

Abstract
Introduction
Galactosialidosis is a glycoprotein storage disease caused by mutations in the CTSA gene, encoding lysosomal protective protein/cathepsin A (PPCA). The enzyme’s catalytic activity is distinct from its protective function towards β-galactosidase (β-GAL) and neuraminidase 1 (NEU1), with which PPCA forms a complex. In this configuration the two glycosidases acquire their full activity and stability in lysosomes. Deficiency of PPCA results in combined NEU1/β-GAL deficiency. Because of its low incidence, galactosialidosis is considered an orphan disorder with no therapy yet available.
Areas covered
This review gives a historic overview on the discovery of PPCA, which defined galactosialidosis as a new clinical entity; the evidence for the existence of the PPCA/NEU1/β-GAL complex; the clinical forms of galactosialidosis and disease-causing CTSA mutations. Ppca−/− mice have proven to be a suitable model to test different therapeutic approaches, paving the way for the development of clinical trials for patients with galactosialidosis.
Expert opinion
Improved understanding of the molecular bases of disease has sparked renewed incentive from clinicians and scientists alike to develop therapies for rare conditions, like GS, and has increased the willingness of biotech companies to invest in the manufacturing of new therapeutics. Both ERT and gene therapy may become available to patients in the near future.
Keywords: PPCA, CTSA, galactosialidosis, therapy, lysosomal storage disease
1. Galactosialidosis and PPCA: historical aspects
Galactosialidosis (GS) was first classified as a variant of GM1-gangliosidosis, a glycosphingolipid storage disease due to an isolated deficiency of the lysosomal β-GAL 1,2. However, the high residual β-GAL activity measured in GS fibroblasts (~15–20% of control values) could not account for the severe, early onset presentation of the symptoms in patients. Subsequently, the identification by Wenger et al. 3 of undetectable NEU1 activity, in addition to the partial deficiency of β-GAL, in fibroblasts from a patient described as variant of GM1-gangliosidosis defined GS as a separate clinical entity. At that time the disease was thought to be caused by a primary deficiency of NEU1. Yet, co-culturing of fibroblasts with a combined NEU1/β-GAL deficiency with fibroblasts with an isolated NEU1 deficiency partially corrected NEU1 activity in the former cells, raising the possibility that a third gene product was involved in GS. The identity of this “corrective factor” secreted in the medium and capable of restoring NEU1 and β-GAL activities in GS fibroblasts was eventually discovered fortuitously because of its physical association with β-GAL 4,5. An antiserum raised against purified β-GAL precipitated this enzyme with three previously unknown proteins of 54-, 32-, 20-kDa from human fibroblasts 5. Remarkably, these three proteins were absent in fibroblasts of a GS patient with a severe clinical presentation. It was soon apparent that the three proteins were the products of a single gene and that the 54-kDa polypeptide was in fact the uncleaved precursor of the 32- and 20-kDa proteins 5. The 54-kDa precursor was also the form present extracellularly that once taken up by GS cells restored NEU1 and β-GAL activities and their lysosomal stability. These findings gave the first proof that deficiency of a “protective protein” secondarily affected both glycosidases, and reinforced the biochemical evidence that the three proteins physically associate to form a multienzyme complex.
This discovery established GS as a disease distinctive from GM1-gangliosidosis and sialidosis. After the cloning of the protective protein cDNA, Galjart et al. 6 demonstrated that some of the early onset cases of GS lacked the protective protein mRNA and identified the homology of the protein with serine proteases. Independently, the group of Ervin Erdös 7 confirmed that the protective protein was indeed a serine protease with carboxypeptidase/deamidase/esterase activity (see below). Together these findings proved unequivocally the nature of the primary defect in GS.
2. GS: Clinical Phenotypes
GS is a prototypical lysosomal storage disease (LSD) of glycoprotein catabolism, which is inherited as an autosomal recessive trait. The disease is rare, although its prevalence is currently unknown. GS is one of the few LSDs caused by a primary defect in one of the lysosomal cathepsins. However, the secondary severe loss of NEU1 activity probably accounts for most of the overt clinical manifestations seen in patients and for the disease pathogenesis. GS primarily affects cells of the reticuloendothelial system and can be suspected in children with features typical of a lysosomal disorder, such as coarse facies, macular cherry-red spots, vertebral changes, foam cells in the bone marrow, and vacuolated lymphocytes in peripheral blood 8. Sialyloligosacchariduria is diagnostic of the disease but it is indistinguishable from that observed in patients with sialidosis, caused by an isolated deficiency of NEU1 (sialidase). In fact, patients with GS and those with sialidosis share clinical and biochemical features that are attributed at least in part to the loss of NEU1 function in both diseases 8. Based on the age of onset and the severity of the symptoms, patients with GS are usually classified into three clinical types.
2.1 Early Infantile GS
The early infantile type of GS presents with signs of the disease between birth and 3-months of age; these include non-immune hydrops fetalis, neonatal edema, coarse facies, inguinal hernias, proteinuria and telangectasias. In addition, these patients develop visceromegaly, skeletal dysplasia, renal and cardiac failure and variable neurological involvement. Ocular abnormalities, including corneal clouding and fundal changes (cherry-red spot), and heart involvement with cardiomegaly and thickening of the septum have been seen in a number of patients 8,11. These very severe patients die within the first year of life likely because of heart and kidney failure 8. Importantly, early infantile type of GS may be associated with fetal loss.
2.2 Late Infantile GS
The late infantile type of GS comprises a distinct group of patients characterized by mild or absent cognitive disability. Patients usually present with phenotypic alterations and symptoms within the first 2 years of life and they slowly progress into adulthood. These include coarse facies, hepatosplenomegaly, dysostosis multiplex, especially of the spine, and growth retardation associated with muscular atrophy. Heart involvement with thickening of the mitral and aortic valves, and hearing loss are recurrent features. Cherry-red spots and corneal clouding can occur in some patients, while seizures and overt neurologic signs are very rare. Many of the patients diagnosed with the late infantile phenotype survive into adulthood and are still alive. Therefore, they may develop additional kidney and pulmonary complications 8,12.
2.3 Juvenile/Adult
The majority of GS patients belong to the juvenile/adult type. The reason for the dual designation is that their age of onset and clinical course vary greatly and patients present with a broad and continuous spectrum of severity of their symptoms 8,13–15. They are mostly of Japanese origin. In contrast to the late infantile types, patients with the juvenile/adult form of GS have a more severe clinical presentation. Besides characteristic features such as coarse facies, vertebral changes, cherry-red spots and corneal clouding, patients develop severe neurologic manifestations, including myoclonus, cerebellar ataxia, generalized seizures, progressive cognitive impairment and mental retardation. Absence of visceromegaly, angiokeratoma and long survival are features frequently observed in this group of patients 8,16
As it is the case for sialidosis 17, a few examples of atypical patients with confirmed diagnosis of GS but no oligosacchariduria have been reported 18,19. These findings highlight the fact that GS should be taken into consideration for some of the undiagnosed cases, even in the absence of this diagnostic marker of the disease.
