

Abstract
Background
The current trend of Plasmodium vivax cases imported from Southeast Asia into China has sharply increased recently, especially from the China–Myanmar border (CMB) area. High recombination rates of P. vivax populations associated with varied transmission intensity might cause distinct local selective pressures. The information on the genetic variability of P. vivax in this area is scant. Hence, this study assessed the genetic diversity of P. vivax genome sequence in CMB area and aimed to provide information on the positive selection of new gene loci.
Results
This study reports a genome-wide survey of P. vivax in CMB area, using blood samples from local patients to identify population-specific selective processes. The result showed that considerable genetic diversity and mean pair-wise divergence among the sequenced P. vivax isolates were higher in some important gene families. Using the standardized integrated haplotype score (|iHS|) for all SNPs in chromosomal regions with SNPs above the top 1% distribution, it was observed that the top score locus involved 356 genes and most of them are associated with red blood cell invasion and immune evasion. The XP-EHH test was also applied and some important genes associated with anti-malarial drug resistance were observed in high positive scores list. This result suggests that P. vivax in CMB area is facing more pressure to survive than any other region and this has led to the strong positive selection of genes that are associated with host-parasite interactions.
Conclusions
This study suggests that greater genetic diversity in P. vivax from CMB area and positive selection signals in invasion and drug resistance genes are consistent with the history of drug use during malaria elimination programme in CMB area. Furthermore, this result also demonstrates that haplotype-based detecting selection can assist the genome-wide methods to identify the determinants of P. vivax diversity.
Electronic supplementary material
The online version of this article (doi:10.1186/s12936-017-1882-0) contains supplementary material, which is available to authorized users.
Keywords: Plasmodium vivax, Haplotype-based detecting, Positive selection, Invasion, Immune evasion, Drug resistance
Background
The Greater Mekong Sub-region is one of the most threatened foci of malaria in Southeast Asia [1, 2] because it accounts for more than half of the malaria cases reported in the region and an estimated 75% of malaria deaths occurred in Myanmar [3]. Moreover, China and Myanmar border region have the highest malaria incidence among the international border regions in Asia [1, 4]. Case management of imported malaria within the context of malaria pre-elimination is increasingly considered to be relevant because of the risk of resurgence [5, 6]. The genetic diversity and evolutionary plasticity of Plasmodium vivax constitute major obstacles for malaria elimination. Several P. vivax isolates genome sequencing projects were completed recently [7, 8], but information is scant about the genetic variability in China–Myanmar border (CMB) area. The lack of reliable and adequate information on the genetic variability of P. vivax genome creates knowledge gap and uncertainty about the preventive strategies for effective malaria control.
The development in recent years has transformed Plasmodium genome sequencing from a complicated task into a well-defined set of procedures and pipelines that are potentially accessible to all researchers [9]. These pipelines provide a deep understanding on the intricacies of parasite population, generate reliable information to enhance monitoring their effects and to raise alert for action in case of emergency [10]. Though, several methods are available but haplotype-based approaches are particularly useful for identifying variants that have undergone a partial or incomplete selective sweep by using metrics that probe such reduced haplotype diversity [11, 12]. The selective sweep results in the rapid rise in frequency of beneficial alleles accompanied by a reduction in haplotype diversity in the neighbourhood of functional mutations due to a hitching effect.
Previous studies on Plasmodium falciparum adapted the haplotype-based XP-EHH (cross-population extended haplotype homozygosity) test on parasites from Senegal and detected that several loci are associated with drug resistance genes, including some known signals at chloroquine resistance transporter gene (crt), bifunctional dihydrofolate reductase gene (dhfr) and multidrug resistance protein 1 gene (mdr1) [13]. Mobegi et al. reported a genome-wide survey in Guinea for positive selection from the standardized integrated haplotype test and identified ten chromosomal loci that had two or more SNPs with high |iHS| score (top 1% of the distribution). It revealed strong signatures around the two major chloroquine resistance genes (crt and mdr1) and weak signatures around the sulfadoxine resistance gene (dhps) [14]. Similar methods were applied to analyse the genomes of P. vivax parasites collected from patients in CMB area and the positive selection signatures on genes associated with invasion, immune evasion and drug resistance were observed. These results showed that the genetic diversity is similar to the global scale even in such small area. The mean pairwise divergence among the sequenced P. vivax isolates is higher in gene families associated with red blood cell invasion and immune evasion. This suggests that P. vivax in CMB area is facing more pressure to survive than any other region.
It is important to identify molecular markers of drug resistance for improvement on drug resistance surveillance and prevention of complications that arises from inadequate therapies to be achieved. Therefore, this study identified sign of hard selective sweep involved in drug resistance genes that are solely consistent with the history of drug use during the national malaria elimination programme in CMB area. The identification of these signatures of positive selection could help us to identify drug resistance genes as well as new vaccine candidates.
Methods
Ethics statement
The study was approved by the Ethics Committee of the National Institute of Parasitic Diseases (NIPD), China CDC. The study protocol, potential risks and potential benefits were explained to the participants. After agreement by the participants to be recruited into the study, the informed consent to participate was given and all the participants provided written informed consent.
Collection of genomic data
Genome data previously published from seven monkey adapted strains: Sal I [15], Belem [16], Chesson [17], Brazil-I, India-VII, North Korean, Peru [18] and Mauritania-I [19] were used for the analyses. Meanwhile, six human clinical isolates genome were referenced: Cambodia (C08, C15, and C127) and Madagascar (M08, M15, M19) [20]. Raw sequences of the strains deposited in the GenBank database under the following SRR number were downloaded, these include; (Sal I resequencing: SRR575089, Madagascar: SRR570031, SRR828416, SRR572651, Cambodia: SRR572648, SRR572650, SRR572649, Brazil: SRR332573, SRR332569, IQ07: SRR064844, SRR073125, India VII: SRR332913, SRR332914, North Korea: SRR332565, SRR332562, Mauritania I: SRR332413, SRR332408, Belem: SRR575087 and Chesson: SRR828528). More so, this study employed recently released genotype calls data set (Variant Call Format file) with several countries throughout the world: Cambodia, China, India, Indonesia, Laos, Malaysia, Myanmar, Thailand, Vietnam and Papua New Guinea [21]. The SNP information and allele frequencies were downloaded from the P. vivax Genome Variation Project [8]. In addition, the annotation of the Sal I reference from PlasmoDB database was downloaded [22].
