

Abstract
Extracts of Alzheimer's disease (AD) brain that contain what appear to be SDS-stable amyloid β-protein (Aβ) dimers potently block LTP and impair memory consolidation. Brain-derived dimers can be physically separated from Aβ monomer, consist primarily of Aβ42 and resist denaturation by powerful chaotropic agents. In nature, covalently cross-linked Aβ dimers could be generated in only one of two different ways - either by the formation of a dityrosine (DiY) or an isopeptide ε-(γ-glutamyl)-lysine (Q-K) bond. We enzymatically cross-linked recombinant Aβ42 monomer to produce DiY and Q-K dimers and then applied a range of biophysical methods to study their aggregation. Both Q-K and DiY dimers aggregate to form soluble assemblies distinct from the fibrillar aggregates formed by Aβ monomer. These results suggest that Aβ dimers allow the formation of soluble aggregates akin to those in aqueous extracts of AD brain. Thus it seems that Aβ dimers may play an important role in determining the formation of soluble rather than insoluble aggregates.
Keywords: Alzheimer's disease, amyloid β-protein, analytical size exclusion chromatography, light scattering, electron microscopy, thioflavin T
Introduction
Alzheimer's disease is a huge personal problem and a growing challenge to healthcare systems throughout the world (1). The molecular changes leading to AD are not well understood, but substantial data indicate that the amyloid β-protein (Aβ) plays a central initiating role (2, 3). Although the forms of Aβ that mediate memory impairment and the toxic pathways activated by Aβ remain unresolved, numerous studies have shown that non-fibrillar, water-soluble Aβ from a variety of sources are potent synaptotoxins (4-8). Studies of post-mortem brain indicate that levels of water-soluble Aβ are elevated in AD (9-13). Moreover, in vitro and in vivo studies show that such material robustly inhibits long-term potentiation, facilitates long-term depression and induces tau hyperphosphorylation and synaptic degeneration (8, 14-17). Aβ from bioactive AD brain extract migrates on SDS-PAGE as two broad bands at ~4 and ~7 kDa – putative monomers and dimers, and recent work suggests that the dimer is a building block of toxic assemblies (18). Intriguingly, such dimers are highly stable, neither their migration on SDS-PAGE nor size exclusion chromatography (SEC) is altered by treatment with strong chaotropes such as formic acid, urea and guanidine hydrochloride (18).
A likely explanation for the high stability of brain-derived dimers is that their component monomers are coupled by a covalent bond. In nature there are only two reactions that could yield such dimers. One mechanism would involve the phenolic coupling of tyrosine residues and the other the formation of an isopeptide bond between Gln15 and Lys16 (Figure 1). Oxidative stress can cause formation of dityrosine (DiY) cross-linking of proteins and in several human diseases a number of different tyrosine-containing proteins have been shown to undergo phenolic coupling (19-21). Indeed there is evidence of increased levels of DiY in AD (22-25) and in vitro, Aβ can readily be induced to form DiY cross-linked dimers (26-29). Cross-linking of Aβ can also be achieved by the enzyme catalyzed formation of isopeptide bonds between glutamine and lysine residues (Figure 1B). In vivo ε-(γ-glutamyl)-lysine bonds (Q-K) are formed by the action of the enzyme transglutaminase (TG) (30). Under normal conditions TG plays an important role in the post-translational stabilization of particular proteins, but in certain pathological conditions the activity of TG is up-regulated, resulting in non-specific cross-linking of proteins (31, 32). Indeed, Q-K formation is known to be increased in AD (33) and synthetic Aβ can serve as a substrate for TG leading to production of isopeptide cross-linked dimers (34, 35).
Figure 1. The primary structure of [Aβ(M1-42)]DiY and [Aβ(M1-42)]Q-K dimers formed from Aβ(M1-42) monomer.
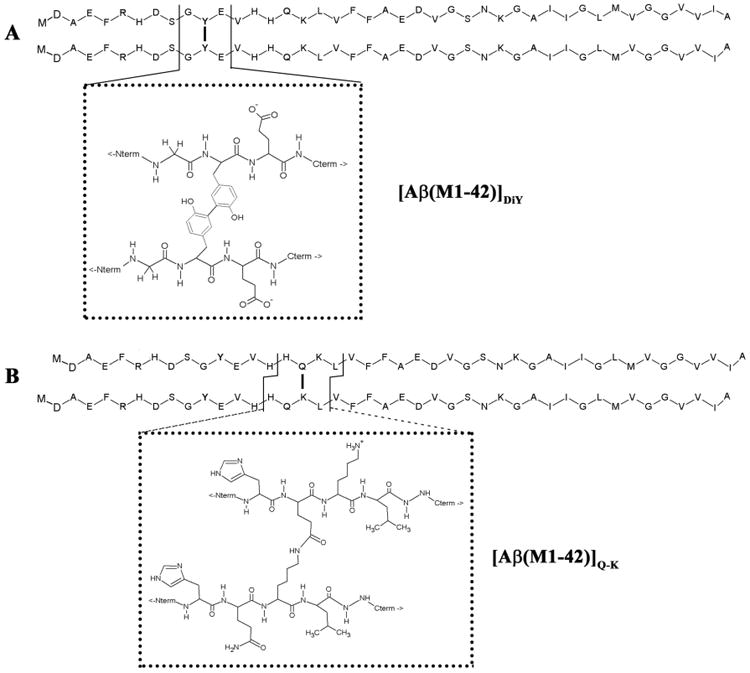
(A) The primary sequence of two Aβ(M1-42) monomers linked at tyrosine (Y) 10 are shown using the single letter amino acid code, with the atomic structures around the phenolic bond shown in the dotted box. (B) The primary sequence of two Aβ(M1-42) monomers linked via an isopeptide bond between glutamine (Q) 16 and lysine (K) 17, with the atomic structure around the isopeptide bond shown in the dotted box. For both dimers individual monomer chains are shown with N-termini in register, but it is also feasible that one chain could run from N-terminal to C-terminal, and the other chain C-terminal to N-terminal.
Heretofore the aggregation of Aβ42 Q-K and DiY dimers was not investigated. However several reports found that cysteine-containing Aβ peptides could be induced to form cystine cross-linked dimers, and that the aggregation of those design dimers was distinct from that of their component monomers (36-39). Although these studies provided the first information about the unique aggregation properties of Aβ dimers, the Aβ sequence does not naturally contain cysteines. The initial attempts to study dimers held together by cross-links that could occur in AD were restricted to dityrosine cross-linked Aβ40 (29, 40). Like cystine cross-linked dimers, the self-assembled structure and aggregation kinetics of Aβ40DiY was found to be distinct from that of Aβ40 monomer. Here we investigated the aggregation kinetics and products of Q-K ([Aβ(M1-42)]Q-K) and DiY ([Aβ(M1-42)]DiY) dimers. Using Thioflavin T (ThT) fluorescence, centrifugation, quasielastic light scattering (QLS), analytical SEC (aSEC) and negative contrast electron microscopy (EM) we compared the aggregation of [Aβ(M1-42)]Q-K and [Aβ(M1-42)]DiY, alongside that of Aβ42 monomer (Aβ(M1-42)). Notably, both dimers assembled to produce soluble aggregates akin to those detected in aqueous extracts of AD brain (18), but unlike Aβ42 monomer they did not readily form insoluble aggregates. These results suggest that in nature dimerization of Aβ may act to produce long-lived diffusible aggregates that may play a role in disease.
Materials and Methods
Reagents
Unless otherwise stated all chemicals and reagents were purchased from Sigma (Sigma-Aldrich, St. Louis, MO) and were of the highest purity available. Recombinant Aβ(M1-42) was produced and purified as described previously (29, 41). Following anion exchange isolation of recombinant Aβ from bacterial extract, approximately 10 mg of Aβ(M1-42) was dialyzed into 50 mM ammonium bicarbonate buffer, pH 8.5 (AmBiC), lyophilized and then dissolved in 5 ml of 50 mM Tris-HCl, pH 8.5 containing 7 M guanidinium HCl and 5 mM EDTA (denaturing buffer) and incubated overnight. Aβ monomer was isolated by gel-filtration chromatography using two Superdex 16/60 columns linked in series, eluted at 0.8 ml/min AmBiC containing 5 mM EDTA. The peak fraction was collected and the concentration determined by measuring the absorbance at 275 nm (Abs275; ε =1361 M-1 cm-1). Thereafter aliquots were prepared and lyophilized. Peptide mass and purity (>99%) were confirmed by MALDI-TOF mass spectrometry, reverse phase HPLC and SDS-PAGE with silver staining (Supplementary Figure 1).
