

Abstract
Idiopathic CD4+ lymphopenia (ICL) predisposes to opportunistic infections (OIs) but can often remain asymptomatic and does not have a strong association with monogenic mutations. Likewise, cryptococcal meningoencephalitis, the most common OI in ICL, is not strongly associated with monogenic mutations. In this study, we describe 2 patients with ICL plus an additional immune defect: one from an E57K genetic mutation in the nuclear factor-κβ essential modulator, and the other with acquired autoantibodies to granulocyte-macrophage colony-stimulating factor. Thus, these cases may exemplify a “multi-hit model” in patients with ICL who acquire OIs.
Keywords: autoantibody, meningoencephalitis, Cryptococcus, idiopathic CD4+ lymphopenia
Idiopathic CD4+ lymphopenia (ICL) is a rare heterogeneous syndrome defined as an absolute CD4+ T-cell count less than 300/mm3 on 2 separate occasions in the absence of identified causes of lymphopenia such as human immunodeficiency virus (HIV)-1/2 [1]. Defects in cellular immunity, including depletion of naive and expansion of memory T-cell subsets as well as reduced T-cell receptor (TCR) diversity and signaling, have been described [2]. However, few monogenic mutations have been described that result in ICL-related opportunistic infections (OIs) such as UNC119 or TNFSF6/Fas ligand mutations [3, 4]. Patients with ICL may acquire OIs including, most commonly, cryptococcal meningoencephalitis (CM) with the acquired immune deficiency syndrome-related fungus, Cryptococcus neoformans (Cn), but can also remain asymptomatic and have no readily identifiable immunodeficiency [5, 6]. Molecular mechanisms that differentiate infected from persistently asymptomatic patients remain elusive. It is interesting to note that unlike HIV, which predisposes to a variety of OIs, often simultaneously, previously healthy patients including those with ICL who develop infections with OIs typically tend to each have only 1 or 2 infective organisms, suggesting other genetic defects that may bias towards a specific pathogen predisposition. In addition, although several monogenic defects have been associated with cell wall-exposed fungi such as Candida albicans [7], only a few associations have been made with infections from encapsulated fungi such as Cn, predominantly those with antibodies to granulocyte-macrophage colony stimulating factor (anti-GMCSF) infected with Cryptococcus gattii, and 1 case with a GATA2 mutation and another with autoantibodies to IFN-γ [8]. This has led us to search for secondary predispositions to OIs with Cn in a cohort of previously healthy patients with ICL.
METHODS
Informed consent was obtained from all patients followed at the National Institutes of Health Clinical Center under an institutional review board-approved protocol (National Institute of Allergy and Infectious Diseases [NIAID] Protocol No. 93-I-0106). We performed whole exome sequencing on deoxyribonucleic acid (DNA) acquired from blood (PAXgene, PreAnalytiX; QIAGEN). We used an ion torrent platform to readily identify mutations that affect certain defined immunologic pathways. All candidate gene polymorphisms were confirmed via Sanger sequencing of polymerase chain reaction products. For example, we used the following primers on complementary DNA for the IKBKG gene that encodes nuclear factor-κβ (NF-κB) essential modulator (NEMO): forward: 5’-ACTTGACTGCGCTCTATCGA-3’and reverse: 5’-TCAACAGCTGAAGCGTAAGG-3’.
