

All cells in the body metabolize oxygen in a series of reactions in the mitochondria to generate energy. A by-product of these reactions is the production of highly reactive oxygen radicals which have the potential to cause irreparable damage to cellular components, and have been linked to mutagenic and neurotoxicity of the nervous system. Under normal conditions, innate molecular systems, glutathione, superoxide dismutases, catalase, etc., have evolved to neutralize these reactive oxygen species (ROS) (Uttara et al., 2009). However, there are many situations which lead to an imbalance in the production of ROS which compromise cellular systems by what is commonly known as oxidative stress (OS). In response, health professionals have typically prescribed the use of hyper doses of antioxidant (AO) supplements as well as foods and teas with high AO concentrations. Furthermore, AOs such as Vitamins C and E (Poljsak et al., 2013) are commonly used as an ongoing remedial, ‘over-the-counter’ treatment to combat OS commonly caused by colds and flues, the occasional bout of strenuous exercise, etc. However, a surplus of AOs at the cellular level will not only neutralize ROS, but will cause the antithesis of OS, which is known as reductive stress (RS).
RS decreases blood-brain barrier (BBB) permeability. Our study on the aggressive use of AOs on brain endothelial cells (BEC) (Mentor and Fisher, 2017) showed that a commonly used fermented tea (Rooibos; Rf) known for its high levels of AOs, produced profound physiological effects by causing RS. Treating confluent layers of BECs, the primary cellular component of the BBB, with Rf increased the impermeability across the monolayer in a dose-related manner. The primary function of the BBB is to regulate the flux of substances into and out of the brain. This is crucial to the homeostatic regulation of the brain parenchyma, that milieu of extracellular fluid which baths the neurons. Any change in permeability will impact the ease at which substances enter and leave the brain, thus impacting the dynamic regulation of both metabolic and ionic permeability. Subtle changes to the BBB permeability may indeed compromise the regulation of ionic concentrations within the brain impacting neuronal thresholds for impulse activity, with concomitant effects on thought processing, psychological status, sensory and motor function, etc. (Zlokovic et al., 2011).
RS attenuates endothelial cell proliferation. The effect of excess AO treatment further impacted the rate of cellular proliferation, also in a dose-related manner. The toxicological study showed that the decrease in cellular proliferation was not as a result of cellular toxicity in the Rf-treated groups of cells. The dose-related effects observed in both the permeability study and the proliferation study, suggests a common mechanism for this effect. Reports in the literature supports these findings in that Apigenin, an AO flavonoid, inhibited cellular proliferation by affecting the cell cycle of keratinocytes, human diploid fibroblasts and neuronal cells (Lepley et al., 1997). Also, Lamosŏvá et al. (1997) reported the inhibition of cellular proliferation in primary cultures of chick skeletal cells after treatment with concentrations of 2–100% of a Rooibos extract, containing high concentrations of AOs.
AO and RS target different cellular mechanisms. These studies all indicate a cellular based mechanism to explain the effect of RS caused by excess AOs. The study by Mentor and Fisher (2017) also showed that AOs cause adecrease in permeability across monolayers of BECs. In contrast, OS, is widely reported to cause increased permeability by compromising the paracellular tight junctions (occluding and claudin), resulting in the observed decrease in transendothelial electrical resistance (i.e. increase permeability) (Haorah et al., 2005; Lochhead et al., 2010). It is, therefore, difficult to conceive that RS, affects transendothelial permeability by increasing the ‘tightness’ or impermeability of the paracellular pathways. The above mentioned evidence and the data reported by Mentor and Fisher (2017) indicate that a more likely explanation would be that RS impacts the serial apical and basolateral endothelial membranes by implicating the open/closed states of membrane ionic channels, statistically favouring the close state of the channel and thereby decreasing the permeability across these endothelial cells. Alternatively, or synergistically, AO-induced RS could suppress the recruitment of membrane ionic channels, and by implication, decrease the permeability across cellular apical and/or basolateral membranes (Figure 1).
Figure 1.