Sampling Plasmodium vivax parasites from malaria patients and genome sequencing
The blood sample were collected from six clinical malaria cases that were microscopically positive for P. vivax with high parasite density (40,000–260,000 parasites/μl) from Tengchong county, an area of China–Myanmar border of Yunnan province in 2010. The samples were confirmed P. vivax mono-species infection by Plasmodium species PCR-based diagnosis [6]. Genomic DNA was extracted from each frozen blood sample using the QIAGEN DNeasy Blood & Tissue Kit (Qiagen, UK), and sheared into 500 bp fragments to construct the Illumina sequencing libraries with insert sizes of 250 bp. Previously, an initial sequencing result of sample CMB-1 and subsequent analysis was reported [23]. Using the same method, all libraries on Illumina HiSeq2000 were sequenced and generated an average of 120 M paired-end reads of 125 bp. All Illumina raw sequencing reads have been submitted to the NCBI Short Read Archive (BioProject no. PRJNA284437). All reads were filtered by removing the adapter sequences and low quality sequences with Trimmomatic-3.0 [24].
Identification of SNPs from Plasmodium vivax parasites
Sequencing reads from 25 samples (6 samples from CMB area and 19 reference samples) were mapped to P. vivax Sal I genome using BWA [25] with default parameters and SNPs called using SAMTOOLS [26]. High-quality single nucleotide polymorphism (SNPs) that met the following criteria were obtained: (1) quality scores >30; (2) not at the extremes of the genomic coverage distribution (<5- or >1000-fold), which normally reflect deletions or copy number variants. Due to the variety of quality in the reference and the CMB samples, the SNPs on 14 chromosomes were retained and the rest was excluded. SNPs were also excluded from analysis if they were positioned within sub-telomeric regions or the repetitive sequences. A total of 188,757 SNPs remained for analysis after filtering. Then, the distribution of high-quality SNPs in each gene was calculated using an in-house Perl script, and a principal component analysis (PCA) of all strains was performed using all the SNPs identified. The majority allele within each infection was identified for use in the analysis of population allele frequencies. In the subsequent analysis, the SNP dataset was further filtered to exclude samples with missing calls at >5% of all positions.
Positive selection tests
For the high-quality SNPs in the CMB area population, the nucleotide diversity () and the Watterson’s estimator ( ω) were estimated for the whole genome mutation rate in 2 kb slide windows across each chromosome in ARLEQUIN-Ver3.5 [27]. Integrated haplotype score (iHS) and cross-population extended haplotype homozygosity (XP-EHH) in Selscan-Ver1.10a were used to detect signals of recent or ongoing positive selection [28]. These statistical analyses are based on the selective sweep model, where a mutation arises on a haplotype that quickly sweeps toward fixation and reduces diversity around the locus.
iHS is the standardized log ratio of the integrated extended-haplotype homozygosity (EHH) [29], which calculated the six CMB clinical samples by tracking the decay of haplotype homozygosity for both the ancestral and derived haplotypes extending from every SNP site [30]. SNPs with inferred ancestral states and a minor allele frequency of at least 5% were used for iHS. Each unstandardized scores were normalized in frequency bins across the entire genome. During the EHH computation of each SNP loci, if the start/end of a chromosome arm is reached before EHH < 0.05 or if a physical distance (kbp) between two markers >200 is encountered, the calculation is aborted. XP-EHH is the standardized log ratio of the integrated site-specific EHH at core SNPs between populations A and B, which is defined in this study as CMB samples and the reference samples from all over the world respectively [11]. Site-specific EHH does not require markers to be polymorphic within the population. Therefore, it can detect selective sweeps for alleles that have risen to fixation. In this calculation, the sums in each locus were truncated at the SNP with EHH value <0.05 or if the computation extends more than 1 Mbp from the core loci. Previous analyses suggested that iHS has maximum capacity to detect selective sweeps that have reached moderate frequency, while XP-EHH has the capacity to detect selective sweeps at high frequency, thus making the two tests complementary.
Results
Whole genome sequencing of P. vivax parasites and mapping
Previously, a direct sequencing approach which requires only high parasitemia for P. vivax sample without leukocytes filtration was reported. The same method was used to sequence the clinical isolates of P. vivax genome sequence obtained in the China–Myanmar border area. Among hundreds of samples collected from CMB area, only six sequenced results were good enough for the standard and provided enough coverage so far. This study generated between 34 and 215 million paired-end reads with an average read length of 125 bp from each of these samples (Table 1).
Table 1.
Sequencing and mapping summary statistics of six samples from CMB area
| Samples | Pv96 | Pv113 | Pv128 | Pv129 | Pv138 | Pv204 |
|---|---|---|---|---|---|---|
| Sequencing and mapping | ||||||
| Number of reads | 34,817,405 | 215,617,516 | 36,303,847 | 71,449,001 | 36,731,899 | 34,210,120 |
| Mapped on P. vivax | 5,316,739 | 32,851,379 | 4,816,957 | 8,108,382 | 5,962,674 | 10,159,389 |
| Mapped (%) | 15.27 | 15.24 | 13.27 | 11.35 | 16.23 | 29.7 |
| Mean mapping quality | 35.38 | 37.63 | 37.01 | 36.72 | 38.28 | 36.05 |
| Coverage | ||||||
| Coverage fold | 17.32 | 108.72 | 14.55 | 21.16 | 19.79 | 40.41 |
| Genome covered (%) | 93.28 | 97.32 | 93.06 | 91.24 | 95.96 | 94.85 |
| Chromosomes covered (%) | 98.50 | 99.58 | 97.29 | 95.49 | 99.02 | 99.24 |
Sequence reads from the 6 CMB samples and 19 reference isolates were aligned to the P. vivax Sal I reference genome. A variable proportion of reads (11–29%) from all the isolate samples were mapped to the reference. High-quality consensus base calls for an average 94.28% coverage on whole genome and 98.18% on chromosomes were generated for each sample. Despite the presence of some poorly covered regions, the mapped reads allows for comparison between multiple individuals for the 14 chromosomes of the P. vivax genome. Of the 188,757 SNPs, 6 CMB samples involved a total of 109,665 SNPs. Excluding the low-frequency SNPs in each population (55,301 SNPs with minor allele frequency <5%), a total of 133,456 SNPs across the 25 isolates were identified.
Single nucleotide polymorphism were identified across the 4653 genes on 14 chromosomes in CMB samples and 2467 genes with more than 5 SNPs. These genes were considered informative for comparisons of polymorphic nucleotide site frequency spectra for such a comprehensive analyses. CMB samples were observed to have genes with higher SNPs than samples from other parts of the globe in each cluster. This suggests a greater genetic diversity and faster evolution in CMB area (Fig. 1a, b).
Fig. 1.
