

Abstract
Mild traumatic brain injury (TBI), also called concussion, initiates sequelae leading to motor deficits, cognitive impairments and subtly compromised neurobehaviors. While the acute phase of TBI is associated with neuroinflammation and nitroxidative burst, the chronic phase shows a lack of stimulation of the neurorepair process and regeneration. The deficiency of nitric oxide (NO), the consequent disturbed NO metabolome, and imbalanced mechanisms of S-nitrosylation are implicated in blocking the mechanisms of neurorepair processes and functional recovery in the both phases. Hypoxia inducible factor-1 alpha (HIF-1α), a master regulator of hypoxia/ischemia, stimulates the process of neurorepair and thus aids in functional recovery after brain trauma. The activity of HIF-1α is regulated by NO via the mechanism of S-nitrosylation of HIF-1α. S-nitrosylation is dynamically regulated by NO metabolites such as S-nitrosoglutathione (GSNO) and peroxynitrite. GSNO stabilizes, and peroxynitrite destabilizes HIF-1α. Exogenously administered GSNO was found not only to stabilize HIF-1α and to induce HIF-1α-dependent genes but also to stimulate the regeneration process and to aid in functional recovery in TBI animals.
Keywords: traumatic brain injury, hypoxia inducible factor-1 alpha, S-nitrosoglutathione, neurorepair, functional recovery
Introduction
The U.S. Centers for Disease Control and Prevention define a traumatic brain injury (TBI) as being caused by a bump, blow, or jolt to the head or a penetrating head injury that disrupts normal brain function (www.cdc.gov/traumaticbraininjury/data). The causes of TBI are extremely diverse, ranging from accidents on the highways, to involvement in sports related injuries, and the effects of improvised explosive devices in the theater of war. Falls are the major cause of TBI in children and the elderly (Blennow et al., 2016). TBI causes neurobehavioral deficits, especially in motor and cognitive functions (Langlois et al., 2006). The observed cognitive changes that follow TBI include decreased mental flexibility, impaired attention, poor planning/judgment, deficits in verbal fluency, dementia, and problems with working memory (Levin and Kraus, 1994; Johnson et al., 2010). Furthermore, TBI is associated with significant morbidity/mortality, pain, and fatigue (Levin and Diaz-Arrastia, 2015; Blennow et al., 2016; Mollayeva et al., 2017). TBI patients are also susceptible to stroke, epilepsy, and Alzheimer's disease (Johnson et al., 2010; Liu et al., 2017).
Over 5.3 million Americans suffer lifelong disabilities due to TBI and 1.7 million Americans meet with TBI-associated accidents annually (Gardner and Zafonte, 2016). Approximately 52,000 Americans die annually as a result of TBI (www.cdc.gov/traumaticbraininjury/data). As estimated by the World Health Organization, TBI will become the leading cause of death and disability worldwide by the year 2020 (Hyder et al., 2007). In terms of TBI-related mortality, the US 2006–2010 data revealed males had an almost three-fold increased risk of TBI-related death than females, and individuals over the age of 64 years had the highest mortality rates (www.cdc.gov/traumaticbraininjury/data; Faul and Coronado, 2015). TBI among children aged 0–14 years is also prevalent due to falls (Langlois et al., 2005). The total (direct and indirect) TBI costs in the USA were approximately $60.43 billion in 2000 (Corso et al., 2006), which has now increased to approximately $76.5 billion (www.cdc.gov/traumaticbraininjury/data). Despite substantial investments in TBI research, the treatment options are limited to manage the sequelae of the injury. The state of TBI science and pharmacotherapy have been thoroughly reviewed recently (Diaz-Arrastia et al., 2014).
Immediately following TBI, the direct trauma and lack of blood flow cause necrotic neuronal death; however, even greater apoptotic neuron loss can occur later from secondary injury caused by hypoxia/ischemia and insults associated with oxidative stress and inflammation (Coles, 2004; Greve and Zink, 2009; Diaz-Arrastia et al., 2014). Focal injury, as a result of TBI, affects not only locomotor function but also cognition, perhaps because damage to brain connectivity is a critical component in the cognitive impairment from TBI. Moreover, cognitive impairment may not be the result of a single event but due to multiple mechanisms originating from secondary injury (Lloyd et al., 2008). The neurorepair process (in the chronic phase) depends on regeneration mechanisms that involve a coordinated integration of angiogenesis, neurogenesis, and remyelination of new and spared axons (Lu et al., 2007). Therapies to increase regeneration activity (angiogenesis, neurogenesis, remyelination) during the chronic phase of TBI therefore hold promise as a treatment strategy for stimulating the recovery of neurological functions.
Approximately 40% of all TBIs are contusions; therefore, animal models of TBI using the focal cortical impact injury (CCI) technique are recognized as physiologically relevant to human TBI (Pennings et al., 1993). The CCI technique was developed by General Motors to model head injuries from automobile accidents and was later adapted for wider experimental use (Lighthall et al., 1989). It reproduces many of the features of brain injuries, including motor deficits, dementia, memory loss, and neuronal loss (Colicos et al., 1996). The severity of injury can be controlled by altering the velocity and depth of the impact and the size of the impact or tip (Dixon et al., 1991). CCI provides an animal model system to evaluate injuries in both the acute and chronic phases. The mechanisms of the injury in the two phases are different and complex. While CCI results in a significant number of necrotic as well as apoptotic neurons in the acute phase, it lacks sufficient regeneration process stimulation (Diaz-Arrastia et al., 2014). Stimulating neurorepair activity by therapeutic modalities, via neurotrophic and growth factors, has been shown to improve motor and cognitive functions (Oyesiku et al., 1999; Kim et al., 2001; Wu et al., 2008; Sun et al., 2009). Our studies show that S-nitrosoglutathione (GSNO)-induced mechanisms stabilize hypoxia-inducible factor-1 alpha (HIF-1α) and stimulate the mechanisms of regeneration and functional recovery in TBI (Khan et al., 2016a). Unlike in stroke, the role of HIF-1α in TBI is less understood. While the activity of HIF-1α is increased immediately after TBI, its expression levels are significantly decreased 24 hours following TBI (Ding et al., 2009). Studies from other laboratories have also reported that neurorepair (stimulation of the expression of vascular endothelial growth factor (VEGF) and brain-derived neurotrophic factor (BDNF)) mechanisms in the chronic phase of TBI are dependent on HIF-1α activity (Sen and Sen, 2016; Thelin et al., 2016).
