

ABSTRACT
Ataxia telangiectasia mutated (ATM) is inactivated in a significant minority of pancreatic ductal adenocarcinomas and may be predictor of treatment response. We determined if ATM deficiency renders pancreatic cancer cells more sensitive to fractionated radiation or commonly used chemotherapeutics. ATM expression was knocked down in three pancreatic cancer cell lines using ATM-targeting shRNA. Isogenic cell lines were tested for sensitivity to several chemotherapeutic agents and radiation. DNA repair kinetics were analyzed in irradiated cells using the comet assay. We find that while rendering pancreatic cancer cells ATM-deficient did not significantly change their sensitivity to several chemotherapeutics, it did render them exquisitely sensitized to radiation. Pancreatic cancer ATM status may help predict response to radiotherapy.
KeyWords: ATM, pancreatic cancer, radiosensitivity
Background
Pancreatic cancer is the third-leading cause of cancer death in the USA, with a 5-year survival rate of 8%.1 Patients with locally advanced or borderline resectable disease commonly receive combination chemotherapy and radiotherapy despite the lack of clear evidence that radiotherapy improves outcomes.2,3 Stereotactic body radiation therapy (SBRT) is currently undergoing evaluation as a therapy for pancreatic cancer.4-6 A recent phase II study using SBRT to treat unresectable pancreatic cancer reported a local control rate of 57% but with significant toxicity (4.5% of patients suffered gastric perforation).6 Thus, SBRT may improve outcomes, but more studies are needed.
It is likely that a pancreatic cancer's molecular profile influences its response to radiotherapy. One predictor of radiation response is status of ATM, a phosphatidylinositol 3-kinase-like kinase (PIKK) that has a primary role in orchestrating the complex DNA damage response (DDR),7 chromatin remodeling, response to oxidative stress,8 and other cellular functions. Biallelic germline inactivation of ATM causes the severe, developmental neurodegenerative disease, Ataxia Telangiectasia,9 which carries a 25% lifetime risk of cancer. Patients with Ataxia Telangiectasia are unable to respond effectively to DNA double-stranded breaks (DSBs) and are hypersensitive to radiation. ATM has been identified as a pancreatic cancer susceptibility gene and biallelic inactivation of the ATM gene has been identified in the pancreatic cancers of patients with heterozygous germline ATM mutations.10,11 Deleterious germline ATM mutations are one of the most common known causes of inherited pancreatic cancer10,11,12,13 and are also identified in patients with apparently sporadic pancreatic cancer.14,15 In addition, a small percentage of pancreatic cancers harbor biallelic, somatic inactivation of ATM.16 Immunohistochemical analysis of a large series of pancreatic cancers found loss of ATM protein expression in 38/347 (11%) of cancers from patients without a family history of the disease and in 12 out of 49 (24.5%) cancers from patients with a family history of the disease.17
Overall, ATM inactivation occurs sufficiently often in pancreatic ductal adenocarcinomas that it could be important to determine if a patient's pancreatic cancer harbors inactivating ATM mutations or if loss of ATM protein expression when making decisions about therapy. Although ATM status is known to be important in predicting radiation and chemotherapy response in certain settings, pancreatic cancer is considered a relatively radio- and chemo-resistant cancer and not much is known about whether or not ATM status has an important role in determining treatment response. One study found that knockdown of either ATM or the ATM-interacting protein APPL in MIA PaCa2 cells results in radiosensitization and defective DNA repair.18 Simeone et al have found that another one of ATM's many downstream effectors, ATDC, influences response to radiotherapy in preclinical models.19 ATM expression is associated with radioresistance in glioblastoma cells.20 ATM status has also been shown to influence response to chemotherapy in preclinical studies and in some clinical trials. For example, suppression of ATM in p53-deficient mouse embryonic fibroblasts sensitizes them to doxorubicin.21 ATM-deficient chronic lymphocytic leukemia cells die when treated with an ATR inhibitor.22 Lymphomas23,24 and gastric cancers25 with ATM deficiency are more likely to respond to the PARP inhibitor, olaparib. In some settings, ATM inactivation can confer resistance to certain chemotherapy,26 perhaps because of other concurrent molecular alterations such as p53 status.27,21,28
In this study, we examined the effect of ATM-depletion in multiple pancreatic cancer cell lines in response to several chemotherapeutic agents and to fractionated radiation. We also examined the effect of radiation on DNA integrity and repair kinetics in ATM-deficient pancreatic cancer cells.
Results
ATM knockdown and chemosensitivity
ATM expression was knocked down using shRNA in two cell lines with wild-type ATM and with mutant TP53 (MIA PaCa2, Panc2.5) and one wild-type ATM cell line with wild-type TP53 (Panc8.13) (Figure 1).