2.4 Disease-causing CTSA Mutations
The human CTSA locus is situated on chromosome 20q13.1 in a complex genomic region with two overlapping genes both at the 3′ and 5′ end. Using genomic DNA, cDNA, or whole-exome sequencing a total of 27 mutations have been identified in the CTSA gene, including small deletions/insertions, missense mutations, splicing variants and only one nonsense mutation (refer to http://www.hgmd.cf.ac.uk/ac/index.php and http://www.ncbi.nlm.nih.gov/clinvar) (Table 1). As foreseeable, the most heterogeneous pool of CTSA mutations have been found in patients with the severe, early infantile form of GS, although some recurrent mutations seem to point to a founder effect in specific ethnic groups 20–23. Instead, the group of patients with the late infantile form of the disease is characterized by two recurrent, missense mutations, resulting in two amino acid substitutions, Phe458Val and Tyr267Asn, that appear to be pathognomonic for this clinical phenotype. The extent of severity and type of symptoms in these patients may vary depending on whether these mutations are present in homozygosity or compound heterozygosity 20.
Table 1.
CTSA disease-causing mutations identified in galactosialidosis patients
| Codon number | Type of mutation | Aminoacid change | Reference |
|---|---|---|---|
| 37 | Deletion | C111delG | 24 |
| 67 | Missense | GLN-ARG | 21 |
| 69 | Missense | SER-TYR | 25 |
| 83 | Missense | TRP-ARG | 21 |
| 95 | Deletion | c.284delC | Unidad de Diagnostico y Tratamiento de Errores Congenitos del Metabolismo |
| 103 | Missense | GLY-SER | 26 |
| 103 | Missense | GLY-VAL | 12 |
| 108 | Base substitution | SER-LEU | 21 |
| 116 | Missense | HIS-ARG | 23 |
| 150 | Missense | VAL-MET | 25 |
| 189 | Deletion | c.564delTT | 27 |
| 217 | Deletion | C649delC | 28 |
| 254 | Missense | LEU-PRO | 25 |
| 259 | Missense | CYS-ARG | 23 |
| 267 | Missense | TYR-ASN | 25 |
| 296 | Deletion | C887_888delAT | 25 |
| 347 | Insertion | C904insC | 26 |
| 406 | Nonsense | GLN-STOP | 23 |
| 413 | Missense | TYR-CYS | 21 |
| 424 | Missense | MET-THR | 25 |
| 442 | Missense | ARG-TRP | 12 |
| 457 | Missense | GLY-SER | 25 |
| 458 | Missense | PHE-VAL | 25 |
| 471 | Missense | LYS-GLU | 20 |
| Splicing | IVS3 ds +1 G-T | 24 | |
| Splicing | IVS7 ds +3 A-G | 22 | |
| Splicing | IVS8 ds +9 C-G | 27 |
By far, the majority of CTSA mutations reported in the literature have been identified in Japanese patients 8,20,21. These patients share a common single base substitution at the donor splice site of intron 7, causing aberrant splicing of the pre-mRNA and the skipping of exon 7 (SpDEx7) 22. This mutation apparently segregates with the juvenile/adult type of the disease. However, the patients’ clinical presentation may be more or less severe, depending if it occurs in compound heterozygosity with missense mutations found in early infantile cases, or in homozygosity, although exceptions have been reported also within this group of patients.
Expression and structural studies have examined the impact of different mutations on the biochemical and structural properties of PPCA 25,29 Recently, using an in silico program Caciotti et al. 23 have predicted the effects of specific CTSA mutations on the functional properties of the PPCA protein, proposing this method as a reliable tool for future studies on novel mutations.
In general, we can assert that there is a strong correlation between the type and combination of amino acid substitutions and the clinical outcome. However, we need to keep in mind that other genetic, environmental and dietary factors could play a crucial role in the expression and penetrance of specific clinical manifestations in patients.
3. PPCA biosynthesis
Human PPCA is synthesized as a 480-amino acid pre-proform containing a canonical hydrophobic signal peptide of ~46 residues and nine cysteines. The signal peptide includes a stretch of leucine repeats that can vary in number from individual to individual, which caused discrepancies in the numbering of the PPCA amino-acids; so a careful assessment of any PPCA amino acid sequence is highly recommended in order to ascertain that the correct number is assigned to individual amino acids, especially when mutations identified in GS patients need to be assigned to the protein sequence (see http://www.hgmd.cf.ac.uk/ac/index.php). After removal of the signal peptide, the precursor is N-linked glycosylated at sites Asn163 and Asn351, and forms a homodimer soon after synthesis in the endoplasmic reticulum. The acquisition of the mannose-6-phosphate recognition marker on the Asn117 oligosaccharide chain allows for the correct routing and lysosomal localization of the PPCA precursor. A small pool of phosphorylated precursor can also be recovered extracellularly, but this secreted form can be re-internalized by cells via mannose-6-phosphate receptor-mediated endocytosis and routed to the lysosomes where it becomes catalytically active as the de novo synthesized enzyme 5,6.
In the endosomal/lysosomal compartment the inactive 54 kDa precursor undergoes an initial endoproteolytic cleavage that generates a 34- and 20-kDa inactive, two-chain intermediate. This proteolytic step is followed by the removal of a 2-kDa “linker” or “excision” peptide from the C-terminus of the large chain, which ultimately gives rise to the mature and fully active, two-chain enzyme of 32- and 20-kDa, covalently linked by disulfide bridges 30. Tryptic peptide sequencing of the mature enzyme purified from placenta revealed that the excision peptide encompasses residues Met331-Arg344 6. The structural basis of PPCA maturation and catalytic activation has been revealed through the determination of the 3D structure of the precursor and the mature enzyme 29,31. Maturation of the PPCA precursor can also be recapitulated in vitro by limited proteolysis with trypsin (Figure 1) 6.
Figure 1.
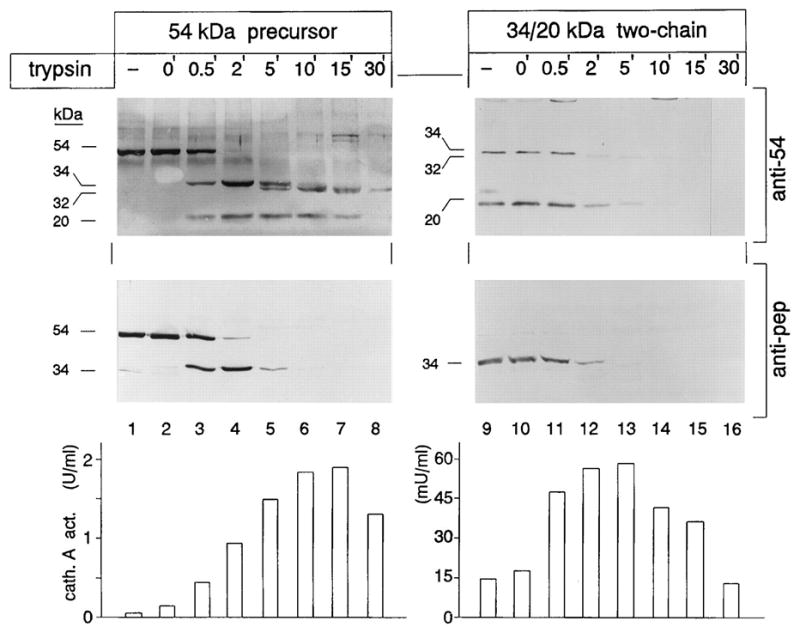
Limited proteolysis with trypsin of 54-kDa precursor and 34- and 20-kDa reconstituted two-chain protein. Aliquots of medium concentrates containing the 54-kDa precursor and 34- and 20-kDa associated protein were incubated at 37 °C with 1 mg of trypsin in the presence of bovine serum albumin (1 mg/ml) for the indicated periods of time. Reactions were stopped with 3 mg of trypsin inhibitor. Samples in lanes 1 and 9 were untreated. At time 0 (lanes 2 and 10), the samples were treated with trypsin inhibitor prior to the addition of trypsin. A portion of each sample was separated by SDS-polyacrylamide gel electrophoresis, followed by electroblotting and immunostaining with anti-54 and anti-pep antibodies. Cathepsin A activity toward the acylated dipeptide benzyloxycarbonyl-phenylalanyl-alanine was measured in each aliquot. One milliunit (mU) of activity is defined as the enzyme activity that releases one nanomole of alanine/minute. Adapted from Bonten et al JBC 1995, 30 with permission of JBC.
