

FIS2 and MEA have diverged in concert after simultaneous gene duplication, resulting in functional divergence of the PRC2 complexes in Brassicaceae, which is a novel fate for duplicated genes whose products act in complexes.
Abstract
Duplicated genes are a major contributor to genome evolution and phenotypic novelty. There are multiple possible evolutionary fates of duplicated genes. Here, we provide an example of concerted divergence of simultaneously duplicated genes whose products function in the same complex. We studied POLYCOMB REPRESSIVE COMPLEX2 (PRC2) in Brassicaceae. The VERNALIZATION (VRN)-PRC2 complex contains VRN2 and SWINGER (SWN), and both genes were duplicated during a whole-genome duplication to generate FERTILIZATION INDEPENDENT SEED2 (FIS2) and MEDEA (MEA), which function in the Brassicaceae-specific FIS-PRC2 complex that regulates seed development. We examined the expression of FIS2, MEA, and their paralogs, compared their cytosine and histone methylation patterns, and analyzed the sequence evolution of the genes. We found that FIS2 and MEA have reproductive-specific expression patterns that are correlated and derived from the broadly expressed VRN2 and SWN in outgroup species. In vegetative tissues of Arabidopsis (Arabidopsis thaliana), repressive methylation marks are enriched in FIS2 and MEA, whereas active marks are associated with their paralogs. We detected comparable accelerated amino acid substitution rates in FIS2 and MEA but not in their paralogs. We also show divergence patterns of the PRC2-associated VERNALIZATION5/VIN3-LIKE2 that are similar to FIS2 and MEA. These lines of evidence indicate that FIS2 and MEA have diverged in concert, resulting in functional divergence of the PRC2 complexes in Brassicaceae. This type of concerted divergence is a previously unreported fate of duplicated genes. In addition, the Brassicaceae-specific FIS-PRC2 complex modified the regulatory pathways in female gametophyte and seed development.
Duplicated genes are continuously formed during evolution by various types of gene duplication events in eukaryotes, and they can have effects on morphological and physiological evolution (for review, see Van de Peer et al., 2009; Soltis and Soltis, 2016). Gene duplication can happen at small scales, such as tandem duplication, segmental duplication, and duplicative retroposition. The largest scale of gene duplication is whole-genome duplication (WGD), which gives rise to thousands of duplicated gene pairs. The genetic model plant Arabidopsis (Arabidopsis thaliana) has experienced five rounds of WGD events in the evolutionary history of seed plants (Jiao et al., 2011; Li et al., 2015). The most recent polyploidy event, the α WGD, is specific to the Brassicaceae family, which took place after the divergence of the closest sister family, Cleomaceae (Schranz and Mitchell-Olds, 2006). There are about 2,500 pairs of duplicated genes retained from this WGD in the Arabidopsis genome (Blanc et al., 2003; Bowers et al., 2003).
Fates of duplicated genes vary during evolutionary history. One duplicate may eventually be lost or become a pseudogene; thus, the once duplicated pair returns to a single-copy status. Several mechanisms drive the retention of both copies. Duplicated pairs could preserve similar functions to maintain dosage balance (Birchler et al., 2005; Coate et al., 2016). Duplicated pairs also can diverge through subfunctionalization or neofunctionalization, where two duplicated genes divide the ancestral function or gain a novel function, respectively (Force et al., 1999; Moore and Purugganan, 2005). These types of divergence also could be inferred from expression patterns. For example, two duplicates that together make up the preduplicate expression profile is referred to as regulatory subfunctionalization, and regulatory neofunctionalization indicates that one or both copies gain a new expression pattern (Duarte et al., 2006; Liu et al., 2011). Sometimes, these processes are difficult to distinguish, and there can be a combination of different mechanisms, such as subneofunctionalization (He and Zhang, 2005).
There are many protein complexes whose members are encoded by different gene families. If multiple components in a complex are duplicated simultaneously, such as in a WGD, the doubled components could redundantly cross-interact or go on to experience subsequent divergence (Capra et al., 2012; Aakre et al., 2015). Thus, a type of coevolution between the interacting gene products is hypothetically possible, but this has not been described in the plant kingdom. Extending the concept of concerted divergence, which is discussed in the context of coexpression patterns of duplicated genes in the same metabolic or regulatory pathways (Blanc and Wolfe, 2004), we here propose the evolutionary scenario that simultaneous duplication of two genes whose products function together in a complex, followed by parallel evolution and the divergence of each derived gene, can lead to functional divergence of the complexes.
In this study, we focus on genes in POLYCOMB REPRESSIVE COMPLEX2 (PRC2) in Brassicaceae species as a potential example to demonstrate the proposed scenario. Those complexes are histone modifiers and regulate gene expression primarily by the trimethylation of Lys-27 on histone H3 (H3K27me3) associated with target genes, which leads to transcriptional repression (Hennig and Derkacheva, 2009; Mozgova et al., 2015). One type of PRC2, the VERNALIZATION (VRN) complex, regulates vegetative tissue differentiation and, more importantly, the vernalization process to control flowering time in Arabidopsis (Chen et al., 2009; Hennig and Derkacheva, 2009; Mozgova et al., 2015). This complex also represses autonomous seed coat development (Roszak and Köhler, 2011), and it is present across rosids. The VRN complex consists of four subunits: REDUCED VERNALIZATION RESPONSE2 (VRN2), SET DOMAIN-CONTAINING PROTEIN10 (SWINGER [SWN]), and two WD-40 repeat proteins that act as the scaffold of the complex assemblies, FERTILIZATION-INDEPENDENT ENDOSPERM (FIE) and MULTICOPY SUPRESSOR OF IRA1 (MSI1). In Brassicaceae, the α WGD gave rise to a duplication of VRN2 to create its paralog FERTILIZATION INDEPENDENT SEED2 (FIS2) and a duplication of SWN to create its paralog SET DOMAIN-CONTAINING PROTEIN5 (MEDEA [MEA]; Fig. 1; Spillane et al., 2007; Luo et al., 2009). Substituting for their paralogous proteins, FIS2 and MEA, together with FIE and MSI1 (the α WGD paralogs of these two genes were lost), make up a new Brassicaceae-specific PRC2, referred to as the FIS complex (Fig. 1). The FIS complex functions in gametophyte and seed development, preventing female gamete proliferation before fertilization and facilitating endosperm cellularization after fertilization (Hennig and Derkacheva, 2009). A typical fis phenotype, caused by nonfunctional mutation in FIS2, MEA (also known as FIS1), or FIE (also known as FIS3), shows fertilization-independent embryogenesis, and other types of mutants have abnormal seed development, even abolished seeds (Hennig and Derkacheva, 2009).
Figure 1.

Two PRC2 complexes in Brassicaceae, the VRN complex and the Brassicaceae-specific FIS complex, arose by the α WGD, where VRN2 duplicated to form FIS2 and SWN duplicated to form MEA.
The observed divergence in the functions of the two kinds of PRC2 complexes leads to the hypothesis that FIS2 and MEA have undergone divergence in a concerted way to give rise to the FIS complex. This study aimed to evaluate this hypothesis by examining expression patterns, DNA and histone methylation, and rates of sequence evolution in both genes compared with their paralogs. We found evidence for the parallel divergence of FIS2 and MEA from their paralogs in multiple ways, which has accompanied functional divergence of the two complexes. This study supports a model of concerted divergence of simultaneously duplicated genes whose products function in a complex. To our knowledge, this is a previously unreported fate of duplicated genes.