Figure 1.
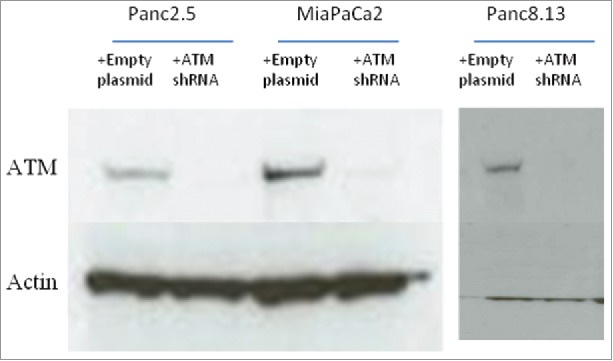
ShRNA-mediated knockdown of ATM.
We first evaluated if ATM knockdown affected the chemosensitivity of pancreatic cancer cells to several commonly used chemotherapeutic agents. We included agents used to treat pancreatic cancer such as the nucleoside gemcitabine, the topoisomerase inhibitors topotecan and doxorubicin, the Parp inhibitor, olaparib, the MEK inhibitor, trametenib, the intercalating agent cisplatin, and the cross-linking agent mitomycin c, as well as two drugs that could in theory potentiate the effects of ATM knockdown, namely, Ve821 (an ATR inhibitor) and Nu7441 (a DNA-Pkcs inhibitor). While most of these drugs were cytotoxic, we observed no significant difference in the chemosensitivity of ATM-deficient cells vs. ATM-wild-type cells for any of the drugs tested (Figure 2). Prior studies have found that evidence that AT-deficient cancer cells are more sensitive to certain drugs such as topoisomerase inhibitors,29 Parp inhibitors23-25 and MEK inhibitors,30 they were not found to be more sensitive to DNA methylating agents or mitomycin C.31 It is likely that the chemosensitivity of ATM-deficient cancer cells is very dependent on the cell type and mutational background. We did find that the BRCA2-mutated pancreatic cancer cell line Capan-1 was exquisitely sensitive to low-dose olaparib as has been previously reported (data not shown).32
Figure 2.
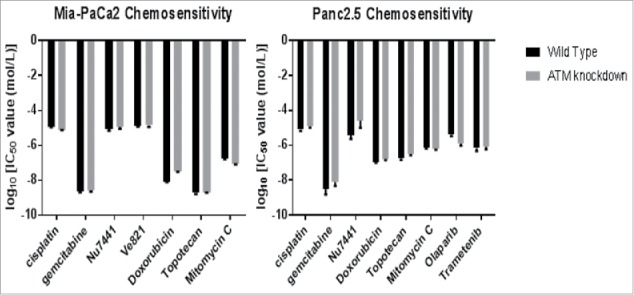
Chemosensitivity of ATM-deficient vs. ATM-wild-type pancreatic cancer cells. Cells were exposed to serial dilutions of each agent from 1 nM to 100 um for three days, then assayed for viability with AlamarBlue. Ic50 values were calculated from the dose-response curve.
Radiosensitivity
We compared the cloning efficiency of irradiated ATM-proficient vs. ATM-knockdown pancreatic cancer cells. Panc2.5, MIA PaCa-2, and Panc8.13 cells all showed markedly reduced cloning efficiency at all radiation doses across a range of plating densities in ATM-deficient cells compared to ATM wild-type cells (representative results shown in Figure 3). In all three cell lines, the difference in cloning efficiency between ATM-deficient and wild-type cells grew with each radiation dose. While surviving wild-type clones generally appeared healthy and stable at lower doses, surviving ATM-deficient cells at any dose frequently exhibited aberrations: cell enlargement, multinucleation, and twisted, malformed cell boundaries consistent with having sustained genotoxic insult.33
Figure 3.
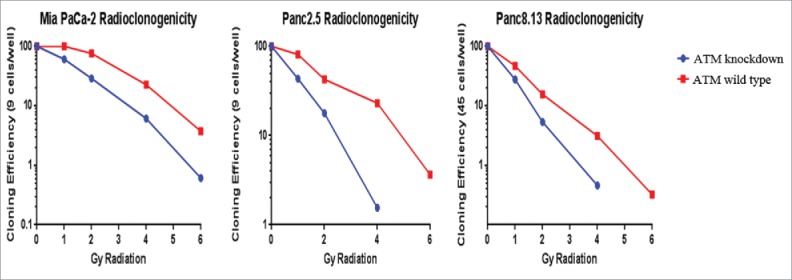
Radiosensitivity of ATM-deficient pancreatic cancer cells. Cells were plated at 96 wells and irradiated. After 14–21 days, positive wells were counted as those with one or more colonies of >50 cells, cloning efficiencies were calculated for each condition, and normalized to the untreated cells of each cell line.