HIF-1α and its regulating enzymes, including prolyl-4-hydroxylases (PHDs), are directly regulated by S-nitrosylation (Metzen et al., 2003), leading to stabilization of HIF-1α and induction of neurorepair mechanisms in the repair phase. S-nitrosylation-mediated stabilization of HIF-1α was also reported to increase angiogenesis and myocardial protection (Lima et al., 2009), indicating a protective role of S-nitrosylated HIF-1α. Therefore, we investigated whether S-nitrosylation-mediated modulation of HIF-1α induces neurorepair, leading to functional recovery in a rat CCI model of TBI.
HIF-1, a nuclear transcription factor, was discovered by Dr. Semenza in 1996 (Semenza, 1996). It was characterized as the master regulator of cellular oxygen homeostasis. It activates the tissue survival pathways by inducing several key enzymes involved in cell metabolism glucose transporter (GLUT), angiogenesis (VEGF, VEGFR1, angiopoietin), and free radical scavenging (heme hydroxylase-1; HO-1) (Ke and Costa, 2006). HIF is a heterodimeric protein composed of α and β subunits. There are three HIF-α isoforms (HIF-1α, HIF-2α and HIF-3α). The beta class includes HIF-1β. HIF-1 is a combination of the HIF-1α (120 kDa) and HIF-1β (91-94 kDa) subunits. The HIF-1β subunit is a constitutively expressed protein, but the expression of the HIF-1α subunit (a cytosolic protein) is largely dependent on oxygen levels. HIF-1α is rapidly up regulated in response to hypoxia and is rapidly degraded upon reoxygenation/reperfusion. Under normoxia, HIF-1α is bound by the von Hippel-Lindau protein (pVHL). pVHL recruits a ubiquitin ligase that targets HIF-1α for the 26S proteasomal degradation. The binding of pVHL is dependent upon hydroxylation of specific proline residues in HIF-1α (pro402 and pro564) by the PHD family of proteins (PHDs), especially HIF-1α-specific PHDs, such as PHD3/PHD2 (please see Ke and Costa, 2006; Harten et al., 2010 for details). These PHD isozymes share maximum homology, and they are implicated in degradation of HIF-1α. PHDs use oxygen as a substrate; therefore, their activity is inhibited under hypoxia. Oxygen can also activate factor-inhibiting HIF (FIH), leading to prevention of the binding of the co-activators p300/CBP, thus down regulating HIF-1-induced transcriptional activity (Figure 1). HIF-1α knockout mice show impaired vascular development and embryonic lethality, indicating HIF-1's protective role in vascular diseases (Iyer et al., 1998).
Figure 1.
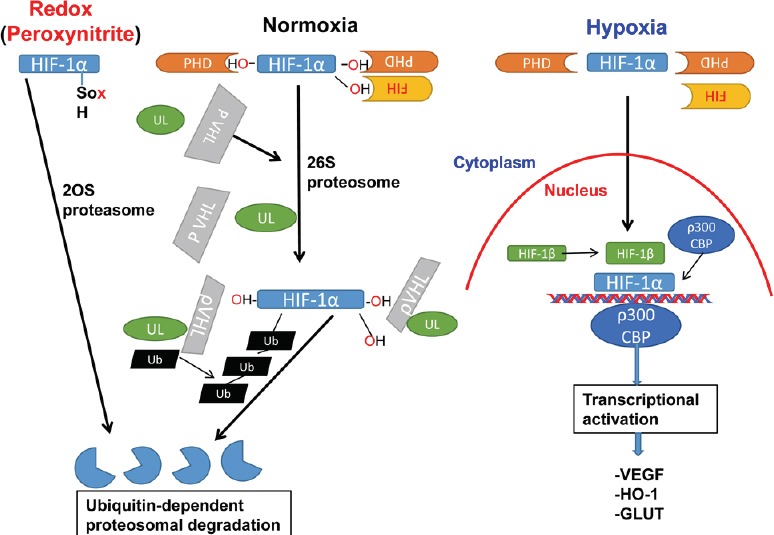
Hypothesized HIF-1α regulation under hypoxia, normoxia, and redox.