Mouse PPCA 32 is 87% identical at the amino-acid level to the human counterpart, and shares similar biochemical and kinetic properties. In fact, murine PPCA can substitute for the human enzyme in correcting both NEU1 and β-GAL activities when taken up by patients’ fibroblasts 32.
4. PPCA - a serine carboxypeptidase identical to cathepsin A
Analysis of the primary structure of human PPCA revealed strong sequence similarity to serine carboxypeptidases, suggesting that the protein had an intrinsic enzymatic activity. By homology with yeast serine carboxypeptidases, the serine in the active site of PPCA was mapped within a domain of six amino acids (GESYA(G)G) found in all serine carboxypeptidases 6. A second conserved domain of four amino acids (HMVP) is located in the 20-kDa chain and includes His475. It was postulated that this histidine and the aspartic acid at position 418 activate the serine in the active site of the protein; hence, the three amino-acids, Ser196, His475 and Asp418, were proposed to make up the catalytic triad of an active PPCA enzyme 29,31.
A serine carboxypeptidase activity for the mature PPCA was first demonstrated in purified preparations of the complex, using a dipeptide substrate Z-Phe-Leu commonly used for the yeast carboxypeptidase CPY 33. These authors named the enzyme carb-L and showed that it was deficient in GS patients 34. However, the biochemical properties of PPCA and its substrate specificity closely correlated with those of a previously identified lysosomal carboxypeptidase, cathepsin A. It was soon demonstrated that indeed PPCA and cathepsin A had the same identity and that an antibody against human PPCA immunoprecipitated a cathepsin A activity that was less than 1% of the control values in GS fibroblasts.
5. PPCA, a pleiotropic enzyme
In spite of its proven cathepsin A activity, the enzyme is still referred to as PPCA because catalytic and protective functions are seemingly independent from one another. PPCA mutants generated by substitution of the Ser196 or His475 active site residues loose cathepsin A activity but still retain their ability to interact and “protect” NEU1 and β-GAL when taken up by GS fibroblasts 35.
In absence of functional PPCA, NEU1 is retained in the endosomal compartment while the pool of β-GAL reaching the lysosomes is rapidly degraded 36. Formation of the PPCA/NEU1/β-GAL complex 5,6,37–41 has a reciprocal advantage for the three enzymes that in such configuration acquire their active and stable conformation in lysosomes 36. However, only a fraction of PPCA and β-GAL activities is found in the complex, which instead contains all of the measurable NEU1 catalytic activity, hinting on potential functions of PPCA outside the multienzyme complex. It is nonetheless genetically and biochemically proven that PPCA functions as an indispensable chaperone/auxiliary transport protein, especially for NEU1, that is catalytically inert without PPCA. The two proteins likely interact already in an early biosynthetic compartment. The mode of their interaction is particularly intriguing because amino acid domains within NEU1, which are important for its interaction with PPCA, also serve as binding sites for other NEU1 molecules. Thus, it appears that PPCA prevents self-aggregation of NEU1 molecules, suggesting that heterodimerization between NEU1 and PPCA helps them to acquire the most suitable conformation for proper intra-organellar routing and full activation/stability in lysosomes 42.
Aside from its cathepsin A activity at acidic pH, PPCA also functions as deamidase and esterase at neutral pH. In vitro, the enzyme can deamidate selected neuropeptides, such as substance P and neurokinin and can function as carboxypeptidase on oxytocin-free acid, bradykinin, and endothelin I 7,43–48. All enzymatic activities of PPCA were shown to be drastically reduced in lymphoblastoid cells and fibroblasts from numerous GS patients of Japanese origin 45. In addition, it was also demonstrated that an enzyme hydrolyzing the C- terminus of endothelin I was deficient in tissues from a GS patient 46, a finding that implicates cathepsin A activity in the degradation of endothelin I in human tissues. A recent study has investigated the impact of cathepsin A upregulation on the murine cardiac proteome, using a mouse model with restricted expression of the protein in cardiomyocytes. Interestingly, increased levels of cathepsin A induced the upregulation of cathepsin B, D and Z, and the downregulation of numerous protease inhibitors, as well as several antioxidative stress proteins 49. The same group has further demonstrated that inhibition of cathepsin A activity has cardioprotective properties and proposed the use of this approach for the treatment of heart failure after myocardial infarction 50.
All together, these data suggest that PPCA has pleiotropic activities that may go beyond its lysosomal functions and, if so, may contribute to the complex phenotype of GS patients. An indication that reinforced this notion came from experiments aimed to purify the lysosomal- associated membrane protein 2a (LAMP2a), one of the isoforms of LAMP2 generated through alternative splicing, which is involved in the process of chaperone-mediated autophagy or CMA 51. CMA entails the chaperone-dependent selection of soluble cytosolic proteins that are destined for degradation in lysosomes. Under specific conditions, mostly associated with cellular stress, cytosolic proteins embedding a specific stretch of amino acids (KFERQ) directly translocate across the lysosomal membrane using LAMP2a as acceptor receptor, and gain access to the lysosomal lumen for degradation. The unique features of this type of autophagy are the selectivity of the protein substrates targeted to lysosomes and their mode of translocation into the organelle that does not require vesicular trafficking 52. All substrates of CMA contain the KFERQ or KFERQ-like target motif that is recognized by the heat shock protein Hsc70, which, in turn, binds to the cytosolic tail of LAMP2a during the translocation across the lysosomal membrane. It was shown that in lysosomal preparations isolated from rat liver PPCA co-purified with LAMP2a, indicating that cathepsin A alone or in complex controls the turnover of LAMP2a 51. In fact, a reduction in the rate of LAMP2a degradation was demonstrated in both human and mouse PPCA-deficient fibroblasts, which in contrast displayed increased levels of CMA 51.
6. Mouse models with PPCA deficiency
6.1 PPCA−/− animal model
A mouse homozygous for a null mutation at the Ppca (Ctsa) locus was one of the first genetically engineered animal models for an LSD 53. These mice have a clinical and pathological presentation that closely recapitulates the early onset forms of GS (Figure 2). Tissues and cells isolated from mutant mice have no cathepsin A activity and severe secondary deficiency of Neu1, while β-Gal is partially reduced only in some cell types 53,54. Extensive morphological changes are recognizable already in the first weeks of life, with severe vacuolation and lysosomal expansion in some cells of most systemic organs and the central and peripheral nervous system. As the disease primarily affects the reticuloendothelial system, pathologic changes are first detected in tissues and organs of epithelial origin. Similarly to patients with the severe form of the disease, mutant mice present with severe early-onset nephropathy, associated with edema and proteinuria, in addition to oligosacchariduria. Time-dependent splenomegaly and heart involvement are also characteristic features of the disease. Homozygous knockouts are infertile, because of structural changes in the blood-epididymal barrier, resulting in altered sperm motility 55, and have a reduced lifespan of ~6–9 months, although gender differences have been documented. The complete loss of Neu1 activity explains why many phenotypic abnormalities in Ppca−/− mice are similar to those seen in the Neu1−/− model, although on close examination features that are unique for one or the other disease model have been identified 56. The most overt difference is seen in the cerebellum; early in life Ppca−/− mice acquire acute and progressive ataxia that is associated with regional loss of cerebellar Purkinje cells and impaired motor coordination (Figure 3) 56. This phenotype is not observed in the Neu1−/− mice at least not until the end of their lifespan (5–7 months). Because the expression levels of PPCA in Purkinje cells are greater than those of Neu1, it is possible that these neurons are more sensitive to the loss of cathepsin A activity than of Neu1 activity, but more rigorous testing is needed to corroborate this hypothesis.