RESULTS
FIS2 and MEA Have Specific and Similar Expression Patterns in Reproductive Organs
FIS2 and MEA formed by the α WGD that is specific to the Brassicaceae family after the divergence of the Brassicaceae lineage from the Caricaceae lineage. After gene duplication, duplicated genes may experience expression divergence. We analyzed microarray data in Arabidopsis to compare the expression profiles of paralogous interacting gene pairs FIS2/VRN2 and MEA/SWN. We obtained two sets of ATH1 microarray data and analyzed them separately: 63 different organ types and developmental stages (Schmid et al., 2005), referred to as the ADA (Arabidopsis developmental atlas) data set hereafter, and 42 different tissue types during seed developmental stages (Le et al., 2010; Belmonte et al., 2013), referred to as the ASA (Arabidopsis seed atlas). We first calculated the expression specificity (τ) of the four genes defined by Yang and Gaut (2011). VRN2 and SWN have expression specificity values of 0.19 and 0.17, respectively, indicating that both genes have relatively broad expression in nearly all organ types included in the ADA data set (Fig. 2). In contrast, FIS2 has an expression specificity value of 0.7 and that of MEA is 0.63, indicating an organ-specific expression pattern. We observed that the expression of FIS2 and MEA is restricted to flowers and siliques, and the absence of vegetative expression explains the high expression specificity. Yang and Gaut (2011) analyzed the ADA data set and found that the recent whole-genome duplicates have a median τ close to 0.2. Thus, what we observed for FIS2 and MEA is quite high, and what we observed for VRN2 and SWN is about average.
Figure 2.

A, Organ/tissue-specific expression indices based on two sets of microarray data. A high value indicates that expression is restricted to fewer organ or tissue types, while a low value indicates broad expression. B, Correlation of the expression profile of each gene pair. Left, ADA set (63 organ types and developmental stages); right, ASA set (42 seed tissue types and developmental stages). Black arrows indicate a positive correlation, and gray arrows indicate a negative correlation. The thickness of the arrows indicates the level of the correlation coefficient. The correlation coefficient and the P value of the expression profile of each gene pair are labeled along the arrows. Boldface values indicate positive correlations.
We also analyzed τ in the ASA data set (Fig. 2A). Similarly, the τ of FIS2 is 0.48 and that of MEA is 0.56, while VRN2 has τ of 0.21 and SWN has τ of 0.22. FIS2 and MEA turn out to show more tissue-specific expression in seed tissues. We broke down the ASA data and observed that FIS2 and MEA tend to be expressed in the triploid endosperm rather than in the diploid embryo or maternally derived seed coat. We did a 1,000-replicate permutation test and gained statistical support that the expression specificity differences in the FIS2-MEA and VRN2-SWN comparisons are not significant (Supplemental Fig. S1), indicative of the concerted divergence in their expression profiles. In contrast, the tissue specificity expression profiles are significantly different in the two duplicated pairs, VRN2-FIS2 and SWN-MEA, indicative of their regulatory divergence.
Not only did we analyze the expression index for those genes individually, we also performed a correlation test to examine the association of the expression profiles of the four genes, as their products function in a complex (Fig. 2B). We found that the expression patterns of FIS2 and MEA are positively correlated in both the ADA and ASA data sets, while broadly expressed VRN2 and SWN are coexpressed. However, the expression of both FIS2 and MEA is negatively correlated to the expression of VRN2 and SWN. The negative coefficients are around −0.5 (Fig. 2B), which is below 1% of the total α whole-genome pairs analyzed by Blanc and Wolfe (2004). Overall, the FIS2-MEA expression patterns indicate parallel divergence from the VRN2-SWN expression patterns in a concerted manner.
FIS2 and MEA Acquired New Expression Patterns
As the microarray data from the ADA indicated, FIS2 and MEA both have expression patterns that are restricted to reproductive organs, such as flowers and siliques, but not vegetative organs, including roots, stems, and leaves. We confirmed this result with reverse transcription (RT)-PCR (Fig. 3). In contrast, their paralogs, VRN2 and SWN, have a broad expression pattern in both vegetative and reproductive organs, and they are expressed ubiquitously in all examined organ types in our RT-PCR results (Fig. 3). To infer the ancestral expression patterns of the two gene pairs, we assayed the expression patterns of orthologs in Tarenaya hassleriana (formerly known as Cleome spinosa), Carica papaya, and Vitis vinifera. Among those species with sequenced genomes, T. hassleriana belongs to Cleomaceae, the most closely related sister group to Brassicaceae. Although T. hassleriana had its own genome triplication after the divergence between Cleomaceae and Brassicaceae (Cheng et al., 2013), only a single copy each of the orthologous VRN2 and SWN has been retained. C. papaya also is in the order Brassicales. V. vinifera was chosen because its lineage has not experienced any WGD events since the γ WGD during early eudicot evolution, which applies to C. papaya as well; thus, genes are frequently single copy in these taxa. These single-copy orthologs can facilitate the inference of ancestral expression patterns. We confirmed that these sequences are true orthologs of FIS2/VRN2 and MEA/SWN by phylogenetic analysis of the gene families.
Figure 3.

RT-PCR assays indicate that FIS2 and MEA have lost the ancestral vegetative expression pattern after duplication. Plus signs indicate reactions with reverse transcriptase, and minus signs indicate controls with no reverse transcriptase. Species abbreviations are as follows: At, Arabidopsis; Th, T. hassleriana; Cp, C. papaya; and Vv, V. vinifera.
For both the FIS2/VRN2 and MEA/SWN pairs, their orthologs in T. hassleriana, C. papaya, and V. vinifera are widely expressed in all examined organ types, which is the same as VRN2 and SWN in Arabidopsis (Fig. 3). The absence of expression in vegetative organs is observed only in FIS2 and MEA. Collectively, we inferred that the preduplicated expression state is likely to be a broad expression pattern, which is reflected by VRN2 and SWN. The Brassicaceae FIS2 and MEA both lost expression in vegetative organs to become expressed specifically in reproductive organs.
FIS2 and MEA Acquired Novel Epigenetic Modifications
The epigenetic features of cytosine methylation and histone methylation often are associated with the expression or silencing of genes. To examine the patterns of cytosine and histone methylation in organ types where the expression of FIS2 and MEA was lost, we investigated the epigenetic variation among these genes in vegetative tissues, including leaves, roots, and seedlings, of Arabidopsis (for details, see “Materials and Methods”). For DNA methylation, we found that cytosine methylation at CpG sites is enriched in the promoter region (defined as 1,500 bp upstream of the transcription start site) of the FIS2 genomic sequence but not in the gene body (Fig. 4). The opposite is found for VRN2, with the promoter region unmarked but the gene body highly methylated (Fig. 4). The same divergence of DNA methylation was found for MEA and SWN (Fig. 4). Cytosine methylation is enriched in the promoter region of MEA but only in the gene body of SWN. The DNA methylation patterns in EMBRYONIC FLOWER 2 (EMF2) and CURLY LEAF (CLF), the more distant paralogs of VRN2 and SWN, respectively, are also gene body enrichment, the same as VRN2 and SWN, suggesting that the pattern of DNA methylation for FIS2 and MEA changed after duplication. As promoter cytosine methylation is associated with transcriptional repression and gene body methylation is indicative of expression activation (Suzuki and Bird, 2008), this finding is consistent with the expression data. We did not examine methylation patterns in whole endosperm, because in the ASA data set, FIS2 and MEA showed variable expression patterns in different parts of the endosperm and different developmental stages.
Figure 4.

DNA methylation at the genomic region of the VEF domain genes and SET domain genes. CLF and EMF2 are ancient paralogs of SWN and VRN2, respectively. For each gene, four rows represent four replicates, and the dashed line separates 1,500 bp upstream of the transcription start site. Vertical bars in each row represent the level of methylation.