To evaluate if ATM-deficient cells were able to survive irradiation by repairing DSBs with compensatory DNA repair mechanisms such as ATR and DNA-PKcs pathways, we repeated our radioclonogenicity assays combining irradiation with an ATR inhibitor or with a DNA-PKcs inhibitor. In each case, while ATM-deficient cells were more sensitive to the drug/radiation combination than wild-type cells, we saw no evidence of a greater effect on cloning efficiency to suggest that the drug was having any synergistic effect (Figure 4).
Figure 4.
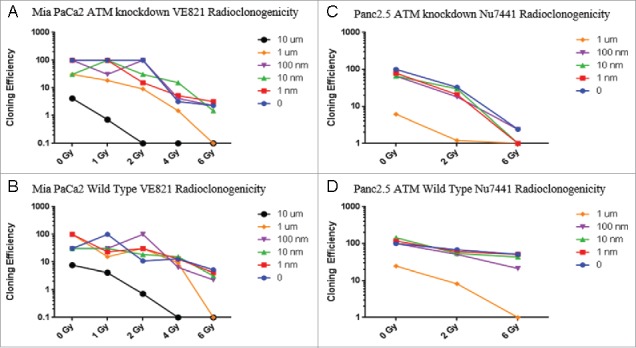
Chemoradioclonogenicity of ATM-deficient cells. Cells were treated with Ve821, an ATR inhibitor (MIA PaCa-2 cells, A, B) or Nu7441, a DNA-PKcs inhibitor (Panc2.5 cells, C, D) one hour before irradiation. After 14–21 days, the number of wells (out of 96) with one or more colonies of >50 cells were determined, cloning efficiencies were calculated for each condition, and normalized to the untreated cells of each cell line.
We also examined the sensitivity of ATM-deficient pancreatic cancer cells to fractionated radiation. Fractionation target more cells at different stages of the cell cycle, helping to overcome the relative resistance of cells during S phase.34 We applied fractionated radiation doses to ATM-knockdown Panc2.5 and Panc8.13 cells. Cells were treated with either 5 Gy or a therapeutically equivalent 6 Gy over 3 doses of 2 Gy each. We find that in both cell lines, the fractionated doses were marginally more effective at inhibiting clonogenic growth than the single 5 Gy dose against ATM-deficient but not ATM wild-type pancreatic cancer cells (Figure 5).
Figure 5.
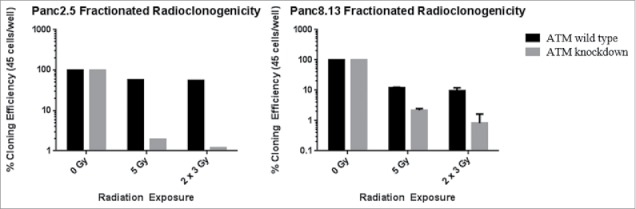
Effect of fractionated radiotherapy on radioclonogenicity. Radiation doses were applied every 24 hours to cells grown in 96-well plates. Cloning efficiency values were calculated from the number of wells out of 96 containing clones of 50 or more cells for each treatment. Cloning efficiency values were normalized to the cloning efficiency values of each untreated cell line.
We were additionally interested in whether any synergistic effect could be observed between treatment with olaparib, a PARP inhibitor, and fractionated radiation. In both tested cell lines, treatment of ATM knockdown pancreatic cancer cells with 100 nM olaparib or an equivalent concentration of DMSO one hour before fractionated radiation resulted in a total loss of clonogenicity (Figure 6). Treatment with 100 nM olaparib one hour before fractionated radiation resulted in a total loss of clonogenicity for ATM wild-type Panc8.13 cells and 2.94% cloning efficiency in ATM wild-type Panc2.5 cells.
Figure 6.
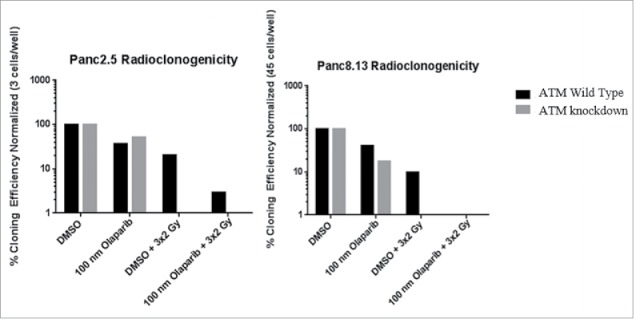
Effect of combining olaparib and fractionated radiation on clonigenic growth. Cells were plated on 96-well plates. Every 24 hours, media was replaced with normal media + 100 nM olaparib or normal media + 0.1% DMSO. Fractionated radiotherapy doses were applied one hour after changing media. Cloning efficiency values were calculated from the number of wells out of 96 containing clones of 50 or more cells for each treatment. Cloning efficiency values were normalized to the cloning efficiency values of each untreated cell line.