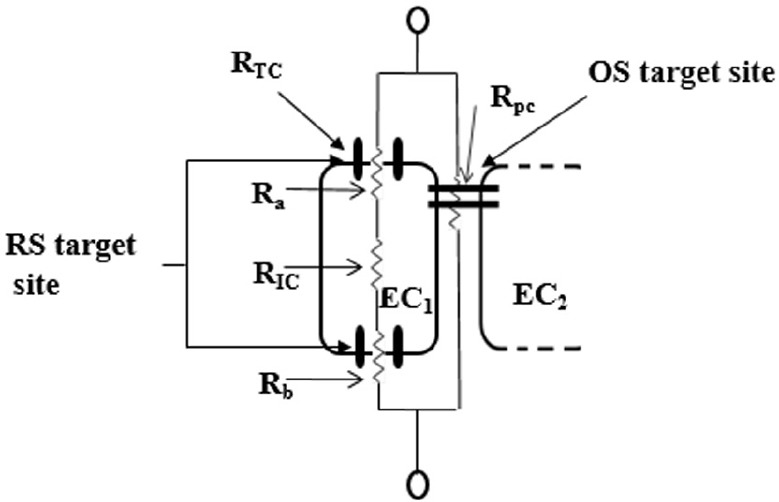
An illustration of the proposed mechanisms for ROS-induced BBB dysfunction in comparison to RS-induced BBB dysfunction.
ROS affects the BBB endothelial cells by compromising the tight junctions which regulate the permeability through the paracellular pathways, resulting in a decreased electrical resistance (increase permeability) across the BBB endothelium. RS affects the BBB endothelial cells by affecting the permeability across their apical and basolateral membranes, by increasing the close states of ionic channels or their insertion into these membranes, causing increase electrical resistance (decrease permeability) across the BBB endothelium. By using an equivalent electrical circuit to represent the in vitro BBB we represent the various components of the transendothelial ionic pathways, and depict the RS and OS target sites across the BBB endothelium. The apical resistance (Ra), basolateral resistance (Rb) and intracellular resistance (RIC), which are located in series, represents the transcellular pathway and the two parallel bars, located apicolaterally, between endothelial cell 1 and 2 (EC1 and EC2) represents tight junctions of the paracellular pathway. BBB: Blood-brain barrier; OS: oxidative stress; ROS: reactive oxygen species; RS: reductive stress. RTC: trancellular resistance; RPC: paracellular resistance.
The postulated altering of transendothelial permeability due to RS by affecting ionic fluxes across the cells is very likely to have attenuated cell proliferation in a dose-related fashion (Hermann et al., 2015). This is a novel postulate in that for the first time it has been reported that the mechanism of OS and RS on cells are implemented via distinctly different cellular routes (Figure 1).
It is, therefore, of no surprise that clinical trials using AO therapy have reported little success and in some cases have increased mortality (Poljsak et al., 2013). It is clear that AO therapy, which is not judiciously used to treat excess ROS, will result in RS which may have detrimental effects for brain homeostasis and the BBB vasculature.
AO-induced RS impacts mitochondrial activity. It is well established that slight excesses in cellular ROS concentrations are essential for the normal homeostatic regulation of cellular pathways. Under normal conditions ROS concentrations are finely regulated by the cells endogenous AO system. Treating BECs with daily low doses (0.003–0.013%) and high doses (0.03–0.1%) of AOs showed that mitochondrial activity (MA) was initially suppressed by the low doses, but not by the high doses, a trend which continued at 48 hours (Mentor and Fisher, 2017). Low doses of AOs neutralized cellular ROS, interfering with the ROS induction of glucose transporters into the apical membrane of the BECs, essentially suffocating the glycolytic pathway of glucose (Liemburg-Apers, et al., 2015), subsequently resulting in a decrease metabolic substrate (pyruvate) for the mitochondria-located tricarboxylic acid (TCA) cycle, thus decreasing mitochondrial function. The mitochondria of BECs seem to be fairly resistant to RS over 72 hours, however at 96 hours, chronic treatment with higher doses of AOs (0.05% and 1%) decreased mitochondrial function. The data reported by Fisher and Mentor (2017) was endorsed in a recent study by Singh et al. (2015) which showed conclusively that RS impaired the mitochondrial function of myoblasts, and addition, they also reported that within 24 hours RS stimulated the activation of mitochondrial biogenesis pathways to adapt to the reduction-oxidation reaction (redox) stress. BEC mitochondria showed that they too are able to adapt to RS after 24 hours, but only at low to mid doses, but succumbed to high doses of AOs after 96 hours exposure.