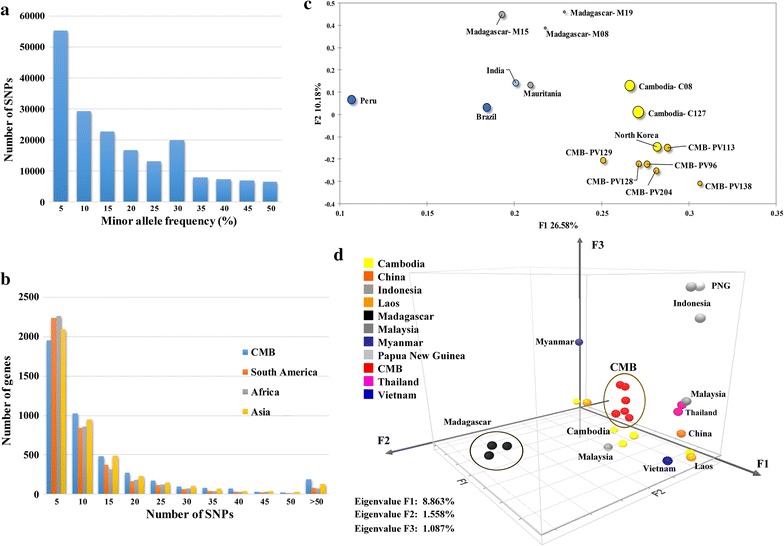
Summary of the SNPs frequency distribution and PCA shows the regional differences. a Frequency distribution of the minor alleles for each of the SNPs scored in a population sample of 6 P. vivax clinical isolates from CMB area. b Distribution numbers of genes (N = 4653 analysed in total) with each given number of SNPs in different population. c Principal component analysis based on 133,456 SNPs in CMB isolates and reference strains. The samples are dyed by their geographic origin: blue for Peru and Brazil from South America, green for India from South Asia and grey for Madagascar and Mauritania from Africa, and yellow for Cambodia, North Korean and CMB. The PCA result shows that P. vivax clustered according to their geographic origin. d Three-eigenvector PCA using variation data from CMB isolates and some Asian reference strains. The clinical isolates outside Asia were used as outgroup indicators: Papua New Guinea (PN0046-C), Indonesia (PJ0007-C and PJ0009-C) and Madagascar (M08, M15 and M19). Each isolate is dyed according to its origin and the percentage contribution of each eigenvector is indicated
Principal component analysis (PCA) of all strains was performed to assess the regional differences. As part of the Asia isolates, the CMB isolates illustrated a higher discrepancies with the Sal I genome (Fig. 1c). PCA result shows that P. vivax clustered generally according to their geographic origin and the host switch in reference was not a major determinant of the genetic diversity. As previously reported [31], the India strain was tagged under the Africa category instead of Asia, and more closely to the South America clades in PCA approach. Another PCA test was performed to explore the Asian population structure of P. vivax (Fig. 1d). The major axis of differentiation clustered the CMB samples together, and divides them from China, Thailand and Vietnam samples. The second and third principal components define a distinct outgroup Madagascar and Papua New Guinea clusters, respectively. It is also important to note that some samples from the same area (Cambodia and Laos) with different periods were also divided in the major axis, suggesting higher diversity from other Asia populations.
Haplotype-based detecting positive selection
This study used integrated haplotype score (iHS) statistic to detect incomplete sweeps and cross-population extended haplotype homozygosity (XP-EHH) in case the sweep is near fixation within population. The two methods are complementary in terms of their scope. Using standardized integrated haplotype score (|iHS|) for all SNPs (MAF > 5%) in the CMB samples, all 14 chromosomal regions were identified with SNPs above the top 5% value of the randomly expected distribution (|iHS| > 4.78), especially the top 1% (|iHS| > 5.93) (see Additional file 1: Table S1 for a complete list of the top 5% score genes). The top 1% SNP locus include 356 gene encoding proteins, most of their families are associated with red blood cell invasion and immune evasion such as merozoite surface protein 1 (MSP1, |iHS| = 6.15), MSP3.1 (|iHS| = 6.35), MSP4 (|iHS| = 6.65), MSP1P (|iHS| = 5.96), tryptophan-rich antigen (PVX_097575, |iHS| = 7.00), rhoptry neck protein 2 (RON2, |iHS| = 6.41), serine-repeat antigen 4 (SERA4, |iHS| = 6.77), merozoite adhesive erythrocytic binding protein (MAEBL, |iHS| = 6.18), reticulocyte binding protein 2b (RBP2b, |iHS| = 6.37), reticulocyte binding protein 2c (RBP2c, |iHS| = 5.80) (Table 2), as well as VIR family, such as variable surface protein 4 (Vir4, |iHS| = 7.31) (Fig. 2). In addition, most of the gene family members shown up in the top 5% list are located close to each other on the chromosome. For example, eight SERA genes are contiguously arranged on chromosome 4, gene PVX_003845 (sera4) involved the top 1% SNP locus (|iHS| = 6.77), others were also in the 5% iHS candidate list. This result is expected because the process of positive natural selection increases the prevalence of both selected variant as well as of nearby variants, generating local regions of extended haplotypes. Elevated |iHS| values were observed in some important individual gene encoding proteins, such as apical membrane antigen 1 (AMA1, |iHS| = 5.15), cysteine-rich protective antigen (CyRPA, |iHS| = 5.23), GPI-anchored micronemal antigen (GAMA, |iHS| = 4.94), and reticulocyte binding protein 2a (RBP2a, |iHS| = 4.79). The positive selection result is similar to results obtained from previous studies [32, 33], and the selection of vaccines targeting polymorphic antigens may explain the hurdle in eliciting cross-protective immune responses.
Table 2.
Notable P. vivax genes with their associated positive selection statistics
| Gene and product description | |iHS| | XP-EHH | |
|---|---|---|---|
| Mosquito transmission | |||
| PVX_095475, circumsporozoite and TRAP-related (CTRP) | 4.9995 | 0.0053 | |
| PVX_118360, TRAP-like protein (TREP) | 6.1642 | 0.0011 | |
| Red blood cell invasion | |||
| PVX_099980, merozoite surface protein 1 (MSP1) | 6.1512 | 0.0185 | |
| PVX_099975, merozoite surface protein 1 paralog (MSP1P) | 5.9582 | 1.9937 | 0.0036 |
| PVX_092975, merozoite adhesive erythrocytic binding (MAEBL) | 6.1795 | 0.0024 | |
| PVX_097575, tryptophan-rich antigen | 6.9579 | 0.0082 | |
| PVX_092275, apical membrane antigen 1 (AMA1) | 5.1501 | 0.0056 | |
| PVX_117880, rhoptry neck protein 2 (RON2) | 6.4102 | 0.0027 | |
| PVX_003845, serine-repeat antigen 4 (SERA4) | 6.7713 | 0.2834 | |
| PVX_121920, reticulocyte binding protein 2a (RBP2a) | 4.7869 | 1.9589 | 0.0004 |
| PVX_081270, reticulocyte binding protein 2b (RBP2b) | 6.3732 | 0.0036 | |
| PVX_090325, reticulocyte binding protein 2c (RBP2c) | 5.7962 | 0.0353 | |
| Drug resistance | |||
| PVX_087980, chloroquine resistance transporter (CRT) | 4.9064 | 0.0024 | |
| PVX_118062, chloroquine resistance marker protein | 7.2833 | 1.8212 | 0.0119 |
| PVX_118100, multidrug resistance protein 2 (MDR2) | 5.3704 | 1.8858 | 0.0093 |
Fig. 2.