HIF-1α is characterized as the master regulator of cellular oxygen homeostasis. It activates the tissue survival pathways by inducing several key enzymes involved in cell metabolism (GLUT), angiogenesis (VEGF, VEGFR1, angiopoietin), and free radical scavenging (heme hydroxylase-1; HO-1). HIF-1 is a combination of the HIF-1α and HIF-1β subunits. The HIF-1β subunit is a constitutively expressed protein, but the expression of the HIF-1α subunit is largely dependent on oxygen levels. HIF-1α is rapidly up regulated, stabilized and moves to the nucleus in response to hypoxia. In contrast, it is rapidly degraded upon reoxygenation/reperfusion and in normoxic conditions. Under normoxia, HIF-1α is bound by the pVHL. pVHL recruits an ubiquitin ligase that targets HIF-1α for 26S proteasomal degradation. The binding of pVHL is dependent upon hydroxylation of specific proline residues in HIF-1α (pro402 and pro564) by the PHD family of proteins (PHDs). PHDs use oxygen as a substrate; therefore, their activity is inhibited under hypoxia. Oxygen can also activate FIH, preventing the binding of the co-activators p300/CBP, thus down regulating HIF-1-induced transcriptional activity. Reactive oxygen species such as peroxynitrite, destabilize HIF-1α by oxidizing its thiol group, which is degraded by 20S proteasome system. CBP: CREB-binding protein; FIH: factor-inhibiting HIF; GLUT: glucose transporter; HIF-1: hypoxia-inducible factor 1; HO-1: heme oxygenase-1; PHD: prolyl-4-hydroxylase; pVHL: von Hippel-Lindau protein; VEGF: vascular endothelial growth factor; VEGFR: VEGF receptor.
Remarkably, the HIF-1α pathway is involved in both pathological (hypoxia) and neurorepair (normoxia) mechanisms following TBI. The HIF-1α stabilizers/inducers, such as desferrioxamine (an iron chelator approved for haemochromatosis treatment), promote a number of survival pathways, including neuroprotection, angiogenesis and neurotrophins, and reduce brain infarctions when administered pre- or post-stroke (Kasivisvanathan et al., 2011). PHD inhibitors, such as FG-4539, are presently in a phase II anemia trial because of their activity to stabilize HIF-1α by preventing degradation with the ubiquitin proteasome system (Harten et al., 2010). However, inhibition of HIF-1α in the acute injury phase of TBI has also been reported to be neuroprotective (Shenaq et al., 2012; Schaible et al., 2014).
Under normoxic conditions, studies are lacking on direct stabilization of HIF-1α by secondary modification and the induction of consequent protective genes. Nevertheless, S-nitrosylation has been shown to stabilize HIF-1 protein expression and activity in normoxic endothelial cells (Palmer et al., 2000). Later, it was confirmed that, while GSNO stabilizes HIF-1α by S-nitrosylation, reactive nitrogen species (peroxynitrite) destabilize HIF-1α (Figure 1) (Wellman et al., 2004). GSNO-mediated stabilization of HIF-1α has been shown to be dependent on PI3K/Akt activity (Carver et al., 2007). A recent study in a mouse model of stroke shows that S-nitrosylation of phosphatase and tensin homolog (PTEN) results in an inhibition of its activity, leading to the activation of Akt (Numajiri et al., 2011). GSNO also activated Akt in a rat model of experimental stroke (Sakakima et al., 2012). Furthermore, GSNO also attenuated PHD activity during normoxia and inhibited proteasomal degradation of HIF-1α (Metzen et al., 2003). S-nitrosylation-mediated stabilization of HIF-1α has been shown to protect against myocardial injury via the VEGF/angiogenesis pathway in GSNO reductase (GSNOR) knockout mice (Lima et al., 2009), indicating that HIF-1 is a key player in the regeneration process. Our TBI studies showing that the HIF-1α/VEGF pathway accelerated functional recovery in a 2-week mouse model of TBI via S-nitrosylation of HIF-1α further support the neuroreparative role of HIF-1α (Khan et al., 2016a) as depicted in Figure 2. S-nitrosylation/GSNO-mediated increased expression HIF-1α and stimulation of neurotrophic factors provide a strong rationale to evaluate the potential of a GSNO-mediated HIF-1α pathway for human therapy in the chronic phase of TBI.
Figure 2.
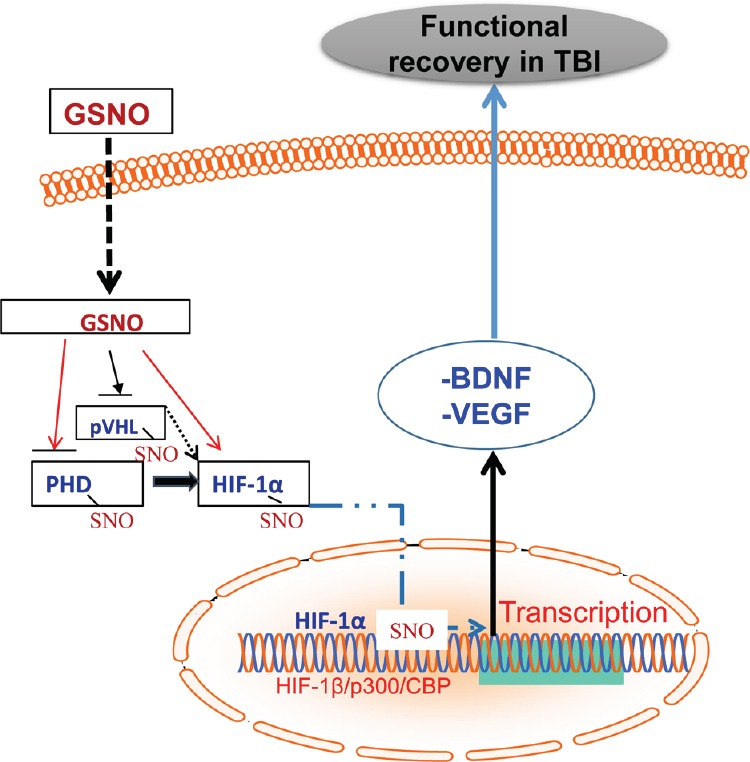
Schematic showing that exogenously administered GSNO stabilizes HIF-1α via S-nitrosylation, leading to the stimulation of neurorepair mediators and functional recovery in TBI.