Peripheral blood mononuclear cells (PBMCs) were obtained via Ficoll isolation of heparinized blood and frozen (90% fetal calf serum/10% dimethyl sulfoxide) at −140°C until use. Experiments were done in Roswell Park Memorial Institute (RPMI) media (see below) and 10% fetal calf serum after thawing. T lymphocyte stimulations of indicated duration were performed with anti-CD3-coated plates at 10 μg/mL, 5 μg/mL, 1 μg/mL, and unstimulated titrations with 2 μg/mL fixed anti-CD28 upon CD3+ negatively selected lymphocytes (eBioscience/Affymetrix, San Diego, CA). We performed flow cytometry to define leukocyte subsets using CD4, APC-eFluor 780; CD8a, Pacific Blue; CD27, PerCP-eFluor 710; CD45RA, Alexa Fluor 700; and interferon (IFN)γ fluorescein isothiocyanate. Intracellular staining for nuclear factor κ-light-chain enhancer of activated B cells – NF-κB (RelA [p65], S529) efluor660 (eBioscience/Affymetrix) expression and IκBα phycoerythrin ([PE] BD Bioscience, San Jose, CA) degradation was performed, as previously described [9]. Data were collected using FACS LSRII or Calibur (BD Biosciences) and analyzed using FlowJo (TreeStar). All flow experiments were performed in at least duplicate.
Plasma samples were screened for anticytokine autoantibodies using a particle-based approach. In brief, differentially fluorescing magnetic beads (Bio-Rad) were covalently coupled to 2.5 μg of recombinant human GMCSF (R&D Systems), IFNα (PBL Biomedical Laboratories), IFNβ (PeproTech), IFNγ, IFNω (PeproTech), interleukin (IL)-1α, IL-4, IL-6, IL-12p70, IL-17A, IL-17F, IL-18, or IL-22 (all from eBioscience). Beads were combined and incubated for 30 minutes with plasma samples (diluted 1:100), washed, then incubated for an additional 30 minutes with PE-labeled goat antihuman immunoglobulin (Ig)G (eBioscience/Affymetrix). Washed beads were run in a multiplex fashion on the BioPlex X200 instrument (Bio-Rad). Data were graphed with Prism 6 (GraphPad). Normal PBMCs (1 × 106) were cultured in complete RPMI 1640 media consisting of 2 mM glutamine, 20 mM Hepes, 100 U/mL penicillin, and 100 μg/mL streptomycin with 10% patient or normal plasma. Cultures were left unstimulated or stimulated with recombinant human GMCSF (10 ng/mL; R&D Systems) for 30 minutes at 37°C. Monocytes were identified by CD14 surface staining before being fixed and permeabilized for intranuclear staining with anti-phosphoSTAT-5 (Y694) antibody (BD Biosciences) [10].
RESULTS
Cryptococcal meningoencephalitis Case 1 was a 53-year-old, previously healthy, HIV-negative male who presented with vertigo, unstable gait, paranoia, disorientation, and short-term memory deficits. Two months after presentation, the patient was diagnosed with CM due to Cn by cerebrospinal fluid (CSF) culture. Clinical immunological evaluation demonstrated persistent CD4+ lymphopenia and expansion of terminal effector memory cells relative to naive T-cell subsets and evidence of elevated activation markers such as increased numbers of HLA-DR% cells (Table 1), as reported previously with ICL [2]. Bone marrow biopsy revealed oligoclonal TCR gene rearrangement but no evidence of hematological malignancy by flow cytometry and cytogenetics. He responded well to 4 weeks of liposomal amphotericin with negative CSF cultures, but his course was subsequently complicated by hemolytic anemia that worsened over time (hemoglobin = 10.7 g/dL [13.7–17.5 normal], mean cell volume = 100.3 femtoliters [79.0–92.2], reticulocyte% = 5.12 [0.51–1.81 normal], %iron saturation = 