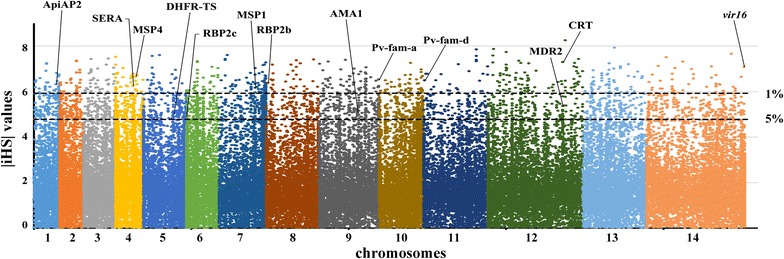
Genome-wide scan of standardized |iHS| for P. vivax SNPs with minor allele frequency of at least 5% in CMB area. Individual chromosomes are identified by alternate colouring of their SNPs, with high scoring SNPs highlighted, indicating loci most likely to have been under recent positive directional selection
As a supplementary approach, XP-EHH test was applied in this study to compare the average haplotype length associated with each SNP between CMB samples and the references from other regions. It identifies areas in the genome where destination samples show much longer haplotypes than the reference, indicative of recent positive selection on the tested population. Some SNP loci were discarded because the physical distance (kbp) between two markers >200 and 80,024 loci were retained. As earlier indicated, the top 1% locus (±0.5%, XP-EHH score >1.7 or <−2.93) were concerned. These SNP loci includes 173 genes, 99 of them were annotated clearly (see Additional file 1: Table S2 for a complete list of the top 1% score genes). Some important gene encoding proteins associated with red blood cell invasion and immune evasion were observed in positive XP-EHH scores list (top 0.5%), such as RBP2a, MSP1P, MSP3, CyRPA and Pv-fam-a (Fig. 3a). The positive selection signals suggest that P. vivax in CMB area are facing more pressure to survive than in any other region. This led to higher ratios of diversity in the genes that are associated with host-parasite interactions. However, the selection signals that are close to some drug resistance genes were less effective when the reference was changed to Asian strains (Fig. 3b). Some genes encoding multidrug resistance protein 2 (MDR2) and chloroquine resistance marker (CRT) were absent on the top list of comparison between CMB and other Asian countries. This suggests that fast evolution in drug resistance genes is a common feature throughout the Southeast Asia region, where the recent selection of genes (dhfr and dhps) resistance to pyrimethamine and sulfadoxine have already been observed [8]. However, when the negative value list of XP-EHH was checked, msp3 and vir families were found in the top list of both comparisons. The result suggests that the CMB samples do not possess such advantageous allele in these gene families, even though the variants were high in the population.
Fig. 3.
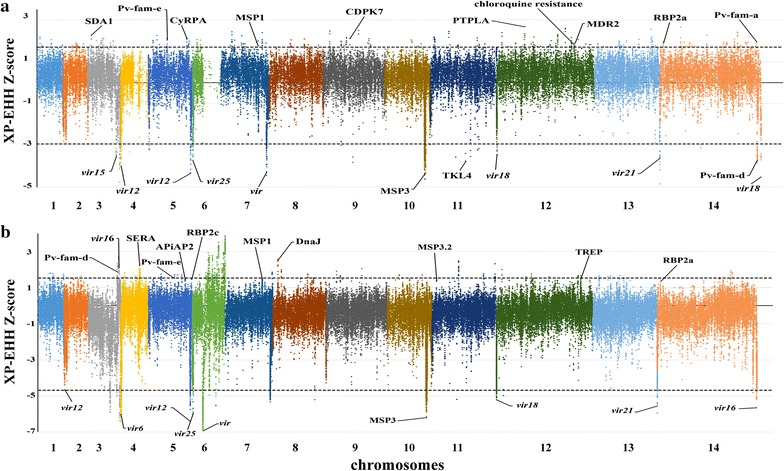
Genome-wide scan of standardized XP-EHH for P. vivax SNPs with minor allele frequency of at least 5% in CMB area. Individual chromosomes are identified by alternate colouring of their SNPs, with high scoring SNPs highlighted, indicating loci most likely to have been under recent positive directional selection. a Using 19 isolates obtained from different continents across the globe as the reference population. Dashed lines indicate the top 1% of XP-EHH values (±0.5%, XP-EHH score >1.7 or <−2.93). b Using 18 isolates from eight Asian countries as the reference population: Cambodia, Malaysia, Thailand, Vietnam, China, Myanmar, Laos, and Indonesia. Dashed lines indicate the top 1% of XP-EHH values (±0.5%, XP-EHH score >1.54 or <−4.68)
Previous studies have claimed that drug resistance is largely driven by positive selection in Plasmodium, and the XP-EHH test can be used to identify areas in the genome where resistant parasites show much longer haplotypes than sensitive parasites, indicating recent positive selection on the resistant population [13]. This study found positive selection signals in 11 drug resistance genes (see Additional file 1: Table S3), including crt (|iHS| = 7.28), mdr2 (|iHS| = 5.37) and bifunctional dihydrofolate reductase-thymidylate synthase gene (dhrf-ts, |iHS| = 5.01) (Table 2). Their higher XP-EHH values indicate that the sweep is young and the signal has not yet been lost through recombination. As earlier noted, the severe changes in XP-EHH values of some drug resistance genes show that the fast evolution is not restricted to CMB area but ubiquitous throughout the Southeast Asia region. In P. falciparum, genes that produce the most abundant proteins in sporozoites have been investigated as vaccine candidates [34, 35]. Also, some of the candidates including circumsporozoite-related antigen and sporozoite invasion-associated protein were found to appear concurrently with higher iHS and XP-EHH scores.