GSNO-mediated S-nitrosylation occurs on pVHL, PHD, and HIF-1α, contributing to the inhibition of HIF-1α degradation. S-nitrosylated HIF-1α moves to the nucleus, where it dimerizes with HIF-1β and interacts with P300 and CBP, leading to transcription of neurorepair mediators such as VEGF and BDNF under TBI conditions. Black broken arrows indicate that these mechanisms are not well understood. BDNF: Brain-derived neurotrophic factor; CBP: CREB-binding protein; GSNO: S-nitrosoglutathione; HIF-1: hypoxia-inducible factor 1; PHD: prolyl-4-hydroxylase; pVHL: von Hippel-Lindau protein; SNO: S-nitrosylation; TBI: traumatic brain injury; VEGF: vascular endothelial growth factor.
GSNO is a natural component of the human body produced by the reaction of nitric oxide (NO) with glutathione (GSH) in the presence of oxygen (Singh et al., 1996). It is sensitive to light, ascorbate, thiols and divalent cations such as Fe2+ and Cu2+ (Broniowska et al., 2013). GSNO is present in the brain and other organs (Kluge et al., 1997). It is directly involved in cell signaling via S-nitrosylation of target proteins, including nuclear factor kappaB (NF-κB), signal transducer and activator of transcription 3 (STAT3), cyclooxygenase-2 (COX-2), caspase-3, calpains, inducible nitric oxide synthase (iNOS), and endothelial NOS (eNOS) and neuronal NOS (nNOS) (Jaffrey et al., 2001; Khan et al., 2005, 2006, 2012, 2016a, b; Kim et al., 2013). Exogenous administration of GSNO (Rassaf et al., 2006) also protects against cardiac ischemic injury (Konorev et al., 1995; Lima et al., 2009), supporting the therapeutic potential of GSNO. Studies have also reported that GSNO inhibits platelet activation in humans (Radomski et al., 1992) and protects both blood-brain barrier integrity and epithelial permeability (Savidge et al., 2007; Khan et al., 2009). Various disease conditions are known to have reduced levels of S-nitrosothiols (-SNO/GSNO) (Snyder et al., 2002; Heiss et al., 2006; Schonhoff et al., 2006) and exogenous administration of GSNO has increased endogenous GSNO and S-NO levels (Khan et al., 2012; Zanini et al., 2012; Hu et al., 2013).
In a microenvironment of TBI, NO released by conventional NO-donors or NO gas itself is anticipated to be inactivated by superoxide, thus forming deleterious peroxynitrite (Singh et al., 2007; Deng-Bryant et al., 2008; Reed et al., 2009). Unlike NO, the disadvantage of inactivation is not associated with the S-nitrosylating agent GSNO (Khan et al., 2006). In addition, S-nitrosylation of cysteine residue (a reversible modification) prevents it from further oxidation to sulfinic and sulfonic acids (an irreversible modification), thereby preventing inactivation of both NO and proteins. The neurorepair effect of GSNO may be mediated by two different mechanisms: 1) S-nitrosylation and 2) maintaining redox by mechanistically reducing the production of oxidants, including peroxynitrite. Such multi-mechanistic functional and therapeutic abilities are not embedded in conventional NO donors as previously reported (Khan et al., 2006), making GSNO a unique candidate to be investigated for the stimulation of functional recovery following TBI.
Several studies showing the efficacy of GSNO in human diseases have been listed by Hornyak et al. (Hornyak et al., 2011). Recently, GSNO was also used in early onset of preeclampsia (Christopher et al.). None of the studies report major or significant side effects associated with the use of GSNO in humans. GSNO-releasing nanoparticles, hydrogel and/or polymers are also used tropically in wound healing and skin diseases (Georgii et al., 2011; Chouake et al., 2012). Microparticles loaded with GSNO have a much longer half-life than free GSNO and show neurovascular protective efficacy in an animal model of embolic stroke (Parent et al., 2015). GSNO-mediated therapeutic effects can also be achieved via the inhibition of GSNO reductase (GSNOR) enzyme. GSNOR is the major GSNO-metabolizing enzyme and thus GSNOR knock out mice store GSNO in excess. GSNOR degrades GSNO into ammonia and oxidized glutathione without releasing free NO. Other enzymes, including carbonyl reductase, formaldehyde dehydrogenase and gamma glutamyl transpeptidase also metabolize GSNO, but their activity is not specific toward GSNO (Foster et al., 2009). Pharmacological inhibition of GSNOR has also been shown to improve endothelial functions (Chen et al., 2013), indicating a protective role of GSNO in neurovascular dysfunction. A recent report shows that GSNOR knock out mice behave normally and GSNO invokes its mechanistic effect via the mechanisms of trans-S-nitrosylation (Moon et al., 2017). However, another study found GSNOR knock mice having compromised neuro-muscular functions (Montagna et al., 2014). Use of GSNOR inhibitors have been found beneficial in animal models of experimental asthma (Ferrini et al., 2013), allergic airway inflammation (Blonder et al., 2014) and endothelial vasodilatory dysfunction (Chen et al., 2013). These results support the association of beneficial activity with GSNO-mediated mechanisms in several diseases.
Conclusion
The potential of GSNO as an HIF-1α stabilization-based therapeutic agent in TBI offers a novel target for further investigation (Figure 2). Mechanistically, GSNO invokes its action mainly via an S-nitrosylation-based mechanism, a physiological secondary protein modification process. Unlike other chemical therapeutics, GSNO is an endogenous neurorepair-inducing agent and its exogenous administration protects against neurodegenerative disease mechanisms in stroke, spinal cord injury, and TBI. The treatment with GSNO accelerated functional recovery and improved overall outcomes in a comparatively long-term TBI study (Khan et al., 2016a). Furthermore, GSNO's administration in humans for other indications resulted in no toxicity or side effects, thus supporting the translational potential of GSNO therapy in TBI. A long term study showing stimulation of neurorepair mechanisms and improvements of neurological functions in humans will determine the overall efficacy and the clinical relevance of GSNO as a rehabilitation therapy in TBI.