17 [15–62 normal], lactate dehydrogenase = 285 U/L [113–226 normal], platelets = 171 K/µL [161–347 normal], white blood cells = 2.62 K/µL [4.23–9.07 normal], absolute neutrophil count = 1.82 K/µL [1.78–5.38 normal], absolute lymphocyte count = 0.48 K/µL [1.32–3.57 normal], absolute monocyte count = 0.27 K/µL [0.30–0.82 normal]) responsive to splenectomy and pulmonary emboli with an associated lupus anticoagulant. The patient demonstrated normal Ig levels but had a suboptimal response to the 23-valent pneumococcal vaccine given 2 months after 13-valent conjugate pneumococcal vaccine with <8 serotypes responding (Table 2). Autoantibodies to GMCSF and IFNγ were negative, but genetic analysis demonstrated a G169A IKBKG (accession number: Q9Y6K9) X-linked hemizygous mutation (E57K NEMO) (Figure 1A), although he possessed no clinical features of anhidrotic ectodermal dysplasia reported with this mutation [11]. The patient recovered from his infectious course, and his hemolytic anemia remained stable at 18 months, but ICL persisted; partial recovery of CD8+ T-cell counts after splenectomy suggested that a direct genetic effect for his lymphopenia was less likely (Figure 1B). Moreover, as shown in Figure 1C, the E57K NEMO mutation of CM Case 1 was associated with decreased IFNγ production by cells enriched for CD4+ T cells after TCR stimulation with anti-CD3 and anti-CD28 compared with healthy controls, but it was similar to that of 2 children (E57K no. 1 and E57K no. 2) carrying E57K NEMO mutations as controls (NIAID Protocol no. 93- I-0119), although the latter were not associated with CD4+ lymphopenias (Table 1) or unusual infections. In addition, upstream NF-κB (RelA [p65]) phospho-S529 expression and IkBα degradation of cells from CM Case 1 enriched for CD3 cells were decreased, as expected for a functional mutation in NEMO (Figure 1D).
Table 1.
Lymphocyte Subpopulations
| Analysis | CM Case 1a | E57K No. 1 | E57K No. 2 | CM Case 2 | Normalb |
|---|---|---|---|---|---|
| Lymphocyte Subpopulations | |||||
| Lymph count | 330 | 1640 | 1900 | 370 | 1320–3570/µL |
| Lymph distribution | |||||
| T cells | 221 | 1260 | 1376 | 168 | 714–2266/µL |
| B cells | 30 | 335 | 397 | 187 | 59–329/µL |
| NK cells | 79 | 51 | 131 | 165 | 126–729/µL |
| T cells (%CD3+) | |||||
| TCRαβ+ | 59.1 | 73 | 67 | 17 | 56%–80% |
| TCRγδ+ | 7.8 | 4 | 6 | 15 | 0.5%–10% |
| HLA-DR% | 17.7 | 8 | 4 | 10 | 0%–20% |
| CD4+CD8− | 14.5 | 38 | 41 | 5 | 32%–62% |
| CD4−CD8+ | 45.9 | 33 | 25 | 23 | 11%–35% |
| CD4−CD8− | 5.9 | 5 | 6 | 5 | 1%–9% |
| CD62L+CD45RA+ (naive) | |||||
| (%CD4) | 23 | 62 | 53 | 20 | 14%–67% |
| (%CD8) | 34 | 69 | 67 | 10 | 25%–73% |
| CD62L+CD45RA− (CM) | |||||
| (%CD4) | 6.3 | 25 | 33 | 42 | 26%–64% |
| (%CD8) | 0 | 9 | 8 | 36 | 6%–40% |
| CD62L−CD45RA− | (EM) | ||||
| (%CD4) | 21 | 7 | 9 | 38 | 5%–30% |
| (%CD8) | 0 | 7 | 13 | 44 | 6%–34% |
| CD62L−CD45RA+ | (TEMRA) | ||||
| (%CD4) | 50 | 6 | 5 | 0 | 0%–4% |
| (%CD8) | 66 | 15 | 13 | 10 | 5%–33% |
| CD4/CD45RA/CD31% | NP | NP | NP | 1 | 2%–20% |
| CD8/CD45RA/CD31% | NP | NP | NP | 2 | 3%–18% |
| Immunoglobulin Assessment | |||||
| IgM | 183 | <21 | <21 | 72 | 34–342 mg/dL |
| IgG | 924 | 398 | 619 | 1190 | 642–1730 mg/dL |
| IgA | 285 | 26 | 43 | 84 | 91–499 mg/dL |
| IgE | 17.7 | NP | NP | 11.8 | 0.0–90.0 IU/mL |
Abbreviations: ANOVA, analysis of variance; CM, cryptococcal meningoencephalitis; EM, effector memory; Ig, immunoglobulin; NK, natural killer; NP, not performed; TEMRA, terminal effector memory expressing CD45RA.