Figure 2.
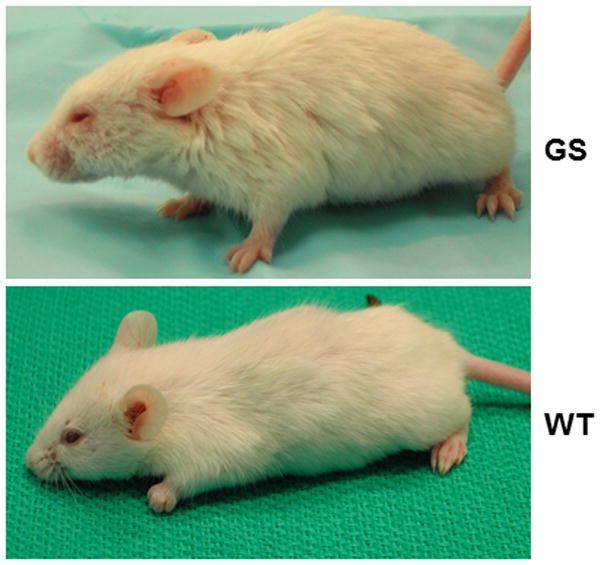
Gross phenotypic appearance of a PPCA −/− mouse at 7 months of age, compared to a wild-type littermate. The affected mouse has a broad face, disheveled coat, and swollen limbs and eyelids. Mouse model generated in Zhou et al 1995 53.
Figure 3.
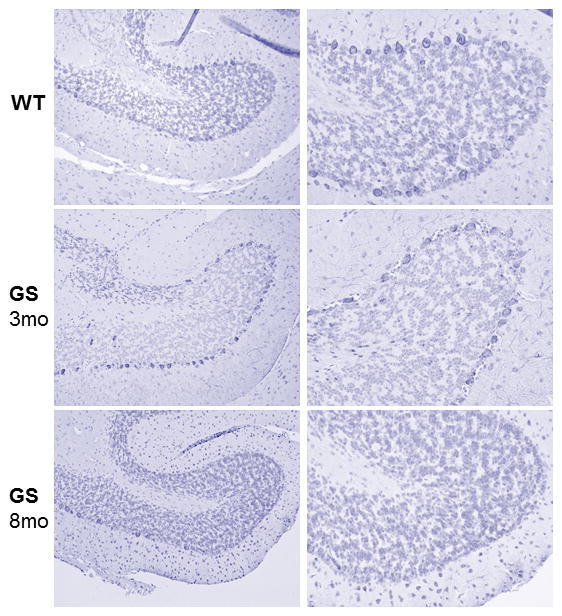
Low magnification images of a cerebellar lobe show progressive loss of Purkinje cells in the PPCA−/− mouse. Sections were immunostained with an antibody against the PEP 19 marker. Size bar 100 μm. Adapted from de Geest et al. Hum Mol Genet 2010, 56 with permission of Oxford Journal.
6.2 CathAS190A-Neo animal model
A knock-in mouse model (CathAS190A-Neo ) was generated, using a targeting construct carrying a point mutation that resulted in serine to alanine amino acid substitution at the catalytic site of the Ppca protein, followed by a PGK-Neo cassette inserted in intron 7 of the Ppca gene 47. Although the mutation targeted the Ppca locus, the CathAS190A-Neo mice displayed a drastically reduced activity of Neu1 in most tissues, due to destabilization of Ppca mRNA by the Neo cassette. Contrary to the Ppca−/− mice, the CathAS190A-Neo mice are apparently vital and fertile, develop normally and have a normal lifespan, suggesting that 10% NEU1 activity is sufficient to promote normal development and growth. Upon removal of the Neo cassette from intron 7, the same authors successfully generated mice (CathAS190A) with an isolated deficiency of cathepsin A, but with intact protective properties toward NEU1; CathAS190A mice have normal Neu1 activity. At the histopathological level these mice show abnormalities in elastin-rich tissues, such as skin, arteries, and lung, and their skin fibroblasts have impaired deposition of insoluble elastin 47. In addition, CathAS190A mice have significantly higher levels of both diastolic and systolic blood pressure than their wild-type siblings. The latter phenotype could be the results of impaired processing of endothelin-1, a potent vasoconstrictive peptide, due to loss of cathepsin A activity. In fact, cultured embryonic brain cells isolated from CathAS190A mice secrete higher levels of endothelin-1 and have reduced degradation rate of endothelin-1 in blood and tissues 47. These results demonstrate that cathepsin A plays an important role in blood pressure regulation and development of the elastic fibers which may influence the complex phenotype observed both in the GS mouse model and GS patients.
7. Investigated and emerging therapies for GS
7.1 Investigated therapies in the GS mouse model
A number of therapeutic approaches have been developed and implemented, using the knockout mouse model of GS. The first experimental therapy was conducted on the newly generated Ppca−/− mice. The strategy, which was very innovative at that time, consisted on transplanting mutant mice with bone marrow isolated from transgenic mice, in which the expression of a human PPCA minigene was driven by the promoter and “locus control region” of the β-globin gene 53. These transgenic mice overexpressed the human PPCA exclusively in their erythroid precursors and secreted it abundantly into the circulation. Remarkably, this secreted precursor was present in sufficient amount in the serum of transgenic mice to correct NEU1 and β-GAL activities when taken up by deficient patients’ fibroblasts 53. Transplantation of Ppca−/− mice with the transgenic bone marrow led to complete reversal of their systemic pathology, normalization of urinary oligosaccharides and increased cathepsin A and Neu1 activities, although correction of the CNS pathology was only partial 53. Treated mice lived longer than affected mice. This unexpectedly positive outcome gave the first indication that bone marrow transplantation (BMT) could be beneficial for GS, provided that a compatible allogeneic donor BM is available.
These initial findings spearheaded the testing of a similar therapeutic approach designed to target more efficiently the brain disease. It was hypothesized that BM-derived monocytes, tissue macrophages and microglia would constitute a more appropriate population of cells to be used as the source of the therapeutic enzyme 57. For this reason, the human colony stimulating factor 1 receptor (CSF-1R) was chosen to drive expression of the human PPCA restricted to the monocyte/macrophage population of transgenic mice 57. Bone marrow isolated from these mice and transplanted into the Ppca−/− mice afforded widespread correction of the systemic phenotype, indicating that cell-specific overexpression of PPCA had sufficient therapeutic potential. Most importantly, 1-year post transplantation in the brain of treated mice numerous PPCA-positive, BM-derived repopulating microglia were likely responsible for the amelioration of the brain morphology and the improved motor coordination 57.
