridge) 2002;10:1249–1260. doi: 10.1016/s0969-2126(02)00835-3. [DOI] [PubMed] [Google Scholar]
- 8.Konarska M., Filipowicz W., Domdey H., Gross H.J. Formation of a 2′-phosphomonoester, 3′,5′-phosphodiester linkage by a novel RNA ligase in wheat germ. Nature. 1981;293:112–116. doi: 10.1038/293112a0. [DOI] [PubMed] [Google Scholar]
- 9.Konarska M., Filipowicz W., Gross H.J. RNA ligation via 2′-phosphomonoester, 3′5′-phosphodiester linkage: requirement of 2′, 3′-cyclic phosphate termini and involvement of a 5′-hydroxyl polynucleotide kinase. Proc. Natl Acad. Sci. USA. 1982;79:1474–1478. doi: 10.1073/pnas.79.5.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greer C.L., Peebles C.L., Gegenheimer P., Abelson J. Mechanism of action of a yeast RNA ligase in tRNA splicing. Cell. 1983;32:537–546. doi: 10.1016/0092-8674(83)90473-7. [DOI] [PubMed] [Google Scholar]
- 11.Goodman H.M., Olson M.V., Hall B.D. Nucleotide sequence of a mutant eukaryotic gene: the yeast tyrosine-insertion ochre suppressor SUP4-o. Proc. Natl Acad. Sci. USA. 1977;74:5453–5457. doi: 10.1073/pnas.74.12.5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abelson J., Trotta C.R., Li H. tRNA splicing. J. Biol. Chem. 1998;273:12685–12688. doi: 10.1074/jbc.273.21.12685. [DOI] [PubMed] [Google Scholar]
- 13.Phizicky E.M., Schwartz R.C., Abelson J. Saccharomyces cerevisiae tRNA ligase. Purification of the protein and isolation of the structural gene. J. Biol. Chem. 1986;261:2978–2986. [PubMed] [Google Scholar]
- 14.Apostol B.L., Westaway S.K., Abelson J., Greer C.L. Deletion analysis of a multifunctional yeast tRNA ligase polypeptide. Identification of essential and dispensable functional domains. J. Biol. Chem. 1991;266:7445–7455. [PubMed] [Google Scholar]
- 15.Sawaya R., Schwer B., Shuman S. Genetic and biochemical analysis of the functional domains of yeast tRNA ligase. J. Biol. Chem. 2003;278:43928–43938. doi: 10.1074/jbc.M307839200. [DOI] [PubMed] [Google Scholar]
- 16.Pick L., Hurwitz J. Purification of wheat germ RNA ligase. I. Characterization of a ligase-associated 5′-hydroxyl polynucleotide kinase activity. J. Biol. Chem. 1986;261:6684–6693. [PubMed] [Google Scholar]
- 17.Pick L., Furneaux H., Hurwitz J. Purification of wheat germ RNA ligase. II Mechanism of action of wheat germ RNA ligase. J. Biol. Chem. 1986;261:6694–6704. [PubMed] [Google Scholar]
- 18.Gegenheimer P., Gabius H.J., Peebles C.L., Abelson J. An RNA ligase from wheat germ which participates in transfer RNA splicing in vitro. J. Biol. Chem. 1983;258:8365–8373. [PubMed] [Google Scholar]
- 19.Stange N., Beier D., Beier H. Intron excision from tRNA precursors by plant splicing endonuclease requires unique features of the mature tRNA domain. Eur. J. Biochem. 1992;210:193–203. doi: 10.1111/j.1432-1033.1992.tb17408.x. [DOI] [PubMed] [Google Scholar]
- 20.Rösch S., Späth B., Schiffer S., Englert M., Beier H., Marchfelder M., Hartmann R.K., Bindereif A., Schön A., Westhof E. Handbook of RNA Biochemistry. Weinheim: Wiley-VCH; 2005. Use of RNA affinity matrices for the isolation of RNA-binding proteins; pp. 667–675. [Google Scholar]
- 21.Fuchs T., Beier D., Beier H. The tRNATyr multigene family of Nicotiana rustica: genome organization, sequence analyses and expression in vitro. Plant Mol. Biol. 1992;20:869–878. doi: 10.1007/BF00027158. [DOI] [PubMed] [Google Scholar]
- 22.Cheong C., Varani G., Tinoco I. Solution structure of an unusually stable RNA hairpin, 5′GGAC(UUCG)GUCC. Nature. 1990;346:680–682. doi: 10.1038/346680a0. [DOI] [PubMed] [Google Scholar]
- 23.Gubler U., Hoffman B.J. A simple and very efficient method for generating cDNA libraries. Gene. 1983;25:263–269. doi: 10.1016/0378-1119(83)90230-5. [DOI] [PubMed] [Google Scholar]
- 24.Kieber J.J., Rothenberg M., Roman G., Feldmann K.A., Ecker J.R. CTR1, a negative regulator of the ethylene response pathway in Arabidopsis, encodes a member of the raf family of protein kinases. Cell. 1993;72:427–441. doi: 10.1016/0092-8674(93)90119-b. [DOI] [PubMed] [Google Scholar]
- 25.Nishimura S. Modified nucleosides in tRNA. In: Schimmel P.R., Söll D., Abelson J.N., editors. Transfer RNA: Structure, Properties and Recognition. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1979. pp. 59–79. [Google Scholar]