aWe performed an ANOVA analysis finding that CM Case 1 had significantly lower total lymphocyte and T-cell counts compared with controls (P < .002 and P < .01, respectively), but further analyses were not conducted due to the small number of values in this initial case report.
bNinety-five percent range of 40 healthy volunteers.
Table 2.
Immunoglobulin (Ig) Assessment
| PneumococcalSerotype | CM Case 1 Random | CM Case 1 Postimmunization | CM Case 2 Random | CM Case 2 Postimmunization | Normal Range(mcg/mL) |
|---|---|---|---|---|---|
| Serotype 1 | <0.5 | 1.1 | 1.1 | 1.9 | ≥2.3 |
| Serotype 2 | <0.2 | 0.2 | 2.9 | 5.4 | ≥1.0 |
| Serotype 3 | <0.2 | 0.3 | 2.5 | 2.6 | ≥1.8 |
| Serotype 4 | <0.2 | 0.2 | 0.2 | 0.2 | ≥0.6 |
| Serotype 5 | 1 | 1.7 | 5.3 | 9.6 | ≥10.7 |
| Serotype 8 | <0.3 | 0.8 | 1.8 | 1.3 | ≥2.9 |
| Serotype 9N | <0.3 | 0.5 | 0.6 | 0.4 | ≥9.2 |
| Serotype 12F | <0.1 | 0.1 | 0.5 | 0.2 | ≥0.6 |
| Serotype 14 | <0.7 | <0.7 | 6.4 | 4.1 | ≥7.0 |
| Serotype 17F | <0.9 | 2 | 2.8 | 1.9 | ≥7.8 |
| Serotype 19F | 0.9 | 1.5 | 1.9 | 1.9 | ≥15.0 |
| Serotype 20 | <0.2 | 0.2 | 1.1 | 1.6 | ≥1.3 |
| Serotype 22F | 1.2 | 2 | 5.7 | 2.6 | ≥7.2 |
| Serotype 23F | <1.2 | 1.6 | 2.3 | 1.7 | ≥8.0 |
| Serotype 6B | <0.6 | <0.6 | 24.6 | 37.4 | ≥4.7 |
| Serotype 10A | <0.5 | 1 | 0.9 | 0.7 | ≥2.9 |
| Serotype 11A | 0.3 | 1 | 2.7 | 2.5 | ≥2.4 |
| Serotype 7F | <0.6 | 0.6 | 5.9 | 3.9 | ≥3.2 |
| Serotype 15B | <0.3 | 1.1 | 1.4 | 1.2 | ≥3.3 |
| Serotype 18C | <0.1 | 0.3 | 2.5 | 5.3 | ≥3.3 |
| Serotype 19A | <0.8 | 0.9 | 9.5 | 6.7 | ≥17.1 |
| Serotype 9V | <0.6 | 1.2 | 1.8 | 2.8 | ≥2.6 |
| Serotype 33F | <0.2 | 0.6 | 0.6 | 0.6 | ≥1.7 |
Abbreviation: CM, cryptococcal meningoencephalitis.
Figure 1.
