

Abstract
Nitric oxide (NO) contributes to protection from tuberculosis (TB). It is generally assumed that this protection is due to direct inhibition of Mycobacterium tuberculosis (Mtb) growth, which prevents subsequent pathological inflammation. In contrast, we report NO primarily protects mice by repressing an interleukin-1 and 12/15-lipoxygenase dependent neutrophil recruitment cascade that promotes bacterial replication. Using Mtb mutants as indicators of the pathogen's environment, we inferred that granulocytic inflammation generates a nutrient-replete niche that supports Mtb growth. Parallel clinical studies indicate that a similar inflammatory pathway promotes TB in patients. The human 12/15 lipoxygenase ortholog, ALOX12, is expressed in cavitary TB lesions, the abundance of its products correlate with the number of airway neutrophils and bacterial burden, and a genetic polymorphism that increases ALOX12 expression is associated with TB risk. These data suggest that Mtb exploits neutrophilic inflammation to preferentially replicate at sites of tissue damage that promote contagion.
Despite Mtb's ability to sustain a persistent infection that can last for decades, most individuals remain asymptomatic because their immune response effectively contains the pathogen. Only a fraction of those infected with Mtb will develop the progressive inflammatory disease, tuberculosis. In these individuals, continual bacterial replication and progressive necrosis produces cavitary lesions contiguous with the airway, which allow bacteria to exit the host and infect others1. While all infected individuals mount a robust immune response to the pathogen, the immune mechanisms that differentiate protection from disease remain unclear.
Protective immunity to TB requires T cell-derived interferon gamma (IFN-γ), which induces expression of nitric oxide synthase 2 (Nos2) required for generating nitric oxide (NO) in macrophages. Animals lacking either of these factors suffer from severe TB disease characterized by high bacterial loads and granulocytic inflammation2. This correlation between neutrophils, Mtb burden, and pathology is a common feature of both animal models and human TB3-5. Since NO can kill Mtb in axenic culture, most models of protective immunity posit that this mediator primarily acts by inhibiting bacterial replication, which limits the subsequent inflammatory tissue damage. However, NO also inhibits inflammation by repressing the Caspase1-dependent processing of pro-IL-1β, and this activity prevents persistent neutrophil recruitment2. We sought to quantify the individual contribution of these two distinct activities of NO.
The primary protective role of NO is anti-inflammatory
Upon infection with Mtb, mice deficient for inducible NO generation, either due to genetic deletion of the Nos2 gene6 or chemical inhibition with aminoguanidine (AG)7, suffer from high bacterial burdens, weight loss, and progressive granulocyte accumulation that correlates with increased IL-1α and IL-1β (Supplementary Fig.1a-e). The granulocytes that accumulate in the lungs and spleens of Nos2-/- animals were predominantly CD11b+, Gr1hi, Ly-6Chi and Ly-6Ghi, and had the nuclear morphology of neutrophils (Fig. 1a and Supplementary Fig. 1f-i). A smaller proportion of Gr1int myeloid cells with more heterogeneous cytological appearances was also present, as shown in other susceptible mice8-10. Bone marrow chimeric mice were created to identify the source of protective NO and determine if this compound's cell-intrinsic antimicrobial activity is responsible for inhibiting disease. Hematopoietic reconstitution of irradiated Nos2-/- mice with wild type, but not Nos2-deficient, bone marrow cells inhibited all metrics of disease; weight loss, neutrophil influx, and bacterial replication (Fig. 1b and Supplementary Fig. 2a). To determine if the loss of Nos2 diminished the cell-autonomous anti-microbial capacity of the macrophage, wild type recipients were reconstituted with a 1:1 mixture of bone marrow cells from congenically marked wild type or mutant mice lacking either Nos2 or the IFNγ receptor (Ifngr). After a month of infection, chimerism of the CD11b+ myeloid compartment was maintained, indicating that both genotypes populated the lung and that no obvious differences in cell death were apparent (Supplementary Fig. 2b-c). At this time point, leukocytes were isolated from the lung, the wild type and mutant cells were separated, and the bacterial burden in each subpopulation was quantified. The relative proportion of Ly6G-positive and –negative cells recovered from each set of chimeric animals varied to some degree, but the purified cells were found to be an accurate representation of the populations found in the lung (Supplementary Fig. 2d-e). While Ifngr- deficient cells harbored significantly more bacteria than wild type, no difference in bacterial burden was observed between wild type and Nos2-/- cells, though both Ifngr-/- and Nos2-/- chimeric mice had relatively higher bacterial load in their lungs than the wild type (Fig. 1c-d, Supplementary figure 2f-g). It is possible that diffusible NO might partially complement an anti-microbial defect in the Nos2-/- cells. However, the greater effect of Ifngr deletion indicates that IFNγ-dependent anti-microbial processes11 are important for the control of Mtb replication, and that these processes are largely independent of Nos2.
Figure 1. Anti-inflammatory activity of Nos2 protects mice from TB disease.
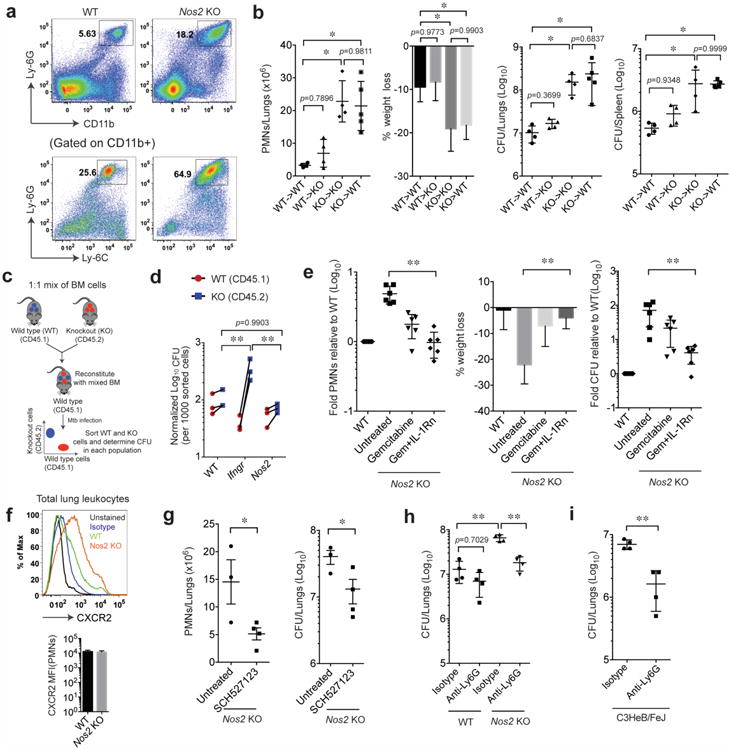
a. CD11b+ and Ly6-Ghi (inset in top panel), Ly-6C+ and Ly-6G+ (inset in bottom panel) neutrophils accumulate in the lungs of Mtb-infected Nos2-/- mice (gated on singlet/live cells). b. Bone marrow chimeric mice were infected with Mtb (notation indicates bone marrow donor genotype -> recipient genotype), and lung neutrophils, % weight loss, and total bacterial burden in the lungs and spleen (expressed in colony forming units, CFU) were assessed 4wks after infection. Values presented as Mean ± SD. **, p<0.01, one-way ANOVA with Tukey's multiple comparison test. c. Schematic for generation of mixed bone marrow chimeric mice. d. CFU were determined in the purified hematopoietic cells of indicated genotypes. **, p<0.01, Two-way ANOVA with Bonferroni multiple comparison test. e. Mtb infected Nos2-/- mice were treated with gemcitabine, either alone or in combination with IL-1Rn. After four weeks of infection, the indicated metrics of disease were quantified. Values shown (Mean ± SD) are pooled from two independent experiments. **, p<0.01, one-way ANOVA with Tukey's multiple comparison test. f. CXCR2 surface expression was determined in CD45+ lung leukocytes (top panel) and mean fluorescence intensity (MFI) of CXCR2 in CD45+ CD11b+ Ly-6G+ F4-80- neutrophils (bottom panel) obtained at 4wks post infection. g. CXCR1/2 signaling was blocked in infected Nos2-/- mice with SCH-527123, and the number of neutrophils and CFU in the lung were determined after 4wks of infection. Values presented as Mean ± SD. **, p<0.05, Two-tailed unpaired t-test with Welch correction. h. Bacterial load in the lungs of anti-Ly-6G- or isotype-treated animals were determined by CFU assay after 14 days of neutrophil depletion. All mice were infected via the aerosol route. **, p<0.001, one-way ANOVA with Tukey's multiple comparison test. i. Bacterial burden in the lungs of C3HeB/FeJ mice was determined in the lungs of anti-Ly-6G- or isotype-treated animals after 14 days of neutrophil depletion. **, p<0.001, Two-tailed unpaired t-test with Welch correction. All the data shown are representative of at least two independent experiments except in “g” and “i” (done only once).