We also examined histone methylation in the region of these genes in the seedlings of Arabidopsis based on the data generated by Roudier et al. (2011). Similar to DNA methylation, we found that VRN2, SWN, EMF2, and CLF have the same types of histone methylation, which are different from FIS2 and MEA (Table I). We noticed that FIS2 and MEA lost trimethylation of Lys-4 on histone H3 (H3K4me3), which is shared by all the other genes. Instead, they gained a novel mark of H3K27me3. H3K4me3 is an activating mark, while its antagonistic mark, H3K27me3, is repressive. This could help explain the expression of VRN2, SWN, EMF2, and CLF in the vegetative tissue but the lack of expression of FIS2 and MEA. It is also notable that, in the fie mutant, where the PRC2 function was supposed to be abolished, FIS2 and MEA lost their H3K27me3 but, instead, VRN2, SWN, EMF2, and CLF were marked by H3K27me3 (Bouyer et al., 2011). As H3K27me3 is regulated by PRC2 complexes, this finding suggests the self- and cross-regulation among these genes. With both DNA and histone modification comparative analyses, we observed the convergent evolution of epigenetic features in FIS2 and MEA, divergent from their preduplicated and postduplicated paralogs.
Table I. Histone methylation of the studied genes.
An x indicates the presence of a particular type of histone methylation.
Gene Structural Changes in FIS2 and MEA
FIS2 formed from VRN2 by duplication, and MEA duplicated from SWN, during the α WGD. FIS2 in Arabidopsis lost three exons, called the E15-E17 region (corresponding to the 15th to 17th exons in Arabidopsis EMF2, not named after VRN2), compared with VRN2 (Supplemental Fig. S2A; Chen et al., 2009). FIS2 has a large Ser-rich domain that is not shared with any other VEF genes in any species, indicating gain of the domain in Brassicaceae (Supplemental Fig. S2A; Chen et al., 2009). Our sequence analysis showed that the Ser-rich domain is highly variable among FIS2 sequences from different Brassicaceae species (Supplemental Fig. S2A). The lost E15-E17 domain and the gained Ser-rich domain are both neighboring the VEF domain that interacts with the C5 domain in MEA.
MEA is about 150 amino acids shorter than SWN, and the deleted region is just downstream of the C5 domain that interacts with the VEF domain in FIS2, due to a large shrinkage in a single exon (the ninth in Arabidopsis MEA and SWN) where Brassicaceae SWN and orthologous SWN-like sequences are not conserved (Supplemental Fig. S2B). How the structural changes affect the physical interaction of FIS2 and MEA remains to be tested. In addition to the rearrangement of functional domains, those shared domains show different levels of amino acid sequence divergence. In contrast, VRN2/EMF2-like sequences and SWN/CLF-like sequences show relative conservation across all flowering plants in amino acid sequences and functional domains (Chen et al., 2009; Qian et al., 2014).
FIS2 and MEA Show Accelerated Amino Acid Substitution Rates and Evidence for Positive Selection
Duplicated genes diverge not only in expression pattern but also in their sequences. We first analyzed by Ka/Ks analysis (the ratio of the number of nonsynonymous substitutions per non- synonymous site to the number of synonymous substitutions per synonymous site) the full-length coding regions of FIS2, VRN2, MEA, and SWN genes (Supplemental Fig. S3). The Brassicaceae FIS2 clade had a much higher average Ka/Ks than VRN2 lineages, 3.5-fold greater than the paralogous Brassicaceae VRN2 clade and 10-fold greater than the orthologous preduplicate VRN2 sequences. Similarly, the Brassicaceae MEA clade had a high average Ka/Ks comparable to the FIS2 clade, which is 3.5-fold greater than the paralogous Brassicaceae SWN clade and 4.5-fold greater than the orthologous preduplicated SWN sequences. We implemented different models assuming similar versus different Ka/Ks ratios in these clades, described in “Materials and Methods,” and the likelihood ratio tests indicated that the divergence in sequence rate is significant (Supplemental Table S1). These analyses indicate that, while the paralogous Brassicaceae VRN2 and SWN lineages are under stronger purifying selection along with the orthologous genes in outgroup species, FIS2 and MEA in the Brassicaceae have experienced relaxation of purifying selection. Asymmetric Ka/Ks ratios are seen in a minority of duplicated gene pairs in Arabidopsis; for example, Gossmann and Schmid (2011) estimated that 7% of the duplicated pairs they analyzed have asymmetric Ka/Ks ratios.
Additionally, among the branch-wise Ka/Ks of specific FIS2 and MEA sequences, we detected possible positive selection, indicated by Ka/Ks greater than 1, acting on the sequences from certain lineages (Supplemental Fig. S3). In order to distinguish certain amino acid sites evolving under positive selection from relaxed purifying selection, we also applied a branch-site model, which suggested that both branches leading to Arabidopsis FIS2 (P < 0.0001) and MEA (P = 0.007) have positively selected amino acid sites across different functional domains (Supplemental Fig. S4).
Thus, we further studied the sequence evolution of characterized functional domains of FIS2/VRN2 and MEA/SWN genes, including the VEF and C2H2 domains in the FIS2 and VRN2 genes and the C5, SET, SANT, and CXC domains in the MEA and SWN genes (Fig. 5; Supplemental Fig. S3). We observed that the trend of acceleration in sequence evolution of FIS2 and MEA, and the evolutionary constraint resulting in the conservation of VRN2 and SWN, were reflected by all the functional domains we analyzed individually. The VEF domain in FIS2/VRN2 genes and the C5 domain in MEA/SWN genes interact physically with each other; thus, the comparison between the two sets of Ka/Ks ratios best describes the coevolution between FIS2 and MEA at the coding sequence level from a protein-protein interaction perspective (Fig. 5). Consistent with the full-length gene analyses, the VEF domain in the FIS2 lineages and the C5 domain in the MEA lineages both have accelerated amino acid substitution rates, with evidence (Ka/Ks > 1) suggesting positive selection on a few branches (Supplemental Fig. S3; Supplemental Table S1). Similar results were found in the DNA binding-related domains, C2H2 in FIS2/VRN2 and CXC and SANT in MEA/SWN genes (Supplemental Fig. S3), indicating that the PRC2 complexes with FIS2 and MEA may have affinity to specific DNA regions, regulating a novel network of gene expression. The SET domain plays the role of methyltransferase in the PRC2 complex and is usually highly conserved across eukaryotes (Baumbusch et al., 2001). This is reflected by the low Ka/Ks ratios detected in the SWN SET domains (Supplemental Fig. S3). Instead, the SET domain in the Brassicaceae MEA shows evidence for positive selection (Supplemental Fig. S4). The rapid amino acid substitution rates in the PRC2 functional domains together likely relate to the functional divergence of the PRC2 complexes containing FIS2 and MEA.
Figure 5.

Ka/Ks values of the interacting domains: the VEF domain in FIS2/VRN2 and the C5 domain in MEA/SWN. The estimated average Ka/Ks ratio of each clade is shown between the two trees. The values above branches are Ka/Ks ratios (where no value suggests the lack of power to detect the accurate Ka/Ks ratio in the PAML analysis). The black dots indicate the α WGD at the base of the Brassicaceae. The scale bars indicate 0.1 substitution per codon. Species abbreviations are as follows: At, Arabidopsis; Al, Arabidopsis lyrata; Cr, Capsella rubella; Sp, Schrenkiella parvula; Es, Eutrema salsugineum; Br, Brassica rapa; Bo, Brassica oleracea; Th, T. hassleriana; Cp, C. papaya; Gr, Gossypium raimondii; Tc, Theobroma cacao; Pt, Populus trichocarpa; Rc, Ricinus communis; Me, Manihot esculenta; and Vv, V. vinifera.