Production of dityrosine (DiY) cross-linked Aβ42 dimer
Dityrosine cross-linking of peptides was performed essentially as described previously (29). Five milligrams of Aβ(M1-42) was dissolved in denaturing buffer to produce a solution of 10 mg/ml and then chromatographed on two Superdex 75 10/300 columns linked in tandem (GE Healthcare Biosciences, Pittsburgh, PA), and eluted in AmBiC. Monomer peak fractions were collected, pooled and concentration determined by Abs275. Peptide was diluted to 40 μM using AmBiC and incubated with 2.2 μM horseradish peroxidase (Thermo Scientific, Waltham, MA) and 250 μM H2O2 at 37°C for ~14 h (23). Thereafter, this material was lyophilized and the lyophilyzate redissolved in denaturing buffer (3 ml), incubated overnight at room temperature and dityrosine cross-linked Aβ dimer ([Aβ(M1-42)]DiY) was isolated from unreacted monomer and aggregates using a Superdex 75 16/60 column eluted with AmBiC. Peak fractions of dimer were pooled and the concentration determined by Abs283 (ε = 6226 M-1 cm-1) (29). Based on starting monomer concentration, this cross-linking and purification protocol typically yielded 25% as much dimer as starting material, i.e. starting with 5 mg of Aβ(M1-42) the protocol yielded ~1.25 mg of [Aβ(M1-42)]DiY.
Production of ε-(γ-glutamyl)-lysine (Q-K) cross-linked of Aβ42 dimer
Aβ(M1-42) monomer was isolated by SEC as described above and lyophilized in 1 mg aliquots. Five mg of Aβ(M1-42) was then dissolved at a concentration of 80 μM in 0.01 mM NaOH, and diluted to 40 μM with 100 mM Tris-HCl, pH 8.5 containing 10 mM CaCl2, 20 mM dithiothreitol (DTT) and incubated with 0.05 μg/μl transglutaminase (Sigma Aldrich, St. Louis, MO) at 37°C for 30 min (42). Thereafter, Q-K dimer ([Aβ(M1-42)]Q-K) was isolated as described for DiY dimer. Peak fractions of dimer were pooled and their concentration determined by UV absorbance at 275 nm (ε = 2722 M-1 cm-1) (29). An extinction coefficient, twice that of Aβ monomer was used to determine the concentration of Q-K dimer as it contains 2 free tyrosine residues and displays a UV absorbance spectrum similar to that of Aβ(M1-42) (Supplementary Figure 2). Typically 5 mg of Aβ(M1-42) yielded ~1.0 mg of [Aβ(M1-42)]Q-K (i.e. a ~20% yield).
Continuous Thioflavin T fluorescence assay
Peptide was dissolved in denaturing buffer, incubated overnight at room temperature and chromatographed on two Superdex 75 10/300 columns linked in tandem and eluted with 20 mM sodium phosphate, pH 8.0 containing 200 μM EDTA and 0.02% NaN3. Only the peak fractions were used for aggregation assays (Supplementary Figure 3). Aggregation was monitored using a continuous ThT fluorescence assay (43). SEC isolated peptide was diluted to 10.1 μM with 20 mM sodium phosphate, pH 8.0 containing 200 μM EDTA and 0.02% NaN3 and combined with a 100 × ThT stock to yield 20 μM ThT and 10 μM Aβ. Serial dilutions of Aβ were prepared by diluting with 20 mM sodium phosphate, pH 8.0 containing 200 μM EDTA, 0.02% NaN3 and 20 μM ThT. Six replicates (100 μl) of each Aβ concentration were transferred to wells of a black, clear bottom, 96 well half-area low-binding polystyrene plate (Corning Life Sciences, Corning, NY). A blank without any Aβ was also prepared and analyzed alongside Aβ containing wells. The outer edge wells of the plate were not used for samples, but were filled with buffer and plates sealed with an adhesive cover (Fischer Scientific, Cambridge, MA). Samples were incubated at 37°C in a POLARstar Omega plate reader (BMG Labtech, Offernburg, Germany) and fluorescence recorded every 5 minutes (Ex440nm; Em480nm). Fluorescence values are shown as continuous lines derived by point-to-point fitting to the value obtained for each 5 min interval. Three criteria (Lag time, maximal aggregation rate and maximal fluorescence) were used to compare aggregation curves produced by different peptides (Supplementary Figure 4). Lag time is defined as the first of two consecutive time points showing a statistically significant increase in fluorescence (Student's t test) compared to the t = 0 reading. The maximal rate of aggregation is given by the maximum slope of the linear phase of aggregation (44). Curves were considered to have reached a plateau once the rate of aggregation had slowed to ≤ 5% of that of the maximal rate of aggregation.
At maximal aggregation (t = max), six, 100 μl replicates were pooled and fluorescence recorded from 2× 120 μl replicates using a SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA; Ex440, Em480). The remaining pool (~360 μl) was then centrifuged at 16,000 g for 30 minutes, the upper 90% of supernatant was removed and the fluorescence determined (37).
Semi-continuous Thioflavin T fluorescence assay
Samples for semi-continuous ThT assay were prepared essentially as described above for the continuous ThT assay, however in the absence of ThT dye. Peptide samples minus ThT (5 μM Aβ monomer or dimer) were transferred to the wells a black, clear bottom, 96 well half-area low-binding polystyrene plate, alongside identical samples containing ThT. Sample plates were incubated at 37°C and the fluorescence recorded every 5 minutes (Ex440nm; Em480nm) using a CLARIOstar Omega plate reader (BMG Labtech, Offernburg, Germany). At specific times during the aggregation assay, (110 min for Aβ(M1-42), 1200 min for [Aβ(M1-42)]DiY and 130 min [Aβ(M1-42)]Q-K) plates were quickly removed from the plate reader, 1 μl of 2 mM ThT (100× stock) added to wells containing sample minus ThT and the plate returned to the plate reader to continue recording fluorescence.
Analytical SEC
Ten micromolar samples (100 μl) without ThT, were prepared and incubated as above and aliquots removed at specific intervals. Twenty five μl of each was injected via a 20 μl loop onto a Superdex 200 3.2/300 PE column, and eluted at 0.1 ml/min with 20 mM sodium phosphate, pH 8.0 containing 200 μM EDTA and 0.02% NaN3. Absorbance at 214 nm was recorded. The time points (t = ¼ max, t = ½ max, t = max) analyzed were based on prior ThT assays and correspond to ¼ maximal, ½ maximal, or maximal fluorescence of a given peptide (see Supplementary Figure 4). At each interval samples were centrifuged at 16,000g for 1 min to remove large aggregates, and then loaded on SEC.
Quasielastic light scattering
Triplicate 10 μM samples (100 μl) were prepared and transferred to the same plates as used for ThT experiments, except no ThT was added. At intervals, ten, 10 second acquisitions were collected using a DynaPro® Plate Reader (Wyatt Technology, Santa Barbara, CA). The intensity correlation function and the distribution of the hydrodynamic radii of the particles contributing to scattering were determined using Dynamics 7 software (Wyatt Technology, Santa Barbara, CA).
Negative stain electron microscopy
Samples (10 μl) were applied to carbon-coated Formvar grids for one min and then cross-linked using 0.5% gluteraldehyde (10 μl). Grids were washed gently with MQ water, stained for two min with 2% uranyl acetate (Electron Microscope Sciences, Fort Washington, PA) and blotted dry. Samples were prepared in duplicate and examined using a Tecnai G2 Spirit BioTWIN electron microscope (FEI, Hillsboro, OR). EM grids were scanned in a serpentine fashion at ~12,000× magnification, then regions of interest were examined at higher magnification and images captured with an AMT 2k CCD camera (AMT Corp., MA).
Circular dichroism spectroscopy
Aβ(M1-42) monomer or dimer samples were analyzed by CD at t = 0 or t = max. Samples (200 μl) were transferred to a 1 mm quartz cuvette (Starna Scientific, Hainault, UK). Spectra were recorded at 4°C between 260 nm and 190 nm with 0.2 nm intervals and 20 nm/min continuous scanning using a J-185 CD spectropolarimeter (JASCO Corp., Tokyo, Japan). Curves generated from the average of three accumulations were manipulated by subtracting the blank buffer signal and smoothened using a means-movement function with a convolution width of 20 data points. Data are shown as mean molar ellipticity (θ).