Radiation-induced DNA damage
Using the comet assay, we observed a reduced fraction of DNA in the comet head immediately after treating MIA PaCa-2 cells with 4 Gy radiation (Figure 7A). This reduction was equivalent between wild-type and ATM-deficient cells, indicating that ATM status did not affect the rate at which DNA damage occurs in the cell, but rather its ability to tolerate and repair that damage.
Figure 7.
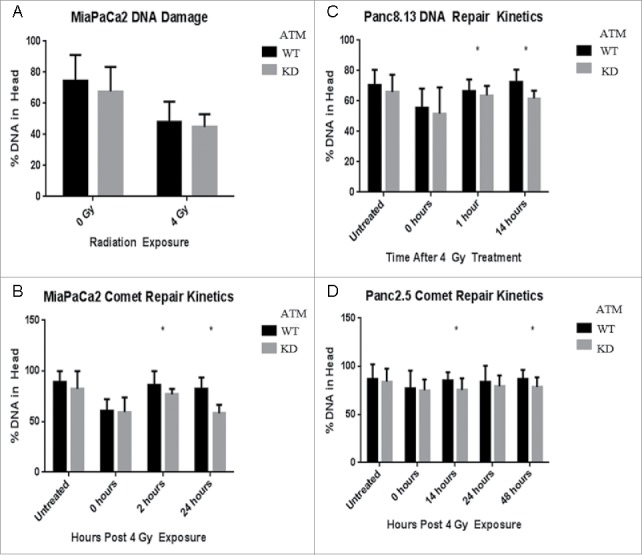
DNA repair kinetics of ATM-deficient cells. The comet assay was used to evaluate DNA damage in cells post-irradiation. For each condition, 100 cells were imaged and analyzed for the proportion of DNA in their head (intact) and tail (damaged). (A) Immediately post-irradiation, there is no significant difference in the proportion of damaged DNA between ATM-deficient and wild-type cells. (B-D) The comet assay was performed on cells at multiple time points post-irradiation.
We next used the comet assay to quantify the repair kinetics of pancreatic cancer cells and the effect of ATM knockdown. After MIA PaCa2, Panc2.5, and Panc8.13 cells were treated with 4 Gy radiation, we observed comparable DNA damage in wild-type and ATM-deficient cells immediately post-exposure, but significantly reduced repair in ATM-deficient cells compared to ATM-wild-type cells (Figure 7). These results are consistent with prior findings that the repair of 10–15% of DSBs is ATM-dependent.35
Discussion
We find that ATM-deficient pancreatic cancer cells are more sensitive to radiation but not to several chemotherapeutic agents. An important question is how well these results might translate into clinical settings. ATM mutations can be detected in fine needle aspirate samples, and loss of tumoral ATM expression could be obtained in fine needle biopsies of pancreatic cancer tumor masses36,37 and so it would be attractive to test the hypothesis that ATM-mutated or otherwise ATM-deficient pancreatic cancers are more sensitive to radiotherapy than ATM-wild-type cancers and if this is the case whether or not the use of radiotherapy for local disease control should take into account the results of tumor molecular profiling.38,39
Interestingly, we did not find any significant differences in the chemosensitivity of ATM-deficient compared to ATM-intact pancreatic cancer cells with intact ATM even for the Parp inhibitor, olaparib. Prior in vitro studies40 and clinical trials have found PARP inhibitors such as olaparib to be especially effective for ATM-deficient breast and prostate cancers.41,42 PARP inhibitors are particularly effective for cancers with defects in homologous repair, such as from BRCA mutations, and there is evidence that these agents can be effective in patients with pancreatic cancer who have BRCA mutations in unpublished observations from a phase I clinical trial (NCT01296763).43
In summary, our results support the hypothesis that ATM-deficient pancreatic cancer cells are more sensitive to fractionated radiation than wild-type pancreatic cancer cells. Our results support the rationale for determining the role using the ATM status of a pancreatic cancer to predict response to fractionated radiotherapy.