VEL2 and VEL1, Which Interact with PRC2 Complexes, Show Corresponding Divergence Patterns to FIS2/VRN2 and MEA/SWN
A family of five PHD finger proteins is necessary for the core PRC2 complex to maintain the repressed status of chromatin (Kim and Sung, 2010, 2013). Among them, VERNALIZATION5/VIN3-LIKE1 (VEL1) and VEL2 are a pair of α whole genome duplicates. VEL2 is a maternally expressed imprinted gene (Wolff et al., 2011). We analyzed their expression profiles in the ADA and ASA microarray data sets and detected that VEL1 shows a coexpression pattern with VRN2 and SWN that is similar to the broadly expressed VEL homologs, whereas VEL2 has a similar expression pattern to FIS2 and MEA due to the loss of vegetative expression (Fig. 6A; Qiu et al., 2014). VEL2 has a higher specificity than its paralog VEL1 (Fig. 6B). Thus, the observed concerted divergence in expression pattern in the FIS complex is not limited to the core complex but also includes other associated proteins.
Figure 6.

VEL2 and VEL1 expression and sequence evolution. A, Organ/tissue specificity of VEL genes. B, Correlation of the expression profile between VEL genes and PRC2 core components. Left, ADA set (63 organ types and developmental stages); right, ASA set (42 seed tissue types and developmental stages). Black arrows indicate positive correlation, and gray arrows indicate negative correlation. The thickness of the arrows indicates the level of the correlation coefficient. The correlation coefficient and the P value of the expression profile of each gene pair are labeled along the arrows. Boldface values indicate positive correlations. C, DNA methylation at the genomic region of VEL genes (as in Fig.4). D, Ka/Ks values of the VEL genes. The average Ka/Ks ratio of each clade is shown. The black dot at the node indicates a gene duplication event.
For cytosine methylation in the vegetative tissue, VEL1 is marked through the coding exons but not the promoter region, whereas VEL2 has cytosine methylation enriched in the upstream promoter region and the first two introns located between the 5′ untranslated region exons (Schmitz et al., 2013; Stroud et al., 2013; Zemach et al., 2013). For histone methylation in the vegetative tissue, VEL1 is marked by activating marks, including H3K4me3, trimethylation of Lys-36 on histone H3 (H3K36me3), and dimethylation of Lys-4 on histone H3 (H3K4me2; Roudier et al., 2011). VEL2 has lost the H3K4me3 and H3K36me3 but gained the repressive mark H3K27me3. Those epigenetic features not only correspond to the vegetative expression level but also are consistent with the divergence of the core PRC2 components FIS2 and MEA (Table I).
We further analyzed the sequence evolution of the VEL genes. The VEL2 sequences have an elevated average Ka/Ks ratio compared with the Brassicaceae VEL1 and orthologous VEL genes (Fig. 6B). While VEL1 and orthologous sequences have a low Ka/Ks ratio close to 0, indicating strong purifying selection, a 3-fold change in VEL2 sequences suggests the relaxation of purifying selection. This coincides with the accelerated amino acid substitution rates of FIS2 and MEA.
DISCUSSION
Concerted Divergence of FIS2 and MEA in the FIS-PRC2 Complex
Upon gene duplication, hypothetically, two duplicates are identical in function as well as expression pattern if the cis-elements also are entirely duplicated. Considering that many proteins function through interactions with other proteins, in a regulatory or metabolic pathway, through protein-protein interaction, or form an integral complex, either the duplicates are redundant or both duplicates could integrate into either complex and affect the function of the complex if they have divergence. A shift in expression pattern would be one way to avoid potentially disadvantageous cross talk between interacting members (Aakre et al., 2015). Blanc and Wolfe (2004) described a process of concerted divergence of gene expression in Arabidopsis, in which pairs of duplicates, whose protein products interact, diverge in a parallel manner in expression pattern. However, as FIS2 and VRN2 were not identified as α whole-genome duplicates by the genome-wide study (Blanc et al., 2003), their concerted divergence in expression pattern with MEA and SWN was not included.
Here, we show that FIS2 and MEA diverged in expression pattern in a concerted manner, modified from coexpressed VRN2 and SWN, whose expression pattern resembles the ancestral status. In addition, we show that cytosine methylation and histone methylation patterns in FIS2 and MEA also diverged in a concerted manner. It is possible that the methylation change contributed to the changes in expression patterns, although mutations in regulatory elements also may have played a role in the expression pattern changes. FIS2 and MEA are marked by H3K27me3 in the vegetative tissue, suggesting they both became the targets of a vegetative PRC2 complex after formation by gene duplication (Bouyer et al., 2011). In addition to the vegetative epigenetic divergence, FIS2 and MEA are well known as imprinted genes during seed development, both of which are maternally expressed genes (Berger and Chaudhury, 2009). Based on the genome-wide data sets from Hsieh et al. (2011) and Gehring et al. (2011), we determined that VRN2 and SWN are not imprinted, while the more distant relatives in their gene families, EMF2 and CLF, also lack evidence for imprinting. Thus, we infer that FIS2 and MEA became imprinted genes after their divergence from VRN2 and SWN. This concerted change in the regulation of both genes ensures the dosage balance between the interacting proteins. The concerted divergence of FIS2 and MEA from their paralogs also is reflected by the elevated Ka/Ks ratios in the coding sequences at comparable levels, suggesting that similar relaxed purifying selection is acting on the two genes. Altogether, these changes indicate that FIS2 and MEA have been diverging in concert in multiple ways, which likely contributed to the divergence in functions between the FIS-PRC2 complex and the VRN-PRC2 complex.
Functional Divergence in the FIS-PRC2 Complex
VRN2, SWN/CLF, FIE, and MSI1 form the VRN complex, which regulates vernalization to control flowering time in Arabidopsis (Fig. 1; Hennig and Derkacheva, 2009). The complex also represses autonomous seed coat development (Roszak and Köhler, 2011). The FIS complex contains FIS2, MEA, FIE, and MSI1. The FIS complex is important in gametophyte and seed development and has two major functions. A prefertilization role for the FIS complex is that it prevents proliferation of the central cell of the female gametophyte until after fertilization, so that seed development does not start until after fertilization (Hennig and Derkacheva, 2009). The FIS complex also acts postfertilization. It is needed for regulating endosperm cellularization during seed development (Hehenberger et al., 2012). FIS2 mutants show a phenotype of abnormal female gametophyte development into embryos and are defective in controlling central cell proliferation in the female gametophyte, suggesting that FIS2 is not redundant with VRN2 in the prefertilization function (Roszak and Köhler, 2011). Thus, the FIS complex function in the female gametophyte is specific to the FIS complex and not the VRN complex. MEA also was shown to not be redundant with SWN (Roszak and Köhler, 2011). Unlike all the key components in the FIS complex, a SWN mutant failed to lead to autonomous seed development in the absence of fertilization, nor to seed abortion with embryo and endosperm overgrowth (Luo et al., 1999); thus, it is possible that MEA is functionally specialized for the prefertilization function of the FIS complex and cannot be complemented by SWN. As for the postfertilization function, SWN was shown to be not essential in seed development (Spillane et al., 2007). Thus, it was proposed that MEA underwent neofunctionalization to gain a postfertilization role in regulating seed development after its duplication from SWN (Spillane et al., 2007).
Taking the two parts of the FIS complex functions together, it appears that the novel PRC2 made up by FIS2 and MEA created a Brassicaceae-specific complex for preventing seed development prior to fertilization and facilitating seed development after fertilization in Brassicaceae. This functional divergence complements the concerted divergence of FIS2 and MEA in other ways that we show in this study. The FIS complex also plays an important role in establishing the imprinted expression of many genes in the endosperm, especially paternally expressed imprinted genes, as the differentially methylated paternal or maternal allele can affect the targeting by this complex (Wolff et al., 2011; Köhler et al., 2012). The concerted divergence of FIS2 and MEA in expression patterns, methylation patterns, and accelerated sequence evolution may have contributed to functional diversification or, potentially, neofunctionalization of the FIS-PRC2 complex. An alternative to neofunctionalization of the FIS-PRC2 complex is subfunctionalization after the formation of FIS2 and MEA from their paralogs. Without knowledge of the ancestral function of the PRC2 complex in plants closely related to the Brassicaceae, discussed below, we cannot say for sure if there has been neofunctionalization or subfunctionalization. We show in this study that there has been regulatory neofunctionalization of FIS2 and MEA, which leads us to favor the possibility of neofunctionalization of the complex. Nonetheless, under a scenario of subfunctionalization, FIS2 and MEA still show concerted divergence in their expression patterns, cytosine and histone methylation, and accelerated sequence evolution. In order to distinguish the two possible hypotheses, more research on VRN complexes in rosid species will provide valuable information to indicate the function of the ancestral rosid PRC2 complex.