Since we were unable to detect a cell-autonomous antimicrobial function for Nos2, we hypothesized that NO's IL-1 modulating anti-inflammatory activity might be responsible for protection. IL-1 plays a complex role in TB. This cytokine is produced early in infection, when it promotes anti-bacterial activity in macrophages12 and is essential for the establishment of immunity13. Later, upon onset of adaptive immunity, IL-1 production is suppressed by NO and the over-production of this cytokine can promote neutrophil influx and disease2,14. Thus, to specifically assess the importance of NO's ability to prevent IL-1 dependent neutrophil recruitment, we allowed the infection to progress for two weeks before inhibiting IL-1 signaling with IL-1 receptor antagonist (IL-1Rn) and/or depleting granulocytic precursors with the cytosine analog gemcitabine. Gemcitabine treatment reduced the number of lung neutrophils, histopathological disease, and wasting observed in Mtb-infected Nos2-/- mice, and the co-administration of IL-1Rn accentuated this effect (Fig. 1e). These treatments specifically affected the accumulation of neutrophils in the Nos2-/- animals, as the numbers of other leukocytes were not altered (Supplementary Fig. 3a). Remarkably, the anti-inflammatory activity of these treatments also restored control of bacterial replication. In this experiment, the number of neutrophils in the tissue correlated with bacterial burden and weight loss, and combination therapy returned these metrics to a level comparable with wild type C57BL/6 mice (Fig. 1e and Supplementary Fig. 3b). This observation suggested that sustained neutrophil recruitment might be the primary cause of both failed anti-mycobacterial immunity and tissue damage in Nos2-/- mice.
To further investigate the relationship between neutrophils and bacterial replication, we employed more specific reagents to inhibit the accumulation of these cells. CXCR2 signaling promotes neutrophil recruitment to the lungs of Mtb-infected mice15. We confirmed that lung-infiltrating neutrophils expressed CXCR2, and that Nos2-/- mice recruited more CXCR2+ cells (Fig. 1f). Like gemcitabine and IL-1Rn, pharmacological inhibition of CXCR2 reduced the number of neutrophils and bacteria in the lungs of Nos2-/- animals (Fig. 1g). The same effect was observed upon administration of the anti-Ly6G antibody, 1A8, which depleted Gr1hi neutrophils (Fig. 1h and Supplementary Fig. 3c-d). We conclude that NO's primary protective function in this model was to restrict the accumulation of neutrophils, and its direct anti-microbial activity played a relatively minor role. To extend the observations beyond the Nos2-/- mouse model, we investigated the role of neutrophils in the susceptible C3HeB/FeJ mouse. These animals develop inflammatory TB lesions due to increased macrophage necrosis, and accumulate a large number of neutrophils in their lungs and spleen, relative to C57BL/6 mice16 (Supplementary Fig. 1h and 3e). As observed in Nos2-/- animals, 1A8 administration depleted Gr-1hi neutrophils, and reduced lung bacterial burden in C3HeB/FeJ mice (Fig. 1i and Supplementary Fig. 3f), verifying that neutrophil influx promoted bacterial replication in multiple models of TB susceptibility.
Neutrophilic inflammation creates a growth-permissive environment for Mtb
The ability of neutrophil influx to increase the burden of Mtb suggested that this generally anti-microbial immune response was paradoxically creating a growth-permissive environment for Mtb. To investigate whether a change in the leukocytes encountered by Mtb could contribute to these environmental alterations, mice were infected with fluorescent Mtb that could be quantified by flow cytometry. We found that the loss of Nos2 altered the distribution of bacteria between myeloid populations. In both lung and spleen, the primary reservoir of Mtb changed from CD11b+, Ly-6G- monocytes/macrophages in wild type mice to CD11b+, Ly-6Ghi neutrophils in Nos2-/- animals (Fig. 2a-d).
Figure 2. Neutrophilic inflammation produces a growth permissive environment for Mtb.
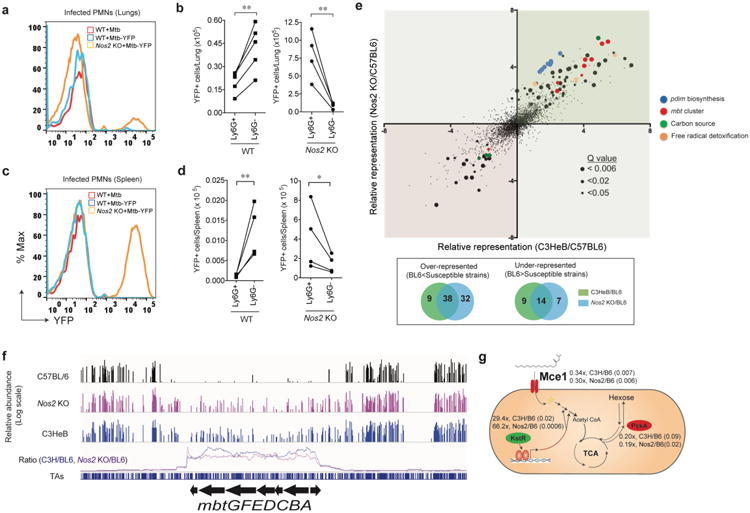
a. Representative histogram of Mtb-YFP fluorescence in CD11b+Ly-6Ghigh lung leukocytes of wild type C57BL/6 (WT) or Nos2-/- mice infected with either Mtb or Mtb-YFP via aerosol. b. Enumeration of infected (YFP+) neutrophils (CD11b+Ly-6G+) or monocytes/macrophages (CD11b+Ly-6G-) in WT and Nos2-/- mouse lungs after aerosol infection. c. Representative histogram of Mtb-YFP fluorescence in CD11b+Ly-6Ghigh cells and d. Enumeration of infected (YFP+) neutrophils (CD11b+Ly-6G+) or monocytes/macrophages (CD11b+Ly-6G-) in WT and Nos2-/- mice spleen after intravenous infection. (b and d). **, p<0.05, two-tailed unpaired t-test with Welch correction. Data presented are representative of two independent experiments. e. Genome-wide mutant fitness analysis in mouse models of different inflammatory potential. Mice were infected with Mtb transposon mutant library by intravenous injection. After four weeks of selection in mice, the relative abundance of individual Mtb mutants in which a single non-essential gene is disrupted (n=3127) was determined using TNseq. Each point represents the relative abundance of mutants lacking an individual gene in Nos2-/- (y-axis) or C3HeB mice (x-axis), each compared to C57BL/6. Point size indicates statistical significance (log of Q value) between C57BL/6 and Nos2-/- mice. Genes of known function are colored as described in the panel. Venn diagram showing the differential representation of the number of mutants in susceptible strains (Nos2-/- and C3HeB) relative to C57BL6. f. Representative TNseq data for the mycobactin gene cluster. Raw data (upper three tracks) displays the abundance of each insertion mutant (height of each bar on log scale) in mutant pools selected in the three indicated mouse strains. ‘Relative Abundance’ represents the number of Illumina reads corresponding to the insertion site as described in Methods. Lower track represents the ratio of relative mutant abundance over a window of 20 insertion sites between each mutant mouse and C57BL/6 control (indicated by color). g. Summary of differential selection on carbon metabolic mutants. The function of each gene or gene cluster is depicted along with the degree of differential abundance and Q value relative to the C57BL/6 control strain (Q val in parentheses). In panels f and g, the color of each protein represents the sign of its differential representation; red and green indicate decreased or increased representation in mutant mice, respectively.
Unlike activated macrophages, neutrophils do not produce significant amounts of NO17 and have a very limited capacity to restrict the replication of Mtb in vitro18. We confirmed that while wild type and Nos2-/- neutrophils were equally capable of killing gram-negative bacterial pathogens in ex vivo culture, these cells did not alter the viability of Mtb (Supplementary Fig. 4a-c). As a result, we hypothesized that neutrophil-rich lesions might represent a relatively benign niche for Mtb. To understand how growth in association with neutrophils altered the bacterial environment, we profiled the behavior of a bacterial mutant library in mice that develop histiocytic (C57BL/6) and granulocytic (Nos2-/-) lesions. To assess the impact of altered host cell association, and not simply the loss of NO per se, we also included the C3HeB/FeJ mouse strain in which neutrophil depletion reduce bacterial burden (Fig. 1i). Each mouse was infected with a saturated library of ∼50,000 independent Mtb transposon mutants, in which every non-essential gene is disrupted by insertion. One month after intravenous infection, bacteria were recovered and the relative abundance of each bacterial mutant in the three differentially selected libraries was compared by deep sequencing the amplified transposon-chromosome junctions. This approach provides a measure of each gene's contribution to the pathogen's fitness under conditions encountered in each mouse strain19. To maintain the complexity of the library in every animal, we assessed the relative fitness of bacterial mutants in the spleen, which represented a tractable model for the differential association of Mtb with macrophages versus neutrophils (Fig. 2c-d). The majority of mutants with significantly altered representation in Nos2-/- mice displayed comparable differential growth in C3HeB/FeJ animals (Fig. 2e), indicating that the bacterium is encountering a similar environment in these two mouse strains.
This analysis predominantly identified genes that were less stringently required for growth in the more permissive, hyper-inflammatory hosts (Fig. 2e, green quadrant). The genes of known function belonged to a small number of pathways related to stress responses and nutrient acquisition. One class of differentially required genes contained functions implicated in NO resistance that were less-stringently required in Nos2-/- mice. These included the ctpC and sseA genes that are necessary for toxic radical resistance20,21, the Mpa subunit of the proteasome that influences NO sensitivity22, and genes required for the synthesis of phthiocerol dimycocerosate (PDIM), a lipid that inhibits the recruitment of Nos2-expressing myeloid cells23 (Fig. 2e and Supplementary Tables 1 and 2). Together, these mechanisms appear to make Mtb resistant to the levels of NO encountered in vivo.
A second class of pathways related to nutrient acquisition was found to be less important in Nos2-/- and C3HeB/FeJ mice. Mutants lacking genes required for Mtb's only iron scavenging siderophore (mbtGFEADCBA)24 were lost specifically from the C57BL/6-passaged pool, indicating scavenging this micronutrient is less important in the susceptible mice (Fig. 2e-f). Neutrophilic inflammation also appeared to alter macronutrient availability. Mutations in central metabolic genes and regulators indicated that genes necessary for long-chain fatty acid uptake (Mce1)25, lipid catabolism (KstR regulon)26, and conversion of lipid substrates to biomass through gluconeogenesis (PckA)27 were more important for bacterial growth in Nos2-/- and C3HeB/FeJ animals (Fig. 2e, g and Supplementary Table 2). We infer that Mtb growing in association with neutrophils encounters a more hospitable environment that is replete with micronutrients, such as iron and lipid carbon sources.