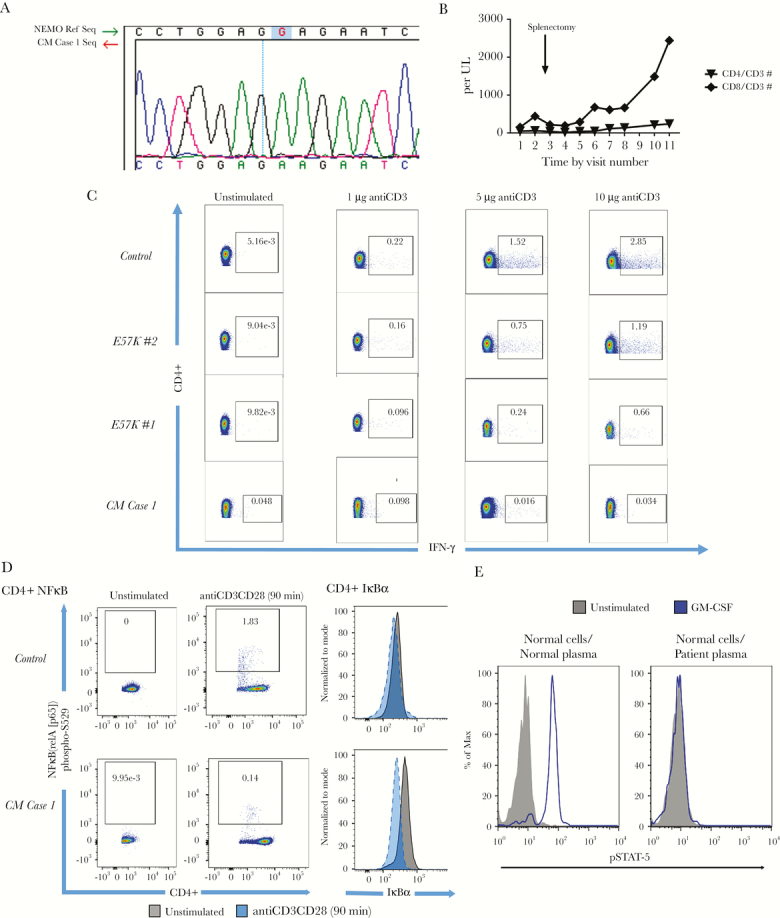
Evaluation of nuclear factor-κβ (NF-κB) essential modulator (NEMO) mutation cryptococcal meningoencephalitis (CM) Case 1 and anti-granulocyte-macrophage colony-stimulating factor (GM-CSF) autoantibodies in CM Case 2 with idiopathic CD4+ lymphopenia. (A) Sanger deoxyribonucleic acid sequencing of polymerase chain reaction product demonstrates X-linked G>A 169 mutation in IKBKG gene, which encodes E57K NEMO in CM Case 1. (B) Trends in CD4+ T lymphocyte count over time for CM Case 1 with E57K NEMO mutation depict persistent CD4+ lymphopenia despite recovery of CD8+ T cells postsplenectomy. (C) Flow cytometry T-cell receptor (TCR) stimulation experiments using different titers of anti-CD3 (10 μg/mL, 5 μg/mL, 1 μg/mL), and unstimulated upon CD3+ negatively selected T lymphocytes with fixed costimulation with anti-CD28 (2 μg/mL) (eBioscience/Affymetrix, San Diego, CA) over 36 hours reveals significant increasing perecentage of interferon (IFN)γ induction among CD4+ T lymphocytes with increasing anti-CD3CD28 titer among controls but less amounts among CM Case 1 with E57K NEMO and CM and other patients with the same mutation but no CM. Numerical values in boxes represent %CD4+ expressing IFNγ. (D) Increased induction of phosphorylated NF-κB (RelA/p65) corresponds to more IkBα degradation among CD4+ T lymphocytes from control compared with CM Case 1 as predicted by NEMO mutation. The IkBα-phosphorylated state in stimulated case is at level of unstimulated control. The TCR stimulation was performed at fixed anti-CD3 (5 μg/mL) and anti-CD28 (2 μg/mL). Numerical values in boxes represent %CD4+ expressing NF-κB (RelA/p65) phosphorylated on serine at position 529. (E) The GM-CSF-induced STAT-5 phosphorylation in normal CD14+ monocytes in the presence of 10% normal or patient plasma from CM Case 2.