Results
Aβ42 dimers form soluble, ThT positive assemblies
We previously reported that certain covalently cross-linked Aβ40 peptides aggregate at different rates and produce different structures than the Aβ40 monomer (29). Here we focused on Aβ42 dimers containing the only cross-links that could occur in nature; and studied these dimers alongside Aβ42 monomer (Figure 2). Homogenous solutions of monomer or dimer were isolated using SEC (Supplementary Figure 3), adjusted to the required concentration and immediately used. It should be noted that relative to globular protein standards both Aβ monomers and dimers elute anomalously from SEC (45), whereas relative to linear dextran standards monomers and dimers elute with predicted molecular weights within 10% of their actual mass. Further, in prior studies QLS analysis of peak fractions such as those in Supplementary Figure 3A-C were shown to be comprised of low molecular weight species consistent with Aβ monomer and dimers (29, 46).
Figure 2. [Aβ(M1-42)]DiY and [Aβ(M1-42)]Q-K form ThT positive assemblies at rates different to that of Aβ(M1-42).
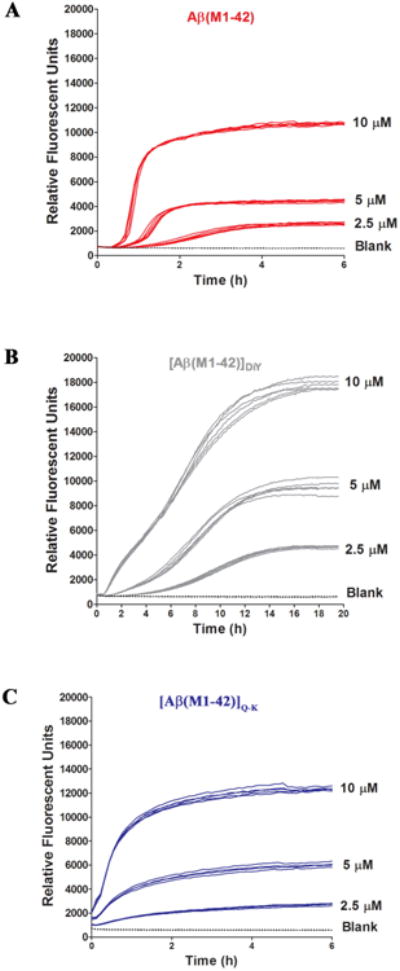
SEC-isolated (A) Aβ(M1-42), (B) [Aβ(M1-42)]DiY and (C) [Aβ(M1-42)]Q-K were diluted to 10.1 μM with 20 mM sodium phosphate pH 8.0 containing 200 μM EDTA and 0.02% NaN3 and combined with 2 mM ThT to yield a solution of 10 μM Aβ and 20 μM ThT. Serial dilutions of peptide (5 μM and 2.5 μM) were prepared in low binding tubes using phosphate buffer containing 20 μM ThT. Six, 100 μl replicates of each concentration for a given peptide were transferred to a 96 well, half-area plate and fluorescence was measured at five minute intervals (Ex440, Em480). Curves show high reproducibility between replicates. There is a concentration dependent effect on lag, rate and extent of ThT fluorescence for all peptides. As a control, buffer alone containing 10 μM ThT but no Aβ (blank) was also analyzed. Data shown are representative of a total of 3 experiments. To better appreciate differences in the early phase of aggregation, data for the first 2 hours are shown in Supplementary Figure 6.
Initially aggregation was investigated by monitoring the evolution of material capable of binding ThT (43). Because our experiments require the simultaneous SEC isolation of peptides and we had access to only 2 FPLCs, experiments were undertaken such that each dimer was directly compared versus monomer, and each dimer was studied alongside monomer in at least 3 separate experiments. The kinetic traces for 6 separate Aβ(M1-42) experiments demonstrate the high reproducibility of this method (Supplementary Figure 5). Indeed, in experiments using different peptide batches analyzed more than 1 year apart the aggregation profiles of Aβ(M1-42) and [Aβ(M1-42)]Q-K were remarkably similar (compare Figure 2 and Supplementary Figure 7).
In all experiments, monomer and dimers aggregated in a highly concentration-dependent manner (Figure 2). Aβ(M1-42) aggregation had 4 distinct phases: (1) an interval during which no change in ThT fluorescence was detected, (2) an initial relatively slow phase of increased ThT fluorescence, (3) a rapid increase in ThT fluorescence, and (4) a plateau during which ThT fluorescence remained relatively constant (Figure 2A and Supplementary Figure 6). [Aβ(M1-42)]Q-K exhibited a similar kinetic profile to Aβ(M1-42), but at the 3 concentrations tested the lag was significantly shorter for Q-K vs. monomer, and the maximal rate of aggregation was slower for Q-K than monomer. The kinetic profile of [Aβ(M1-42)]DiY was more variable than that of the other peptides. At 2.5 μM and 5 μM, [Aβ(M1-42)]DiY had a 4-phase profile reminiscent of Aβ(M1-42), but at 10 μM at least 5 phases were obvious (Figure 2B). As with Aβ40 peptides (29), [Aβ(M1-42)]DiY had the longest lag and slowest growth phases, but attained the highest maximal fluorescence (Table 1). These results indicate that end products of the 3 peptides are significantly different and that the relative rates of the underlying microscopic steps is affected by the cross-links.
Table 1. Quantitative analysis of ThT-monitored aggregation.
| Aβ(M1-42) | [Aβ(M1-42)]DiY | [Aβ (M1-42)]Q-k | |||||||
|---|---|---|---|---|---|---|---|---|---|
| [Aβ] (μM) | 10 | 5 | 2.5 | 10 | 5 | 2.5 | 10 | 5 | 2.5 |
| Lag Time (min) | 33 ± 6 | 43 ± 3 | 80 ± 5 | 33 ± 11 | 75 ± 7 | 180 ± 28 | 2 ± 3 | 13 ± 6 | 23 ± 6 |
| Rate (RFU/min) | 185.5 ± 29.4 | 68.0 ± 18.3 | 11.0 ± 1.3 | 22.5 ± 0.4 | 15.4 ± 1.3 | 1.7 ± 1.3 | 136.0 ± 14.5 | 44.9 ±12.8 | 10.5 ± 2.2 |
| Max Fluor. (RFU) | 10869 ± 990 | 9963 ± 363 | 2618 ± 90 | 18660 ± 990 | 9963 ± 343 | 4400 ± 226 | 11110 ± 1512 | 4810 ± 1463 | 1733 ± 339 |
| t = max | ~ 5 hrs | - | - | ~ 17 hrs | - | - | ~ 5.5 hrs | - | - |
| t = ½ max | ~ 1 hr | - | - | ~ 6.5 hrs | - | - | ~1 hr | - | - |
| t = ¼ max | ~ 30 min | - | - | ~ 3 hrs | - | - | ~ 30 min | - | - |
Time points were calculated based on criteria described in Supplementary Figure 4 and are the averaged from at least two separate experiments.
Values are mean ± standard deviation are given.
Although widely used to monitor protein aggregation, the molecular interactions between ThT and protein aggregates, and how this results in a shift in ThT fluorescence are not fully understood (47-50). Thus the differences in intensities observed for the 3 peptides could reflect different modes of ThT binding to different types of aggregates (51, 52) and differences in fluorescence quantum yield due to the local environment of the bound dye. To exclude the possibility that the presence of ThT interferes with the aggregation reaction and contributed to the differences in intensity between Aβ(M1-42), [Aβ(M1-42)]DiY and [Aβ(M1-42)]Q-K, we directly compared the aggregation of peptides using both our standard continuous ThT assay versus a semi-continuous ThT assay. For the latter, samples were incubated without dye and only mixed with dye after certain incubation periods (Supplementary Figure 7). Both the continuous and semi-continuous assays produced similar kinetic curves indicating that the continuous presence of ThT did not unduly influence the aggregation of Aβ monomers and dimers. In addition, we used CD to investigate the secondary structure of aggregates formed in the absence of ThT. In all cases the aggregates contained appreciable extents of β-structure (Supplementary Figure 8).