AO-induced RS may impact endogenous AO capacity. The elegant interplay of neuronal activity driven ROS production, astrocyte support of endogenous cellular AO systems and the transcriptional control of the glutathione based AO capacity (Baxter et al., 2015), suggests that chronic overuse of AOs on a daily basis may indeed lead to the cellular suppression of their endogenous synthesis of AOs, and thus precariously compromise their endogenous AO capacity. The addition of daily copious amounts of exogenous AOs may deplete the BEC's AO stores and its capacity, making it vulnerable to respond innately to transient increases in ROS. Furthermore, the cell's natural AO-ROS balance is tipped slightly in favour of ROS, which is critical for normal cell function, such as cell division, glucose transport into the cell, etc. (Poljak et al., 2013; Liemburg-Apers et al., 2015). Thus, AO-induced-RS, removes the stimulatory effects of ROS, creating a redox disequilibrium which is detrimental to the homeostatic status and normal physiological function of the BEC.
Conclusion. The data of Mentor and Fisher (2017) conclusively shows that the use of excess AOs causes RS, which perturbs the BBB functionality and angiogenic properties, both of which has adverse implications on the regulation of the homeostatic environment of the brain paramecium, while the suppression in cellular proliferation impacts both the maintenance and repair function of capillaries within the brain. Unjudicial treatment with AOs tend to blunt ROS-induced stimulation of normal cellular mechanisms including cell proliferation, permeability and membrane transport and mitochondrial function. The latter implication infers that excess AOs will lead to an impaired response to mechanical-induced injury (e.g., stroke) and pathogenic infection of the BBB, resulting in the subsequent compromised patient recovery.
References
- Baxter PS, Bell KFS, Hasel P, Kaindl AM, Fricker M, Thomson D, Cregan SP, Gillingwater TH, Hardingham GE. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat Commun. 2015;6:6761. doi: 10.1038/ncomms7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haorah J, Knipe B, Leibhart J, Ghorpade A, and Persidsky Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J Leukoc Biol. 2005;78:1223–1232. doi: 10.1189/jlb.0605340. [DOI] [PubMed] [Google Scholar]
- Hermann A, Sitdikova GF, Weiger TM. Oxidative stress and maxi calcium-activated potassium (BK) channels. Biomolecules. 2015;5:1870–1911. doi: 10.3390/biom5031870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamosová D, Juráni M, Greksák M, Nakano M, Vaneková M. Effect of Rooibos tea (Aspalathus linearis) on chick skeletal muscle cell growth in culture. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol. 1997;116:39–45. doi: 10.1016/s0742-8413(96)00138-7. [DOI] [PubMed] [Google Scholar]
- Lepley DM, Pelling JC. Induction of p21/WAFI and G1 cell-cycle arrest by the chemopreventative agent apigenin. Mol Carcinog. 1997;19:74–82. doi: 10.1002/(sici)1098-2744(199707)19:2<74::aid-mc2>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Liemburg-Apers DC, Willems PHGM, Koopman WJH, Grefte S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch Toxicol. 2015;89:1209–1226. doi: 10.1007/s00204-015-1520-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead JJ, McCaffrey G, Quigley CE, Finch J, DeMarco KM, Nametz N, Davis TP. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab. 2010;30:1625–1636. doi: 10.1038/jcbfm.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentor S, Fisher D. Aggressive antioxidant reductive stress impairs brain endothelial cell angiogenesis and blood brain barrier function. Curr Neurovasc Res. 2017;14:71–81. doi: 10.2174/1567202613666161129113950. [DOI] [PubMed] [Google Scholar]
- Poljsak B, Suput D, Milisav I. Achieving the balance between ROS and antioxidants: when to use the synthetic antioxidants. Oxid Med Cell Longev. 2013;2013:956792. doi: 10.1155/2013/956792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh F, Charles A, Schlagowski A, Bouitbir J, Bonifacio A, Piquard F, Krähenbühl, Geny B, Zoll J. Reductive stress impairs myoblasts mitochondrial function and triggers mitochondrial hormesis. Biochim Biophys Acta. 2015;1853:1574–1585. doi: 10.1016/j.bbamcr.2015.03.006. [DOI] [PubMed] [Google Scholar]
- Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]