IL-1 dependent 12/15-lipoxygenase expression promotes neutrophil recruitment and disease
We hypothesized that the pathological neutrophil recruitment cascade that is derepressed in Nos2-/- mice might be a more general mechanism promoting TB susceptibility in some individuals. Since previous work found that neutrophil influx into the lungs of Nos2-/- mice required NLRP3 inflammasome dependent IL-1 production2, we sought to define the inflammatory pathway downstream of IL-1 that is responsible for disease in these animals. To do this, we profiled the expression of a panel of proinflammatory mediators in animals that were lacking either Nlrp3 or the Il1r1. This panel focused on TNF as a known inflammatory cytokine regulated by IL-1, and the cyclooxygenase (COX) and lipoxygenase (LOX) enzymes, which produce potent inflammation-modulating eicosanoids28 (Supplementary Fig. 5a). To eliminate the contribution of differential bacterial replication in these mice, we used an auxotrophic strain of Mtb that is unable to replicate during infection, but remains viable and promotes an inflammatory reaction2,29. As expected, deletion of either Nlrp3 or Il1r1 abolished neutrophil recruitment to the lungs of infected animals, even when Nos2 was inhibited (Fig. 3a). Of the eicosanoid-biosynthetic enzymes that we profiled, only the expression of Alox15, which encodes the 12/15-lipoxygenase (12/15-LOX) enzyme, correlated with neutrophil influx (Fig. 3b and Supplementary Fig. 5b). Alox15 mRNA levels were significantly increased upon Nos2 inhibition, and this induction depended upon both Nlrp3 and the ll1r1. Alox15 protein was predominately associated with the leukocytic infiltrate in mouse lung, and was particularly abundant in the context of Nos2 inhibition (Fig. 3c).
Figure 3. IL-1 dependent 12/15-LOX products contribute to TB susceptibility in Nos2-/- mice.
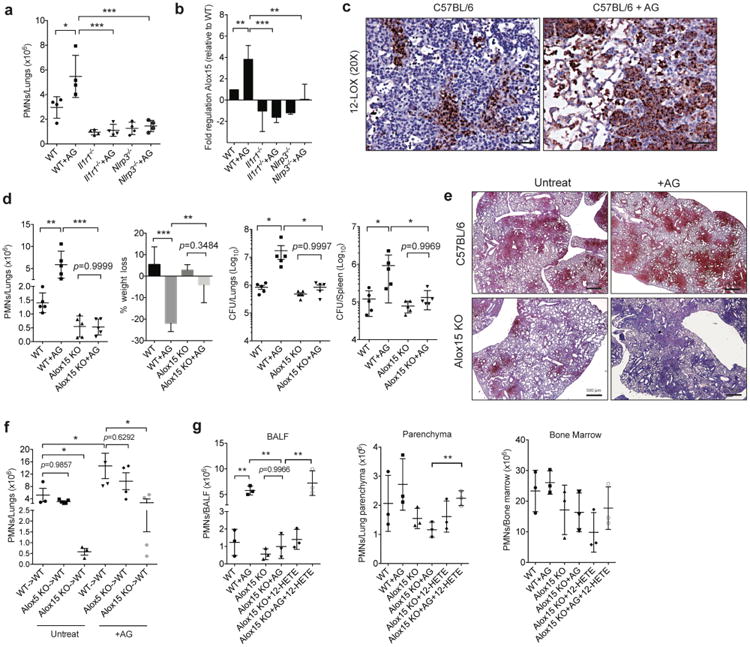
a. Neutrophil infiltration into the lungs was quantified in each mouse strain 4 weeks after intratracheal infection with ∼1×105 Mtb strain 18b. Aminoguanidine treated groups are indicated (“AG”). b. Relative abundance of Alox15 mRNA in the lungs of animals from panel “a” was quantified by qRT-PCR. Data shown (Mean ± SD) are representative of two independent experiments, *, p<0.05; **, p<0.001, ***, p<0.0001, one-way ANOVA with Tukey's multiple comparison test. c. WT and Alox15-/- mice were infected with ∼200 CFU of Mtb H37Rv via the aerosol route. 4 wks after infection, 12/15-LOX expression in the lungs was quantified by immunohistochemical staining of lung sections of untreated- and AG-treated WT mice (Scale bars represent 500um). d. Lung neutrophils, % weight loss, and total bacterial burden in the lung and spleen were quantified in untreated and AG treated groups of WTs and Alox15-/- after 4 wks of Mtb infection. Data shown (Mean ± SD) are representative of six independent experiments. *, p<0.05; **, p<0.001, ***, p<0.0001, one-way ANOVA with Tukey's multiple comparison test. e. Immunohistochemical staining for anti-Ly6G in lung sections of Mtb infected WT and Alox15-/- mice treated with/without AG (Scale bars represent 500um). f. Lung neutrophils in cohorts of the indicated bone marrow chimeric mice were determined five weeks after infection. Notation indicates bone marrow donor genotype -> recipient genotype. Aminoguanidine treated groups are indicated (“AG”). g. Neutrophil numbers in bronchoalveolar lavage fluid (BALF), lung parenchyma after lavage, and bone marrow were determined in Mtb infected WT and Alox15-/- mice, which were treated with 12-HETE and/or AG. (f-g), Data shown (Mean ± SD) are from one experiment. *, p<0.05; **, p<0.001, one-way ANOVA with Tukey's multiple comparison test. All mice were infected via the aerosol route, except as indicated in panel “a”.
Consistent with these observations, Alox15-/- mice recruited very few neutrophils to their lungs upon Mtb infection, when compared to wild type controls. Even Nos2 inhibition did not promote neutrophil recruitment in Alox15-/- animals (Fig. 3d-e). In contrast, deletion of Alox5, which encodes the 5-lipoxygenase (5-LOX), had no effect on neutrophil recruitment (Supplementary Fig. 6a). Similarly, mice lacking the leukotriene B4 receptor 1 (Ltb4r1-/-), which governs neutrophil chemotaxis in response to 5-LOX derived LTB430, had a relatively minor impairment in neutrophil influx when compared to Alox15 deletion (Supplementary Fig. 6b). As we observed with neutrophil depletion in Nos2-/- mice, the reduction in lung neutrophils in Alox15-/- mice correlated with a reversal in weight loss, improvement of histopathological disease, and restoration of bacterial control after Nos2 inhibition (Fig. 3d-e). Neither neutrophil depletion or Alox15 deletion significantly affected CFU numbers in Nos2-sufficient animals, likely because relatively few neutrophils infiltrate the lung in this situation. Pharmacological blockade of 12/15-LOX31, had a qualitatively similar effect as genetic deletion of Alox15, ruling out unappreciated developmental effects of the Alox15 mutation (Supplementary Fig. 6c). By these metrics of disease, Alox15-/- mice were essentially resistant to the exacerbated TB disease caused by Nos2 inhibition (Fig. 3d). Reconstitution of wild type mice with Alox15-/-, but not Alox5-/-, bone marrow reduced neutrophil influx into the lung. While the effect of Alox15 deletion on neutrophil numbers and CFU was less robust in these chimeric animals than intact Alox15-/- animals, these data support an important role for hematopoetically-derived 12/15-LOX in neutrophil recruitment (Fig. 3f and Supplementary Fig. 6b).
To determine which eicosanoids correlated with 12/15-LOX-dependent neutrophil recruitment, we quantified the levels of relevant lipid mediators in Alox15-/- and wild type animals in the presence or absence of Nos2 inhibitor. No significant alterations in COX products were observed between these cohorts. The abundance of both the 5-LOX product, LTB4, and the 12/15-LOX product, 12-hydroxyeicosatetraenoic acid (12-HETE), correlated with the degree of inflammation (Supplementary Fig. 7). As both of these products are produced by inflammatory leukocytes, we concluded that LOX products are markers of neutrophil influx. However, our genetic data specifically implicated 12/15-LOX products as the primary drivers of inflammation.
12/15-LOX produces 12-hydroperoxyeicosateraenoic acid (12-HpETE) and 12-HETE which could be responsible for pro-inflammatory effects either by acting directly on neutrophils32 or through conversion into more potent chemotactic mediators33. To verify that 12/15-LOX products could contribute to neutrophil recruitment during Mtb infection, we supplemented Alox15-/- animals with 12-HETE. This treatment restored neutrophil numbers in the bronchoalveolar lavage (BAL) to wild type levels (Fig. 3g). The opposing effects of Nos2 and Alox15 were manifested predominantly in the airway, modulation of these mediators had only a modest effect on the number of neutrophils remaining in the lung after lavage, and no significant effect on the number of CD11b+/Ly-6G+ neutrophil precursors in the bone marrow (Fig. 3g). 12-HETE supplementation of both wild type and Alox15-/- mice increased the total number of lung neutrophils, and the numbers of these cells correlated with bacterial burden throughout this experiment (Supplementary Fig. 6c). Thus, a single 12/15-LOX product was sufficient to promote neutrophil recruitment to the lung.