Materials and methods
Cell culture
Human pancreatic cancer cell lines MIA PaCa2, Panc 2.5, and Panc 08.13 were obtained from the American Type Culture Collection (Rockville, MD, USA). These cancer cell lines were recently authenticated using genetic markers by the Johns Hopkins Genetics Core facility. All cell lines, were cultured in Dulbecco's modified Eagle's medium (Life Technologies, Inc., Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (Mediatech, Inc., Manassas, VA, USA) and 1% antibiotics (Pen/Strep; Life Technologies) and incubated at 37°C in a humidified atmosphere of 5% CO2 in air.
shRNA inhibition of ATM
Hairpin shRNA sequences targeting ATM were designed with the Oligoengine software (www.oligoengine.com) and ordered from Integrated DNA Technologies (www.idtdna.com):
Pair 1:
Forward: 5’-AGCTTAAAAAGAGGTCAAACCTAGAAAGCTCTCTTGAAGCTTTCTA GGTTTGACCTCGGG
Reverse: 5’-GATCCCCGAGGTCAAACCTAGAAAGCTTCAAGAGAGCTTTCTAGG TTTGACCTCTTTTTA
Pair 2:
Forward:5’-GATCCCCGCTGCAGAGTCAATCAATATTCAAGAGATATTGATTGACTCTGCAGCTTTTTA
Reverse:5’-AGCTTAAAAAGCTGCAGAGTCAATCAATATCTCTTGAATATTGATTGACTCTGCAGCGGG
Sequences were annealed and ligated into linearized pSuper.retro.puro vector. Subcloning Efficiency DH5α Competent Cells (ThermoFisher) were transformed with constructs and selected with 100 µg/ml ampicillin LB agar plates. Isolated bacteria were grown overnight in liquid LB broth with 100 µg/ml ampicillin and processed for plasmid using a GeneJET Plasmid Miniprep Kit (ThermoFisher). Harvested plasmid was checked for the correct insertion by Sanger sequencing (sequencing primer: 5’-GGAAGCCTTGGCTTTTG).
Panc2.5 and Mia PaCa2 cells were transfected with shRNA Pair 1. Panc8.13 cells were transfected with shRNA Pair 2. 1.5 µg of pSuper construct and 1 µg of amphotropic envelope expression vector (pVSV-G) were used to transfect GP-293 cells (ATCC) in 6-well dishes using Lipofectamine 2000 (Life Technologies) according to the manufacturer's protocol. Medium was changed after 24 hours. After an additional 24 hours, cell culture supernatants were harvested, filtered with 0.22 µM filters, and diluted 1:2 in fresh medium. Polybrene was added to dilute viral supernatant at 6 µg/mL which was then added to cells at 30% confluency in 6-well plates. After 24 hours, viral media was replaced with fresh media. After an additional 48 hours, media was replaced with fresh media containing Puromycin. Selection continued for 7 days.
Western blot
Total protein lysates were extracted in RIPA buffer (Roche Diagnostics, Indianapolis, IN) with cOmplete Mini tablets (Roche Diagnostics) and homogenized with a Bioruptor (Diagenode, Denville, NJ) for 8 cycles (30s high, 30S off). Protein concentrations were determined using the Pierce BCA Protein Assay Kit (ThermoScientific, Waltham, MA). Membranes were incubated overnight at 4º C with primary antibodies: rabbit anti-ATM (Abcam, Cambridge, MA) or goat anti-Actin (Santa Cruz Biotechnology). Membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody in 5% dry milk for 1 hour. Bound antibody was detected with a Pierce ECL Plus kit (ThermoScientific).
Chemosensitivity
Chemosensitivity assays were performed using a previously described protocol.44 In brief, cells were plated at a density of 3,000 cells/well in the 60 center wells of 96-well plates. Media was aspirated 24 hours later and replaced with 200 µl/well drug-supplemented media. Drugs were serially diluted 10-fold in standard growth medium at concentrations ranging from 1 nM to 100 µM. For drugs dissolved in dimethyl sulfoxide (DMSO) vehicle, untreated cells were incubated in DMSO-supplemented media as a negative control. Plates were incubated for 72 hours. 10% AlamarBlue Cell Viability reagent (ThermoFisher Scientific) was added to each well as per the manufacturer's protocol. Readings were performed using a BMG FluoStar Galaxy instrument (BMG Labtechnologies; excitation at 544 nM, reading at 590 nM). Statistical analyses were performed and ic50 curves were generated using Graphpad Prism 6.0.
Radioclonogenicity
Cells were plated at low density (9 cells/well for Panc2.5 and MIA PaCa2 cells, 45 cells/well for Panc8.13) and irradiated in a Gammacell 40A (cesium-137 source) at doses of 0, 2, 4, or 6 Gy, then allowed to grow until distinct colonies of >50 cells were countable in untreated plates (8-21 days, depending on the cell line). For fractionated radiation experiments, doses were delivered every 24 hours. Staining was performed with Crystal Violet solution (5% Crystal Violet, 25% methanol) and wells with 1 or more colonies of >50 cells were counted. Cloning efficiency was calculated as: (-ln (total wells-clone wells/total wells))/(number of cells/well) normalized to the number of clone-positive wells on untreated plates.