How are the FIS complex functions performed in other angiosperms outside of Brassicaceae? Some clues come from studies of FIE, which is a member of the FIS complex, in Hieracium piloselloides (Asteraceae). The central cell proliferation phenotype of Arabidopsis fie mutants is not seen in sexual H. piloselloides FIE RNA interference lines; thus, a PRC2 complex does not regulate central cell proliferation in the female gametophyte of H. piloselloides, in contrast to Arabidopsis (Rodrigues et al., 2008). This might indicate that parts of the prefertilization function of FIS-PRC2 in Brassicaceae is an evolutionary innovation; at the same time, it is possible that the unknown mechanism repressing central cell proliferation is specific to the H. piloselloides lineage. FIE down-regulation in H. piloselloides leads to seed abortion (Rodrigues et al., 2008); thus, FIE is important for seed development, presumably as part of a PRC2 complex. Asterids do not contain FIS2, VRN2, or MEA. Thus, if there is a PRC2 complex regulating seed development in asterids, it probably contains the product of lineage-specific polycomb proteins and a mechanism independently evolved from Brassicaceae. In maize (Zea mays) and rice (Oryza sativa), there has been duplication of FIE (Luo et al., 2009; Li et al., 2014). Thus, the grasses may have PRC2 complexes that are divergent from the ancestral state. The requirement of H3K27me3 in rice and maize endosperm for the establishment of imprinting suggests the functional conservation or convergence of a PRC2 complex in Brassicaceae and Poaceae (Makarevitch et al., 2013; Zhang et al., 2014).
Evolution of Protein Complexes after the Duplication of Components
We propose a model of simultaneous gene duplication and concerted divergence of one copy of each duplicated pair (Fig. 7). Following formation by duplication, two genes whose products function together in a complex diverge in similar ways, and the complex diverges in function. This divergence pattern is not limited to neofunctionalization/subfunctionalization but includes some other modifications of these scenarios, such as escape from adaptive conflict. To our knowledge, the PRC2 complexes in Brassicaceae we examined in this study provide the first example of this type of divergence of duplicated genes. We contrast this scenario with single-gene duplication and divergence, where one component in the complex undergoes gene duplication and then the paralog diverges, driving the two complexes with either paralog to diverge in function as a result. Intuitively, many described functionally divergent paralogs may contribute to this type of divergence of their protein complexes. One example is the centromere-defining histone variant CENH3 in the histone core octamers that show duplication specific to the genus Mimulus and sequence divergence, whereas other components in the histone core octamers do not show duplications specific to Mimulus (Finseth et al., 2015). Another case is the telomere-associated proteins POT1a and POT1b in the telomerase RNP complexes in Brassicaceae, where POT1a experienced positive selection that enhanced its affinity with interacting proteins (Beilstein et al., 2015). A variation on this model is when there is a subsequent gene duplication at a later time of another gene whose product functions in the complex, followed by divergence. An example is the plant-specific RNA polymerase IV and V, where rounds of independent lineage-specific duplications and subsequent divergence of varying kinds of subunits have increased RNA polymerase complexity and specificity among different plant groups (Wang and Ma, 2015).
Figure 7.

Schematic diagrams illustrating models of protein complex divergence. Colors indicate conservation versus divergence (could be neofunctionalization, subfunctionalization, loss of partial function, and other types of divergence). A, Single-gene duplication and divergence: a single gene (dark blue) in a complex is duplicated. After duplication, there is subsequent divergence (light blue versus red) of the ancestral gene (dark blue) to give rise to divergent protein complexes. B, Simultaneous gene duplication and concerted divergence: two (or more) genes (dark green + dark blue) were duplicated simultaneously. After duplication, there is parallel divergence (light green + light blue versus yellow + red) to give rise to divergent protein complexes. C, The PRC2 complexes in this study are an example of simultaneous gene duplication and concerted divergence.
Concerted Divergence of the Functionally Associated VELs and Some PRC2 Targets
The VEL genes, VEL1 and VEL2, which are required to maintain and facilitate polycomb transcriptional repression, interact with the PRC2 complex but are not part of the complex itself. Our expression, methylation, and sequence analysis results indicate that VEL2 has similar patterns to FIS2 and MEA, whereas VEL1 has similar patterns to VRN2 and SWN. Thus, VEL2 appears to be diverging in concert with FIS2 and MEA. VEL2 also is a maternally expressed gene and regulated by the FIS complex in the endosperm (Wolff et al., 2011), and VEL2 works together with the FIS core complex to impose maternal regulation in seed development similar to FIS2 and MEA.
Several PRC2 targets duplicated through the α WGD show similar patterns of divergence as well. PKR2 and JMJ15 are FIS-PRC2-regulated imprinted genes (Hsieh et al., 2011; Wolff et al., 2011), whereas their paralogs, PKL and JMJ18, show broad expression, are not imprinted, and are associated with a vegetative PRC2 complex (Aichinger et al., 2011; Yang et al., 2012; Zhang et al., 2012). Out of 46 imprinted genes regulated by FIS2 (Wolff et al., 2011), we identified 41 Brassicaceae-specific duplicated genes. Some of those genes have roles in seed development, such as PHERES1 (Köhler et al., 2003; Villar et al., 2009) and ADMETOS (Kradolfer et al., 2013). Thus, there are new Brassicaceae-specific genes involved in seed development that are regulated by the FIS-PRC2 complex. The functional innovation of the FIS complex appears to have rewired, to some extent, the regulatory pathway of seed development specific to Brassicaceae.
Simultaneous gene duplication events, such as polyploidy, give rise to pairs of duplicated genes that can then codiverge (Shan et al., 2009). Many of the genes that are PRC2 targets, included in the previous paragraph, were derived by the α WGD. FIS2, MEA, and VEL2 also were derived from that WGD. Thus, this study illustrates the potential of concerted divergence after simultaneous gene duplication to affect the functions as well as the regulation of other genes.
MATERIALS AND METHODS
Comparing Expression Specificity and Detecting Coexpression Using Microarray Data Analyses
Two sets of ATH1 microarray data from Arabidopsis (Arabidopsis thaliana) were obtained: the ADA from the TAIR Web site (http://www.Arabidopsis.org/), which included 63 different organ types and developmental stages (Schmid et al., 2005), and the ASA from the Goldberg Lab Arabidopsis thaliana Gene Chip Database (http://seedgenenetwork.net/arabidopsis), which included 42 different tissue types from seed developmental stages (Le et al., 2010; Belmonte et al., 2013). The data were GC-RMA normalized using the gcrma package in R. We used the expression specificity (τ) defined by Yang and Gaut (2011) to describe the expression patterns of FIS2, VRN2, MEA, and SWN:
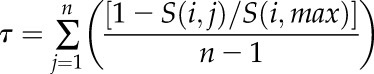 |
where n is the total number of samples (63 or 42) and S(i,max) is the highest log2-transformed expression value for gene i across the n organ types. High values of expression specificity indicate genes with expression limited to few organ or tissue types or developmental stages, while low values of expression specificity indicate broad expression of genes with similar expression levels in most of the organ or tissue types and developmental stages. To test if there is any significant difference of expression specificity between any two of the four genes, we applied 1,000 Monte Carlo randomization tests to each two-gene comparison. For the Monte Carlo randomization test, we computed the following statistic: DIF = |τGENE1 − τGENE2|, where DIF indicates the absolute difference of expression specificity between two genes. Then, we compared the observed value (DIFobs) against the null distribution of simulated DIF value (DIFsim) from 1,000 randomized data. If the null hypothesis is rejected, the expression specificity of any two compared genes is significantly different. The cutoff of the significant P value was set to 0.05.