Increased 12-LOX activity is associated with inflammatory TB in humans
To understand if the inflammatory pathway delineated in Nos2-/- mice represents a mechanism of TB susceptibility in natural populations, we investigated whether polymorphisms in orthologous genes were associated with human TB. Significant differences in eicosanoid pathways exist between mice and humans. For example, the 12- and 15-LOX activities possessed by murine 12/15-LOX are expressed by separate human proteins. To account for these differences, a total of 112 single nucleotide polymorphisms (SNPs) in six eicosanoid-modifying genes (Supplementary Table 3) were genotyped in a cohort of TB patients and healthy controls. These SNPs were prioritized based on their minor allele frequency (>10%) and their position in 5′ or 3′ untranslated regions that could alter gene expression.
Polymorphic alleles of the leukotriene A4 hydrolase (LTA4H) gene were previously associated with TB susceptibility34. In general support of these findings, our diseased group was enriched for the minor alleles of two LTA4H SNPs. However, these associations did not reach statistical significance after multiple testing correction (Supplementary Table 4). Similarly, no significant association was found for SNPs in genes encoding COX-1 (PTGS1), COX -2 (PTGS2), or 15-LOX1 (ALOX15). In contrast, among 12 SNPs in the 5-LOX gene (ALOX5), one was significantly associated with TB. More notably, two SNPs (rs3840880 and rs9904779) near the myeloid/platelet-expressed 12-LOX (ALOX12) gene, which produces 12-HETE in humans, were associated with TB (Table 1).
Table 1. The distribution of alleles and genotypes for two ALOX12 SNPs between tuberculosis patients (TB) and healthy controls (HC).
| SNP ID | Trait | No. | Allele (frequency%) | Allelic comparison | |||||
|---|---|---|---|---|---|---|---|---|---|
| DEL | G | χ2 | P * | P adj # | OR | 95%CI | |||
| rs3840880 | TB | 943 | 1027(54.5) | 859(45.5) | |||||
| HC | 934 | 1119(59.9) | 749(40.1) | 11.38 | 0.0007 | 0.001 | 1.25 | 1.098-1.422 | |
| C | G | ||||||||
| rs9904779 | TB | 943 | 961(51.0) | 925(49.0) | |||||
| HC | 934 | 856(45.8) | 1012(54.2) | 9.89 | 0.0017 | 0.027 | 0.814 | 0.716-0.925 | |
The genotypes of SNP rs3840880 and rs9904779, which are located at the promoter region of ALOX12 gene, were determined using Massarray platform in TB patients (n=943) and healthy controls (n=934).
Compares the difference in allele frequency between TB cases and healthy controls.
P value adjusted by Bonferroni correction for multiple tests.
Both rs3840880 and rs9904779 are located in the promoter region of the ALOX12 gene. Using a luciferase reporter system, the TB-associated allele of rs3840880 produced significantly higher transcriptional activity than its allelic variant, indicating a direct effect of this polymorphism on ALOX12 expression. No significant difference in transcription was observed between allelic variants at position rs9904779 (Fig. 4a-b). These results provided genetic evidence that increased ALOX12 expression could promote TB.
Figure 4. Increased expression and activity of 12-LOX are associated with active tuberculosis in humans.
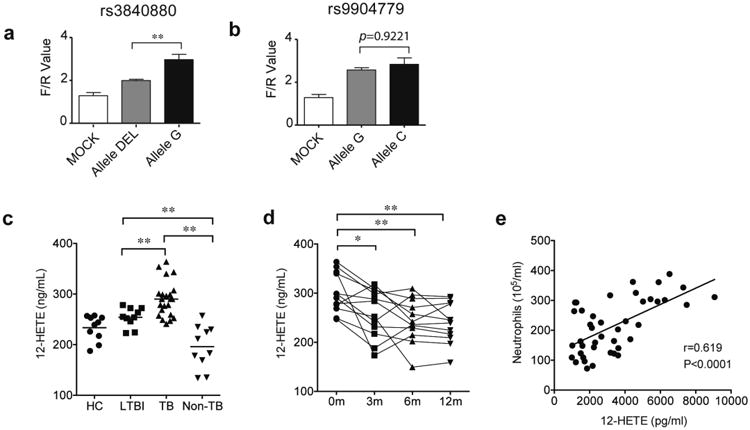
a-b. ALOX12-promoter luciferase reporter plasmids carrying different alleles at rs3840880 or rs9904779 were transfected into HeLa cells. After 48h, the indicated luciferase activities were quantified. “F/R value” indicates the ratio of Firefly/Renilla luciferase. Values shown as Mean ± SD. **, p<0.001, one-way ANOVA with Tukey's multiple comparison test. c. The concentrations of 12-HETE in the plasma from healthy controls, ‘HC’ (n = 10), latent TB infection, ‘LTBI’ (n = 10), pulmonary tuberculosis, ‘TB’ (n = 20), and subjects suffering from non-TB lung disease, ‘non-TB’ (n = 10) were determined by ELISA. d. 12-HETE levels in the plasma of active TB patients were determined by ELISA at the indicated time points after initiation of anti-TB chemotherapy. e. 12-HETE levels were significantly correlated with the neutrophil counts in the BALF of patients with pulmonary TB, at the time of diagnosis (n=42). The difference among groups were compared by using one-way ANOVA followed by Tukey's multiple comparison test *, p<0.05, **, p<0.001. Pearson's correlation coefficient (r) and P value of correlation (P) are indicated.
To further investigate whether human 12-LOX activity correlates with inflammatory TB, we assessed the concentration of its product, 12-HETE, in relation to disease, bacterial burden, and neutrophil infiltration. The 12-HETE concentration in peripheral blood of TB patients was significantly higher than in healthy cohorts (HC) or patients with non-TB lung diseases. (Fig. 4c). Plasma 12-HETE levels decreased significantly over the first three months of TB chemotherapy (Fig. 4d) mirroring the clearance of bacteria. Moreover, in bronchoalveolar lavage fluid (BALF) of TB patients, the level of 12-HETE positively correlated with the abundance of neutrophils (Fig. 4e). This association was similar to that observed previously for IL-1β and neutrophils in BALF of TB patients14.
The association between 12-LOX and granulocytic inflammation was further investigated in human cavitary TB lesions using quantitative immunohistochemistry. These lesions consist of an acellular necrotic center (caseum), which is sequentially surrounded by a myeloid cell-rich region (‘macrophage rim’) and a fibrotic capsule. Quantification of multiple lesions indicated that inflammatory markers, such as TNF-α and myeloperoxidase (MPO) expressing neutrophils, were found in the region adjacent to the caseum (Fig. 5a-g). This region encompassed both the cellular and fibrotic outer edge of the cavity and the macrophage rim. Consistent with the pro-inflammatory role played by its murine ortholog, we found that human 12-LOX expression was concentrated in this TNF-α and MPO-rich region of the cavitary granuloma (Fig. 5h) which was previously defined as an inflammatory region35. Collectively, these clinical and histopathological observations indicate that ALOX12 expression and 12-HETE levels correlate with TB disease, the burden of bacteria, and the recruitment of neutrophils. More generally, these findings support a coordinated role for IL-1 and 12-LOX products in the generation of inflammatory lesions that promote the replication and dissemination of Mtb.
Figure 5. ALOX12 in expressed in inflammatory areas of cavitary TB lesions.
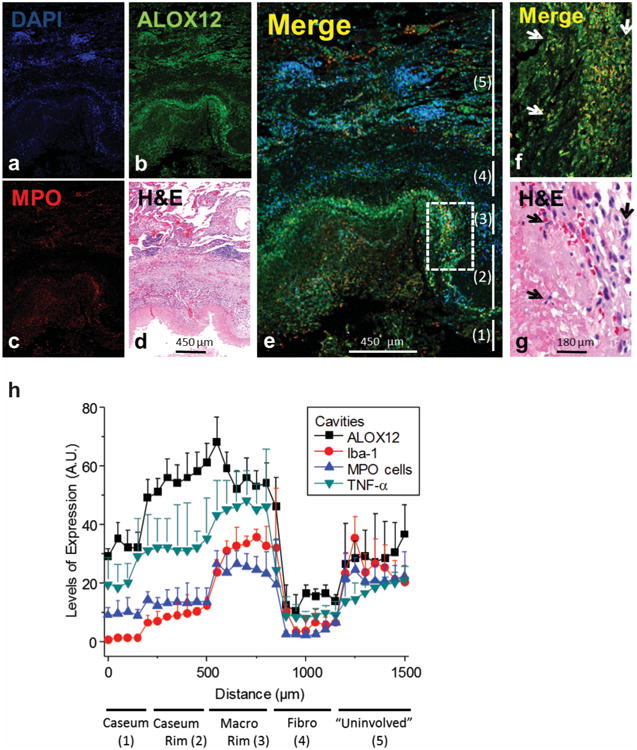
(a-e) Representative single channel images of a lung cavity, including the caseous center at the bottom and adjacent cellular/fibrotic layers, stained for DAPI (a), ALOX12 (b) MPO (c) and a merged image of all three (e). d. Hematoxylin-eosin (H&E) staining of the region shown in (a-e). f. Magnification of the inset outlined in (e) showing the co-localization of MPO positive cells and ALOX12 at the interface between the caseous region and cellular rim. The numbers to the right refer to the 5 regions of the cavitary lesion delineated in panel (h), from the caseum and outward into the uninvolved lung tissue. (g) H&E staining of the region shown in (f). Arrows in (f-g) denote MPO- and ALOX12-positive neutrophils with characteristic multi-lobular nuclei. h. Quantification of ALOX12, TNF-α, Iba-1 and MPO positive pixels in regions extending from the caseous center to the outer rim and into the cellular and fibrotic layers, performed in 4 different cavitary tissues and averaged. Arbitrary units (A.U.) on y-axis indicates the average number of positive pixels in the indicated region. X-axis indicates the ‘distance’ from a fixed point in the caseum. The analysis is described in detail in the Methods section.