Because of the successful outcome of BMT with PPCA overexpressing BM cells, an ex-vivo BM-mediated gene therapy strategy was later tested for the treatment of GS. Stable hematopoietic progenitors cells (HPCs) isolated from Ppca−/− animals were transduced with a murine stem cell virus (MSCV)-based bicistronic retroviral vector, overexpressing PPCA and the GFP marker, and transplanted back into Ppca−/− mice 58. Transplanted mice showed sustained and long-term expression (up to 10 months) of the transgene, which was accompanied by a marked reduction of disease pathology in all visceral organs (Figure 4) and resolution of the edematous phenotype characteristic of the disease. In the brain, this therapeutic approach resulted in a general improvement of the brain architecture with significant reduction of the number of neural cells containing storage material. In addition, treated mice showed delayed loss of cerebellar Purkinje cells and, in turn, enhanced motor coordination 58.
Figure 4. Histology of systemic organs and cerebellum from BM-transplanted GS mice.
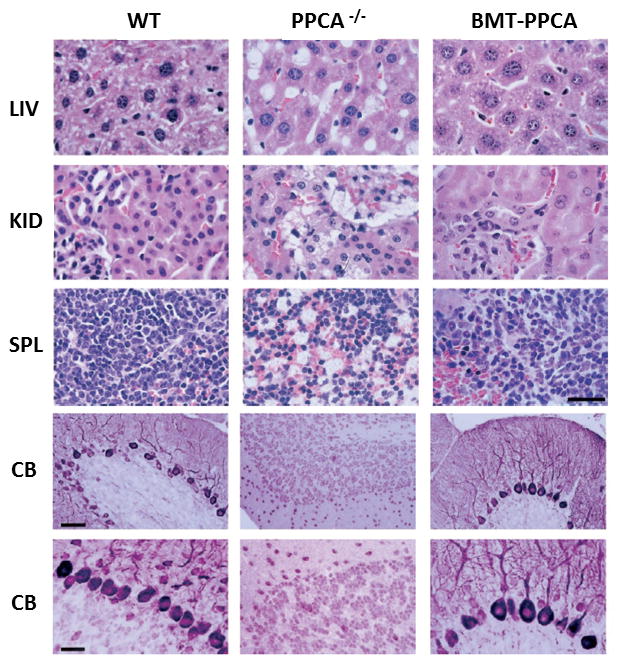
Top panels: Organs from PPCA −/− mice transplanted with total −/− BM transduced with the MSCV-PPCA (BMT-PPCA) retrovirus were isolated at different time points after treatment. Hematoxylin and eosin-stained tissue sections of the liver (LIV), kidney (KID), and spleen (SP) from a BMT-PPCA–treated PPCA −/− mouse sacrificed 9 months after treatment, and from age-matched wild-type and PPCA −/− mice revealed the complete restoration of normal tissue morphology with BM expressing PPCA, compared to the extensive vacuolation present in the PPCA −/− control mouse. Size bar corresponds to 30 μm. Lower panels: Serial sections of the cerebellum from a 9-month-old GS mouse transplanted with MSCV-PPCA–marked BM cells were stained with anti-PEP19 antibody. Note the dramatic loss of Purkinje cells in an age-matched GS mouse and the significant number of these cells that are retained in the treated animal. Size bars correspond to 60 μm and 30 μm Adapted from Leimig et al 2002 Blood, 58 with permission of the American Society of Hematology.
Together these results suggest that BM-mediated ex vivo gene therapy could be sought for the treatment of GS.
The beneficial therapeutic effect of targeting monocyte/macrophage cells for reverting or ameliorating the systemic disease in Ppca−/− instigated the testing of enzyme replacement therapy (ERT), addressing specifically monocyte/macrophage population of cells, one of the primary affected cells in GS. For this approach a recombinant human PPCA was generated in insect cells using a baculovirus expression system 59. Glycoproteins expressed in insect cells carry truncated trimannosyl core glycans, which can bind to cells, such as macrophages, that express the mannose receptor. Insect cell-produced recombinant enzyme can be used effectively to treat LSDs with primary involvement of the reticuloendothelial system like GS. Intravenous administration of either PPCA alone or a combination of PPCA and NEU1, 1 month old Ppca mice resulted in complete correction of the systemic phenotype. Two weeks after ERT, treated mice showed normalized cathepsin A activity in many of the systemic organs and reduction of lysosomal storage (Figure 5) 59.
Figure 5. Immunostaining of tissue sections from mice after dual-ERT.
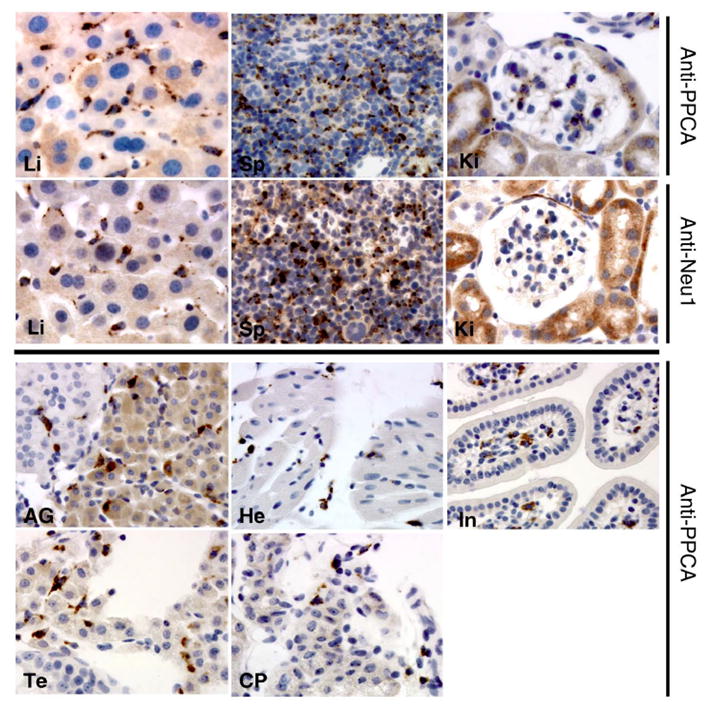
A) Numerous cells expressing both PPCA and Neu1 were detected with anti-PPCA and anti-Neu1 antibodies in liver (Li), spleen (Sp), and kidney (Ki). B) Cells expressing only PPCA were detected in adrenal gland (AG), heart (He), intestine (In), testis (Te), and choroid plexus (CP). Punctuated staining demonstrates internalization of corrective enzyme mostly by Kuppfer cells in the liver and resident macrophages in other tissues. Magnification = x400 Adapted from Bonten et al. Faseb Journal 2004, 59 with permission of FASEB.