Genetic diversity demonstrate the selective sweep events in CMB samples
The nucleotide diversity () and Watterson’s estimator ( ω) were calculated to find the genome regions with unusually high or low genetic diversity in CMB area. On genome scale, was estimated to be 0.0082 and ω to be 0.0067, and genetic diversity is lower in exonic regions but higher in intronic and intergenic regions (Fig. 4a). Mean pair-wise divergence among the sequenced P. vivax isolates is higher in gene families encoding protein associated with red blood cell invasion and immune evasion (e.g. MSP3, VIR, MSP7, RBP, SERA) (Fig. 4b). On those regions which associated with the invasion genes, numerous genes were found at the centre of the low diversity region. The phenomenon in which functional mutations is surrounded by low diversity is the classic sign of hard selective sweep and accompanying beneficial alleles are rapidly raised. Selective sweep events were checked in the genes that were already chosen in iHS or XP-EHH calculation, and extended homozygosity regions with extremely low polymorphic SNPs were observed.
Fig. 4.
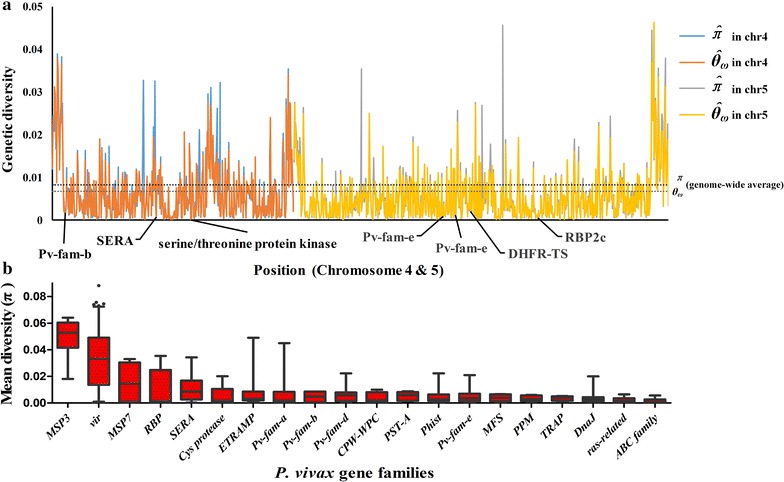
Diversity of P. vivax gene families and examples of selective sweep around some important genes in Chromosome 4 and 5. a Example of genetic diversity (y-axis) calculated in slide windows across Chromosome 4 and 5 (positions in x-axis). Dashed lines represent the genome-wide average of each diversity measure. b Mean pair-wise SNP diversity in P. vivax gene families. Gene families associated with merozoite invasion or immune response modulation show the highest diversity. Red bars on the box plots represent the 25th–75th percentile range, and dots indicate outlier genes
Lastly, higher XP-EHH or |iHS| scores were found on most of those genes which involved in drug resistance in P. vivax, especially the chloroquine resistance marker (PVX_118062) and mdr2 (PVX_118100) (Table 2). These genes are known to confer resistance to chloroquine and pyrimethamine. The ongoing positive selective process suggests that the malaria control programme in Myanmar imposed huge pressure on P. vivax in CMB area and play an important role in the process of diversification.
Discussion
Genetic diversity in CMB area
Sequencing P. vivax genome gave us insights into the parasites’ biology but this has also raised some challenging questions. P. vivax diversity is affected by sampling size, previous work [36] showed the complex geographical pattern of P. vivax variation in Asia where human migration increases local genetic diversity and the Americas due to repeated introduction of P. vivax from many European countries. This study report a genome-wide survey of P. vivax isolates in CMB area, using blood samples from local patients with malaria to identify population-specific selective processes.
The result of this study showed that the genetic diversity estimated from CMB P. vivax isolates ( ω = 0.0067) is higher than the global sample of P. falciparum (where ω is estimated to be 1.03 × 10−3 using isolates from Africa, America, Asia and Oceania) [37]. However, it is still difficult to compare the genetic diversity value to other P. vivax populations, although it is well known that P. vivax is more genetically diverse and less structured than P. falciparum [38]. For example, the ω of P. vivax in Colombia population is estimated to be 7.0 × 10−4 [32] and even less than P. falciparum. Another study using 5.6 kb of non-coding DNA from P. vivax isolates across India reported ω values ranging (from 1.3 × 10−3 to 3 × 10−3) [39]. These values are still lower than the diversity estimates from whole genome of P. vivax isolates from CMB area. Exploring the profile of variation in individual genes and gene families to evaluate the potential functional consequences of the extremely high genomic diversity, it showed that the mean pair-wise divergence among the CMB isolates is highest in gene families associated with red blood cell invasion and immune evasion [21, 40]. The top five families with diversity are genes encoding MSP3, VIR, MSP7, SERA and RBP, which correlates with previous studies [18]. It was reported in a previous study [41] that the genetic diversity in MSP3 ranges from 0.033 (MSP3H) to 0.088 (MSP3E) in Thailand, which is close to the results from CMB isolates (0.018–0.064). The result of this study exhibited large differences in this family, which allow the parasite to invade host cells. Furthermore, studies have reported that the genetic diversity in MSP7 family ranges from 0.0004 (MSP7F) to 0.039 (MSP7E) in Colombia [42]. Compared with result from this study with CMB isolates which ranges from (0.0004 to 0.033), there were close disparity. Study done by [31] found that very few reads could map 130 kb region at the subtelomeric end of chromosome 7. This is as a result of the sharp decline of the GC content along the subtelomeric region and the accompany enrichment of repeated sequences. The presence of artifacts is predicted due to poor coverage in regions with low complexity and this is typical of the vir, sera and msp3 gene families. The early de-novo study with one of these samples (PV113) confirmed that independent deletion event exists but it does not appear in the CMB samples [43]. In conclusion, the CMB isolates show greater genetic diversity than isolates from other parts of the world at whole genome level. This evidence is consistent with their intense transmission level.