Discussion
The protective roles of IFNγ and IFNγ-inducible NO are often attributed to a direct anti-microbial activity, and our data confirm that IFNγ signaling is essential for the control of Mtb intracellular growth (Fig. 1d). In contrast, the basis for susceptibility Nos2-/- mice was not related to cell-autonomous anti-microbial defect. Instead, NO's primary role was anti-inflammatory, and the lack of Nos2 increased susceptibility by providing a growth-permissive milieu for the pathogen in association with neutrophils. Since NO cannot account for the observed anti-microbial activity of IFNγ signaling, other effector mechanisms induced by this cytokine11, must be responsible for IFNγ's ability to control the replication of Mtb.
While neutrophil depletion can extend the survival of Mtb-susceptible mouse strains other than Nos2-/- mice, the relative contributions of neutrophil-mediated tissue damage versus the amplification of bacterial replication have not been determined in these models. As a result, it is currently unclear if the neutrophils in other mouse models are functionally similar to those found in Nos2-/- mice. Indeed, Ly6G+ granulocytes are now appreciated to encompass a variety of subsets that serve both phagocytic and immunomodulatory roles9. Further functional characterization of the neutrophils that promote Mtb replication in Nos2-/- and other mouse strains could provide additional markers of susceptibility to be investigated in clinical cohorts.
While other intracellular pathogens transiently infect neutrophils during the establishment of infection41 or use these cells as a primary replication site42, the preferential growth of Mtb in neutrophil-rich lesions is most reminiscent of Salmonella enterica subspecies Typhimurium. This pathogen thrives at sites of inflammation by using oxidized byproducts of neutrophil activation as substrates for anaerobic respiration43. Our global phenotypic analysis of bacterial mutants showed no indication of altered usage of analogous trimethylamine-N-oxide (TMAO) or nitrate reductases in Nos2-/- mice (Supplementary Table 1 and 2). Instead, Mtb appears to exploit the increased accessibility of nutrients in granulocyte-rich lesions (Supplementary Fig. 9). We speculate that the inferred abundance of nutrients is due to the cell death observed in advanced TB lesions, which are rich in the lipid carbon substrates preferred by Mtb44, and are likely to contain iron sources such as heme that Mtb can acquire without mycobactin45. A remarkable similarity between these two pathogens is the common reliance on inflammation for efficient transmission, salmonella via generation of bacteria-rich inflammatory diarrhea, and Mtb via generation of cavitary lesions that facilitate the production of infectious aerosols. Enhancement of transmission provides strong selection for these pathogens to thrive at sites of intense inflammation that would be lethal to other microbes.
Methods
Mice
C57BL/6 (stock no. 000664), Nos2-/- (B6.129P2-Nos2™1Lau/J, stock no. 002609), Alox15-/- (B6.129S2-Alox15™1Fun/J, stock no. 002778) and Alox5-/- (B6.129S2-Alox5™1Fun/J, stock no. 004155), C3HeB/FeJ (stock no. 00658), B6.SJL-Ptprca Pepcb/BoyJ carrying the pan leukocyte marker CD45.1 or Ly-5.1 (stock no. 002014), Ltb4r1-/- (B6.129S4-Ltb4r1™1Adl/J, stock no. 008102), Il1r1-/- (B6.129S7-Il1r1™1Imx/J, stock no. 003245and Nlrp3-/- (B6.129S6-Nlrp3™1Bhk/J, stock no. 021302) were purchased from the Jackson Laboratory. Mice were housed under specific pathogen-free conditions, and in accordance with the University of Massachusetts (UMASS) Medical School, IACUC guidelines. All mouse strains used in this study were of C57BL/6 background unless otherwise indicated.
Mouse infection
The wild type strain of M. tuberculosis (Mtb) used in these studies was PDIM positive H37Rv. Bacteria were cultured in 7H9 medium containing 0.05% Tween 80 and OADC enrichment (Becton Dickinson). Bacteria expressing yellow fluorescent protein (msfYFP) were generated by transformation of H37Rv with plasmid PMV261 that constitutively expresses msfYFP under the control of the hsp60 promoter. For infections, mycobacteria were suspended in phosphate-buffered saline (PBS)-Tween 80 (0.05%); clumps were dissociated by sonication, and ∼200 CFU were delivered via the respiratory route using an aerosol generation device (Glas-Col) or 1×106 CFU by intravenous route. Mouse infections with the streptomycin auxotrophic strain of M.tuberculosis, 18b, were carried out as described previously2.
Immunohistochemistry (IHC)
Lung tissues were fixed in 10% buffered formalin and embedded in paraffin. Five micrometer-thick sections were stained with hematoxylin and eosin. All staining was done by the Diabetes and Endocrinology Research Center histopathology core facility at the University of Massachusetts Medical School. IHC for 12/15 LOX in the lung sections were done by staining the lung sections with 15-LO antibody clone H-235 (sc-32940). Lung sections from Alox15-/- mice were used as negative control for these staining.
Drug treatment of animals
Aminoguanidine treatment
Mice were supplied with drinking water containing 2.5% aminoguanidine hemisulfate (CAS no. 996-19-0, Sigma) 7 days prior to Mtb infection. Water was replaced every week.
Gemcitabine treatment
Nos2-/- mice were treated with gemcitabine hydrochloride (Cat. no. G6423, Sigma-Aldrich, St. Louis, MO) via intra-peritoneal route, 10 days' post Mtb infection. Gemcitabine treatment was repeated in every three days until the 25th day post infection in a total of four doses of 100mg/kg/mouse.
Anakinra treatment
IL-1Rn (Kineret™) was given at 25mg/kg via Alzet osmotic pumps, implanted in each animal subcutaneously near the trunk.
12/15 LOX inhibitors
Baicalein (Cat. no.1761), from TOCRIS, was given to Nos2-/- mouse at 25mg/kg through Alzet osmotic pumps from 2 days to 28 days' post infection.
12-HETE treatment
12-HETE (Cayman Chemicals) was administered to mice via alzet osmotic mini pumps (50ug/kg) for four weeks starting on the day of infection.
CXCR2 inhibitor
CXCR1/2 antagonist SCH527123 (Cat. no. CS-0609, ChemScene) was administered to animals via Alzet osmotic mini pumps at 5mg/kg, for four weeks, beginning one day after infection.
Neutrophil depletion
Anti-Ly-6G depleting antibody, clone 1A8 (BioXcell) was administered as described by Nandi et al46. 200 ug of 1A8 or isotype control (clone 2A3, BioXcell) antibody was administered to animals via intra-peritoneal injection starting 10 days' post-infection, and then every other day for 11 days. Ly-6G+ neutrophil depletion was verified by FACS analysis of CD11b+ Gr-1+ Ly-6C- cell populations and animals were euthanized at day 24 of infection to determine bacterial burden and cytokines in the lungs.
Flow Cytometry
Single cell suspensions were prepared from the bronchoalveolar lavage fluid (BALF) obtained from the mouse lung. Lung tissue was digested with Collagenase type IV/DNaseI and passed through 40 μm cell strainers to obtain single cell suspension. Non-specific antibody binding sites were blocked by Fc-Block CD16/32 (Clone 93, cat. no. 101319) and the cells were stained with anti-CD3-PE (Clone 17A2, cat. no. 100205), anti-CD11b-PerCP Cy5.5 (Clone M1/70, cat. no.101227), anti-Ly-6G-FITC (Clone 1A8, cat. no.127605), anti-Ly-6C-PE (Clone HK1.4, cat. no.128007), anti-Gr1-APC (Clone RB6-8C5, cat. no.108411), anti-CXCR2/CD182-PE (Clone SA04484, cat. no. 149303), anti-CD11c-Alexa 700 (Clone N418, cat. no. 117319), anti-F4/80-PE-Cy7 (Clone BM8, cat. no.), anti-CD45-Pacific Blue (Clone 30-F11, cat. no. 103126), anti-CD45.1-PE (Clone A20, cat. no. 110707), anti-CD45.2-FITC (Clone 104, cat. no. 109804). Antibodies were purchased from Bio legend. All analyses were performed on live cells only after staining them with fixable live dead stain conjugated with eFlour780, purchased from eBiosciences. For infections with fluorescent H37Rv, lung tissue was prepared as described above and stained with anti-Ly-6G-Pacific Blue (Clone 1A8 cat. no. 127612), anti-CD11b-APC-Cy7 (Clone M1/70 cat. no. 101226) and Live/Dead Fixable Violet Dead Cell Stain (Molecular Probes, cat. no. L-34963), and compared against lung tissue infected with non-fluorescent H37Rv. All the staining was done according to the manufacturer's instructions. Lung or spleen cells were surface stained for 30 minutes at room temperature, fixed for 20 minutes at 4°C using the Cytofix buffer (BD-Biosciences, cat. no. 554655).
Cell sorting and morphological analysis
To sort Gr-1high an Gr-1int cells, single cell suspensions were prepared from M. tuberculosis Erdman infected lungs of mice as described above. After incubating with 5ug/ml of Fc-Block (eBiosciences) for 15 minutes at 4°C, cells were stained with anti-CD11b (clone M1/70) and Gr-1(clone RB6-8C5) as described above. A live-dead stain was used to exclude the dead cells. Cells were then fixed with Cytofix (BD biosciences). Fixed cells were further fractionated into CD11b+Gr1high and CD11b+Gr-1int by a FACS Aria IIu Cell sorter (BD biosciences). Sorted cells were analyzed for purity using FACS Aria and both the Gr-1high and Gr-1int cells were obtained at >90% purity. FACS sorted cells were cytospun onto cytoslides (Thermo-scientific), stained with DAPI and images were acquired in a Delta Vision deconvolution microscope. The images were taken with a 60× objective and the Delta Vision SoftWorx software was used to deconvolve the images.