- 26.Furneaux H., Pick L., Hurwitz J. Isolation and characterization of RNA ligase from wheat germ. Proc. Natl Acad. Sci. USA. 1983;80:3933–3937. doi: 10.1073/pnas.80.13.3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stange N., Beier H. A gene for the major cytoplasmic tRNATyr from Nicotiana rustica contains a 13 nucleotides long intron. Nucleic Acids Res. 1986;14:8691. doi: 10.1093/nar/14.21.8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akama K., Kashihara M. Plant nuclear tRNAMet genes are ubiquitously interrupted by introns. Plant Mol. Biol. 1996;32:427–434. doi: 10.1007/BF00019094. [DOI] [PubMed] [Google Scholar]
- 29.Arends S., Kraus J., Beier H. The tRNATyr multigene family of Triticum aestivum: genome organization, sequence analyses and maturation of intron-containing pre-tRNAs in wheat germ extract. FEBS Lett. 1996;384:222–226. doi: 10.1016/0014-5793(96)00313-4. [DOI] [PubMed] [Google Scholar]
- 30.Akama K., Nass A., Junker V., Beier H. Characterization of nuclear tRNATyr introns: their evolution from red algae to higher plants. FEBS Lett. 1997;417:213–218. doi: 10.1016/s0014-5793(97)01288-x. [DOI] [PubMed] [Google Scholar]
- 31.Stange N., Beier H. A cell-free plant extract for accurate pre-tRNA processing, splicing and modification. EMBO J. 1987;6:2811–2818. doi: 10.1002/j.1460-2075.1987.tb02577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akama K., Junker V., Yukawa Y., Sugiura M., Beier H. Splicing of Arabidopsis tRNAMet precursors in tobacco cell and wheat germ extracts. Plant Mol. Biol. 2000;44:155–165. doi: 10.1023/a:1006486315360. [DOI] [PubMed] [Google Scholar]
- 33.Axelos M., Bardet C., Liboz T., Le Van Thai A., Curie C., Lescure B. The gene family encoding the Arabidopsis thaliana translation elongation factor EF-1 alpha: molecular cloning, characterization and expression. Mol. Gen. Genet. 1989;19:106–112. doi: 10.1007/BF00261164. [DOI] [PubMed] [Google Scholar]
- 34.Westaway S.K., Phizicky E.M., Abelson J. Structure and function of the yeast tRNA ligase gene. J. Biol. Chem. 1988;263:3171–3176. [PubMed] [Google Scholar]
- 35.Shuman S., Schwer B. RNA capping enzyme and DNA ligase: a superfamily of covalent nucleotidyl transferases. Mol. Microbiol. 1995;17:405–410. doi: 10.1111/j.1365-2958.1995.mmi_17030405.x. [DOI] [PubMed] [Google Scholar]
- 36.Yin S., Ho C.K., Shuman S. Structure–function analysis of T4 RNA ligase 2. J. Biol. Chem. 2003;278:17601–17608. doi: 10.1074/jbc.M300817200. [DOI] [PubMed] [Google Scholar]
- 37.Xu Q., Phizicky E.M., Greer C.L., Abelson J.N. Purification of yeast transfer RNA ligase. Methods Enzymol. 1990;181:463–471. doi: 10.1016/0076-6879(90)81144-j. [DOI] [PubMed] [Google Scholar]
- 38.Wang L.K., Shuman S. Mutational analysis defines the 5′-kinase and 3′-phosphatase active sites of T4 polynucleotide kinase. Nucleic Acids Res. 2002;30:1073–1080. doi: 10.1093/nar/30.4.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nasr F., Filipowicz W. Characterization of the Saccharomyces cerevisiae cyclic nucleotide phosphodiesterase involved in the metabolism of ADP-ribose 1′,2′-cyclic phosphate. Nucleic Acids Res. 2000;28:1676–1683. doi: 10.1093/nar/28.8.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Englert M., Beier H. Efficient overexpression of functionally active yeast tRNA ligase in the RTS 100 wheat germ lysate. Biochemica. 2004;3:27–29. [Google Scholar]
- 41.Lykke-Andersen J., Garrett R.A. RNA–protein interaction of an archaeal homotetrameric splicing endoribonuclease with an exceptional evolutionary history. EMBO J. 1997;16:6290–6300. doi: 10.1093/emboj/16.20.6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H., Trotta C.R., Abelson J. Crystal structure and evolution of a transfer RNA splicing enzyme. Science. 1998;280:279–284. doi: 10.1126/science.280.5361.279. [DOI] [PubMed] [Google Scholar]
- 43.Kleman-Leyer K., Armbruster D.W., Daniels C.J. Properties of H. volcanii tRNA intron endonuclease reveals a relationship between the archaeal and eucaryal tRNA intron processing systems. Cell. 1997;89:839–847. doi: 10.1016/s0092-8674(00)80269-x. [DOI] [PubMed] [Google Scholar]