Thereafter we investigated the physical properties of both end-point and intermediate-time point aggregates. An issue of particular importance is relative solubility. In the context of amyloid proteins all Aβ species, from monomers through large fibrils, are soluble. However, Aβ species are often differentiated based on an operational definition, that is, whether they can be sedimented at a chosen centrifugal force (2). In these experiments we determined if the end-point aggregates that give rise to maximal ThT fluorescence could be sedimented by centrifugation at 16,000 g for 30 min. As expected, centrifugation dramatically reduced the fluorescence signal of Aβ(M1-42) t = max. For instance, when the t = max 10 μM samples from 3 separate experiments were analyzed before and after centrifugation, ThT fluorescence was reduced by a massive ~96% (Figure 3A), in line with the low concentration of monomer remaining in solution at the end of the process (43). In contrast, the fluorescence of end-point dimer samples were not significantly altered by centrifugation (Figure 3B and C). Thus it appears that ThT-positive aggregates formed by dimers have relatively lower sedimentation coefficients than those produced from monomer. In separate experiments samples were spun at 16,000 g for 30 min and then for longer duration and at higher speeds (100,000 g for 2 hrs). As before when end-stage Aβ(M1-42) aggregates were centrifuged at 16,000 g for 30 min the majority of the ThT signal was sedimented (Supplementary Figure 7D). In contrast, when end-stage Aβ(M1-42)]Q-K was centrifuged at 100,000 g for 2 hrs the ThT signal in the supernatant was unchanged. For end-stage [Aβ(M1-42)]DiY the ThT signal in the supernatant was unchanged after centrifugation at 16,000 g, but ~80% of the ThT signal was removed by centrifugation. These results suggest that aggregates formed by [Aβ(M1-42)]DiY tend to be larger than those formed by Aβ(M1-42)]Q-K, but are much smaller than those formed by Aβ(M1-42).
Figure 3. ThT-positive assemblies formed by [Aβ(M1-42)]DiY and [Aβ(M1-42)]Q-K remain in solution following centrifugation.
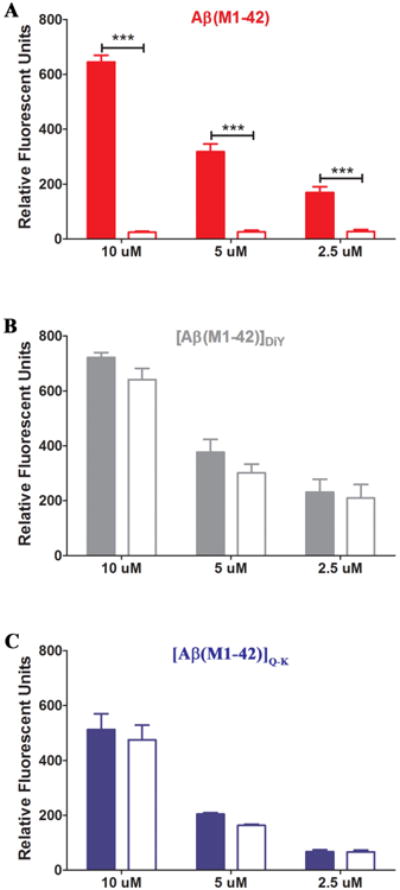
At the end of each ThT assay, six, 100 μl replicates from each concentration of a given peptide were pooled and the fluorescence measured (2× 100 μl replicated; Ex440, Em480) before (colored bars) and after (empty bars) centrifugation at 16,000 g for 30 minutes. Data displayed are the average of three separate experiments ± standard deviation. Signals before and after centrifugation were analyzed by Student t test and *** represents p < 0.0001.
Aβ42 dimers form soluble intermediate molecular weight assemblies, whereas Aβ42 monomer forms meshworks of amyloid fibrils
To examine the extent to which monomer or dimer is consumed and to monitor the evolution of soluble aggregates we employed analytical SEC (aSEC). Since the 3 peptides aggregated at different rates we used the results of our ThT experiments to define and study equivalent intermediate and end-stage points in the aggregation processes. The times used correspond to t = 0 - immediately following SEC isolation of monomer/dimer, t = ¼ max - the time required to achieve ~25% of the maximal ThT fluorescence attainable, t = ½ max - the time required to achieve half of maximal ThT fluorescence, and t = max - the time required to achieve maximal ThT fluorescence (Supplementary Figure 4). Prior to aSEC, samples were briefly centrifuged to remove insoluble aggregates that might clog the column, but otherwise samples were not manipulated in any way. All peptides exhibited a symmetrical peak at t = 0, that eluted within the included volume. When compared to the elution of unbranched linear dextran standards Aβ monomers (Figure 4A-C) and dimers (Figure 4B and C) eluted as one would expect from unfolded proteins of ~4.6 and ~9.2 kDa.
Figure 4. Time course analytical-SEC reveals that dimers, but not monomer form soluble aggregates.
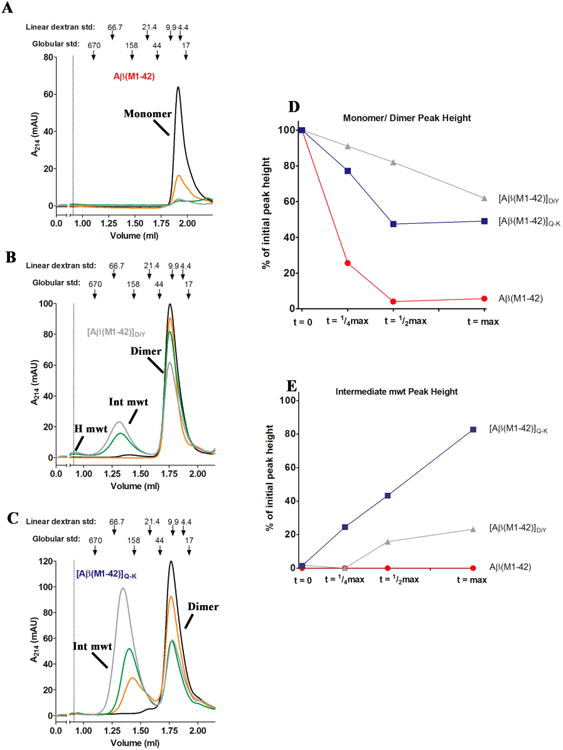
Time points determined relative to the degree of ThT fluorescence (see Supplementary Figure 4 and Table 1) were used to sample and analyze 10 μM preparations of (A) Aβ(M1-42), (B) [Aβ(M1-42)]DiY and (C) [Aβ(M1-42)]Q-K. Samples at t = 0 (black line), ¼ max (orange line), ½ max (green line) and max (gray line) were centrifuged at 16,000 g for 1 min prior to aSEC. Absorbance at 214 nm was monitored. For all samples, monomer and dimer peak decreased over time, and for Aβ(M1-42)]DiY and [Aβ(M1-42)]Q-K, a new peak appeared in the (~0.9 ml) or near (~1.3 ml) the void volume. The elution of blue dextran (at 1.1 ml) is indicated by a dashed vertical line. The positions where 66,700, 21,400, 9,890 and 4,440 linear dextran standards elute is indicate with down-pointing arrows, as is the elution of 17, 44, 158 and 670 kDa globular protein standards. Peak height of monomer/dimer (D) and intermediate molecular weight aggregates (E) are plotted as a percentage of the monomer/dimer levels at time zero, which were normalized to a value of 100%. For (D) and (E) Aβ(M1-42) is red, [Aβ(M1-42)]DiY is gray and [Aβ(M1-42)]Q-K is blue.