Cytokine measurement and immunoblotting in tissue homogenates
Murine cytokine concentrations in culture supernatants and cell free lung homogenates were quantified using commercial ELISA kits (BD Opt EIA). All samples were normalized for total protein content. 12/15 LOX expression in macrophage lysates was determined by western blotting with 12-LOX antibody H-100 (sc-32939) purchased from Santa Cruz biotechnology Inc.
Generation of Bone marrow chimeric mice
B6.SJL-Ptprca Pepcb/BoyJ (CD45.1/Ly-5.1) mice were lethally irradiated (900-1000 rads or 90-100Gy) and reconstituted with a total of 107 donor bone marrow (BM) cells from C57BL/6 CD45.2 or Alox15-/- or Alox5-/- or Nos2-/- in CD45.2 background. Mice were allowed to reconstitute for 8 weeks before aerosol infection with H37Rv was performed. Approximately 85-95% reconstitution of donor bone marrow was achieved in this experiment. Surface expression of congenic markers CD45.1 and CD45.2 antigens was checked by flow cytometry before (blood) and after infection (infected lung) in each cohort.
Mixed bone marrow chimera generation and cell sorting
Wild type CD45.1+ mice were lethally irradiated with two doses of 600 rads. The following day bone marrow from CD45.1+ wild type mice and CD45.2+ knockout mice (wild type, Ifngr-/-, or Nos2-/-) was isolated, red blood cells were lysed using Tris-buffered Ammonium Chloride (ACT), and the remaining cells were quantified using a hemocytometer. CD45.1+ and CD45.2+ cells were then mixed equally at a 1:1 ratio and 107 cells from this mixture were injected intravenously into lethally irradiated hosts. Reconstitution of host mice lasted 6-8 weeks with preventative sulfatrim treatment for the first 4 weeks. Mixed bone marrow chimera mice were then infected by low dose aerosol with M. tuberculosis H37Rv. 4 weeks following infection the lungs of chimera mice were harvested and single cell suspensions were made following collagenase treatment and tissue disruption. An aliquot of this suspension was saved for flow cytometry analysis of the lung population. The remaining cells were split equally and stained with either Anti-CD45.1 conjugated to APC or Anti-CD45.2 conjugated to PE. Stained populations were then incubated with either Anti-APC or Anti-PE magnetic beads (Miltenyi) following manufacturer's instructions. Stained populations were then sorted individually by positive selection using LS-columns following manufacturer's instructions (Miltenyi). Following elution of each population, purified cells were divided equally and then plated for M. tuberculosis on 7H10 agar or counted and stained for analysis of cellular purity. Cells from the input homogenate, flow through and the positive were stained with fixable Live/Dead Aqua (Life Tech), Anti-CD11b-FITC, Anti-CD45.2 PE, Anti-CD45.1 APC, Anti-CD4 PerCP, Ly6G-Pacific Blue (Biolegend) and analyzed on a MacsQuant flow cytometer (Miltenyi). Samples with >90% were used for subsequent analysis. 21 days after plating colonies were enumerated and the M. tuberculosis levels per sorted cell were determined.
Isolation of neutrophils from mouse bone marrow
Bone marrow cells were isolated from C57BL/6J and Nos2-/- mice femurs, red blood cells were lysed using Tris-buffered Ammonium Chloride (ACT), and single cell suspension was prepared. Mouse neutrophils were prepared by negative selection using the neutrophil isolation kit from Miltenyi biotec (Cat. no. 130-097-658) according to the manufacturer's protocol. Cell isolation was performed with an LS column. Purity of the neutrophils were tested by counterstaining the isolated cell population with anti-Ly-6G-APC (Clone 1A8, cat. no. 127613) and further analyzed in flow cytometer. Isolated neutrophils were incubated with Yersinia pestis strains (JG102A with an intact Type III secretion system and JG102B, that lacks the type III secretion system) and Enterohemorragic E. coli strain lacking the hemolytic toxin. Cells were either activated with phorbol myristate acetate (PMA) or left unactivated during the assay. Yersinia pestis strains were grown in a custom made serum nutritional medium47. M.tuberculosis Erdman strain was incubated for 16h with media alone or bone marrow derived neutrophils from WT or Nos2-/- mice. The % survival in the presence of neutrophils was calculated relative to media control.
Sample preparation for TnSeq analysis
For infection with Mtb transposon mutants, bacteria were grown in 7H9 medium containing 0.05% Tween 80, OADC, and kanamycin (25ug/mL) to an OD600=1.0. Bacterial cells were pelleted and resuspended in PBS containing 0.05% Tween 80 and sonicated. 0.2 ml of single cell suspension was used for infecting different mouse strains via the lateral tail vein at a concentration of 5×106 bacilli/ml. At indicated times, animals were sacked, and spleen homogenates were plated on 7H10 plates supplemented with 0.05% Tween 80, OADC and 25ug/mL of Kanamycin. Colonies were scraped off the plates by a rubber policeman and bacterial genomic DNA was prepared as described in Long JE et al19. Transposon insertion sites tagged with digital barcodes, amplified, sequenced, and quantified as described. To determine relative abundance of the number of independent transposon-chromosome junctions corresponding to a given gene were divided by the number of possible insertions sites in that gene that contained insertions (‘Hits/TA”). This metric was compared between samples to determine ‘Relative abundance’. Statistical significance was determined using a nonparametric permutation test to calculate a P value and adjusting for multiple testing using the Benjamin–Hochberg correction to obtain the Q value.
qRT-PCR
Total RNA from lung tissue was isolated by homogenizing the lung tissue in QIAZOL (QIAGEN) and then using the Qiagen RNeasy Plus universal kit with genomic DNA eliminator solution. Individual RNA samples (100ng each) were subjected to quantitative RT-PCR using the QuantiTect SYBR Green RT-PCR kit. In this method, mRNA levels for each sample were normalized to Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA levels and then expressed as fold regulation in gene expression using the SABiosciences RT2 profiler PCR data analysis software (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php). The sequences of the specific primers are as follows: Lta4h: Forward: 5′-gctactggcaggtgacaagt-3′ and reverse: 5′-tttccccaaacaaccgtcca-3′, Alox5: Forward: 5′-ctacgatgtcaccgtggatg-3′ and reverse: 5′-gtgctgcttgaggatgtgaa-3′, Ptgs1/Cox1: Forward: 5′-cttctccacgatctggcttcgtga-3′ and reverse: 5′-cttgagctgcaggaaatagccact-3′, Ptgs2/Cox2: Forward: 5′-acacactctatcactggcacc-3′ and reverse: 5′-ttcagggagaagcgtttgc-3′, Alox15: Forward: 5′-gtctccctgcccgcctggta-3′ and reverse: 5′-gggggatccaagggcgtga-3′, Tnf: Forward: 5′-cacagaaagcatgatccgcgacgt-3′ and reverse: 5′-cggcagagaggaggttgactttct-3′. Gapdh: Forward: 5′-aactttggcattgtggaagg-3′ and reverse: 5′-acacattgggggtaggaaca-3′.
Lipid quantification by LC-MS
Lung homogenates were prepared in 50% methanol containing 0.02% bromohydroxytoluene (BHT) by homogenizing the infected lung samples first in cold PBS+0.04% BHT, followed by equal volume of 100% chilled methanol+0.04% BHT. The homogenates were then kept at 4C for 60 minutes, centrifuged at 4000 rpm for 20 minutes at 4C, and the supernatants were filtered through 0.2um PTFE filters. Extracted lung homogenates were reduced to near dryness, resuspended in 1 mL of water, and applied to 3 mL, 500 mg Bond Elute C18 cartridges (Agilent Technologies) pre-conditioned with 10 mL methanol followed by 10 mL water. The bound material was washed with 4 mL water, 5 mL ice-cold hexane, and eicosanoids eluted with 5 mL methanol. Samples were then dried by nitrogen and reconstituted in 30 μL of 50% methanol for LC-MS/MS analysis.
Deuterium-labeled or unlabeled internal standards (listed in Supplementary Table 5), Cayman Chemical), were suspended in 100% ethanol and serially diluted to generate an 11-point standard curve. Briefly, each standard solution was added 100 μL (10 ng) of the working internal standard solution. Final working concentrations of the standard curve ranged from 6.25 ng/mL to 10 μg/mL for each eicosanoid.
Samples were analyzed on a Dionex UltiMate 3000 UPLC (Thermo Scientific) configured to a TSQ Quantiva (Thermo) triple quadrupole mass spectrometer operating in negative ion mode. Samples and calibrants were injected (10 uL) at 200 μL/min onto a 100 × 2.1 mm BEH (Waters Corporation) C18 1.7 μm UPLC column heated at 40°C and fitted with a guard column of similar chemistry (Waters). Mobile phase A was 10 mM aqueous ammonium acetate (pH 5.0) and B was 100% acetonitrile. The gradient program was as follows: 0-4 min (20-40 %B); 4-14 min (40-80 %B); 14-14.1 min (80-95 %B); 14.1-16 min (95% B); 16-16.1 min (95-20 %B); 16.1-25 min (20 %B). Eicosanoid molecular anions were analyzed by scheduled single reaction monitoring (SRM) with parent to fragment ion transitions and optimized collision energies previously determined by direct infusion of eicosanoid standards (Supplementary Table 5). Transitions highlighted in bold were used for quantitative analysis while others were used for secondary confirmation.