Cryptococcal meningoencephalitis Case 2 was a 73-year-old, HIV-seronegative male who presented with a 7-week history of low-grade fever, frontal headaches, myalgia, and diplopia as well as 2 days of left hearing loss and disequilibrium. The patient was diagnosed with CM due to Cn growth from his CSF and responded well to antifungals. Immunological studies revealed a CD4+ and CD8+ lymphopenia but without distortions in T-cell subsets as in CM Case 1 (Table 1) and normal response to (at least 8 serotypes) pneumococcal immunizations (Table 2). Analysis of serum demonstrated anti-GMCSF autoantibodies that blocked phosphorylation of STAT5 (Figure 1E). It is interesting to note that the patient did not show any evidence of pulmonary alveolar proteinosis as demonstrated by normal computed tomography (CT) chest imaging and pulmonary function testing including pulmonary diffusing capacity. Neither CM Case 1 nor CM Case 2 carry mutations known to be associated with ICL including LCK, RAG1, UNC119, or FAS. In addition, we screened NEMO exons of 42 additional patients with ICL, some of whom also had CM (n = 18), and found no other patients with mutations in this gene.
DISCUSSION
The present cases illustrate 2 aspects related to ICL and infections with Cryptococcus, both without strong associations with Mendelian inheritance patterns. In the first case, a functional NEMO mutation E57K led to a TCR defect. Control patients with the same mutation as well as that in the reported literature [11] suggest little OI susceptibility from the E57K mutation alone and no relationship with ICL. The NEMO defect was likely carried through the patient’s adult life until late adulthood, where a quantitative T-cell defect of ICL presumably increased susceptibility to CM. Indeed, clinical manifestations of CM, in many cases, are thought to represent a reactivation disease, originally acquired as an asymptomatic infection during early life [12], which suggests that ICL could have developed later in life.
Because most patients with ICL do not develop CM, despite high exposures of the individuals beginning after their second birthday [13], we posit that a “multi-hit model” is a possible scenario in ICL susceptibility to CM, possibly by digenic inheritance [14] or additional acquired defect(s). This includes a NEMO germline mutation plus ICL as in CM Case 1 or ICL plus anti-GMCSF antibodies as in CM Case 2. In both cases, ICL would appear to have been necessary but not sufficient to increase susceptibility to CM. Upon TCR activation, NEMO canonically regulates the release of the transcription factor, NF-κB (RelA-p50), by permitting the degradation of IkBα to which it has been bound. However, the E57K NEMO mutation results in defects in binding to TRAF6—an E3 ligase that ubiquitinates NEMO; it is this ubiquitination that usually leads to phosphorylation of IkBα bound to NF-κB (RelA-p50), triggering the release of the latter and subsequent downstream effects, including production of Th1-biasing cytokines IFNγ [11], which is important in protection against CM [6]. Although defects in the NF-κB pathway have been reported with intracellular pathogens such as mycobacteria, such defects have not been previously reported in patients with cryptococcosis [15]. In recent studies, however, investigators have reported a differential effect on NF-κB pathway signaling between intracellular intact Cn and extracellular, heat-killed organism or glucuronyxlomannan (GXM)—the major constituent of the yeast capsule. Specifically, translational interference of IκBα, increasing the duration of RelA nuclear localization activity, occurred in intracellular infection, whereas impairment of RelA nuclear translocation occurred during extracellular presentation, and these effects were GXM dependent [15]. Therefore, our finding of an NF-κB signaling defect in CM is biologically plausible and expands the spectrum of infectious diseases that can occur via impairment of this pathway.
CONCLUSIONS
In the same way, acquisition of an anti-GMCSF autoantibody, typically associated with susceptibility to the reportedly highly virulent C gattii and not Cn [8, 10], in combination with ICL, facilitated Cn disease development. Susceptibility to CM due to Cn occurred, despite a milder form of anti-GMCSF disease that did not result in typical pulmonary alveolar proteinosis, which was evident by a lack of pulmonary symptoms and clear lung imaging by CT. Larger epidemiological studies are needed to determine whether CM susceptibility in ICL can be validated in such a multi-hit model.