In addition to the comparison of expression specificity among gene pairs, we applied the Pearson correlation analysis to determine if the expression profile between any two genes showed any evidence of coexpression (i.e. correlated expression across different organ types or tissue types). Coexpression is determined when the Pearson correlation coefficient (r) is significantly positive, and vice versa.
Inferring the Ancestral Expression States Using RT-PCR
Total RNA samples of Arabidopsis, Tarenaya hassleriana (formerly known as Cleome spinosa), Carica papaya, and Vitis vinifera were extracted from liquid N2-frozen tissue of five organ types: root, stem, leaf (rosette leaves in Arabidopsis), flower, and seed (whole siliques in Arabidopsis and T. hassleriana). A modified CTAB method was used for RNA extraction (Zhou et al., 2011). The quality of each RNA sample was checked on 2% agarose gels by electrophoresis, and the amount of each RNA sample was determined by a Nanodrop spectrophotometer. After DNaseI (Invitrogen) treatment to remove residual DNA, M-MLV reverse transcriptase (Invitrogen) was applied to the RNA samples to generate cDNA, according to the manufacturer’s instructions. PCR was performed with cDNA templates to detect the organ-specific expression of Arabidopsis FIS2/VRN2 and MEA/SWN paralogous pairs as well as orthologous genes in outgroup species for inference of the ancestral, preduplication expression states. Gene-specific primers were designed to amplify 250 to 1,000 bp of the cDNA of targeted genes (Supplemental Table S2). For PCR, the cycling program was as follows: preheating at 94°C for 3 min; 30 to 35 cycles of denaturing at 94°C for 30 s, annealing at 53°C to 56°C for 30 s, and elongation at 72°C for 30 s or 1 min; and a final elongation at 72°C for 7 min. PCR products were checked on 1% agarose gels and sequenced to confirm identity.
Identifying Epigenetic Marks Associated with the Studied Genes
We investigated the epigenetic modifications around the genomic regions of Arabidopsis FIS2, VRN2, MEA, and SWN. We also used EMF2 and CLF, which are members of the FIS2/VRN2 and MEA/SWN families, respectively, to help assess the ancestral state. For DNA methylation, we obtained data from Schmitz et al. (2013), Stroud et al. (2013), and Zemach et al. (2013) from CoGe (https://genomevolution.org/CoGe/), visualized by JBrowse in Araport (https://www.araport.org/). Analyzed data included assayed genomic DNAs from leaves from Schmitz et al. (2013) and Stroud et al. (2013) and assayed genomic DNAs from seedlings and roots from Zemach et al. (2013), which were all vegetative organs. Cytosine methylation at CpG sites was analyzed along the genomic region of a target gene. For histone methylation, we extracted tiling array data from seedlings from Roudier et al. (2011) and chromatin immunoprecipitation-on-chip data from wild-type and fie mutant seedlings from Bouyer et al. (2011). Four histone marks were analyzed: H3K27me3, H3K4me3, H3K4me2, and H3K36me3. The epigenetic features in Arabidopsis seedlings were compared among the paralogous genes in a family and between the two interacting gene families.
Detecting Accelerated Sequence Evolution and Positive Selection by Ka/Ks Analyses
To analyze the selection acting on the gene pairs FIS2/VRN2 and MEA/SWN, several rate analyses were performed using Codeml in the PAML package (Yang, 2007). We obtained the sequences of the four genes from Arabidopsis as well as some other Brassicaceae species, including Arabidopsis lyrata, Arabidopsis halleri, Capsella rubella, Brassica rapa, Brassica oleracea, Eutrema salsugineum (formerly known as Thellungiella halophila), and Schrenkiella parvula (formerly known as Thellungiella parvula). We also identified orthologous sequences, by reciprocal best BLAST hits, from species outside of the Brassicaceae, including T. hassleriana (formerly known as C. spinosa), C. papaya, Gossypium raimondii, Theobroma cacao, Citrus sinensis, Populus trichocarpa, Ricinus communis, Manihot esculenta, and V. vinifera, from PLAZA version 3.0 Dicots (http://bioinformatics.psb.ugent.be/plaza/versions/plaza_v3_dicots/; Proost et al., 2015), Phytozome version 10 (http://phytozome.jgi.doe.gov/pz/portal.html; Goodstein et al., 2012), the BRAD database (http://brassicadb.org/brad/; Cheng et al., 2011), and NCBI’s GenBank. Gene orthology was later confirmed by comparing the topology of the gene phylogeny with the species tree. Alignments of amino acid sequences were generated using MUSCLE under default parameters (Edgar, 2004) and then reverse translated into codon alignments using the customized Perl script. We generated the alignments for the full length of the two gene families as well as some documented functional domains, including the VEF and C2H2 domains in the FIS2 and VRN2 genes and the C5, SET, SANT, and CXC domains in the MEA and SWN genes. Phylogenies of the two gene families were analyzed by RAxML version 7.0.3 with GTR as the substitution matrix (Stamatakis, 2006). Maximum likelihood trees of the two gene families were generated based on codon alignments.
We first used a phylogeny-based free-ratio test to estimate branch-wise Ka/Ks ratios along the phylogenetic tree branches. For the full-length FIS2/VRN2 genes, we implemented four different models to test if the Ka/Ks ratios of the Brassicaceae FIS2 clade and the Brassicaceae VRN2 clade display an asymmetric pattern and how conserved they are compared with the orthologous genes. The first model (model I, one-ratio model) assumes that all the genes have the same Ka/Ks ratio, bearing the hypothesis that all genes are under the same level of selection. The second model (model II, two-ratio model-1) assumes that the Brassicaceae VRN2 clade and the orthologous genes have the same Ka/Ks ratio but the Brassicaceae FIS2 clade can have a different one, suggesting that the Brassicaceae VRN2 clade reflects the ancestral selection but FIS2 evolved in a different manner. The third model (model III, two-ratio model-2) assumes that the duplicated FIS2 and VRN2 clades in Brassicaceae have the same Ka/Ks ratio while the orthologs can have a different ratio, which is a hypothesis that the two Brassicaceae copies evolved at the same rate. The fourth model (model IV, three-ratio model) assumes that the two Brassicaceae branches have different Ka/Ks ratios and, thus, the two genes evolved at different rates, with the third Ka/Ks ratio for the orthologous branches. A set of likelihood ratio tests was applied, where twice the difference of likelihood values was calculated and compared against a χ2 distribution with the degrees of freedom set at 1: comparison between model II and model IV can tell if the selection on the Brassicaceae VRN2 is significantly different from the orthologous genes; and comparisons between model I and model II, as well as between model III and model IV, are used to see if the selection on the Brassicaceae FIS2 is different from the Brassicaceae VRN2 and/or the orthologous genes. When model II fits better than model I and model IV fits better than model III with statistical support, the evolutionary rate of the duplicated pair in Brassicaceae is considered to evolve asymmetrically. The same analyses were performed on the functional domains of the FIS2/VRN2 genes and the full-length MEA/SWN genes and their functional domains (Supplemental Table S1). We also applied a branch-site model to detect positively selected sites along FIS2 as well as MEA. Test 2 of model A with the Bayes Empirical Bayes analysis was applied to identify amino acid sites with a high posterior probability of positive selection (Zhang et al., 2005).
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Permutation test for microarray data to detect the difference in expression profile for all sets of comparisons of gene pairs in the ADA and ASA data sets.
Supplemental Figure S2. Structures of FIS2 and VRN2, along with MEA and SWN, in Brassicaceae and other eurosids.