The height of each t = 0 peak was set to 100% and changes in monomer or dimer at subsequent time points expressed relative to this value (Figure 4D). By t = ¼ max the Aβ(M1-42) monomer peak was reduced by ~80% (Figure 4A). Over time, Aβ42 monomer decreased further such that it could not be accurately measured at t = ½ max and t = max, and no other peaks were detected. These results imply that the ThT-positive material evident at t = ¼ max, ½ max and max were pelleted by a 1 min centrifugation, or less likely, adhered to the gel-filtration matrix. Notably, time-course aSEC demonstrated that the aggregation of DiY and Q-K dimers proceed in a manner distinct from that of Aβ42 monomer (Figure 4). Both dimers evinced temporal evolution of at least 2 species of higher molecular weight than dimer: one which eluted in the included volume of the column and another which eluted in the void volume of the column (Figure 4B and C). At t = ¼ max the [Aβ(M1-42)]DiY peak showed only a very modest decrease and no higher molecular weight species were detected. By t = ½ max a peak with a molecular weight greater than the 150 kDa globular standard was readily detected. At t = max the [Aβ(M1-42)]DiY peak was decreased by ~40% of its starting t =0 value and the height of the intermediate molecular weight peak was further increased (Figure 4B, D and E). aSEC analysis revealed a pattern of aggregation for [Aβ(M1-42)]Q-K that was distinct from both the Aβ42 monomer and [Aβ(M1-42)]DiY. Specifically, [Aβ(M1-42)]Q-K was more rapidly consumed than [Aβ(M1-42)]DiY (Figure 4C and D) and more rapidly gave rise to higher molecular weight aggregates (Figure 4C and E). Interestingly, the elution of the [Aβ(M1-42)]Q-K intermediate molecular weight species shifted to the left with increasing time – indicating that soluble aggregates detected at t max were larger than those detected at t = ½ which in turn were larger than those detected at t = ¼ (Figure 4C – compare the orange, green and grey chromatograms).
To gain insight about the size of aggregates present at t = 0, ¼ max, ½ max and max, samples were also analyzed using QLS. This technique is best suited for determining the size of homogenous solutions, and can only provide qualitative measures of the size distribution of polydisperse aggregates (50). The auto-correlation function (Supplementary Figure 9) was used to determine the size distribution of aggregates expressed as the hydrodynamic radii of scattering particles, and the abundance of each sized particle plotted as % intensity (Figure 5). As expected, buffer alone produced only small and variable amount of scattering (Figure 5A and Supplementary Figure 9) – a pattern consistent with the presence of small amounts of relatively large dust particles that infrequently enter the illuminated volume. At t = 0 little scattering was evident in any of the peptide solutions and the autocorrelation functions for the t = 0 peptide samples were indistinguishable from the buffer control. (Supplementary Figure 9A-C). For Aβ(M1-42), QLS measurements at t = ¼ max indicated the presence of structures with average Rh values of ~70 nm and even larger assemblies at t = ½ max and t = max (Figure 5 and Supplementary Figure 9). Moreover, negative contrast EM revealed extensive lattice works of fibrils in the t = max sample (Figure 6A). Thus, aSEC, QLS and EM demonstrate that Aβ(M1-42) aggregates rapidly to form large superstructures of aggregated fibrils.
Figure 5. QLS reveals that dimers form lower molecular weight aggregates than monomer.
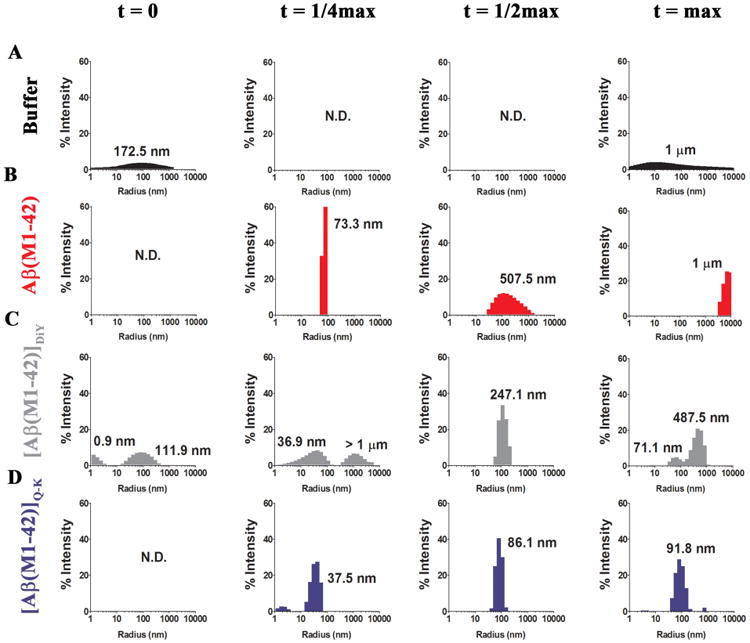
QLS was used as a non-invasive method to determine the population distribution of detectable aggregates for each peptide. At t = 0, t = ¼ max, t = ½ max, t = max triplicate sample recordings were made using 10× 10 sec acquisitions in a DynaPro® plate reader. The average hydrodynamic radii (RH) for species detected in (A) buffer and preparations of (B) Aβ(M1-42), (C) [Aβ(M1-42)]DiY and (D) [Aβ(M1-42)]Q-K are expressed as percentage intensity detected and labelled with the average RH is indicated. ND means no signals were detected.
Figure 6. Visualization of end-point aggregates of Aβ(M1-42), [Aβ(M1-42)]DiY and [Aβ(M1-42)]Q-K display distinct morphologies from one another.
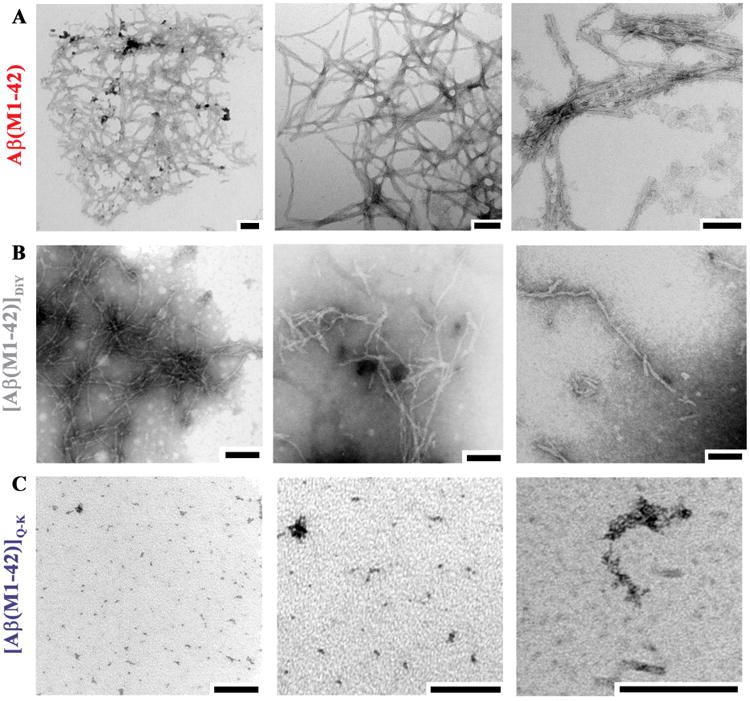
End-point samples of (A) Aβ(M1-42), (B) [Aβ(M1-42)]DiY and (C) [Aβ(M1-42)]Q-K were used for negative contrast EM. Electron micrographs are representative of 4 - 5 fields of view from at least two grids per sample. Size bars = 100 nm.
As with ThT and aSEC, QLS indicates that [Aβ(M1-42)]Q-K and [Aβ(M1-42)]DiY aggregate in ways decidedly different from Aβ42 monomer (Figure 5 and Supplementary Figure 9) with both dimers aggregating to form structures much smaller than the aggregates formed by monomer (Figure 4 and 5, and Supplementary Figure 9). For [Aβ(M1-42)]DiY at t = ¼ max the most abundant scattering species had an average RH of ~37 nm. At t = ½ this shifted to an Rh ~250 nm and at t = max this increased further to ~500 nm (Figure 5B). In contrast the aggregates formed by [Aβ(M1-42)]Q-K tended to be of smaller size than those formed by [Aβ(M1-42)]DiY (compare Figure 5C and D) – even at t max the average RH produced by [Aβ(M1-42)]Q-K aggregates was only ~92 nm (Figure 5D).
Negative contrast EM on t = max (end-point) samples detected a variety of assemblies. Aβ(M1-42) aggregated to form meshworks of fibrils, the extent of which is best appreciated at low magnification (Figure 6A, left panel). Individual fibril mats often had diameters of >1 μM (Figure 6A) consistent with very high RH estimates for Aβ(M1-42) aggregates (Figure 5). At higher magnification lateral association of fibrils into bundles and twisted ribbons are obvious (Figure 6A, middle and right panels). In contrast, [Aβ(M1-42)]DiY formed relatively long (500+ nm) fibrils that showed a lower propensity for lateral association, bundling or formation of mats (Figure 6B), a finding in keeping with our QLS results which indicated the presence of aggregates considerably smaller than those detected in the Aβ(M1-42) sample. Significantly, [Aβ(M1-42)]Q-K didn't produce fibrils, rather samples contained numerous small (10-20 nm) amorphous aggregates (Figure 6C).