Signatures of positive selection
Malaria transmission intensity and parasite genetic diversity are known to vary greatly in different parts of Southeast Asia due to variation in rainfall abundance and seasonality [44]. Since, positive selection is a type of natural selection where environment factors apply constant pressure over generations in favor of specific beneficial trait. Then, it is advantageous to apply selective sweep analyses at population level to study CMB populations because the parasite has a high rate of recombination and gene flow throughout the region. This study has identified parasite loci evidently under distinct processes of selection in a highly endemic population and the positive selection that cause the allele frequency to shift over time mainly in the gene families (Table 2). There are 18 vir, 4 msp and 6 ApiAP2 genes in the top 1% iHS list (|iHS| > 5.93). The sera and rbp genes are also in the top list, with more of their paralog genes in the 5% list. Few genes appeared in the XP-EHH selection candidate list when compared with samples from other continents, but occurred only in individuals, such as: msp1p (PVX_099975), rbp2a (PVX_121920), CyRPA (PVX_090240) and Pv-fam-a (PVX_101515). Previous studies have shown different candidate genes under selection using different approaches. Hupalo et al. [7] explored the genomic profile of divergence and found some loci associated with drug resistance. Hupalo et al. findings are similar to the drug resistance candidates in iHS and XP-EHH test such as crt (PVX_087980) and PI3K (PVX_080480). Strong signals of selection on notable genes were also found in McDonald–Kreitman test, including those associated with red blood cell invasion and with the potentials of adapting to different regions in the host. Also, their list is very close to the prediction of the haplotype-based selection models. These include ama1 (PVX_092275), sera3 (PVX_003840), msp7 (PVX_082650), sera5 (PVX_003830), maebl (PVX_092975) and Pv-fam-a (PVX_092990). More so, using different comparative analysis study [45], Cornejo et al. identified 38 annotated genes with positive selection signature. Despite the application of different methodology, some of the results obtained from their findings were consistent with this study. It includes genes encoding DnaJ (PVX_084650) and helicase (PVX_088190).
Meanwhile, some gene families with high iHS value like sera, PST-A and Phist were totally absent in XP-EHH candidates. Findings show that 8 out of 11 genes in the MSP3 gene family have higher iHS value (top 5%) but lower negative XP-EHH value (bottom 0.5%). The evolution of msp3 should be driven by multi-allelic diversifying selection and this will provide functional redundancy in terms of increasing antigenic diversity [46]. This suggests that the genetic diversity of msp3 depends on sample size, and the CMB isolates would show negative XP-EHH value when compared on larger regional basis. Previous studies show that P. vivax strengthens the invasion and immune evasion capacities to meet the environmental stressors in demand [47]. Finding from this study is sympathetic to this view and exhibits even stronger local characteristics.
Drug resistance of Plasmodium vivax in the China–Myanmar border area
In a recent study of P. falciparum in Southeast Asia, positive selection analysis using haplotype-based method identified loci involved in resistance to chloroquine (crt) in Thailand, Cambodia and Gambia, sulfadoxine–pyrimethamine (dhfr and dhps) in Cambodia, and artemisinin (kelch13) in Cambodia [34]. Decay of these signatures following changes in drug use appears to be rapid, facilitated by the high recombination rate of Plasmodium. In Myanmar, primaquine is used for radical treatment of P. vivax since 1951. Single dose of primaquine is used as gametocidal medicine for P. falciparum since 2002, and artemisinin-based combination therapy (ACT) is free of charge for all ages in public sector. More specifically, the chloroquine and primaquine combination therapy was used for vivax malaria in the China–Myanmar border area in recent years. The results obtained from the application of both iHS and the XP-EHH tests in this study, provide evidence of subtle differences in local selection signatures in the P. vivax isolates from CMB area that are likely to represent ongoing or very recent selection events. This pattern of low diversity that surrounds a fixed substitution is the classic sign of a selective sweep, in which the mutant allele is rapidly fixed by selection. Among the 11 drug resistance genes with positive selection signals, six genes are involved in chloroquine resistance, and three other genes are associated with multidrug resistance. The dihydrofolate reductase gene which confer resistance to pyrimethamine (dhfr-ts, PVX_089950) was also found [48]. A recent study in Peru found haplotypes in drug-resistance genes including dhfr and mdr, suggesting that resistance mutations have arisen independently and might be directly linked to the widespread use of chloroquine, artemisinin and primaquine [49]. Similarly, an abundance of chloroquine and multidrug resistance genes under strong positive selection should be considered as an inevitable consequence of enormous use of drug in Myanmar, where the drugs of choice are still chloroquine and primaquine combination therapy [50].
Plasmodium vivax isolates from CMB area exhibit stronger regional features
Assessing and characterizing the genetic diversity of parasites known to constitute public health burden is important because it provides the platform to assess the direct effects of diversity on clinical disease and also generates reliable information that will help improve therapeutic efficacy and further enhance effective vaccine development [14]. These results further add to the growing evidence that P. vivax populations are genetically diverse. The genetic diversity of P. vivax obtained in this study is similar to findings in other (continents) where they are more transmitted. Also, this study has buttressed previous reports that P. vivax originated from Asia and that human migration increases local diversity [51]. The extreme diversity indicate the persistent synergy and prolonged association of P. vivax with humans in CMB area and this might pose serious challenge to the effective control of malaria cases in the area.
Furthermore, the census of genomic diversity in CMB P. vivax isolates shows high degree of genetic polymorphism, this may translate into important functional variation. The investigation of the evidence for selective sweeps due to drug pressure or other mechanisms was done and exhibited a number of genes with strong signatures of positive selection. It was observed that most of them are associated with red blood cell invasion and immune evasion. Also, some relatively low diversity genes such as Pv-fam-e (PVX_089475), DnaJ (PVX_092765) and PST-A (PVX_118700) also showed positive selection. This suggests that the Indoor-residual-spraying (IRS), larval control plan and treatment complexity subjects CMB’s P. vivax to more pressure for survival.
Conclusion
This study assessed the genetic diversity of P. vivax genome sequence in CMB area to provide information on the positive selection of gene loci involved in red blood cell invasion and immune evasion, as well as drug resistance. It has also given insight into the genetic basis of drug resistance which can be applied to examine genomes using haplotype-based selection detecting methods. The signs of hard selective sweep involved in drug resistance genes were consistent with the history of drug use during national malaria elimination programme in this area, and they have also been identified. Identification of these signatures of positive selection allows for monitoring the emergence of drug resistance in parasite populations.
Authors’ contributions
HS, JC conceived and designed the experiments. SC, YW, BX conducted the experiments. HS, JC analysed the data. SC, EA contributed the reagents/materials/analysis tools. HS, EA, JC drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank the staff of the Yunnan Institute of Parasitic Diseases for collection of the blood samples from P. vivax infected individuals.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data supporting these findings is contained within the manuscript and Additional file 1: Tables S1, S2, S3. All Illumina raw sequencing reads have been submitted to the NCBI Short Read Archive (BioProject No. PRJNA284437).
Ethics approval and consent to participate
The study was approved by the ethics committee at National Institute of Parasitic Diseases, Chinese Center for Disease Control and Prevention.
Funding
This work was supported by the National Research and Development Plan of China (Grant Nos. 2016YFC1200500 and 2016YFC1202000), the fourth round of Three-Year Public Health Action Plan (2015–2017) (Grant No. GWTD2015S06 and GWIV-29), the National Natural Science Foundation of China (Grant No. 81101266). The funding bodies had no role in the design of the study, in collection, analysis, and interpretation of data, or in writing the manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1: Table S1. 485 genes with top 5% integrated haplotype score (|iHS| > 4.78). Table S2. 173 genes with top 1% (±0.5%) cross-population extended haplotype homozygosity value. Table S3. 11 drug resistance genes with high |iHS| and/or XP-EHH value and sweep feature.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12936-017-1882-0) contains supplementary material, which is available to authorized users.