Supplemental Figure S3. Ka/Ks ratios of full-length FIS2/VRN2 and MEA/SWN genes and functional domains.
Supplemental Figure S4. Positive selection on specific sites of MEA and FIS2 genes.
Supplemental Table S1. Ka/Ks ratios under different branch models for full-length FIS2/VRN2 and MEA/SWN genes and functional domains.
Supplemental Table S2. Gene-specific primers used in this study.
Supplementary Material
Glossary
- WGD
whole-genome duplication
- ADA
Arabidopsis developmental atlas
- ASA
Arabidopsis seed atlas
- RT
reverse transcription
- H3K27me3
trimethylation of Lys-27 on histone H3
- H3K4me3
trimethylation of Lys-4 on histone H3
- H3K4me2
dimethylation of Lys-4 on histone H3
- H3K36me3
trimethylation of Lys-36 on histone H3
Footnotes
This work was supported by the Natural Science and Engineering Research Council of Canada (Discovery Grant to K.L.A. and postgraduate fellowship to Y.Q.) and by the Ministry of Science and Technology, Taiwan (to S.-L.L.).
Articles can be viewed without a subscription.
References
- Aakre CD, Herrou J, Phung TN, Perchuk BS, Crosson S, Laub MT (2015) Evolving new protein-protein interaction specificity through promiscuous intermediates. Cell 163: 594–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aichinger E, Villar CB, Di Mambro R, Sabatini S, Köhler C (2011) The CHD3 chromatin remodeler PICKLE and polycomb group proteins antagonistically regulate meristem activity in the Arabidopsis root. Plant Cell 23: 1047–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumbusch LO, Thorstensen T, Krauss V, Fischer A, Naumann K, Assalkhou R, Schulz I, Reuter G, Aalen RB (2001) The Arabidopsis thaliana genome contains at least 29 active genes encoding SET domain proteins that can be assigned to four evolutionarily conserved classes. Nucleic Acids Res 29: 4319–4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilstein MA, Renfrew KB, Song X, Shakirov EV, Zanis MJ, Shippen DE (2015) Evolution of the telomere-associated protein POT1a in Arabidopsis thaliana is characterized by positive selection to reinforce protein-protein interaction. Mol Biol Evol 32: 1329–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte MF, Kirkbride RC, Stone SL, Pelletier JM, Bui AQ, Yeung EC, Hashimoto M, Fei J, Harada CM, Munoz MD, et al. (2013) Comprehensive developmental profiles of gene activity in regions and subregions of the Arabidopsis seed. Proc Natl Acad Sci USA 110: E435–E444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger F, Chaudhury A (2009) Parental memories shape seeds. Trends Plant Sci 14: 550–556 [DOI] [PubMed] [Google Scholar]
- Birchler JA, Riddle NC, Auger DL, Veitia RA (2005) Dosage balance in gene regulation: biological implications. Trends Genet 21: 219–226 [DOI] [PubMed] [Google Scholar]
- Blanc G, Hokamp K, Wolfe KH (2003) A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res 13: 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G, Wolfe KH (2004) Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 16: 1679–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouyer D, Roudier F, Heese M, Andersen ED, Gey D, Nowack MK, Goodrich J, Renou JP, Grini PE, Colot V, et al. (2011) Polycomb repressive complex 2 controls the embryo-to-seedling phase transition. PLoS Genet 7: e1002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers JE, Chapman BA, Rong J, Paterson AH (2003) Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422: 433–438 [DOI] [PubMed] [Google Scholar]
- Capra EJ, Perchuk BS, Skerker JM, Laub MT (2012) Adaptive mutations that prevent crosstalk enable the expansion of paralogous signaling protein families. Cell 150: 222–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LJ, Diao ZY, Specht C, Sung ZR (2009) Molecular evolution of VEF-domain-containing PcG genes in plants. Mol Plant 2: 738–754 [DOI] [PubMed] [Google Scholar]
- Cheng F, Liu S, Wu J, Fang L, Sun S, Liu B, Li P, Hua W, Wang X (2011) BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol 11: 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, van den Bergh E, Zeng P, Zhong X, Xu J, Liu X, Hofberger J, de Bruijn S, Bhide AS, Kuelahoglu C, et al. (2013) The Tarenaya hassleriana genome provides insight into reproductive trait and genome evolution of crucifers. Plant Cell 25: 2813–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate JE, Song MJ, Bombarely A, Doyle JJ (2016) Expression-level support for gene dosage sensitivity in three Glycine subgenus Glycine polyploids and their diploid progenitors. New Phytol 212: 1083–1093 [DOI] [PubMed] [Google Scholar]
- Duarte JM, Cui L, Wall PK, Zhang Q, Zhang X, Leebens-Mack J, Ma H, Altman N, dePamphilis CW (2006) Expression pattern shifts following duplication indicative of subfunctionalization and neofunctionalization in regulatory genes of Arabidopsis. Mol Biol Evol 23: 469–478 [DOI] [PubMed] [Google Scholar]
- Edgar RC. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finseth FR, Dong Y, Saunders A, Fishman L (2015) Duplication and adaptive evolution of a key centromeric protein in Mimulus, a genus with female meiotic drive. Mol Biol Evol 32: 2694–2706 [DOI] [PubMed] [Google Scholar]
- Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J (1999) Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151: 1531–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring M, Missirian V, Henikoff S (2011) Genomic analysis of parent-of-origin allelic expression in Arabidopsis thaliana seeds. PLoS ONE 6: e23687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, et al. (2012) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40: D1178–D1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossmann TI, Schmid KJ (2011) Selection-driven divergence after gene duplication in Arabidopsis thaliana. J Mol Evol 73: 153–165 [DOI] [PubMed] [Google Scholar]
- He X, Zhang J (2005) Rapid subfunctionalization accompanied by prolonged and substantial neofunctionalization in duplicate gene evolution. Genetics 169: 1157–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehenberger E, Kradolfer D, Köhler C (2012) Endosperm cellularization defines an important developmental transition for embryo development. Development 139: 2031–2039 [DOI] [PubMed] [Google Scholar]
- Hennig L, Derkacheva M (2009) Diversity of Polycomb group complexes in plants: same rules, different players? Trends Genet 25: 414–423 [DOI] [PubMed] [Google Scholar]
- Hsieh TF, Shin J, Uzawa R, Silva P, Cohen S, Bauer MJ, Hashimoto M, Kirkbride RC, Harada JJ, Zilberman D, et al. (2011) Regulation of imprinted gene expression in Arabidopsis endosperm. Proc Natl Acad Sci USA 108: 1755–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, et al. (2011) Ancestral polyploidy in seed plants and angiosperms. Nature 473: 97–100 [DOI] [PubMed] [Google Scholar]
- Kim DH, Sung S (2010) The Plant Homeo Domain finger protein, VIN3-LIKE 2, is necessary for photoperiod-mediated epigenetic regulation of the floral repressor, MAF5. Proc Natl Acad Sci USA 107: 17029–17034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Sung S (2013) Coordination of the vernalization response through a VIN3 and FLC gene family regulatory network in Arabidopsis. Plant Cell 25: 454–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler C, Hennig L, Spillane C, Pien S, Gruissem W, Grossniklaus U (2003) The Polycomb-group protein MEDEA regulates seed development by controlling expression of the MADS-box gene PHERES1. Genes Dev 17: 1540–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler C, Wolff P, Spillane C (2012) Epigenetic mechanisms underlying genomic imprinting in plants. Annu Rev Plant Biol 63: 331–352 [DOI] [PubMed] [Google Scholar]