In sum, these results indicate that cross-linking of Aβ monomer to form covalent dimers not only has a major effect on the rate of peptide aggregation, but also on the size and structure of the assemblies formed. Importantly, these conclusions do not rest on a single readout, but are based on results from multiple orthogonal readouts. Specifically, aSEC, QLS, EM and ThT/centrifugation experiments confirmed that dimers form aggregates with structures and solubilities (sedimentation properties) distinct from the mats of amyloid fibrils formed by Aβ(M1-42) monomer. In regard to the wider study of protein aggregation our results illustrate the fact that ThT fluorescence is not specific for fibrils, but that pre-fibrillar structures can also bind to ThT (53, 54).
Discussion
Dimers of Aβ are thought to play a role in AD (8, 18, 55). In vivo there are only two known mechanisms by which Aβ monomers can be covalently cross-linked to form dimers – generation of an isopeptide bond between Gln15 and Lys16, or by phenolic coupling of opposing tyrosine residues. Here we show that Aβ42 dimers bearing these links aggregate to form relatively soluble assemblies, but not characteristic amyloid aggregates. As expected, Aβ monomers form insoluble aggregates of amyloid fibrils, whereas [Aβ(M1-42)]Q-K forms small amorphous aggregates and [Aβ(M1-42)]DiY produce soluble individual fibrils.
Aggregation monitored by ThT indicates significant differences in the rates of dimers and monomer. However, changes in ThT fluorescence provides little insight about the types of aggregates formed (56, 57). For example, all of the peptides analyzed in this study produced aggregates with appreciable ThT binding, but only Aβ(M1-42) formed mats of amyloid fibrils. Furthermore, aggregates produced by [Aβ(M1-42)]DiY caused the greatest degree of change in ThT fluorescence yet even at the highest concentration studied, only ~40% of [Aβ(M1-42)]DiY dimer was incorporated into aggregates. The higher ThT fluorescence intensity produced by assemblies formed from lower molar quantities of “substrate” indicate either that ThT bound to [Aβ(M1-42)]DiY displays a higher quantum yield, or more ThT was bound per unit of aggregate than for Aβ(M1-42) aggregates.
EM, QLS and aSEC demonstrate that [Aβ(M1-42)]DiY forms long individual fibrils and occasional laterally associated fibrils, similar to those produced by [Aβ(M1-40)]DiY (29). The only other covalent dimer that could exist in nature, Aβ Q-K, has never been studied. Here we show that SEC-isolated [Aβ(M1-42)]Q-K aggregates (as measured by ThT) in a manner similar to Aβ(M1-42), exhibiting a sigmoidal profile and attaining a maximal level of ThT fluorescence comparable to Aβ(M1-42). However, Q-K did not form fibrils. EM, QLS, aSEC and sedimentation demonstrate that [Aβ(M1-42)]Q-K aggregates to form structures much smaller than those produced by either Aβ(M1-42) or [Aβ(M1-42)]DiY. Specifically, by aSEC the estimated size based on globular standards of [Aβ(M1-42)]Q-K aggregates were ~158-670 kDa, QLS indicated an RH of ~90 nm and structures visible by EM had dimensions in the order of ~10-20 nm. Interestingly, a prior study found that incubation of Aβ monomer with transglutaminase led to rapid aggregation, but not formation of amyloid fibrils (34). Our finding that Q-K does not readily form amyloid fibrils yet Q-K aggregates produce significant ThT fluorescence has important implications for the use and interpretation of ThT binding and is congruent with prior studies that found ThT capable of binding to non-fibrillar protein aggregates (54) and that ThT fluorescence can vary substantial for fibrils formed by different Aβ peptides and by the same peptides under different conditions (52).
Under the conditions used, Q-K and DiY dimers formed soluble aggregates, but Aβ monomer did not yield detectable amounts of soluble aggregates on aSEC. A myriad of publications have reported that both Aβ40 and Aβ42 can form protofibrils (46, 53, 58-62) and ADDLs (6, 15, 63). However, prior studies that detected protofibrils and ADDLs did not begin with homogenous solutions of monomer. Thus, it is possible that trace amounts of aggregates or chemical impurities (64) retarded aggregation and facilitated formation of protofibrils/ADDLs. In this regard it is important to note that in all reports using well-defined SEC-isolated monomer, aggregation proceeds much more rapidly and at lower concentration (29, 41, 43, 65-69) than in experiments in which Aβ was solubilized using organic solvents. For instance, in all published reports on protofibrils/ADDLs, peptide powder was directly dissolved in solvent or aqueous buffer and incubated at high concentrations (usually ≥100 μM) for 1–7 days (6, 15, 46, 53, 58-62). An explanation for the detection of protofibrils/ADDLs in such solutions is that certain impurities either accelerate nucleation or retard elongation – either of which could increase the quantity of smaller aggregates. In contrast, when homogenous solutions of monomer are used only small amounts of soluble aggregates are detected (66).
When homogenous solutions of SEC-isolated dimers were used they formed soluble aggregates. [Aβ(M1-42)]Q-K shows no lag phase and quickly forms small yet very abundant aggregates. [Aβ(M1-42)]DiY aggregates more slowly and forms long individual fibrils, whereas Aβ(M1-42) exhibits a lag phase intermediate between [Aβ(M1-42)]Q-K and [Aβ(M1-42)]DiY, and forms a meshwork of fibrils consuming all of the monomer during its very rapid elongation phase. The rank order of aggregation was the same for Aβ(M1-42) and [Aβ(M1-42)]DiY as we observed for the corresponding Aβ40 peptides (29), this despite the fact that the prior Aβ40 experiments used polystyrene plates and agitation (29). Moreover, in preliminary experiments (not shown) conducted under conditions identical to those in the present study, [Aβ(M1-40)]Q-K aggregated faster than Aβ(M1-40) whereas [Aβ(M1-40)]DiY aggregated more slowly. As expected the rate of aggregation was faster for Aβ42 peptides than their Aβ40 equivalents.
Although we did not investigate the biological activity of [Aβ(M1-42)]Q-K and [Aβ(M1-42)]DiY, is has been widely promulgated that small soluble Aβ aggregates are likely to be more toxic than large insoluble aggregates (70, 71). Hence, it is tempting to speculate that Q-K and/or DiY dimers may play a role in AD by producing aggregates that can readily diffuse throughout the brain. In this regard it is important to note that while we have taken the first step in investigating dimer aggregation by focusing on structures formed from a single molecular preparation, future studies should investigate the aggregation of mixtures of monomers and dimers and should include monomers and dimers with a variety of disease relevant N- and C-termini (18, 72-74).
Acknowledgments
We are grateful to Dr. John Champagne and Wyatt Technologies for the use of the DynaPro plate reader. We thank Dr. Gunnar Brinkmalm and Ms. M. Ericsson and for technical assistance and Dr. B. O'Nuallain for useful discussions. Supporting information available.
Funding Information: This work was supported by grants to DMW from National Institutes of Health (AG046275), Science Foundation Ireland (Grant 0.8/1N.1/B2033) and by MedImmune.