7.2 Emerging therapies and preclinical trials
In recent years, a preclinical study was designed for the treatment of GS, targeting specifically the late infantile group of patients with the non-neuropathic form of the disease. The strategy was based on the use of a recombinant adeno associated viral (rAAV) vector expressing human PPCA under the control of a liver-specific promoter (scAAV2/8LP1PPCA) 60. The assumption was that overexpression of the enzyme restricted to the liver would minimize potential toxicity and still afford correction of systemic disease via sustained secretion of the precursor protein into the circulation followed by its uptake by cells of neighboring or distant organs. A large cohort of 30 day-old Ppca−/− mice was tail vain injected with increasing doses (low dose, intermediate dose and high dose) of the rAAV and analyzed 4-month after treatment. Regardless of the viral dose used, treated mice appeared indistinguishable from WT mice; they had normal fur, coordination and gait, moved actively and were no longer edematous. Interestingly, treated mice showed reversal of their infertility because they were able to produce normal size litters. Immunohistochemical and biochemical analysis of tissues showed that the levels of PPCA expression in treated mice correlated well with the dose of administered virus. Increased expression of PPCA was paralleled by rescue of the endogenous activity of Neu1 and normalization of urinary oligosaccharides. The rescue of PPCA and Neu1 activities resulted in a dramatic improvement of tissue architecture in all visceral organs tested. Remarkably, mice injected with intermediate or high dose of rAAV showed normal PPCA expression in the choroid plexus, accompanied by clearance of lysosomal storage 60. No adverse side effects have been identified and no difference in heart and lung morphology has been observed. So far, all investigative and preclinical therapeutic approaches applied to the GS mouse model have been extremely successful and have encouraged the development of an AAV-mediated gene therapy for the treatment of the mild late infantile group of GS patients with no neuropathic signs. These findings also hold promise for the treatment of GS with an ERT regimen.
8. Expert opinion
PPCA is an unusual lysosomal enzyme, which functions as a molecular chaperone for NEU1 and β-GAL, while maintaining an intrinsic carboxypeptidase/deamidase/esterase activity towards a selected number of bioactive peptides. Its differential tissue distribution and level of expression in different cell types likely reflect the physiological need for the protein to either complex with the two glycosidases or to remain a free dimer, depending on the range and type of substrates to be degraded or processed. Obviously, the availability of pools of free and assembled enzymes that are committed to diverse functions and potentially be differentially regulated creates a highly dynamic degradative/processing system. Although, based on current evidence, it is the PPCA’s protective function, not its enzymatic activity, at the basis of lysosomal dysfunction in GS patients, the possibility remains that some of the patients’ clinical symptoms and complications are related to the loss of cathepsin A activity. Thus, in spite of recent advancements in the field, a full understanding of PPCA’s pleiotropic functions awaits the identification of the complete range of substrates that are the physiological targets of this enzyme in vivo and could be exploited as prognostic or diagnostic biomarkers.
The availability of a faithful animal model for GS has been instrumental for testing investigative therapies that have been overall very successful in correcting the clinical manifestations characteristic of the disease in patients, including partial reversal of the brain pathology. The favorable outcome of these preclinical studies supports the notion that glycoprotein storage diseases, like GS, are particularly responsive to different types of treatment from ERT to gene therapy. In addition, the biochemical characteristics of PPCA, its long half life as secreted inactive precursor (zymogen) in circulation, its efficient internalization from cells at multiple sites and its scalable purification make it an extremely versatile therapeutic product. Based on these considerations and the renewed interest of pharmaceutical companies to develop therapy for orphan diseases, it is foreseeable that a clinical therapeutic protocol for GS will become available in the near future with a good chance of being successful.
Article highlights.
History of the discovery of galactosialidosis primary defect
Galactosialidosis clinical phenotypes and CTSA mutations
Protective protein cathepsin A as a multifunctional enzyme
Mouse models with CTSA deficiency
Experimental and preclinical therapies in galactosialidosis
Acknowledgments
We apologize if we have omitted some of the outstanding contributions to this field of research because of space constraints.
Funding
This work was funded in part by NIH [grants RO1DK095169 and NIH RO1GM104981], the Assisi Foundation of Memphis, Ultragenyx Pharmaceutical and the American Lebanese Syrian Associated Charities (ALSAC).
Abbreviations
- NEU1
Neuraminidase 1
- PPCA
Protective protein cathesin A
- β-GAL
β-galactosidase
- GS
galactosialidosis
- LSD
lysosomal storage disease
- BMT
bone marrow transplantation
- ERT
enzyme replacement therapy
- rAAV
recombinant adeno associated virus
Footnotes
Declaration of interest
A.d’A. holds the Jewelers for Children Endowed Chair in Genetics and Gene Therapy. The authors have no other relevant affiliations or financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Derry DM, Fawcett JS, Andermann F, et al. Late infantile systemic lipidosis. Major monosialogangliosidosis. Delineation of two types. Neurology. 1968 Apr;18(4):340–348. doi: 10.1212/wnl.18.4.340. [DOI] [PubMed] [Google Scholar]
- 2.Wenger DA, Goodman SI, Myers GG. Letter: beta-galactosidase deficiency in young adults. Lancet. 1974 Nov 30;2(7892):1319–1320. doi: 10.1016/s0140-6736(74)90173-1. [DOI] [PubMed] [Google Scholar]
- 3.Wenger DA, Tarby TJ, Wharton C. Macular cherry-red spots and myoclonus with dementia: coexistent neuraminidase and beta galactosidase deficiencies. Biochem Biophys Res Commun. 1978 May 30;82(2):589–595. doi: 10.1016/0006-291x(78)90915-4. [DOI] [PubMed] [Google Scholar]
- 4.Hoogeveen A, d’Azzo A, Brossmer R, et al. Correction of combined beta-galactosidase/neuraminidase deficiency in human fibroblasts. Biochem Biophys Res Commun. 1981 Nov 16;103(1):292–300. doi: 10.1016/0006-291x(81)91692-2. [DOI] [PubMed] [Google Scholar]
- 5•.D’Azzo A, Hoogeveen A, Reuser AJ, et al. Molecular defect in combined beta-galactosidase and neuraminidase deficiency in man. Proc Natl Acad Sci USA. 1982 Aug;79(15):4535–4539. doi: 10.1073/pnas.79.15.4535. This is the first report referring a combined deficiency of beta galactosidase and neuraminidase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6••.Galjart NJ, Gillemans N, Harris A, et al. Expression of cDNA encoding the human “protective protein” associated with lysosomal beta galactosidase and neuraminidase: homology to yeast proteases. Cell. 1988 Sep 9;54(6):755–764. doi: 10.1016/s0092-8674(88)90999-3. This paper describes the cloning of the human protective protein/Cathepsin A. [DOI] [PubMed] [Google Scholar]