Eicosanoids were analyzed by generating extracted ion chromatograms from the quantitative SRM transitions (Supplementary Table 5) and integrating the peak areas using the Xcalibur (version 3.0.63, Thermo) Quan Browser software module. Chromatograms were background subtracted (INCOS noise) and smoothed (9 points, gaussian) prior to integration. For each analyte, a standard curve was constructed over the 6.25 ng/mL – 10 ug/mL range by plotting the ratio of the peak areas of the analyte to corresponding deuterium labeled internal standard. Exceptions to this were 11-, 8-, and 9-HETE which were expressed relative to 12-, 12-, and 5-HETE-d8, respectively. Standard curves were fit using a “1/x” weighting and omitting the origin. Data from study samples were processed in a similar way to the standards and eicosanoid concentrations were determined by relation to the standard curves. All measured sample concentrations fell within the standard curve with the exception of 12-HETE and PGD2. For the latter analytes, standard curves were extrapolated 5× beyond the top point to allow a concentration estimate.
Human subjects and samples
We used a case-control cohort involving 943 pulmonary TB patients and 934 healthy controls (HC) from the Shenzhen Third People's Hospital that was previously described14,48. Diagnosis of active TB was based on clinical symptoms, chest radiography, and microscopy for acid fast bacilli (AFB), sputum and/or BALF Mtb culture and response to anti-TB chemotherapy. Healthy controls with normal chest radiographic findings and without a clinical history of TB were recruited. Mtb-specific interferon gamma release assays (IGRA) were used to differentiate individuals with LTBI from healthy controls (HC) without infection as described previously49 All subjects were genetically unrelated members of the Chinese Han population. The ages of each group (years, mean±SD) were: pulmonary TB, 38.74±12.33; and HC, 36.46±11.52. The ratio of male to female for TB and HC group is 709/234 and 576/358, respectively. All SNPs were in Hardy-Weinberg equilibrium in both diseased and healthy groups (P>0.05).
Plasma samples were collected from HC (n=10), LTBI (n=10), Mtb cultured confirmed TB (n=20), and patients with non-TB lung diseases (non-TB, n=10). The non-TB lung diseases group included patients with pneumonia caused by Streptococcus pneumoniae (n=3), Mycoplasma pneumoniae (n=2), Cytomegalovirus (n=2), and Klebsiella pneumoniae (n=3). The whole blood and plasma samples were collected and stored at -80C. Unless otherwise indicated, all samples from patients were collected before initiation of antibiotic treatment. Aliquots of bronchoalveolar lavage fluid (BALF) samples from pulmonary TB patients (n= 42), which were described previously, used to correlate 12-HETE levels and neutrophil numbers. The levels of 12-HETE in the BALF and plasma were measured with a kit from Enzo Life Sciences (Farmingdale, NY).
All protocols involved human samples had been approved by the Research Ethics Committee of Shenzhen Third People's Hospital with informed consent obtained from all participants. The use, for research purposes, of excess BALF left over from clinically indicated bronchoscopies was deemed exempt from a requirement for informed consent beyond the consent normally obtained for this clinical procedure, as previously described.
SNP selection and genotyping
Genomic DNA was prepared from peripheral whole blood according to the standard protocols of QIAamp DNA Blood Mini kit (Qiagen, Hilden, Germany). SNPs were selected according to methods described previously, with an additional focus on potential regulatory regions, such as transcription factor binding sites in the promoter region and microRNA target sites in the 3′-untranslated region (UTR). SNPs were genotyped using the MassARRAY system (Sequenom, San Diego, CA) as described elsewhere.
12-LOX promoter plasmid construction and dual-luciferase assay
A 1081 base-pair sequence of the 12-LOX promoter region, from -1029 to +52, was cloned into the pGL3-Basic vector (Promega, Madison, WI) upstream of the firefly luciferase coding region. Positive clones were subjected to site-directed mutagenesis using Quick Change Site-Directed mutagenesis kit (Stratagene, La Jolla, CA) to obtain the desired alleles. Transient transfections into HeLa cell were performed using lipofectamine 2000 reagent (Life Technologies, Carlsbad, CA). Briefly, 2×105 HeLa cells were seeded into 24-well plates and incubated overnight, the cultured cells were co-transfected with 0.7ug of constructed vectors, or pGL3-Basic original vector, and 0.1ug of internal control Renilla luciferase reporter plasmid pRL-CMV. After additional 48h incubation, luciferase activity was measured using Dual-Glo luciferase assay system (Promega, Madison, WI). Firefly luciferase was normalized against Renilla luciferase activity to account for variation in transfection assay. All experiments were performed at least twice, with each transfection in triplicate.
Immunohistochemistry for human lungs
Paraffin lung tissue sections were cut at 6 to 7 μm thickness, mounted on ultraclean glass slides covered in silane, deparaffinized, then dehydrated and rehydrated using the following steps: Ethanol solutions (30, 50, 70, 90, 95 and 100 % for 3 min each), xylenes (2 different solutions for 10 min each) and ethanol solutions (100, 95, 90, 70, 50 and 30 for 3 min each). The slides were washed once in Tris buffer saline (TBS) for 5 min. Slices were subjected to antigen retrieval by boiling in sodium citrate buffer at pH=6.0 for 20 min and incubated in Triton-X 0.1% for 5 min. Slices were removed and allowed to equilibrate to room temperature for at least 20 min, and rinsed with distilled water. Tissue sections were blocked (blocking solution; 0.5 M EDTA, 1% horse serum, 1% Ig free BSA, 4% human serum, and 1% fish gelatin in PBS) and incubated overnight in primary antibodies against the proteins related to our studies. Sections were stained for nuclei (DAPI, blue staining), ALOX12 (Alexa488, green staining), myeloperoxidase, MPO, to identify granulocytes (Cy3, red staining), and Iba-1, a macrophage marker (Cy5, cyan staining). As controls, pre-immune serum and isotype matched controls were used (Supplementary figure 8). After incubation, the tissues were washed several times with sterile TBS at room temperature and incubated in the respective secondary antibodies (anti-mouse, anti-goat or anti-rabbit conjugated to Alexa-488, Alexa588, or Alexa-647) and streptavidin conjugated to Texas-red for mycobacterium detection for at least 1h at room temperature. Tissue sections were mounted using Prolong Gold Antifade reagent (Invitrogen, grand Island, NY) with DAPI, and the tissue sections were examined in an A1 confocal microscope equipped with a spectrum detection and unmixing systems (Nikon Instruments Inc., Japan). Antibody specificity was confirmed by replacing the primary antibody with a non-specific myeloma protein of the same isotype or non-immune serum.
Three-dimensional reconstruction and deconvolution of areas of interest was performed from 12 to 25 optical sections obtained at 0.250 or 0.150 μm intervals50. To analyze and quantify abundance of the study proteins, the number of positive pixels as well as their intensity in macrophages were measured in specific regions of interest. In addition, for each cell population, a linear intensity histogram was generated to examine the expression of each enzyme inside defined cell types or in caseum. To measure the distance from the center of the granuloma or cavity in a manner that was consistent across lesions with caseous foci of varying diameters, we used the macrophage layer directly subtending the caseum as the ‘anchor point’ and computed pixel density both in the inward and outward directions from the anchor point. To compare protein abundance across tissues, similar numbers of cells and areas were included in the 3D deconvoluted optical reconstructions. This analysis avoids problems associated with inflammatory versus normal tissues where density and structure of the tissue are different. Adjacent sections were stained with hematoxylin & eosin (H & E), correlate the confocal findings with histopathology of the same tissue sections. Co-localization and numbers of positive pixels for each color was quantified by using the imaging software NIS Elements (Nikon, Japan). Analysis of nuclei, macrophage, and H & E staining identified at least 5 different layers from the center of the cavity going outward: caseum (acellular); caseum outer rim (interface between caseum and macrophages); macrophage rim (area enriched for Iba-1 positive cells); and uninvolved lung containing minimal infiltration of monocytes and granulocytes. A student's t-test was used for comparing fluorescence average intensities in different regions. Individual pixels with an intensity equal or greater than 8% of the maximum intensity were considered positive (P < 0.00042).
Statistical analysis
The statistical significance of differences between data groups was determined using the Unpaired two tailed student's t-test, or one-way ANOVA using Tukey's multiple comparison test using Graphpad Prism 6; * (p<0.05) ** (p<0.001) *** (p < 0.0001) denotes significant differences compared as indicated in figures.
Supplementary Material
Acknowledgments
This work was supported by grants from the Natural Science Foundation of China to XC (81525016,81501714, 81500004); the NIH to CMS (AI064282 and AI107774), RRL (AI120556), SGP (AI003749), and EE (MH096625); and the Arnold and Mabel Beckman Foundation to AJO. The authors thank Caitlin Moss for providing technical help, Dr. Samuel M Behar for critical review of the manuscript, and the UMMS department of animal medicine animal care.
Footnotes
Data Availability: The data that support the findings of this study are available from the corresponding author upon request.
Author contributions: BBM and CMS conceived and designed the study. BBM, RRL, AJO performed mouse experiments. GZ, WW and XC designed and performed the SNPs study in Chinese cohorts. EE and VD designed and performed the IHC study in the lung biopsies from TB patients and analyzed data. SGP, SN, CMS, MGB and JYP provided technical help during various experiments. BBM, RRL and CMS analyzed the data. BBM and CMS wrote the manuscript and CMS supervised the overall study.
Author information: The authors declare no competing financial interest.