References
- 1.Takizawa C, Thompson PL, van Walsem A, Faure C, Maier WC. Epidemiological and economic burden of Alzheimer's disease: a systematic literature review of data across Europe and the United States of America. J Alz Dis. 2015;43:1271–1284. doi: 10.3233/JAD-141134. [DOI] [PubMed] [Google Scholar]
- 2.Walsh DM, Teplow DB. Alzheimer's disease and the amyloid β-protein. Prog Mol Biol Trans Sci. 2012;107:101–124. doi: 10.1016/B978-0-12-385883-2.00012-6. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 5.Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ. Amyloid β protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welzel AT, Maggio JE, Shankar GM, Walker DE, Ostaszewski BL, Li S, Klyubin I, Rowan MJ, Seubert P, Walsh DM, Selkoe DJ. Secreted amyloid beta-proteins in a cell culture model include N-terminally extended peptides that impair synaptic plasticity. Biochemistry. 2014;53:3908–3921. doi: 10.1021/bi5003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE. Water-soluble Aβ (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271:4077–4081. doi: 10.1074/jbc.271.8.4077. [DOI] [PubMed] [Google Scholar]
- 10.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 12.Mc Donald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, Selkoe DJ, Ince PG, Walsh DM. The presence of sodium dodecyl sulphate-stable Aβ dimers is strongly associated with Alzheimer-type dementia. Brain. 2010;133:1328–1341. doi: 10.1093/brain/awq065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esparza TJ, Zhao H, Cirrito JR, Cairns NJ, Bateman RJ, Holtzman DM, Brody DL. Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol. 2013;73:104–119. doi: 10.1002/ana.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, Clarke AR, Rowan MJ, Walsh DM, Collinge J. Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat Commun. 2011;2:336. doi: 10.1038/ncomms1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid β -protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borlikova GG, Trejo M, Mably AJ, Mc Donald JM, Sala Frigerio C, Regan CM, Murphy KJ, Masliah E, Walsh DM. Alzheimer brain-derived amyloid beta-protein impairs synaptic remodeling and memory consolidation. Neurobiol Aging. 2013;34:1315–1327. doi: 10.1016/j.neurobiolaging.2012.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mc Donald JM, O'Malley TT, Liu W, Mably AJ, Brinkmalm G, Portelius E, Wittbold WM, 3rd, Frosch MP, Walsh DM. The aqueous phase of Alzheimer's disease brain contains assemblies built from approximately 4 and approximately 7 kDa Abeta species. Alzheimer Dement. 2015;11:1286–1305. doi: 10.1016/j.jalz.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balasubramanian D, Kanwar R. Molecular pathology of dityrosine crosslinks in proteins: structural and functional analysis of four proteins. Mol Cell Biochem. 2002;234-235:27–38. [PubMed] [Google Scholar]
- 20.DiMarco T, Giulivi C. Current analytical methods for the detection of dityrosine, a biomarker of oxidative stress, in biological samples. Mass Spectrom Rev. 2007;26:108–120. doi: 10.1002/mas.20109. [DOI] [PubMed] [Google Scholar]
- 21.Malencik DA, Anderson SR. Dityrosine as a product of oxidative stress and fluorescent probe. Amino Acids. 2003;25:233–247. doi: 10.1007/s00726-003-0014-z. [DOI] [PubMed] [Google Scholar]
- 22.Hensley K, Maidt ML, Yu Z, Sang H, Markesbery WR, Floyd RA. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J Neurosci. 1998;18:8126–8132. doi: 10.1523/JNEUROSCI.18-20-08126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moir RD, Tseitlin KA, Soscia S, Hyman BT, Irizarry MC, Tanzi RE. Autoantibodies to redox-modified oligomeric Aβ are attenuated in the plasma of Alzheimer's disease patients. J Biol Chem. 2005;280:17458–17463. doi: 10.1074/jbc.M414176200. [DOI] [PubMed] [Google Scholar]
- 24.Villemagne VL, Perez KA, Pike KE, Kok WM, Rowe CC, White AR, Bourgeat P, Salvado O, Bedo J, Hutton CA, Faux NG, Masters CL, Barnham KJ. Blood-borne amyloid-beta dimer correlates with clinical markers of Alzheimer's disease. J Neurosci. 2010;30:6315–6322. doi: 10.1523/JNEUROSCI.5180-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Hilaly YK, Williams TL, Stewart-Parker M, Ford L, Skaria E, Cole M, Bucher WG, Morris KL, Sada AA, Thorpe JR, Serpell LC. A central role for dityrosine crosslinking of Amyloid-beta in Alzheimer's disease. Acta Neuropathol Commun. 2013;1:83. doi: 10.1186/2051-5960-1-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galeazzi L, Ronchi P, Franceschi C, Giunta S. In vitro peroxidase oxidation induces stable dimers of β-amyloid (1-42) through dityrosine bridge formation. Amyloid. 1999;6:7–13. doi: 10.3109/13506129908993282. [DOI] [PubMed] [Google Scholar]
- 27.Yoburn JC, Tian W, Brower JO, Nowick JS, Glabe CG, Van Vranken DL. Dityrosine cross-linked Aβ peptides: fibrillar beta-structure in Aβ(1-40) is conducive to formation of dityrosine cross-links but a dityrosine cross-link in Aβ(8-14) does not induce β-structure. Chem Res Toxicol. 2003;16:531–535. doi: 10.1021/tx025666g. [DOI] [PubMed] [Google Scholar]
- 28.Atwood CS, Huang X, Khatri A, Scarpa RC, Kim YS, Moir RD, Tanzi RE, Roher AE, Bush AI. Copper catalyzed oxidation of Alzheimer Abeta. Cell Mol Biol. 2000;46:777–783. [PubMed] [Google Scholar]
- 29.O'Malley TT, Oktaviani NA, Zhang D, Lomakin A, O'Nuallain B, Linse S, Benedek GB, Rowan MJ, Mulder FAA, Walsh DW. Aβ dimers differ from monomers in structural propensity aggregation paths, and population of synaptotoxic assemblies. Biochem J. 2014;461:413–426. doi: 10.1042/BJ20140219. [DOI] [PubMed] [Google Scholar]
- 30.Kornguth SE, Waelsch H. Protein modification catalysed by transglutaminase. Nature. 1963;198:188–189. doi: 10.1038/198188a0. [DOI] [PubMed] [Google Scholar]
- 31.Selkoe DJ, Abraham C, Ihara Y. Brain transglutaminase: in vitro crosslinking of human neurofilament proteins into insoluble polymers. Proc Natl Acad Sci U S A. 1982;79:6070–6074. doi: 10.1073/pnas.79.19.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang DS, Dickson DW, Malter JS. Tissue transglutaminase, protein cross-linking and Alzheimer's disease: review and views. Int J Clin Exp Pathol. 2008;1:5–18. [PMC free article] [PubMed] [Google Scholar]
- 33.Wilhelmus MM, Grunberg SC, Bol JG, van Dam AM, Hoozemans JJ, Rozemuller AJ, Drukarch B. Transglutaminases and transglutaminase-catalyzed cross-links colocalize with the pathological lesions in Alzheimer's disease brain. Brain Pathol. 2009;19:612–622. doi: 10.1111/j.1750-3639.2008.00197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartley DM, Zhao C, Speier AC, Woodard GA, Li S, Li Z, Walz T. Transglutaminase induces protofibril-like amyloid β -protein assemblies that are protease-resistant and inhibit long-term potentiation. J Biol Chem. 2008;283:16790–16800. doi: 10.1074/jbc.M802215200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ikura K, Takahata K, Sasaki R. Cross-linking of a synthetic partial-length (1-28) peptide of the Alzheimer β/A4 amyloid protein by transglutaminase. FEBS Lett. 1993;326:109–111. doi: 10.1016/0014-5793(93)81772-r. [DOI] [PubMed] [Google Scholar]
- 36.Hard T. Protein engineering to stabilize soluble amyloid β-protein aggregates for structural and functional studies. FEBS J. 2011;278:3884–3892. doi: 10.1111/j.1742-4658.2011.08295.x. [DOI] [PubMed] [Google Scholar]
- 37.O'Nuallain B, Freir DB, Nicoll AJ, Risse E, Ferguson N, Herron CE, Collinge J, Walsh DM. Amyloid β-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010;30:14411–14419. doi: 10.1523/JNEUROSCI.3537-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muller-Schiffmann A, Andreyeva A, Horn AH, Gottmann K, Korth C, Sticht H. Molecular engineering of a secreted, highly homogeneous, and neurotoxic abeta dimer. ACS Chem Neurosci. 2011;2:242–248. doi: 10.1021/cn200011h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muller-Schiffmann A, Herring A, Abdel-Hafiz L, Chepkova AN, Schable S, Wedel D, Horn AH, Sticht H, de Souza Silva MA, Gottmann K, Sergeeva OA, Huston JP, Keyvani K, Korth C. Amyloid-beta dimers in the absence of plaque pathology impair learning and synaptic plasticity. Brain. 2016;139:509–525. doi: 10.1093/brain/awv355. [DOI] [PubMed] [Google Scholar]