Acknowledgments
Disclaimer. The views herein do not reflect the official opinions of the Uniformed Services University or the Department of Defense.
Financial support. This research was funded by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases (NIAID). This research was also funded by extramural grants AI001123-01 and AI001124-01 from the NIAID. Funding support was also obtained via U01 AI109657 from the NIAID.
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Smith DK, Neal JJ, Holmberg SD. Unexplained opportunistic infections and CD4+ T-lymphocytopenia without HIV infection. An investigation of cases in the United States. The Centers for Disease Control Idiopathic CD4+ T-lymphocytopenia Task Force. N Engl J Med 1993; 328:373–9. [DOI] [PubMed] [Google Scholar]
- 2. Bignon A, Régent A, Klipfel L et al. . DUSP4-mediated accelerated T-cell senescence in idiopathic CD4 lymphopenia. Blood 2015; 125:2507–18. [DOI] [PubMed] [Google Scholar]
- 3. Gorska MM, Alam R. Consequences of a mutation in the UNC119 gene for T cell function in idiopathic CD4 lymphopenia. Curr Allergy Asthma Rep 2012; 12:396–401. [DOI] [PubMed] [Google Scholar]
- 4. Roger PM, Bernard-Pomier G, Counillon E et al. . Overexpression of Fas/CD95 and Fas-induced apoptosis in a patient with idiopathic CD4+ T lymphocytopenia. Clin Infect Dis 1999; 28:1012–6. [DOI] [PubMed] [Google Scholar]
- 5. Zonios DI, Falloon J, Huang CY et al. . Cryptococcosis and idiopathic CD4 lymphocytopenia. Medicine 2007; 86:78–92. [DOI] [PubMed] [Google Scholar]
- 6. Panackal AA, Wuest SC, Lin YC et al. . Paradoxical immune responses in non-HIV cryptococcal meningitis. PLoS Pathog 2015; 11:e1004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang X, van de Veerdonk FL, Netea MG. Basic genetics and immunology of candida infections. Infect Dis Clin North Am 2016; 30:85–102. [DOI] [PubMed] [Google Scholar]
- 8. Byrnes EJ 3rd, Li W, Lewit Y et al. . Emergence and pathogenicity of highly virulent Cryptococcus gattii genotypes in the northwest United States. PLoS Pathog 2010; 6:e1000850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kingeter LM, Paul S, Maynard SK et al. . Cutting edge: TCR ligation triggers digital activation of NF-kappaB. J Immunol 2010; 185:4520–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosen LB, Freeman AF, Yang LM et al. . Anti-GM-CSF autoantibodies in patients with cryptococcal meningitis. J Immunol 2013; 190:3959–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gautheron J, Pescatore A, Fusco F et al. . Identification of a new NEMO/TRAF6 interface affected in incontinentia pigmenti pathology. Hum Mol Genet 2010; 19:3138–49. [DOI] [PubMed] [Google Scholar]
- 12. Garcia-Hermoso D, Janbon G, Dromer F. Epidemiological evidence for dormant Cryptococcus neoformans infection. J Clin Microbiol 1999; 37:3204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goldman DL, Khine H, Abadi J et al. . Serologic evidence for Cryptococcus neoformans infection in early childhood. Pediatrics 2001; 107:E66. [DOI] [PubMed] [Google Scholar]
- 14. Posey JE, Harel T, Liu P et al. . Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med 2017; 376:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hayes JB, Sircy LM, Heusinkveld LE et al. . Modulation of macrophage inflammatory nuclear factor κB (NF-κB) signaling by intracellular Cryptococcus neoformans. J Biol Chem 2016; 291:15614–27. [DOI] [PMC free article] [PubMed] [Google Scholar]