- 7•.Jackman HL, Tan FL, Tamei H, et al. A peptidase in human platelets that deamidates tachykinins. Probable identity with the lysosomal “protective protein”. J Biol Chem. 1990 Jul 5;265(19):11265–11272. This paper describes the deamidase function of Cathepsin A. [PubMed] [Google Scholar]
- 8•.d’Azzo A, Andria G, Bonten E, et al. The online metabolic & molecular bases of inherited diseases. New York (NY): McGraw-Hill Publishing Co; 2013. This chapter includes the most up to date information on the clinical, molecular and biochemical aspects of Galactosialidosis. [Google Scholar]
- 9.Zammarchi E, Donati MA, Morrone A, et al. Early-infantile galactosialidosis: clinical, biochemical, and molecular observations in a new patient. Am J Med Genet. 1996 Aug 23;64(3):453–458. doi: 10.1002/(SICI)1096-8628(19960823)64:3<453::AID-AJMG2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 10.Gravel RA, Lowden JA, Callahan JW, et al. Infantile sialidosis: a phenocopy of type 1 GM1 gangliosidosis distinguished by genetic complementation and urinary oligosaccharides. Am J Hum Genet. 1979 Nov;31(6):669–679. [PMC free article] [PubMed] [Google Scholar]
- 11.Sewell AC, Pontz BF, Weitzel D, et al. Clinical heterogeneity in infantile galactosialidosis. Eur J Pediatr. 1987 Sep;146(5):528–531. doi: 10.1007/BF00441610. [DOI] [PubMed] [Google Scholar]
- 12.Kiss A, Zen PR, Bittencourt V, et al. A Brazilian galactosialidosis patient given renal transplantation: a case report. J Inherit Metab Dis. 2008 Dec;31(Suppl 2):S205–8. doi: 10.1007/s10545-008-0730-3. [DOI] [PubMed] [Google Scholar]
- 13.Mochizuki A, Motoyoshi Y, Takeuchi M, et al. A case of adult type galactosialidosis with involvement of peripheral nerves. J Neurol. 2000 Sep;247(9):708–710. doi: 10.1007/s004150070116. [DOI] [PubMed] [Google Scholar]
- 14.Lehman A, Mattman A, Sin D, et al. Emphysema in an adult with galactosialidosis linked to a defect in primary elastic fiber assembly. Mol Genet Metab. 2012 May;106(1):99–103. doi: 10.1016/j.ymgme.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Usui T, Abe H, Takagi M, et al. Electroretinogram and visual evoked potential in two siblings with adult form galactosialidosis. Metab Pediatr Syst Ophthalmol (1985) 1993;16(1–2):19–22. [PubMed] [Google Scholar]
- 16.d’Azzo A, Kolodny EH, Bonten E, et al. Storage disease of the reticuloendothelial system. 7. New York (NY): Nathan and Oski’s; 2009. [Google Scholar]
- 17.Canafoglia L, Robbiano A, Pareyson D, et al. Expanding sialidosis spectrum by genome-wide screening: NEU1 mutations in adult onset myoclonus. Neurology. 2014 Jun 3;82(22):2003–2006. doi: 10.1212/WNL.0000000000000482. [DOI] [PubMed] [Google Scholar]
- 18.Prada CE, Gonzaga-Jauregui C, Tannenbaum R, et al. Clinical utility of whole-exome sequencing in rare diseases: galactosialidosis. Eur J Med Genet. 2014 Jul;57(7):339–344. doi: 10.1016/j.ejmg.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darin N, Kyllerman M, Hard AL, et al. Juvenile galactosialidosis with attacks of neuropathic pain and absence of sialyl-oligosacchariduria. Eur J Paediatr Neurol. 2009 Nov;13(6):553–555. doi: 10.1016/j.ejpn.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Takiguchi K, Itoh K, Shimmoto M, et al. Structural and functional study of K453E mutant protective protein/cathepsin A causing the late infantile form of galactosialidosis. J Hum Genet. 2000;45(4):200–206. doi: 10.1007/s100380070027. [DOI] [PubMed] [Google Scholar]
- 21.Shimmoto M, Fukuhara Y, Itoh K, et al. Protective protein gene mutations in galactosialidosis. J Clin Invest. 1993 Jun;91(6):2393–2398. doi: 10.1172/JCI116472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimmoto M, Takano T, Fukuhara Y, et al. Japanese-type adult galactosialidosis a unique and common splice junction mutation causing exon skipping in the protective protein/carboxypeptidase gene. Proc Jpn Acad. 1990;66B(10):217–222. [Google Scholar]
- 23•.Caciotti A, Catarzi S, Tonin R, et al. Galactosialidosis: review and analysis of CTSA gene mutations. Orphanet J Rare Dis. 2013;8:114. doi: 10.1186/1750-1172-8-114. This is a recent comprehensive review of mutations occurring in Galactosialidosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malvagia S, Morrone A, Caciotti A, et al. New mutations in the PPBG gene lead to loss of PPCA protein which affects the level of the beta-galactosidase/neuraminidase complex and the EBP-receptor. Mol Genet Metab. 2004 May;82(1):48–55. doi: 10.1016/j.ymgme.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 25.Zhou XY, Van Der Spoel A, Rottier R, et al. Molecular and biochemical analysis of protective protein/cathepsin A mutations: correlation with clinical severity in galactosialidosis. Hum Mol Genet. 1996 Dec;5(12):1977–1987. doi: 10.1093/hmg/5.12.1977. [DOI] [PubMed] [Google Scholar]
- 26.Groener J, Maaswinkel-Mooy P, Smit V, et al. New mutations in two Dutch patients with early infantile galactosialidosis. Mol Genet Metab. 2003 Mar;78(3):222–228. doi: 10.1016/s1096-7192(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 27.Richard C, Tranchemontagne J, Elsliger MA, et al. Molecular pathology of galactosialidosis in a patient affected with two new frameshift mutations in the cathepsin A/protective protein gene. Hum Mutat. 1998;11(6):461–469. doi: 10.1002/(SICI)1098-1004(1998)11:6<461::AID-HUMU7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 28.Shamseldin HE, Tulbah M, Kurdi W, et al. Identification of embryonic lethal genes in humans by autozygosity mapping and exome sequencing in consanguineous families. Genome Biol. 2015;16:116. doi: 10.1186/s13059-015-0681-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rudenko G, Bonten E, Hol WG, et al. The atomic model of the human protective protein/cathepsin A suggests a structural basis for galactosialidosis. Proc Natl Acad Sci USA. 1998 Jan 20;95(2):621–625. doi: 10.1073/pnas.95.2.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonten EJ, Galjart NJ, Willemsen R, et al. Lysosomal protective protein/cathepsin A: role of the “linker” domain in catalytic activation. J Biol Chem. 1995;270(44):26441–26445. doi: 10.1074/jbc.270.44.26441. [DOI] [PubMed] [Google Scholar]
- 31.Rudenko G, Bonten E, d’Azzo A, et al. Three-dimensional structure of the human ‘protective protein’: structure of the precursor form suggests a complex activation mechanism. Structure. 1995 Nov 15;3(11):1249–1259. doi: 10.1016/s0969-2126(01)00260-x. [DOI] [PubMed] [Google Scholar]
- 32.Galjart NJ, Gillemans N, Meijer D, et al. Mouse “protective protein”. cDNA cloning, sequence comparison, and expression. J Biol Chem. 1990 Mar 15;265(8):4678–4684. [PubMed] [Google Scholar]
- 33.Elsliger MA, Potier M. Homologous modeling of the lysosomal protective protein/carboxypeptidase L: structural and functional implications of mutations identified in galactosialidosis patients. Proteins. 1994 Jan;18(1):81–93. doi: 10.1002/prot.340180110. [DOI] [PubMed] [Google Scholar]
- 34.Tranchemontagne J, Michaud L, Potier M. Deficient lysosomal carboxypeptidase activity in galactosialidosis. Biochem Biophys Res Commun. 1990 Apr 16;168(1):22–29. doi: 10.1016/0006-291x(90)91669-j. [DOI] [PubMed] [Google Scholar]