Additional file (179.5KB, pdf) : Open peer review report 1.
Open peer review report 1 on “Hypoxia inducible factor-1 alpha stabilization for regenerative therapy in traumatic brain injury”.
Acknowledgments
We acknowledge Dr. Tom Smith, Ph.D., from the MUSC Writing Center for his valuable editing of the manuscript. We also thank Dr. Tajinder S. Dhammu for his input in understanding the role of HIFs in TBI.
Footnotes
Funding: This work was supported by grants from VA merit awards (BX3401 and RX2090).
Conflicts of interest: The authors declare that they have no competing interests.
Open peer reviewer: Eric Peter Thelin.
References
- Blennow K, Brody DL, Kochanek PM, Levin H, McKee A, Ribbers GM, Yaffe K, Zetterberg H. Traumatic brain injuries. Nat Rev Dis Primers. 2016;2:16084. doi: 10.1038/nrdp.2016.84. [DOI] [PubMed] [Google Scholar]
- Blonder JP, Mutka SC, Sun X, Qiu J, Green LH, Mehra NK, Boyanapalli R, Suniga M, Look K, Delany C, Richards JP, Looker D, Scoggin C, Rosenthal GJ. Pharmacologic inhibition of S-nitrosoglutathione reductase protects against experimental asthma in BALB/c mice through attenuation of both bronchoconstriction and inflammation. BMC Pulm Med. 2014;14:3. doi: 10.1186/1471-2466-14-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broniowska KA, Diers AR, Hogg N. S-nitrosoglutathione. Biochim Biophys Acta. 2013;1830:3173–3181. doi: 10.1016/j.bbagen.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver DJ, Gaston B, Deronde K, Palmer LA. Akt-mediated activation of HIF-1 in pulmonary vascular endothelial cells by S-nitrosoglutathione. Am J Respir Cell Mol Biol. 2007;37:255–263. doi: 10.1165/rcmb.2006-0289SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Sievers RE, Varga M, Kharait S, Haddad DJ, Patton AK, Delany CS, Mutka SC, Blonder JP, Dube GP, Rosenthal GJ, Springer ML. Pharmacological inhibition of S-nitrosoglutathione reductase improves endothelial vasodilatory function in rats in vivo. J Appl Physiol (1985) 2013;114:752–760. doi: 10.1152/japplphysiol.01302.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouake J, Schairer D, Kutner A, Sanchez DA, Makdisi J, Blecher-Paz K, Nacharaju P, Tuckman-Vernon C, Gialanella P, Friedman JM, Nosanchuk JD, Friedman AJ. Nitrosoglutathione generating nitric oxide nanoparticles as an improved strategy for combating Pseudomonas aeruginosa-infected wounds. J Drugs Dermatol. 2012;11:1471–1477. [PubMed] [Google Scholar]
- Coles JP. Regional ischemia after head injury. Curr Opin Crit Care. 2004;10:120–125. doi: 10.1097/00075198-200404000-00008. [DOI] [PubMed] [Google Scholar]
- Colicos MA, Dixon CE, Dash PK. Delayed, selective neuronal death following experimental cortical impact injury in rats: possible role in memory deficits. Brain Res. 1996;739:111–119. doi: 10.1016/s0006-8993(96)00819-0. [DOI] [PubMed] [Google Scholar]
- Corso P, Finkelstein E, Miller T, Fiebelkorn I, Zaloshnja E. Incidence and lifetime costs of injuries in the United States. Inj Prev. 2006;12:212–218. doi: 10.1136/ip.2005.010983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng-Bryant Y, Singh IN, Carrico KM, Hall ED. Neuroprotective effects of tempol, a catalytic scavenger of peroxynitrite-derived free radicals, in a mouse traumatic brain injury model. J Cereb Blood Flow Metab. 2008;28:1114–1126. doi: 10.1038/jcbfm.2008.10. [DOI] [PubMed] [Google Scholar]
- Diaz-Arrastia R, Kochanek PM, Bergold P, Kenney K, Marx CE, Grimes CJ, Loh LT, Adam LT, Oskvig D, Curley KC, Salzer W. Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report of the Department of Defense Neurotrauma Pharmacology Workgroup. J Neurotrauma. 2014;31:135–158. doi: 10.1089/neu.2013.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JY, Kreipke CW, Speirs SL, Schafer P, Schafer S, Rafols JA. Hypoxia-inducible factor-1alpha signaling in aquaporin upregulation after traumatic brain injury. Neurosci Lett. 2009;453:68–72. doi: 10.1016/j.neulet.2009.01.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- Faul M, Coronado V. Epidemiology of traumatic brain injury. Handb Clin Neurol. 2015;127:3–13. doi: 10.1016/B978-0-444-52892-6.00001-5. [DOI] [PubMed] [Google Scholar]
- Ferrini ME, Simons BJ, Bassett DJ, Bradley MO, Roberts K, Jaffar Z. S-nitrosoglutathione reductase inhibition regulates allergen-induced lung inflammation and airway hyperreactivity. PLoS One. 2013;8:e70351. doi: 10.1371/journal.pone.0070351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner AJ, Zafonte R. Neuroepidemiology of traumatic brain injury. Handb Clin Neurol. 2016;138:207–223. doi: 10.1016/B978-0-12-802973-2.00012-4. [DOI] [PubMed] [Google Scholar]
- Georgii JL, Amadeu TP, Seabra AB, de Oliveira MG, Monte-Alto-Costa A. Topical S-nitrosoglutathione-releasing hydrogel improves healing of rat ischaemic wounds. J Tissue Eng Regen Med. 2011;5:612–619. doi: 10.1002/term.353. [DOI] [PubMed] [Google Scholar]