- Kradolfer D, Wolff P, Jiang H, Siretskiy A, Köhler C (2013) An imprinted gene underlies postzygotic reproductive isolation in Arabidopsis thaliana. Dev Cell 26: 525–535 [DOI] [PubMed] [Google Scholar]
- Le BH, Cheng C, Bui AQ, Wagmaister JA, Henry KF, Pelletier J, Kwong L, Belmonte M, Kirkbride R, Horvath S, et al. (2010) Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc Natl Acad Sci USA 107: 8063–8070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zhou B, Peng X, Kuang Q, Huang X, Yao J, Du B, Sun MX (2014) OsFIE2 plays an essential role in the regulation of rice vegetative and reproductive development. New Phytol 201: 66–79 [DOI] [PubMed] [Google Scholar]
- Li Z, Baniaga AE, Sessa EB, Scascitelli M, Graham SW, Rieseberg LH, Barker MS (2015) Early genome duplications in conifers and other seed plants. Sci Adv 1: e1501084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SL, Baute GJ, Adams KL (2011) Organ and cell type-specific complementary expression patterns and regulatory neofunctionalization between duplicated genes in Arabidopsis thaliana. Genome Biol Evol 3: 1419–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M, Bilodeau P, Koltunow A, Dennis ES, Peacock WJ, Chaudhury AM (1999) Genes controlling fertilization-independent seed development in Arabidopsis thaliana. Proc Natl Acad Sci USA 96: 296–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M, Platten D, Chaudhury A, Peacock WJ, Dennis ES (2009) Expression, imprinting, and evolution of rice homologs of the polycomb group genes. Mol Plant 2: 711–723 [DOI] [PubMed] [Google Scholar]
- Makarevitch I, Eichten SR, Briskine R, Waters AJ, Danilevskaya ON, Meeley RB, Myers CL, Vaughn MW, Springer NM (2013) Genomic distribution of maize facultative heterochromatin marked by trimethylation of H3K27. Plant Cell 25: 780–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RC, Purugganan MD (2005) The evolutionary dynamics of plant duplicate genes. Curr Opin Plant Biol 8: 122–128 [DOI] [PubMed] [Google Scholar]
- Mozgova I, Köhler C, Hennig L (2015) Keeping the gate closed: functions of the polycomb repressive complex PRC2 in development. Plant J 83: 121–132 [DOI] [PubMed] [Google Scholar]
- Proost S, Van Bel M, Vaneechoutte D, Van de Peer Y, Inzé D, Mueller-Roeber B, Vandepoele K (2015) PLAZA 3.0: an access point for plant comparative genomics. Nucleic Acids Res 43: D974–D981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Xi Y, Cheng B, Zhu S, Kan X (2014) Identification and characterization of the SET domain gene family in maize. Mol Biol Rep 41: 1341–1354 [DOI] [PubMed] [Google Scholar]
- Qiu Y, Liu SL, Adams KL (2014) Frequent changes in expression profile and accelerated sequence evolution of duplicated imprinted genes in Arabidopsis. Genome Biol Evol 6: 1830–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues JC, Tucker MR, Johnson SD, Hrmova M, Koltunow AM (2008) Sexual and apomictic seed formation in Hieracium requires the plant polycomb-group gene FERTILIZATION INDEPENDENT ENDOSPERM. Plant Cell 20: 2372–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roszak P, Köhler C (2011) Polycomb group proteins are required to couple seed coat initiation to fertilization. Proc Natl Acad Sci USA 108: 20826–20831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudier F, Ahmed I, Bérard C, Sarazin A, Mary-Huard T, Cortijo S, Bouyer D, Caillieux E, Duvernois-Berthet E, Al-Shikhley L, et al. (2011) Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J 30: 1928–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Schölkopf B, Weigel D, Lohmann JU (2005) A gene expression map of Arabidopsis thaliana development. Nat Genet 37: 501–506 [DOI] [PubMed] [Google Scholar]
- Schmitz RJ, Schultz MD, Urich MA, Nery JR, Pelizzola M, Libiger O, Alix A, McCosh RB, Chen H, Schork NJ, et al. (2013) Patterns of population epigenomic diversity. Nature 495: 193–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schranz ME, Mitchell-Olds T (2006) Independent ancient polyploidy events in the sister families Brassicaceae and Cleomaceae. Plant Cell 18: 1152–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan H, Zahn L, Guindon S, Wall PK, Kong H, Ma H, DePamphilis CW, Leebens-Mack J (2009) Evolution of plant MADS box transcription factors: evidence for shifts in selection associated with early angiosperm diversification and concerted gene duplications. Mol Biol Evol 26: 2229–2244 [DOI] [PubMed] [Google Scholar]
- Soltis PS, Soltis DE (2016) Ancient WGD events as drivers of key innovations in angiosperms. Curr Opin Plant Biol 30: 159–165 [DOI] [PubMed] [Google Scholar]
- Spillane C, Schmid KJ, Laoueillé-Duprat S, Pien S, Escobar-Restrepo JM, Baroux C, Gagliardini V, Page DR, Wolfe KH, Grossniklaus U (2007) Positive darwinian selection at the imprinted MEDEA locus in plants. Nature 448: 349–352 [DOI] [PubMed] [Google Scholar]
- Stamatakis A. (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690 [DOI] [PubMed] [Google Scholar]
- Stroud H, Greenberg MV, Feng S, Bernatavichute YV, Jacobsen SE (2013) Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152: 352–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9: 465–476 [DOI] [PubMed] [Google Scholar]
- Van de Peer Y, Maere S, Meyer A (2009) The evolutionary significance of ancient genome duplications. Nat Rev Genet 10: 725–732 [DOI] [PubMed] [Google Scholar]
- Villar CB, Erilova A, Makarevich G, Trösch R, Köhler C (2009) Control of PHERES1 imprinting in Arabidopsis by direct tandem repeats. Mol Plant 2: 654–660 [DOI] [PubMed] [Google Scholar]
- Wang Y, Ma H (2015) Step-wise and lineage-specific diversification of plant RNA polymerase genes and origin of the largest plant-specific subunits. New Phytol 207: 1198–1212 [DOI] [PubMed] [Google Scholar]
- Wolff P, Weinhofer I, Seguin J, Roszak P, Beisel C, Donoghue MT, Spillane C, Nordborg M, Rehmsmeier M, Köhler C (2011) High-resolution analysis of parent-of-origin allelic expression in the Arabidopsis endosperm. PLoS Genet 7: e1002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Han Z, Cao Y, Fan D, Li H, Mo H, Feng Y, Liu L, Wang Z, Yue Y, et al. (2012) A companion cell-dominant and developmentally regulated H3K4 demethylase controls flowering time in Arabidopsis via the repression of FLC expression. PLoS Genet 8: e1002664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Gaut BS (2011) Factors that contribute to variation in evolutionary rate among Arabidopsis genes. Mol Biol Evol 28: 2359–2369 [DOI] [PubMed] [Google Scholar]
- Yang Z. (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24: 1586–1591 [DOI] [PubMed] [Google Scholar]
- Zemach A, Kim MY, Hsieh PH, Coleman-Derr D, Eshed-Williams L, Thao K, Harmer SL, Zilberman D (2013) The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153: 193–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bishop B, Ringenberg W, Muir WM, Ogas J (2012) The CHD3 remodeler PICKLE associates with genes enriched for trimethylation of histone H3 lysine 27. Plant Physiol 159: 418–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Nielsen R, Yang Z (2005) Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol 22: 2472–2479 [DOI] [PubMed] [Google Scholar]
- Zhang M, Xie S, Dong X, Zhao X, Zeng B, Chen J, Li H, Yang W, Zhao H, Wang G, et al. (2014) Genome-wide high resolution parental-specific DNA and histone methylation maps uncover patterns of imprinting regulation in maize. Genome Res 24: 167–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Moshgabadi N, Adams KL (2011) Extensive changes to alternative splicing patterns following allopolyploidy in natural and resynthesized polyploids. Proc Natl Acad Sci USA 108: 16122–16127 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.