Chemoradioclonogenicity
Cells were plated at low density (9 cells/well) and allowed to attach overnight. Media was replaced by serially-diluted drugs at 16 wells/dose: MIA PaCa2 (1 nM-10 µM Ve821 ATR inhibitor) and Panc2.5 (1 nM-1 µM Nu7441 DNA-PKcs inhibitor). Plates were irradiated in a Gammacell 40A at 0, 1, 2, 4, or 6 Gy (MIA Paca2) and 0, 2, or 4 Gy (Panc2.5). After 14 days, staining was performed with Crystal Violet solution and wells with 1 or more colonies of >50 cells were counted. Cloning efficiency was calculated as previously.
Comet assay
Comet assay was performed as previously described.45 Cells were detached and resuspended in 5 mL media at 2 × 105 cells/mL stored on ice to minimize DNA damage from handling. CometAssay LMAgarose (Trevigen, Gaithersburg, MD) was kept ready in a 37º water bath. On ice, cells were irradiated in a Gammacell 40A at 4 Gy.
50 µL of cells were combined with 400 µl of LMAgarose and gently mixed. 50 µL of mix was added to each well of a Flare Slide (Trevigen) and spread evenly with a pipet tip. Slides were immediately incubated on a 4º metal surface to cool. Chilled slides were immersed in a glass dish filled with CometAssay Lysis Solution (Trevigen) for 1 hour at 4º C. Slides were washed twice for 15 min in TBE solution and electrophoresed for 40 minutes at 4º C in TBE buffer using 1 V/cm length of the box. DNA was stained with DAPI ProLong Gold Antifade Mountant (ThermoFisher Scientific) and imaged on a Confocal Eclipse Ti-E microscope (Nikon Instruments, San Quirico, Italy). Pictures were taken of 50 cells for each sample and analyzed with CometScore software (www.autocomet.com).
Statistics
Descriptive statistical values and plots were generated using Graphpad Prism 6.0. Chemosensitivity ic50 values were calculated using a log (inhibitor) vs. normalized response equation with a variable slope. Goodness of fit was determined by adjusted R2 value. Radioclonogenicity was calculated as: (-ln (total wells-clone wells/total wells))/ (number of cells/well) normalized to the number of clone-positive wells on untreated plates. Clone-positive wells were defined as any well with one or more clones of 50 or more cells. For comet assays, CometScore software was used to manually delineate tail and head regions of photographed comets by an operator blinded to sample group. The proportion of DNA localized to the head region was averaged for 50 comets per sample group and compared at each time point using a student's t-test.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by Susan Wojcicki and Dennis Troper, NIH grants (CA62924, R01CA176828 and U01CA210170), and the Rolfe Pancreatic Cancer Foundation. MG is the Sol Goldman Professor of Pancreatic Cancer Research.
Author contributions
MA performed experiments, generated and interpreted data; drafted and revised manuscript JRE provided expertize on radiosensitivity and chemosensitivity assays, interpreted data; revised manuscript 12 MG study concept and design; study supervision; interpretation of data; drafting and revision of manuscript; obtained funding.
References
- [1].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians 2016; 66:7–30. [DOI] [PubMed] [Google Scholar]
- [2].Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, Beger H, Fernandez-Cruz L, Dervenis C, Lacaine F, et al. . A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. The New England journal of medicine 2004; 350:1200–10. [DOI] [PubMed] [Google Scholar]
- [3].Engelke CG, Parsels LA, Qian Y, Zhang Q, Karnak D, Robertson JR, Tanska DM, Wei D, Davis MA, Parsels JD, et al. . Sensitization of pancreatic cancer to chemoradiation by the Chk1 inhibitor MK8776. Clin Cancer Res 2013; 19:4412–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chuong MD, Springett GM, Freilich JM, Park CK, Weber JM, Mellon EA, Hodul PJ, Malafa MP, Meredith KL, Hoffe SE, et al. . Stereotactic body radiation therapy for locally advanced and borderline resectable pancreatic cancer is effective and well tolerated. Int J Radiat Oncol Biol Phys 2013; 86:516–22. [DOI] [PubMed] [Google Scholar]
- [5].Herman JM, Chang DT, Goodman KA, Dholakia AS, Raman SP, Hacker-Prietz A, Iacobuzio-Donahue CA, Griffith ME, Pawlik TM, Pai JS, et al. . Phase 2 multi-institutional trial evaluating gemcitabine and stereotactic body radiotherapy for patients with locally advanced unresectable pancreatic adenocarcinoma. Cancer 2015; 121:1128–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hoyer M, Roed H, Traberg Hansen A, Ohlhuis L, Petersen J, Nellemann H, Kiil Berthelsen A, Grau C, Aage Engelholm S, Von der Maase H. Phase II study on stereotactic body radiotherapy of colorectal metastases. Acta Oncol 2006; 45:823–30. [DOI] [PubMed] [Google Scholar]