- Greve MW, Zink BJ. Pathophysiology of traumatic brain injury. Mt Sinai J Med. 2009;76:97–104. doi: 10.1002/msj.20104. [DOI] [PubMed] [Google Scholar]
- Harten SK, Ashcroft M, Maxwell PH. Prolyl hydroxylase domain inhibitors: a route to HIF activation and neuroprotection. Antioxid Redox Signal. 2010;12:459–480. doi: 10.1089/ars.2009.2870. [DOI] [PubMed] [Google Scholar]
- Heiss C, Lauer T, Dejam A, Kleinbongard P, Hamada S, Rassaf T, Matern S, Feelisch M, Kelm M. Plasma nitroso compounds are decreased in patients with endothelial dysfunction. J Am Coll Cardiol. 2006;47:573–579. doi: 10.1016/j.jacc.2005.06.089. [DOI] [PubMed] [Google Scholar]
- Hornyak I, Pankotai E, Kiss L, Lacza Z. Current developments in the therapeutic potential of S-nitrosoglutathione, an endogenous NO-donor molecule. Curr Pharm Biotechnol. 2011;12:1368–1374. doi: 10.2174/138920111798280983. [DOI] [PubMed] [Google Scholar]
- Hu Z, Bian X, Liu X, Zhu Y, Zhang X, Chen S, Wang K, Wang Y. Honokiol protects brain against ischemia-reperfusion injury in rats through disrupting PSD95-nNOS interaction. Brain Res. 2013;1491:204–212. doi: 10.1016/j.brainres.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC. The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation. 2007;22:341–353. [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nat Rev Neurosci. 2010;11:361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasivisvanathan V, Shalhoub J, Lim CS, Shepherd AC, Thapar A, Davies AH. Hypoxia-inducible factor-1 in arterial disease: a putative therapeutic target. Curr Vasc Pharmacol. 2011;9:333–349. doi: 10.2174/157016111795495602. [DOI] [PubMed] [Google Scholar]
- Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1) Mol Pharmacol. 2006;70:1469–1480. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- Khan M, Jatana M, Elango C, Paintlia AS, Singh AK, Singh I. Cerebrovascular protection by various nitric oxide donors in rats after experimental stroke. Nitric Oxide. 2006;15:114–124. doi: 10.1016/j.niox.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Khan M, Im YB, Shunmugavel A, Gilg AG, Dhindsa RK, Singh AK, Singh I. Administration of S-nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact. J Neuroinflammation. 2009;6:32. doi: 10.1186/1742-2094-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Dhammu TS, Sakakima H, Shunmugavel A, Gilg AG, Singh AK, Singh I. The inhibitory effect of S-nitrosoglutathione on blood-brain barrier disruption and peroxynitrite formation in a rat model of experimental stroke. J Neurochem 123 Suppl. 2012;2:86–97. doi: 10.1111/j.1471-4159.2012.07947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Dhammu TS, Baarine M, Kim J, Paintlia MK, Singh I, Singh AK. GSNO promotes functional recovery in experimental TBI by stabilizing HIF-1alpha. Behav Brain Res. 2016a doi: 10.1016/j.bbr.2016.10.037. doi: 10.1016/j.bbr.2016.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Dhammu TS, Matsuda F, Annamalai B, Dhindsa TS, Singh I, Singh AK. Targeting the nNOS/peroxynitrite/calpain system to confer neuroprotection and aid functional recovery in a mouse model of TBI. Brain Res. 2016b;1630:159–170. doi: 10.1016/j.brainres.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Sekhon B, Giri S, Jatana M, Gilg AG, Ayasolla K, Elango C, Singh AK, Singh I. S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J Cereb Blood Flow Metab. 2005;25:177–192. doi: 10.1038/sj.jcbfm.9600012. [DOI] [PubMed] [Google Scholar]
- Kim BT, Rao VL, Sailor KA, Bowen KK, Dempsey RJ. Protective effects of glial cell line-derived neurotrophic factor on hippocampal neurons after traumatic brain injury in rats. J Neurosurg. 2001;95:674–679. doi: 10.3171/jns.2001.95.4.0674. [DOI] [PubMed] [Google Scholar]
- Kim J, Won JS, Singh AK, Sharma AK, Singh I. STAT3 regulation by S-nitrosylation: implication for inflammatory disease. Antioxid Redox Signal. 2013;20:2514–2527. doi: 10.1089/ars.2013.5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluge I, Gutteck-Amsler U, Zollinger M, Do KQ. S-nitrosoglutathione in rat cerebellum: identification and quantification by liquid chromatography-mass spectrometry. J Neurochem. 1997;69:2599–2607. doi: 10.1046/j.1471-4159.1997.69062599.x. [DOI] [PubMed] [Google Scholar]
- Konorev EA, Tarpey MM, Joseph J, Baker JE, Kalyanaraman B. S-nitrosoglutathione improves functional recovery in the isolated rat heart after cardioplegic ischemic arrest-evidence for a cardioprotective effect of nitric oxide. J Pharmacol Exp Ther. 1995;274:200–206. [PubMed] [Google Scholar]
- Langlois JA, Rutland-Brown W, Thomas KE. The incidence of traumatic brain injury among children in the United States: differences by race. J Head Trauma Rehabil. 2005;20:229–238. doi: 10.1097/00001199-200505000-00006. [DOI] [PubMed] [Google Scholar]
- Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- Levin H, Kraus MF. The frontal lobes and traumatic brain injury. J Neuropsychiatry Clin Neurosci. 1994;6:443–454. doi: 10.1176/jnp.6.4.443. [DOI] [PubMed] [Google Scholar]