- 40.Kok WM, Cottam JM, Ciccotosto GD, Miles LA, Karas JA, Scanlon DB, Roberts BR, Parker MW, Cappai R, Barnham KJ, Hutton CA. Synthetic dityrosine-linked β-amyloid dimers form stable, soluble, neurotoxic oligomers. Chem Sci. 2013:4449. [Google Scholar]
- 41.Walsh DM, Thulin E, Minogue AM, Gustavsson N, Pang E, Teplow DB, Linse S. A facile method for expression and purification of the Alzheimer's disease-associated amyloid β-peptide. FEBS J. 2009;276:1266–1281. doi: 10.1111/j.1742-4658.2008.06862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dudek SM, Johnson GV. Transglutaminase facilitates the formation of polymers of the β-amyloid peptide. Brain Res. 1994;651:129–133. doi: 10.1016/0006-8993(94)90688-2. [DOI] [PubMed] [Google Scholar]
- 43.Hellstrand E, Boland B, Walsh DM, Linse S. Amyloid β -Protein Aggregation Produces Highly Reproducible Kinetic Data and Occurs by a Two-Phase Process. Chem Neurosci. 2010;1:13–18. doi: 10.1021/cn900015v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Betts V, Leissring MA, Dolios G, Wang R, Selkoe DJ, Walsh DM. Aggregation and catabolism of disease-associated intra-Aβ mutations: reduced proteolysis of AβA21G by neprilysin. Neurobiol Dis. 2008;31:442–450. doi: 10.1016/j.nbd.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paivio A, Jarvet J, Graslund A, Lannfelt L, Westlind-Danielsson A. Unique physicochemical profile of beta-amyloid peptide variant Abeta1-40E22G protofibrils: conceivable neuropathogen in arctic mutant carriers. J Mol Biol. 2004;339:145–159. doi: 10.1016/j.jmb.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 46.Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid β-protein fibrillogenesis Detection of a protofibrillar intermediate. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 47.Biancalana M, Koide S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochem Biophys Acta. 2010;1804:1405–1412. doi: 10.1016/j.bbapap.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu C, Biancalana M, Koide S, Shea JE. Binding modes of thioflavin-T to the single-layer beta-sheet of the peptide self-assembly mimics. J Mol Biol. 2009;394:627–633. doi: 10.1016/j.jmb.2009.09.056. [DOI] [PubMed] [Google Scholar]
- 49.Goransson AL, Nilsson KP, Kagedal K, Brorsson AC. Identification of distinct physiochemical properties of toxic prefibrillar species formed by Abeta peptide variants. Biochem Biophys Res Commun. 2012;420:895–900. doi: 10.1016/j.bbrc.2012.03.097. [DOI] [PubMed] [Google Scholar]
- 50.Lomakin A, Benedek GB, Teplow DB. Monitoring protein assembly using quasielastic light scattering spectroscopy. Methods Enzymol. 1999;309:429–459. doi: 10.1016/s0076-6879(99)09029-1. [DOI] [PubMed] [Google Scholar]
- 51.Krebs MR, Macphee CE, Miller AF, Dunlop IE, Dobson CM, Donald AM. The formation of spherulites by amyloid fibrils of bovine insulin. Proc Natl Acad Sci U S A. 2004;101:14420–14424. doi: 10.1073/pnas.0405933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lindberg DJ, Wranne MS, Gilbert Gatty M, Westerlund F, Esbjorner EK. Steady-state and time-resolved Thioflavin-T fluorescence can report on morphological differences in amyloid fibrils formed by Abeta(1-40) and Abeta(1-42) Biochem Biophys Res Commun. 2015;458:418–423. doi: 10.1016/j.bbrc.2015.01.132. [DOI] [PubMed] [Google Scholar]
- 53.Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid β -protein fibrillogenesis Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 54.Maezawa I, Hong HS, Liu R, Wu CY, Cheng RH, Kung MP, Kung HF, Lam KS, Oddo S, Laferla FM, Jin LW. Congo red and thioflavin-T analogs detect Abeta oligomers. J Neurochem. 2008;104:457–468. doi: 10.1111/j.1471-4159.2007.04972.x. [DOI] [PubMed] [Google Scholar]
- 55.Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE. Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271:4077–4081. doi: 10.1074/jbc.271.8.4077. [DOI] [PubMed] [Google Scholar]
- 56.Biancalana M, Makabe K, Koide A, Koide S. Molecular mechanism of thioflavin-T binding to the surface of beta-rich peptide self-assemblies. J Mol Biol. 2009;385:1052–1063. doi: 10.1016/j.jmb.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khurana R, Coleman C, Ionescu-Zanetti C, Carter SA, Krishna V, Grover RK, Roy R, Singh S. Mechanism of thioflavin T binding to amyloid fibrils. J Struct Biol. 2005;151:229–238. doi: 10.1016/j.jsb.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 58.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harper JD, Wong SS, Lieber CM, Lansbury PT. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 60.Harper JD, Wong SS, Lieber CM, Lansbury PT., Jr Assembly of Aβ amyloid protofibrils: an in vitro model for a possible early event in Alzheimer's disease. Biochemistry. 1999;38:8972–8980. doi: 10.1021/bi9904149. [DOI] [PubMed] [Google Scholar]
- 61.Jan A, Hartley DM, Lashuel HA. Preparation and characterization of toxic Aβ aggregates for structural and functional studies in Alzheimer's disease research. Nat Protoc. 2010;5:1186–1209. doi: 10.1038/nprot.2010.72. [DOI] [PubMed] [Google Scholar]
- 62.Lashuel HA, Grillo-Bosch D. In vitro preparation of prefibrillar intermediates of amyloid-beta and alpha-synuclein. Methods Mol Biol. 2005;299:19–33. doi: 10.1385/1-59259-874-9:019. [DOI] [PubMed] [Google Scholar]
- 63.Hepler RW, Grimm KM, Nahas DD, Breese R, Dodson EC, Acton P, Keller PM, Yeager M, Wang H, Shughrue P, Kinney G, Joyce JG. Solution state characterization of amyloid β-derived diffusible ligands. Biochemistry. 2006;45:15157–15167. doi: 10.1021/bi061850f. [DOI] [PubMed] [Google Scholar]
- 64.Finder VH, Vodopivec I, Nitsch RM, Glockshuber R. The recombinant amyloid-beta peptide Abeta1-42 aggregates faster and is more neurotoxic than synthetic Abeta1-42. J Mol Biol. 2010;396:9–18. doi: 10.1016/j.jmb.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 65.Arosio P, Cukalevski R, Frohm B, Knowles TP, Linse S. Quantification of the concentration of Abeta42 propagons during the lag phase by an amyloid chain reaction assay. J Am Chem Soc. 2014;136:219–225. doi: 10.1021/ja408765u. [DOI] [PubMed] [Google Scholar]
- 66.Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TP. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A. 2013;110:9758–9763. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cukalevski R, Boland B, Frohm B, Thulin E, Walsh D, Linse S. Role of aromatic side chains in amyloid beta-protein aggregation. ACS Chem Neurosci. 2012;3:1008–1016. doi: 10.1021/cn300073s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arosio P, Michaels TC, Linse S, Mansson C, Emanuelsson C, Presto J, Johansson J, Vendruscolo M, Dobson CM, Knowles TP. Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat Commun. 2016;7:10948. doi: 10.1038/ncomms10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meisl G, Yang X, Frohm B, Knowles TP, Linse S. Quantitative analysis of intrinsic and extrinsic factors in the aggregation mechanism of Alzheimer-associated Abeta-peptide. Sci Rep. 2016;6:18728. doi: 10.1038/srep18728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 71.Walsh DM, Selkoe DJ. Aβ oligomers - a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 72.Portelius E, Brinkmalm G, Tran A, Andreasson U, Zetterberg H, Westman-Brinkmalm A, Blennow K, Ohrfelt A. Identification of novel N-terminal fragments of amyloid precursor protein in cerebrospinal fluid. Exp Neurol. 2010;223:351–358. doi: 10.1016/j.expneurol.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 73.Roberts BR, Ryan TM, Bush AI, Masters CL, Duce JA. The role of metallobiology and amyloid-beta peptides in Alzheimer's disease. J Neurochem. 2012;120:149–166. doi: 10.1111/j.1471-4159.2011.07500.x. [DOI] [PubMed] [Google Scholar]
- 74.Sergeant N, Bombois S, Ghestem A, Drobecq H, Kostanjevecki V, Missiaen C, Wattez A, David JP, Vanmechelen E, Sergheraert C, Delacourte A. Truncated beta-amyloid peptide species in pre-clinical Alzheimer's disease as new targets for the vaccination approach. J Neurochem. 2003;85:1581–1591. doi: 10.1046/j.1471-4159.2003.01818.x. [DOI] [PubMed] [Google Scholar]