References
- 1.Canetti G. The Tubercle Bacillus in the Pulmonary Lesion of Man. The American Journal of the Medical Sciences. 1956;231:480. [Google Scholar]
- 2.Mishra BB, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat Immunol. 2013;14:52–60. doi: 10.1038/ni.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niazi MKK, et al. Lung necrosis and neutrophils reflect common pathways of susceptibility to Mycobacterium tuberculosis in genetically diverse, immune-competent mice. Dis Model Mech. 2015;8:1141–1153. doi: 10.1242/dmm.020867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mattila JT, Maiello P, Sun T, Via LE, Flynn JL. Granzyme B-expressing neutrophils correlate with bacterial load in granulomas from Mycobacterium tuberculosis-infected cynomolgus macaques. Cell Microbiol. 2015;17:1085–1097. doi: 10.1111/cmi.12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berry MPR, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scanga CA, et al. The Inducible Nitric Oxide Synthase Locus Confers Protection against Aerogenic Challenge of Both Clinical and Laboratory Strains of Mycobacterium tuberculosis in Mice. Infect Immun. 2001;69:7711–7717. doi: 10.1128/IAI.69.12.7711-7717.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flynn JL, Scanga CA, Tanaka KE, Chan J. Effects of aminoguanidine on latent murine tuberculosis. J Immunol. 1998;160:1796–1803. [PubMed] [Google Scholar]
- 8.Tsiganov EN, et al. Gr-1dimCD11b+ immature myeloid-derived suppressor cells but not neutrophils are markers of lethal tuberculosis infection in mice. J Immunol. 2014;192:4718–4727. doi: 10.4049/jimmunol.1301365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knaul JK, et al. Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am J Respir Crit Care Med. 2014;190:1053–1066. doi: 10.1164/rccm.201405-0828OC. [DOI] [PubMed] [Google Scholar]
- 10.Obregón-Henao A, Henao-Tamayo M, Orme IM, Ordway DJ. Gr1(int)CD11b+ myeloid-derived suppressor cells in Mycobacterium tuberculosis infection. PLoS ONE. 2013;8:e80669. doi: 10.1371/journal.pone.0080669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol. 2012;12:367–382. doi: 10.1038/nri3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jayaraman P, et al. IL-1β promotes antimicrobial immunity in macrophages by regulating TNFR signaling and caspase-3 activation. J Immunol. 2013;190:4196–4204. doi: 10.4049/jimmunol.1202688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fremond CM, et al. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol. 2007;179:1178–1189. doi: 10.4049/jimmunol.179.2.1178. [DOI] [PubMed] [Google Scholar]
- 14.Zhang G, et al. Allele-specific induction of IL-1β expression by C/EBPβ and PU.1 contributes to increased tuberculosis susceptibility. PLoS Pathog. 2014;10:e1004426. doi: 10.1371/journal.ppat.1004426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nouailles G, et al. CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J Clin Invest. 2014;124:1268–1282. doi: 10.1172/JCI72030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irwin SM, et al. Presence of multiple lesion types with vastly different microenvironments in C3HeB/FeJ mice following aerosol infection with Mycobacterium tuberculosis. Dis Model Mech. 2015;8:591–602. doi: 10.1242/dmm.019570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Padgett EL, Pruett SB. Rat, mouse and human neutrophils stimulated by a variety of activating agents produce much less nitrite than rodent macrophages. Immunology. 1995;84:135–141. [PMC free article] [PubMed] [Google Scholar]
- 18.Denis M. Human Neutrophils, Activated with Cytokines or Not, Do Not Kill Virulent Mycobacterium tuberculosis. Journal of Infectious Diseases. 1991;163:919–920. doi: 10.1093/infdis/163.4.919. [DOI] [PubMed] [Google Scholar]
- 19.Long JE, et al. Identifying essential genes in Mycobacterium tuberculosis by global phenotypic profiling. Methods Mol Biol. 2015;1279:79–95. doi: 10.1007/978-1-4939-2398-4_6. [DOI] [PubMed] [Google Scholar]
- 20.Padilla-Benavides T, Long JE, Raimunda D, Sassetti CM, Argüello JM. A novel P(1B)-type Mn2+-transporting ATPase is required for secreted protein metallation in mycobacteria. J Biol Chem. 2013;288:11334–11347. doi: 10.1074/jbc.M112.448175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nambi S, et al. The Oxidative Stress Network of Mycobacterium tuberculosis Reveals Coordination between Radical Detoxification Systems. Cell Host Microbe. 2015;17:829–837. doi: 10.1016/j.chom.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 23.Cambier CJ, et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature. 2014;505:218–222. doi: 10.1038/nature12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quadri LE, Sello J, Keating TA, Weinreb PH, Walsh CT. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem Biol. 1998;5:631–645. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]
- 25.Forrellad MA, et al. Role of the Mce1 transporter in the lipid homeostasis of Mycobacterium tuberculosis. Tuberculosis (Edinb) 2014;94:170–177. doi: 10.1016/j.tube.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kendall SL, et al. A highly conserved transcriptional repressor controls a large regulon involved in lipid degradation in Mycobacterium smegmatis and Mycobacterium tuberculosis. Mol Microbiol. 2007;65:684–699. doi: 10.1111/j.1365-2958.2007.05827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marrero J, Rhee KY, Schnappinger D, Pethe K, Ehrt S. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc Natl Acad Sci USA. 2010;107:9819–9824. doi: 10.1073/pnas.1000715107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. 2015;15:511–523. doi: 10.1038/nri3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kashino SS, Ovendale P, Izzo A, Campos-Neto A. Unique model of dormant infection for tuberculosis vaccine development. Clin Vaccine Immunol. 2006;13:1014–1021. doi: 10.1128/CVI.00120-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lämmermann T, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 2013;498:371–375. doi: 10.1038/nature12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Leyen K, et al. Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke. 2006;37:3014–3018. doi: 10.1161/01.STR.0000249004.25444.a5. [DOI] [PubMed] [Google Scholar]
- 32.Reynaud D, Pace-Asciak CR. 12-HETE and 12-HPETE potently stimulate intracellular release of calcium in intact human neutrophils. Prostaglandins Leukot Essent Fatty Acids. 1997;56:9–12. doi: 10.1016/s0952-3278(97)90518-4. [DOI] [PubMed] [Google Scholar]
- 33.Mrsny RJ, et al. Identification of hepoxilin A3 in inflammatory events: a required role in neutrophil migration across intestinal epithelia. Proceedings of the National Academy of Sciences. 2004;101:7421–7426. doi: 10.1073/pnas.0400832101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tobin DM, et al. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell. 2010;140:717–730. doi: 10.1016/j.cell.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marakalala MJ, et al. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat Med. 2016;22:531–538. doi: 10.1038/nm.4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDonald B, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 37.Hurley BP, Pirzai W, Mumy KL, Gronert K, McCormick BA. Selective eicosanoid-generating capacity of cytoplasmic phospholipase A2 in Pseudomonas aeruginosa-infected epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2011;300:L286–94. doi: 10.1152/ajplung.00147.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamang DL, et al. Hepoxilin A(3) facilitates neutrophilic breach of lipoxygenase-expressing airway epithelial barriers. J Immunol. 2012;189:4960–4969. doi: 10.4049/jimmunol.1201922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhowmick R, et al. Systemic disease during Streptococcus pneumoniae acute lung infection requires 12-lipoxygenase-dependent inflammation. J Immunol. 2013;191:5115–5123. doi: 10.4049/jimmunol.1300522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mayer-Barber KD, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. 2014;511:99–103. doi: 10.1038/nature13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laskay T, van Zandbergen G, Solbach W. Neutrophil granulocytes – Trojan horses for Leishmania major and other intracellular microbes? Trends in Microbiology. 2003;11:210–214. doi: 10.1016/s0966-842x(03)00075-1. [DOI] [PubMed] [Google Scholar]
- 42.Herron MJ, et al. Intracellular parasitism by the human granulocytic ehrlichiosis bacterium through the P-selectin ligand, PSGL-1. Science. 2000;288:1653–1656. doi: 10.1126/science.288.5471.1653. [DOI] [PubMed] [Google Scholar]
- 43.Winter SE, Lopez CA, Bäumler AJ. The dynamics of gut-associated microbial communities during inflammation. EMBO Rep. 2013;14:319–327. doi: 10.1038/embor.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim MJ, et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med. 2010;2:258–274. doi: 10.1002/emmm.201000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Owens CP, et al. The Mycobacterium tuberculosis secreted protein Rv0203 transfers heme to membrane proteins MmpL3 and MmpL11. J Biol Chem. 2013;288:21714–21728. doi: 10.1074/jbc.M113.453076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nandi B, Behar SM. Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J Exp Med. 2011;208:2251–2262. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palace SG, Proulx MK, Lu S, Baker RE, Goguen JD. Genome-wide mutant fitness profiling identifies nutritional requirements for optimal growth of Yersinia pestis in deep tissue. MBio. 2014;5:e01385–14–e01385–14. doi: 10.1128/mBio.01385-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang G, et al. An SNP selection strategy identified IL-22 associating with susceptibility to tuberculosis in Chinese. Sci Rep. 2011;1:20. doi: 10.1038/srep00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen X, et al. Diagnosis of active tuberculosis in China using an in-house gamma interferon enzyme-linked immunospot assay. Clin Vaccine Immunol. 2009;16:879–884. doi: 10.1128/CVI.00044-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guan YQ, Cai YY, Zhang X, Lee YT, Opas M. Adaptive correction technique for 3D reconstruction of fluorescence microscopy images. Microsc Res Tech. 2008;71:146–157. doi: 10.1002/jemt.20536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.