- Levin HS, Diaz-Arrastia RR. Diagnosis, prognosis, and clinical management of mild traumatic brain injury. Lancet Neurol. 2015;14:506–517. doi: 10.1016/S1474-4422(15)00002-2. [DOI] [PubMed] [Google Scholar]
- Lighthall JW, Dixon CE, Anderson TE. Experimental models of brain injury. J Neurotrauma. 1989;6:83–97. doi: 10.1089/neu.1989.6.83. [DOI] [PubMed] [Google Scholar]
- Lima B, Lam GK, Xie L, Diesen DL, Villamizar N, Nienaber J, Messina E, Bowles D, Kontos CD, Hare JM, Stamler JS, Rockman HA. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci U S A. 2009;106:6297–6302. doi: 10.1073/pnas.0901043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SW, Huang LC, Chung WF, Chang HK, Wu JC, Chen LF, Chen YC, Huang WC, Cheng H, Lo SS. Increased risk of stroke in patients of concussion: a nationwide cohort study. Int J Environ Res Public Health. 2017;14:E230. doi: 10.3390/ijerph14030230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd E, Somera-Molina K, Van Eldik LJ, Watterson DM, Wainwright MS. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J Neuroinflammation. 2008;5:28. doi: 10.1186/1742-2094-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Qu C, Goussev A, Jiang H, Lu C, Schallert T, Mahmood A, Chen J, Li Y, Chopp M. Statins increase neurogenesis in the dentate gyrus, reduce delayed neuronal death in the hippocampal CA3 region, and improve spatial learning in rat after traumatic brain injury. J Neurotrauma. 2007;24:1132–1146. doi: 10.1089/neu.2007.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell. 2003;14:3470–3481. doi: 10.1091/mbc.E02-12-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollayeva T, Cassidy JD, Shapiro CM, Mollayeva S, Colantonio A. Concussion/mild traumatic brain injury-related chronic pain in males and females: A diagnostic modelling study. Medicine (Baltimore) 2017;96:e5917. doi: 10.1097/MD.0000000000005917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagna C, Di Giacomo G, Rizza S, Cardaci S, Ferraro E, Grumati P, De Zio D, Maiani E, Muscoli C, Lauro F, Ilari S, Bernardini S, Cannata S, Gargioli C, Ciriolo MR, Cecconi F, Bonaldo P, Filomeni G. S-nitrosoglutathione reductase deficiency-induced S-nitrosylation results in neuromuscular dysfunction. Antioxid Redox Signal. 2014;21:570–587. doi: 10.1089/ars.2013.5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon Y, Cao Y, Zhu J, Xu Y, Balkan W, Buys ES, Diaz F, Kerrick WG, Hare JM, Percival JM. GSNOR deficiency enhances in situ skeletal muscle strength, fatigue resistance, and RyR1 S-nitrosylation without impacting mitochondrial content and activity. Antioxid Redox Signal. 2017;26:165–181. doi: 10.1089/ars.2015.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numajiri N, Takasawa K, Nishiya T, Tanaka H, Ohno K, Hayakawa W, Asada M, Matsuda H, Azumi K, Kamata H, Nakamura T, Hara H, Minami M, Lipton SA, Uehara T. On-off system for PI3-kinase-Akt signaling through S-nitrosylation of phosphatase with sequence homology to tensin (PTEN) Proc Natl Acad Sci U S A. 2011;108:10349–10354. doi: 10.1073/pnas.1103503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyesiku NM, Evans CO, Houston S, Darrell RS, Smith JS, Fulop ZL, Dixon CE, Stein DG. Regional changes in the expression of neurotrophic factors and their receptors following acute traumatic brain injury in the adult rat brain. Brain Res. 1999;833:161–172. doi: 10.1016/s0006-8993(99)01501-2. [DOI] [PubMed] [Google Scholar]
- Palmer LA, Gaston B, Johns RA. Normoxic stabilization of hypoxia-inducible factor-1 expression and activity: redox-dependent effect of nitrogen oxides. Mol Pharmacol. 2000;58:1197–1203. doi: 10.1124/mol.58.6.1197. [DOI] [PubMed] [Google Scholar]
- Parent M, Boudier A, Perrin J, Vigneron C, Maincent P, Violle N, Bisson JF, Lartaud I, Dupuis F. In situ microparticles loaded with S-nitrosoglutathione protect from stroke. PLoS One. 2015;10:e0144659. doi: 10.1371/journal.pone.0144659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennings JL, Bachulis BL, Simons CT, Slazinski T. Survival after severe brain injury in the aged. Arch Surg. 1993;128:787–793. doi: 10.1001/archsurg.1993.01420190083011. discussion 793-784. [DOI] [PubMed] [Google Scholar]
- Radomski MW, Rees DD, Dutra A, Moncada S. S-nitroso-glutathione inhibits platelet activation in vitro and in vivo. Br J Pharmacol. 1992;107:745–749. doi: 10.1111/j.1476-5381.1992.tb14517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassaf T, Poll LW, Brouzos P, Lauer T, Totzeck M, Kleinbongard P, Gharini P, Andersen K, Schulz R, Heusch G, Modder U, Kelm M. Positive effects of nitric oxide on left ventricular function in humans. Eur Heart J. 2006;27:1699–1705. doi: 10.1093/eurheartj/ehl096. [DOI] [PubMed] [Google Scholar]