Contributor Information
Hai-Mo Shen, Email: kasumi1979@msn.com.
Shen-Bo Chen, Email: boboanses@126.com.
Yue Wang, Email: wangyuerr@hotmail.com.
Bin Xu, Email: xubin677@hotmail.com.
Eniola Michael Abe, Email: abeeniola11@126.com.
Jun-Hu Chen, Email: junhuchen@hotmail.com, Email: hzjunhuchen@163.com.
References
- 1.Cui L, Yan G, Sattabongkot J, Cao Y, Chen B, Chen X, et al. Malaria in the Greater Mekong Subregion: heterogeneity and complexity. Acta Trop. 2012;121:227–239. doi: 10.1016/j.actatropica.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Delacollette C, D’Souza C, Christophel E, Thimasarn K, Abdur R, Bell D, et al. Malaria trends and challenges in the Greater Mekong Subregion. Southeast Asian J Trop Med Publ Health. 2009;40:674. [PubMed] [Google Scholar]
- 3.Wang Y, Zhong D, Cui L, Lee MC, Yang Z, Yan G, et al. Population dynamics and community structure of Anopheles mosquitoes along the China–Myanmar border. Parasites Vectors. 2015;8:445. doi: 10.1186/s13071-015-1057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen SB, Ju C, Chen JH, Zheng B, Huang F, Xiao N, et al. Operational research needs toward malaria elimination in China. Adv Parasitol. 2014;86:109–133. doi: 10.1016/B978-0-12-800869-0.00005-6. [DOI] [PubMed] [Google Scholar]
- 5.Feng J, Xiao H, Zhang L, Yan H, Feng X, Fang W, et al. The Plasmodium vivax in China: decreased in local cases but increased imported cases from Southeast Asia and Africa. Sci Rep. 2015;5:8847. doi: 10.1038/srep08847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou X, Huang JL, Njuabe MT, Li SG, Chen JH, Zhou XN. A molecular survey of febrile cases in malaria-endemic areas along China–Myanmar border in Yunnan province, People’s Republic of China. Parasite. 2014;21:27. doi: 10.1051/parasite/2014030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hupalo DN, Luo Z, Melnikov A, Sutton PL, Rogov P, Escalante A, et al. Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nat Genet. 2016;48:953–958. doi: 10.1038/ng.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pearson RD, Amato R, Auburn S, Miotto O, Almagro-Garcia J, Amaratunga C, et al. Genomic analysis of local variation and recent evolution in Plasmodium vivax. Nat Genet. 2016;48:959–964. doi: 10.1038/ng.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Volkman SK, Neafsey DE, Schaffner SF, Park DJ, Wirth DF. Harnessing genomics and genome biology to understand malaria biology. Nat Rev Genet. 2012;13:315–328. doi: 10.1038/nrg3187. [DOI] [PubMed] [Google Scholar]
- 10.Kwiatkowski D. Malaria genomics: tracking a diverse and evolving parasite population. Int Health. 2015;7:82–84. doi: 10.1093/inthealth/ihv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amambua-Ngwa A, Park DJ, Volkman SK, Barnes KG, Bei AK, Lukens AK, et al. SNP genotyping identifies new signatures of selection in a deep sample of West African Plasmodium falciparum malaria parasites. Mol Biol Evol. 2012;29:3249–3253. doi: 10.1093/molbev/mss151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park DJ, Lukens AK, Neafsey DE, Schaffner SF, Chang HH, Valim C, et al. Sequence-based association and selection scans identify drug resistance loci in the Plasmodium falciparum malaria parasite. Proc Natl Acad Sci USA. 2012;109:13052–13057. doi: 10.1073/pnas.1210585109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mobegi VA, Duffy CW, Amambua-Ngwa A, Loua KM, Laman E, Nwakanma DC, et al. Genome-wide analysis of selection on the malaria parasite Plasmodium falciparum in West African populations of differing infection endemicity. Mol Biol Evol. 2014;31:1490–1499. doi: 10.1093/molbev/msu106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carlton JM, Adams JH, Silva JC, Bidwell SL, Lorenzi H, Caler E, et al. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature. 2008;455:757–763. doi: 10.1038/nature07327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan ER, Menard D, David PH, Ratsimbasoa A, Kim S, Chim P, et al. Whole genome sequencing of field isolates provides robust characterization of genetic diversity in Plasmodium vivax. PLoS Negl Trop Dis. 2012;6:e1811. doi: 10.1371/journal.pntd.0001811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hester J, Chan ER, Menard D, Mercereau-Puijalon O, Barnwell J, Zimmerman PA, et al. De novo assembly of a field isolate genome reveals novel Plasmodium vivax erythrocyte invasion genes. PLoS Negl Trop Dis. 2013;7:e2569. doi: 10.1371/journal.pntd.0002569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neafsey DE, Galinsky K, Jiang RH, Young L, Sykes SM, Saif S, et al. The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat Genet. 2012;44:1046–1050. doi: 10.1038/ng.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collins WE, Nguyen-Dinh P, Sullivan JS, Morris CL, Galland GG, Richardson BB, et al. Adaptation of a strain of Plasmodium vivax from Mauritania to New World monkeys and anopheline mosquitoes. J Parasitol. 1998;84:619–621. doi: 10.2307/3284734. [DOI] [PubMed] [Google Scholar]
- 20.Menard D, Chan ER, Benedet C, Ratsimbasoa A, Kim S, Chim P, et al. Whole genome sequencing of field isolates reveals a common duplication of the Duffy binding protein gene in Malagasy Plasmodium vivax strains. PLoS Negl Trop Dis. 2013;7:e2489. doi: 10.1371/journal.pntd.0002489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen JH, Chen SB, Wang Y, Ju C, Zhang T, Xu B, et al. An immunomics approach for the analysis of natural antibody responses to Plasmodium vivax infection. Mol BioSyst. 2015;11:2354–2363. doi: 10.1039/C5MB00330J. [DOI] [PubMed] [Google Scholar]