- [7].Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003; 421:499–506. [DOI] [PubMed] [Google Scholar]
- [8].Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14:197–210. [PubMed] [Google Scholar]
- [9].Perlman SL, Boder Deceased E, Sedgewick RP, Gatti RA. Ataxia-telangiectasia. Handb Clin Neurol 2012; 103:307–32. [DOI] [PubMed] [Google Scholar]
- [10].FitzGerald MG, Bean JM, Hegde SR, Unsal H, MacDonald DJ, Harkin DP, Finkelstein DM, Isselbacher KJ, Haber DA. Heterozygous ATM mutations do not contribute to early onset of breast cancer. Nat Genet 1997; 15:307–10. [DOI] [PubMed] [Google Scholar]
- [11].Roberts NJ, Norris AL, Petersen GM, Bondy ML, Brand R, Gallinger S, Kurtz RC, Olson SH, Rustgi AK, Schwartz AG, et al. . Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer discovery 2016; 6:166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Takai E, Yachida S, Shimizu K, Furuse J, Kubo E, Ohmoto A, Suzuki M, Hruban RH, Okusaka T, Morizane C, et al. . Germline mutations in Japanese familial pancreatic cancer patients. Oncotarget 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Grant RC, Al-Sukhni W, Borgida AE, Holter S, Kanji ZS, McPherson T, Whelan E, Serra S, Trinh QM, Peltekova V, et al. . Exome sequencing identifies nonsegregating nonsense ATM and PALB2 variants in familial pancreatic cancer. Human genomics 2013; 7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Grant RC, Selander I, Connor AA, Selvarajah S, Borgida A, Briollais L, Petersen GM, Lerner-Ellis J, Holter S, Gallinger S. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology 2015; 148:556–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hu C, Hart SN, Bamlet WR, Moore RM, Nandakumar K, Eckloff BW, Lee YK, Petersen GM, McWilliams RR, Couch FJ. Prevalence of Pathogenic Mutations in Cancer Predisposition Genes among Pancreatic Cancer Patients. Cancer epidemiology, biomarkers & prevention: A publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 2016; 25:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, et al. . Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012; 491:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kim H, Saka B, Knight S, Borges M, Childs E, Klein A, Wolfgang C, Herman J, Adsay VN, Hruban RH, et al. . Having pancreatic cancer with tumoral loss of ATM and normal TP53 protein expression is associated with a poorer prognosis. Clin Cancer Res 2014; 20:1865–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hennig J, McShane MP, Cordes N, Eke I. APPL proteins modulate DNA repair and radiation survival of pancreatic carcinoma cells by regulating ATM. Cell Death Dis 2014; 5:e1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang L, Yang H, Palmbos PL, Ney G, Detzler TA, Coleman D, Leflein J, Davis M, Zhang M, Tang W, et al. . ATDC/TRIM29 phosphorylation by ATM/MAPKAP kinase 2 mediates radioresistance in pancreatic cancer cells. Cancer research 2014; 74:1778–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tribius S, Pidel A, Casper D. ATM protein expression correlates with radioresistance in primary glioblastoma cells in culture. Int J Radiat Oncol Biol Phys 2001; 50:511–23. [DOI] [PubMed] [Google Scholar]
- [21].Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, Bartek J, Yaffe MB, Hemann MT. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev 2009; 23:1895–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, Stewart G, Brown J, Lau A, Pratt G, et al. . ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016; 127:582–95. [DOI] [PubMed] [Google Scholar]
- [23].Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, Smith G, Powell JE, Rudzki Z, Kearns P, et al. . The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 2010; 116:4578–87. [DOI] [PubMed] [Google Scholar]
- [24].Williamson CT, Kubota E, Hamill JD, Klimowicz A, Ye R, Muzik H, Dean M, Tu L, Gilley D, Magliocco AM, et al. . Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol Med 2012; 4:515–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bang YJ, Im SA, Lee KW, Cho JY, Song EK, Lee KH, Kim YH, Park JO, Chun HG, Zang DY, et al. . Randomized, Double-Blind Phase II Trial With Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients With Recurrent or Metastatic Gastric Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015; 33:3858–65. [DOI] [PubMed] [Google Scholar]
- [26].Austen B, Skowronska A, Baker C, Powell JE, Gardiner A, Oscier D, Majid A, Dyer M, Siebert R, Taylor AM, et al. . Mutation status of the residual ATM allele is an important determinant of the cellular response to chemotherapy and survival in patients with chronic lymphocytic leukemia containing an 11q deletion. J Clin Oncol 2007; 25:5448–57. [DOI] [PubMed] [Google Scholar]