- Reed TT, Owen J, Pierce WM, Sebastian A, Sullivan PG, Butterfield DA. Proteomic identification of nitrated brain proteins in traumatic brain-injured rats treated postinjury with gamma-glutamylcysteine ethyl ester: insights into the role of elevation of glutathione as a potential therapeutic strategy for traumatic brain injury. J Neurosci Res. 2009;87:408–417. doi: 10.1002/jnr.21872. [DOI] [PubMed] [Google Scholar]
- Sakakima H, Khan M, Dhammu TS, Shunmugavel A, Yoshida Y, Singh I, Singh AK. Stimulation of functional recovery via the mechanisms of neurorepair by S-nitrosoglutathione and motor exercise in a rat model of transient cerebral ischemia and reperfusion. Restor Neurol Neurosci. 2012;30:383–396. doi: 10.3233/RNN-2012-110209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidge TC, Newman P, Pothoulakis C, Ruhl A, Neunlist M, Bourreille A, Hurst R, Sofroniew MV. Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology. 2007;132:1344–1358. doi: 10.1053/j.gastro.2007.01.051. [DOI] [PubMed] [Google Scholar]
- Schaible EV, Windschugl J, Bobkiewicz W, Kaburov Y, Dangel L, Kramer T, Huang C, Sebastiani A, Luh C, Werner C, Engelhard K, Thal SC, Schafer MK. 2-Methoxyestradiol confers neuroprotection and inhibits a maladaptive HIF-1alpha response after traumatic brain injury in mice. J Neurochem. 2014;129:940–954. doi: 10.1111/jnc.12708. [DOI] [PubMed] [Google Scholar]
- Schonhoff CM, Matsuoka M, Tummala H, Johnson MA, Estevez AG, Wu R, Kamaid A, Ricart KC, Hashimoto Y, Gaston B, Macdonald TL, Xu Z, Mannick JB. S-nitrosothiol depletion in amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:2404–2409. doi: 10.1073/pnas.0507243103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Transcriptional regulation by hypoxia-inducible factor 1 molecular mechanisms of oxygen homeostasis. Trends Cardiovasc Med. 1996;6:151–157. doi: 10.1016/1050-1738(96)00039-4. [DOI] [PubMed] [Google Scholar]
- Sen T, Sen N. Treatment with an activator of hypoxia-inducible factor 1, DMOG provides neuroprotection after traumatic brain injury. Neuropharmacology. 2016;107:79–88. doi: 10.1016/j.neuropharm.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenaq M, Kassem H, Peng C, Schafer S, Ding JY, Fredrickson V, Guthikonda M, Kreipke CW, Rafols JA, Ding Y. Neuronal damage and functional deficits are ameliorated by inhibition of aquaporin and HIF1alpha after traumatic brain injury (TBI) J Neurol Sci. 2012;323:134–140. doi: 10.1016/j.jns.2012.08.036. [DOI] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Hall ED. Peroxynitrite-mediated oxidative damage to brain mitochondria: Protective effects of peroxynitrite scavengers. J Neurosci Res. 2007;85:2216–2223. doi: 10.1002/jnr.21360. [DOI] [PubMed] [Google Scholar]
- Singh SP, Wishnok JS, Keshive M, Deen WM, Tannenbaum SR. The chemistry of the S-nitrosoglutathione/glutathione system. Proc Natl Acad Sci U S A. 1996;93:14428–14433. doi: 10.1073/pnas.93.25.14428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder AH, McPherson ME, Hunt JF, Johnson M, Stamler JS, Gaston B. Acute effects of aerosolized S-nitrosoglutathione in cystic fibrosis. Am J Respir Crit Care Med. 2002;165:922–926. doi: 10.1164/ajrccm.165.7.2105032. [DOI] [PubMed] [Google Scholar]
- Sun D, Bullock MR, McGinn MJ, Zhou Z, Altememi N, Hagood S, Hamm R, Colello RJ. Basic fibroblast growth factor-enhanced neurogenesis contributes to cognitive recovery in rats following traumatic brain injury. Exp Neurol. 2009;216:56–65. doi: 10.1016/j.expneurol.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelin EP, Frostell A, Mulder J, Mitsios N, Damberg P, Aski SN, Risling M, Svensson M, Morganti-Kossmann MC, Bellander BM. Lesion size is exacerbated in hypoxic rats whereas hypoxia-inducible factor-1 alpha and vascular endothelial growth factor increase in injured normoxic rats: a prospective cohort study of secondary hypoxia in focal traumatic brain injury. Front Neurol. 2016;7:23. doi: 10.3389/fneur.2016.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman TL, Jenkins J, Penar PL, Tranmer B, Zahr R, Lounsbury KM. Nitric oxide and reactive oxygen species exert opposing effects on the stability of hypoxia-inducible factor-1alpha (HIF-1alpha) in explants of human pial arteries. Faseb J. 2004;18:379–381. doi: 10.1096/fj.03-0143fje. [DOI] [PubMed] [Google Scholar]
- Wu H, Lu D, Jiang H, Xiong Y, Qu C, Li B, Mahmood A, Zhou D, Chopp M. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J Neurotrauma. 2008;25:130–139. doi: 10.1089/neu.2007.0369. [DOI] [PubMed] [Google Scholar]
- The US centers for disease control and prevention. Atlanta, GA: www.cdc.gov/traumaticbraininjury/data . [Google Scholar]
- Zanini GM, Martins YC, Cabrales P, Frangos JA, Carvalho LJ. S-nitrosoglutathione prevents experimental cerebral malaria. J Neuroimmune Pharmacol. 2012;7:477–487. doi: 10.1007/s11481-012-9343-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Open peer review report 1 on “Hypoxia inducible factor-1 alpha stabilization for regenerative therapy in traumatic brain injury”.