- 22.Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, et al. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 2009;37:D539–D543. doi: 10.1093/nar/gkn814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen HM, Chen SB, Wang Y, Chen JH. Whole-genome sequencing of a Plasmodium vivax isolate from the China–Myanmar border area. Mem Inst Oswaldo Cruz. 2015;110:814–816. doi: 10.1590/0074-02760150216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 28.Szpiech ZA, Hernandez RD. Selscan: an efficient multi-threaded program to perform EHH-based scans for positive selection. Mol Biol Evol. 2014;31:2824–2827. doi: 10.1093/molbev/msu211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, Schaffner SF, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 30.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan ER, Barnwell JW, Zimmerman PA, Serre D. Comparative analysis of field-isolate and monkey-adapted Plasmodium vivax genomes. PLoS Negl Trop Dis. 2015;9:e0003666. doi: 10.1371/journal.pntd.0003666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winter DJ, Pacheco MA, Vallejo AF, Schwartz RS, Arevalo-Herrera M, Herrera S, et al. Whole genome sequencing of field isolates reveals extensive genetic diversity in Plasmodium vivax from Colombia. PLoS Negl Trop Dis. 2015;9:e0004252. doi: 10.1371/journal.pntd.0004252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedrich LR, Popovici J, Kim S, Dysoley L, Zimmerman PA, Menard D, et al. Complexity of infection and genetic diversity in Cambodian Plasmodium vivax. PLoS Negl Trop Dis. 2016;10:e0004526. doi: 10.1371/journal.pntd.0004526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Samad H, Coll F, Preston MD, Ocholla H, Fairhurst RM, Clark TG. Imputation-based population genetics analysis of Plasmodium falciparum malaria parasites. PLoS Genet. 2015;11:e1005131. doi: 10.1371/journal.pgen.1005131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan L, Zhao H, Wu L, Li X, Parker D, Xu S, et al. Plasmodium falciparum populations from northeastern Myanmar display high levels of genetic diversity at multiple antigenic loci. Acta Trop. 2013;125:53–59. doi: 10.1016/j.actatropica.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor JE, Pacheco MA, Bacon DJ, Beg MA, Machado RLD, Fairhurst RM, et al. The evolutionary history of Plasmodium vivax as inferred from mitochondrial genomes: parasite genetic diversity in the Americas. Mol Biol Evol. 2013;30:2050–2064. doi: 10.1093/molbev/mst104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nygaard S, Braunstein A, Malsen G, Van Dongen S, Gardner PP, Krogh A, et al. Long-and short-term selective forces on malaria parasite genomes. PLoS Genet. 2010;6:e1001099. doi: 10.1371/journal.pgen.1001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jennison C, Arnott A, Tessier N, Tavul L, Koepfli C, Felger I, et al. Plasmodium vivax populations are more genetically diverse and less structured than sympatric Plasmodium falciparum populations. PLoS Negl Trop Dis. 2015;9:e0003634. doi: 10.1371/journal.pntd.0003634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta B, Srivastava N, Das A. Inferring the evolutionary history of Indian Plasmodium vivax from population genetic analyses of multilocus nuclear DNA fragments. Mol Ecol. 2012;21(7):1597–1616. doi: 10.1111/j.1365-294X.2012.05480.x. [DOI] [PubMed] [Google Scholar]
- 40.Chen JH, Jung JW, Wang Y, Ha KS, Lu F, Lim CS, et al. Immunoproteomics profiling of blood stage Plasmodium vivax infection by high-throughput screening assays. J Proteome Res. 2010;9:6479–6489. doi: 10.1021/pr100705g. [DOI] [PubMed] [Google Scholar]
- 41.Mascorro C, Zhao K, Khuntirat B, Sattabongkot J, Yan G, Escalante A, et al. Molecular evolution and intragenic recombination of the merozoite surface protein MSP-3α from the malaria parasite Plasmodium vivax in Thailand. Parasitology. 2005;131:25–35. doi: 10.1017/S0031182005007547. [DOI] [PubMed] [Google Scholar]
- 42.Garzón-Ospina D, Forero-Rodríguez J, Patarroyo MA. Heterogeneous genetic diversity pattern in Plasmodium vivax genes encoding merozoite surface proteins (MSP)-7E, -7F and -7L. Malar J. 2014;13:1. doi: 10.1186/1475-2875-13-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen SB, Wang Y, Kassegne K, Xu B, Shen HM, Chen JH. Whole-genome sequencing of a Plasmodium vivax clinical isolate exhibits geographical characteristics and high genetic variation in China–Myanmar border area. BMC Genom. 2017;18:131. doi: 10.1186/s12864-017-3523-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu G, Yan G, Zhang N, Zhong D, Wang Y, He Z, et al. The Anopheles community and the role of Anopheles minimus on malaria transmission on the China–Myanmar border. Parasit Vectors. 2013;6:264. doi: 10.1186/1756-3305-6-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cornejo OE, Fisher D, Escalante AA. Genome-wide patterns of genetic polymorphism and signatures of selection in Plasmodium vivax. Genome Biol Evol. 2015;7:106–119. doi: 10.1093/gbe/evu267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rice BL, Acosta MM, Pacheco MA, Carlton JM, Barnwell JW, Escalante AA. The origin and diversification of the merozoite surface protein 3 (msp3) multi-gene family in Plasmodium vivax and related parasites. Mol Phylogenet Evol. 2014;78:172–184. doi: 10.1016/j.ympev.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hupalo DN, Bradic M, Carlton JM. The impact of genomics on population genetics of parasitic diseases. Curr Opin Microbiol. 2015;23:49–54. doi: 10.1016/j.mib.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chanama M, Chanama S, Shaw PJ, Chitnumsub P, Leartsakulpanich U, Yuthavong Y. Formation of catalytically active cross-species heterodimers of thymidylate synthase from Plasmodium falciparum and Plasmodium vivax. Mol Biol Rep. 2011;38:1029–1037. doi: 10.1007/s11033-010-0199-7. [DOI] [PubMed] [Google Scholar]
- 49.Flannery EL, Wang T, Akbari A, Corey VC, Gunawan F, Bright AT, et al. Next-generation sequencing of Plasmodium vivax patient samples shows evidence of direct evolution in drug-resistance genes. ACS Infect Dis. 2015;1:367–379. doi: 10.1021/acsinfecdis.5b00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gething PW, Elyazar IR, Moyes CL, Smith DL, Battle KE, Guerra CA, et al. A long neglected world malaria map: Plasmodium vivax endemicity in 2010. PLoS Negl Trop Dis. 2012;6:e1814. doi: 10.1371/journal.pntd.0001814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cornejo OE, Escalante AA. The origin and age of Plasmodium vivax. Trends Parasitol. 2006;22:558–563. doi: 10.1016/j.pt.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data supporting these findings is contained within the manuscript and Additional file 1: Tables S1, S2, S3. All Illumina raw sequencing reads have been submitted to the NCBI Short Read Archive (BioProject No. PRJNA284437).