- [27].Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007; 11:175–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol 2007; 9:573–80. [DOI] [PubMed] [Google Scholar]
- [29].Smith PJ, Makinson TA, Watson JV. Enhanced sensitivity to camptothecin in ataxia-telangiectasia cells and its relationship with the expression of DNA topoisomerase I. International journal of radiation biology 1989; 55:217–31. [DOI] [PubMed] [Google Scholar]
- [30].Smida M, Fece de la Cruz F, Kerzendorfer C, Uras IZ, Mair B, Mazouzi A, Suchankova T, Konopka T, Katz AM, Paz K, et al. . MEK inhibitors block growth of lung tumours with mutations in ataxia-telangiectasia mutated. Nature communications 2016; 7:13701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jaspers NG, de Wit J, Regulski MR, Bootsma D. Abnormal regulation of DNA replication and increased lethality in ataxia telangiectasia cells exposed to carcinogenic agents. Cancer research 1982; 42:335–41. [PubMed] [Google Scholar]
- [32].Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005; 434:913–7. [DOI] [PubMed] [Google Scholar]
- [33].Radford IR, Murphy TK. Radiation response of mouse lymphoid and myeloid cell lines. Part III. Different signals can lead to apoptosis and may influence sensitivity to killing by DNA double-strand breakage. Int J Radiat Biol 1994; 65:229–39. [DOI] [PubMed] [Google Scholar]
- [34].Gregoire V, Van NT, Stephens LC, Brock WA, Milas L, Plunkett W, Hittelman WN. The role of fludarabine-induced apoptosis and cell cycle synchronization in enhanced murine tumor radiation response in vivo. Cancer Res 1994; 54:6201–9. [PubMed] [Google Scholar]
- [35].Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, et al. . A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell 2004; 16:715–24. [DOI] [PubMed] [Google Scholar]
- [36].DeWitt J, Cho CM, Lin J, Al-Haddad M, Canto MI, Salamone A, Hruban RH, Messallam AA, Khashab MA. Comparison of EUS-guided tissue acquisition using two different 19-gauge core biopsy needles: a multicenter, prospective, randomized, and blinded study. Endoscopy international open 2015; 3:E471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kim H, Saka B, Knight S, Borges M, Childs E, Klein AP, Wolfgang CL, Herman JM, Adsay V, Hruban RH, et al. . Having pancreatic cancer with tumoral loss of ATM and normal TP53 protein expression is associated with a poorer prognosis. Clinical cancer research : an official journal of the American Association for Cancer Research 2014; 20:1865–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gleeson FC, Kerr SE, Kipp BR, Voss JS, Minot DM, Tu ZJ, Henry MR, Graham RP, Vasmatzis G, Cheville JC, et al. . Targeted next generation sequencing of endoscopic ultrasound acquired cytology from ampullary and pancreatic adenocarcinoma has the potential to aid patient stratification for optimal therapy selection. Oncotarget 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Valero V, 3rd, Saunders TJ, He J, Weiss MJ, Cameron JL, Dholakia A, Wild AT, Shin EJ, Khashab MA, O'Broin-Lennon AM, et al. . Reliable Detection of Somatic Mutations in Fine Needle Aspirates of Pancreatic Cancer With Next-generation Sequencing: Implications for Surgical Management. Ann Surg 2016; 263:153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lord CJ, McDonald S, Swift S, Turner NC, Ashworth A. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair (Amst) 2008; 7:2010–9. Epub 08 Oct 15. [DOI] [PubMed] [Google Scholar]
- [41].Gilardini Montani MS, Prodosmo A, Stagni V, Merli D, Monteonofrio L, Gatti V, Gentileschi MP, Barila D, Soddu S. ATM-depletion in breast cancer cells confers sensitivity to PARP inhibition. J Exp Clin Cancer Res 2013; 32:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, et al. . DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. The New England journal of medicine 2015; 373:1697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, et al. . Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015; 33:244–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cui Y, Brosnan JA, Blackford AL, Sur S, Hruban RH, Kinzler KW, Vogelstein B, Maitra A, Diaz LA, Jr., Iacobuzio-Donahue CA, et al. . Genetically defined subsets of human pancreatic cancer show unique in vitro chemosensitivity. Clin Cancer Res 2012; 18:6519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hedayati M, Haffner MC, Coulter JB, Raval RR, Zhang Y, Zhou H, Mian O, Knight EJ, Razavi N, Dalrymple S, et al. . Androgen Deprivation Followed by Acute Androgen Stimulation Selectively Sensitizes AR-Positive Prostate Cancer Cells to Ionizing Radiation. Clin Cancer Res 2016; 22:3310–9. [DOI] [PMC free article] [PubMed] [Google Scholar]