- 44.Diener J.L., Moore P.B. Solution structure of a substrate for the archaeal pre-tRNA splicing endonuclease: the bulge–helix–bulge motif. Mol. Cell. 1998;1:883–894. [PubMed] [Google Scholar]
- 45.Marck C., Grosjean H. Identification of BHB splicing motifs in intron-containing tRNAs from 18 archaea: evolutionary implications. RNA. 2003;9:1516–1531. doi: 10.1261/rna.5132503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Filipowicz W., Gross H.J. RNA ligation in eukaryotes. Trends Biochem. Sci. 1984;9:68–71. [Google Scholar]
- 47.Jilani A., Ramotar D., Slack C., Ong C., Yang X.M., Scherer S.W., Lasko D.D. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3′-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage. J. Biol. Chem. 1999;274:24176–24186. doi: 10.1074/jbc.274.34.24176. [DOI] [PubMed] [Google Scholar]
- 48.Filipowicz W., Shatkin A.J. Origin of splice junction phosphate in tRNAs processed by HeLa cell extract. Cell. 1983;32:547–557. doi: 10.1016/0092-8674(83)90474-9. [DOI] [PubMed] [Google Scholar]
- 49.Zillmann M., Gorovsky M.A., Phizicky E.M. Conserved mechanism of tRNA splicing in eukaryotes. Mol. Cell. Biol. 1991;11:5410–5416. doi: 10.1128/mcb.11.11.5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sidrauski C., Chapman R., Walter P. The unfolded protein response: an intracellular signalling pathway with many surprising features. Trends Cell Biol. 1998;8:245–249. doi: 10.1016/s0962-8924(98)01267-7. [DOI] [PubMed] [Google Scholar]
- 51.Tyc K., Kikuchi Y., Konarska M., Filipowicz W., Gross H.J. Ligation of endogenous tRNA 3′ half molecules to their corresponding 5′ halves via 2′-phosphomonoester, 3′,5′-phosphodiester bonds in extracts of Chlamydomonas. EMBO J. 1983;2:605–610. doi: 10.1002/j.1460-2075.1983.tb01470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reid C.E., Lazinski D.W. A host-specific function is required for ligation of a wide variety of ribozyme-processed RNAs. Proc. Natl Acad. Sci. USA. 2000;97:424–429. doi: 10.1073/pnas.97.1.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diener T.O. The viroid: biological oddity or evolutionary fossil? Adv. Virus Res. 2001;57:137–184. doi: 10.1016/s0065-3527(01)57003-7. [DOI] [PubMed] [Google Scholar]
- 54.Flores R., Delgado S., Gas M.E., Carbonell A., Molina D., Gago S., De la Pena M. Viroids: the minimal non-coding RNAs with autonomous replication. FEBS Lett. 2004;567:42–48. doi: 10.1016/j.febslet.2004.03.118. [DOI] [PubMed] [Google Scholar]
- 55.Tabler M., Tsagris M. Viroids: petite RNA pathogens with distinguished talents. Trends Plant Sci. 2004;9:339–348. doi: 10.1016/j.tplants.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 56.Branch A.D., Robertson H.D. A replication cycle for viroids and other small infectious RNA's. Science. 1984;223:450–455. doi: 10.1126/science.6197756. [DOI] [PubMed] [Google Scholar]
- 57.Kikuchi Y., Tyc K., Filipowicz W., Sänger H.L., Gross H.J. Circularization of linear viroid RNA via 2′-phosphomonoester,3′,5′-phosphodiester bonds by a novel type of RNA ligase from wheat germ and Chlamydomonas. Nucleic Acids Res. 1982;10:7521–7529. doi: 10.1093/nar/10.23.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daròs J.A., Flores R. Arabidopsis thaliana has the enzymatic machinery for replicating representative viroid species of the family Pospiviroidae. Proc. Natl Acad. Sci. USA. 2004;101:6792–6797. doi: 10.1073/pnas.0401090101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwartz R.C., Greer C.L., Gegenheimer P., Abelson J. Enzymatic mechanism of an RNA ligase from wheat germ. J. Biol. Chem. 1983;258:8374–8383. [PubMed] [Google Scholar]
- 60.Knapp G., Ogden R.C., Pleebles C.L., Abelson J. Splicing of yeast tRNA precursors: structure of the reaction intermediates. Cell. 1979;18:37–45. doi: 10.1016/0092-8674(79)90351-9. [DOI] [PubMed] [Google Scholar]
- 61.Laskey R.A., Mills A.D. Quantitative film detection of 3H and 14C in polyacrylamide gels by fluorography. Eur. J. Biochem. 1975;56:335–341. doi: 10.1111/j.1432-1033.1975.tb02238.x. [DOI] [PubMed] [Google Scholar]