- 35.Galjart NJ, Morreau H, Willemsen R, et al. Human lysosomal protective protein has cathepsin A-like activity distinct from its protective function. J Biol Chem. 1991 Aug 5;266(22):14754–14762. [PubMed] [Google Scholar]
- 36.Bonten EJ, Annunziata I, d’Azzo A. Lysosomal multienzyme complex: pros and cons of working together. Cell Mol Life Sci. 2014 Jun;71(11):2017–2032. doi: 10.1007/s00018-013-1538-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoogeveen AT, Verheijen FW, Galjaard H. The relation between human lysosomal beta-galactosidase and its protective protein. J Biol Chem. 1983 Oct 25;258(20):12143–12146. [PubMed] [Google Scholar]
- 38.Verheijen F, Brossmer R, Galjaard H. Purification of acid beta-galactosidase and acid neuraminidase from bovine testis: evidence for an enzyme complex. Biochem Biophys Res Commun. 1982 Sep 30;108(2):868–875. doi: 10.1016/0006-291x(82)90911-1. [DOI] [PubMed] [Google Scholar]
- 39.van der Spoel A, Bonten E, d’Azzo A. Transport of human lysosomal neuraminidase to mature lysosomes requires protective protein/ cathepsin. A Embo J. 1998 Mar 16;17(6):1588–1597. doi: 10.1093/emboj/17.6.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pshezhetsky AV, Potier M. Direct affinity purification and supramolecular organization of human lysosomal cathepsin A. Arch Biochem Biophys. 1994 Aug 15;313(1):64–70. doi: 10.1006/abbi.1994.1359. [DOI] [PubMed] [Google Scholar]
- 41.Pshezhetsky AV, Elsliger MA, Vinogradova MV, et al. Human lysosomal beta-galactosidase-cathepsin A complex: definition of the beta-galactosidase-binding interface on cathepsin A. Biochemistry. 1995 Feb 28;34(8):2431–2440. doi: 10.1021/bi00008a005. [DOI] [PubMed] [Google Scholar]
- 42••.Bonten EJ, Campos Y, Zaitsev V, et al. Heterodimerization of the sialidase NEU1 with the chaperone protective protein/cathepsin A prevents its premature oligomerization. J Biol Chem. 2009 Oct 9;284(41):28430–28441. doi: 10.1074/jbc.M109.031419. This paper identifies binding sites on NEU1 and PPCA responsible for their interaction and proposes a novel mechanism of binding between the two proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jackman HL, Morris PW, Deddish PA, et al. Inactivation of endothelin I by deamidase (lysosomal protective protein) J Biol Chem. 1992 Feb 15;267(5):2872–2875. [PubMed] [Google Scholar]
- 44.Hanna WL, Turbov JM, Jackman HL, et al. Dominant chymotrypsin like esterase activity in human lymphocyte granules is mediated by the serine carboxypeptidase called cathepsin A-like protective protein. J Immunol. 1994 Nov 15;153(10):4663–4672. [PubMed] [Google Scholar]
- 45.Kase R, Itoh K, Takiyama N, et al. Galactosialidosis: simultaneous deficiency of esterase, carboxy-terminal deamidase and acid carboxypeptidase activities. Biochem Biophys Res Commun. 1990 Nov 15;172(3):1175–1179. doi: 10.1016/0006-291x(90)91572-a. [DOI] [PubMed] [Google Scholar]
- 46.Itoh K, Kase R, Shimmoto M, et al. Protective protein as an endogenous endothelin degradation enzyme in human tissues. J Biol Chem. 1995 Jan 13;270(2):515–518. doi: 10.1074/jbc.270.2.515. [DOI] [PubMed] [Google Scholar]
- 47.Seyrantepe V, Hinek A, Peng J, et al. Enzymatic activity of lysosomal carboxypeptidase (cathepsin) A is required for proper elastic fiber formation and inactivation of endothelin-1. Circulation. 2008 Apr 15;117(15):1973–1981. doi: 10.1161/CIRCULATIONAHA.107.733212. [DOI] [PubMed] [Google Scholar]
- 48.Pan X, Grigoryeva L, Seyrantepe V, et al. Serine carboxypeptidase SCPEP1 and cathepsin A play complementary roles in regulation of vasoconstriction via inactivation of endothelin-1. Plos Genet. 2014 Feb;10(8):e1004146. doi: 10.1371/journal.pgen.1004146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petrera A, Kern U, Linz D, et al. Proteomic profiling of cardiomyocyte specific cathepsin A overexpression links cathepsin A to the oxidative stress response. J Proteome Res. 2016 Sep 2;15(9):3188–3195. doi: 10.1021/acs.jproteome.6b00413. [DOI] [PubMed] [Google Scholar]
- 50.Petrera A, Gassenhuber J, Ruf S, et al. Cathepsin A inhibition attenuates myocardial infarction-induced heart failure on the functional and proteomic levels. J Transl Med. 2016 May 31;14(1):153. doi: 10.1186/s12967-016-0907-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cuervo AM, Mann L, Bonten EJ, et al. Cathepsin A regulates chaperone- mediated autophagy through cleavage of the lysosomal receptor. Embo J. 2003 Jan 2;22(1):47–59. doi: 10.1093/emboj/cdg002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tasset I, Cuervo AM. Role of chaperone-mediated autophagy in metabolism. Febs J. 2016 Jul;283(13):2403–2413. doi: 10.1111/febs.13677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53••.Zhou XY, Morreau H, Rottier R, et al. Mouse model for the lysosomal disorder galactosialidosis and correction of the phenotype with overexpressing erythroid precursor cells. Genes Dev. 1995 Nov 1;9(21):2623–2634. doi: 10.1101/gad.9.21.2623. This paper describes the first mouse model of Galactosialidosis. [DOI] [PubMed] [Google Scholar]
- 54.Rottier RJ, Hahn CN, Mann LW, et al. Lack of PPCA expression only partially coincides with lysosomal storage in galactosialidosis mice: indirect evidence for spatial requirement of the catalytic rather than the protective function of PPCA. Hum Mol Genet. 1998 Oct;7( 11):1787–1794. doi: 10.1093/hmg/7.11.1787. [DOI] [PubMed] [Google Scholar]
- 55.Hermo L, Korah N, Gregory M, et al. Structural alterations of epididymal epithelial cells in cathepsin A-deficient mice affect the blood-epididymal barrier and lead to altered sperm motility. J Androl. 2007 Sep-Oct;28(5):784–797. doi: 10.2164/jandrol.107.002980. [DOI] [PubMed] [Google Scholar]
- 56.de Geest N, Bonten E, Mann L, et al. Systemic and neurologic abnormalities distinguish the lysosomal disorders sialidosis and galactosialidosis in mice. Hum Mol Genet. 2002 Jun 1;11( 12):1455–1464. doi: 10.1093/hmg/11.12.1455. [DOI] [PubMed] [Google Scholar]
- 57.Hahn CN, del Pilar Martin M, Zhou XY, et al. Correction of murine galactosialidosis by bone marrow-derived macrophages overexpressing human protective protein/cathepsin A under control of the colony-stimulating factor-1 receptor promoter. Proc Natl Acad Sci USA. 1998 Dec 8;95(25):14880–14885. doi: 10.1073/pnas.95.25.14880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leimig T, Mann L, del Martin MP, et al. Functional amelioration of murine galactosialidosis by genetically modified bone marrow hematopoietic progenitor cells. Blood. 2002 May 1;99(9):3169–3178. doi: 10.1182/blood.v99.9.3169. [DOI] [PubMed] [Google Scholar]
- 59.Bonten EJ, Wang D, Toy JN, et al. Targeting macrophages with baculovirus-produced lysosomal enzymes: implications for enzyme replacement therapy of the glycoprotein storage disorder galactosialidosis. Faseb J. 2004 Jun;18(9):971–973. doi: 10.1096/fj.03-0941fje. [DOI] [PubMed] [Google Scholar]
- 60••.Hu H, Gomero E, Bonten E, et al. Preclinical dose-finding study with a liver-tropic, recombinant AAV-2/8 vector in the mouse model of galactosialidosis. Mol Ther. 2012 Feb;20(2):267–274. doi: 10.1038/mt.2011.227. This paper reports the first preclinical studies for the treatment of Galactosialidosis using an in-vivo gene therapy approach. [DOI] [PMC free article] [PubMed] [Google Scholar]