

Abstract
Objective
To identify novel risk factors and corroborate previously identified risk factors for mean annual decline in FEV1 percent predicted in a large, contemporary, U.S. cohort of young cystic fibrosis patients.
Methods
Retrospective observational study of participants in the EPIC Observational Study, who were Pseudomonas–negative and ≤12 years of age at enrollment in 2004-2006. The associations between potential demographic, clinical and environmental risk factors evaluated during the baseline year and subsequent mean annual decline in FEV1 percent predicted were evaluated using generalized estimating equations.
Results
The 946 participants in the current analysis were followed for a mean of 6.2 (SD 1.3) years. Mean annual decline in FEV1 % predicted was 1.01% (95% CI 0.85-1.17%). Children with 1 or no F508del mutations had a significantly smaller annual decline in FEV1 compared to F508del homozygotes. In a multivariable model, risk factors during the baseline year associated with a larger subsequent mean annual lung function decline included female gender, frequent or productive cough, low BMI (<66th percentile, median in the cohort), ≥1 pulmonary exacerbation, high FEV1 (≥115% predicted, in the top quartile), and respiratory culture positive for methicillin-sensitive Staphylococcus aureus, methicillin-resistant Staphylococcus aureus or Stenotrophomonas maltophilia.
Conclusions
We have identified a range of risk factors for FEV1 decline in a large cohort of young, CF patients who were Pa negative at enrollment, including novel as well as previously identified characteristics. These results could inform the design of a clinical trial in which rate of FEV1 decline is the primary endpoint and identify high-risk groups that may benefit from closer monitoring.
Keywords: cystic fibrosis, pediatric, lung function, decline, Pseudomonas aeruginosa, EPIC Observational Study
Introduction
The rate of decline in the forced expiratory volume in one second (FEV1) is widely used as a measure of lung disease progression among persons with cystic fibrosis (CF). The rate of FEV1 decline has been shown to predict mortality and to improve with chronic therapies1-5. FEV1 decline has slowed with progressive birth cohorts6, possibly due to more aggressive and standardized care including widespread use of Pseudomonas (Pa) eradication regimens. Risk factors for rate of FEV1 decline have been identified, including CF transmembrane conductance regulator (CFTR) genotype, female sex, age, meconium ileus, pancreatic insufficiency, pulmonary exacerbations, respiratory microbiology, high initial FEV1, diabetes, and presence of wheeze or crackles7-12. Some of these studies were limited by small sample size, conduct at one or a small number of sites, conflicting results or changes in clinical care since publication. In this exciting era of advances in CFTR modulator therapy, there is renewed interest in rate of FEV1 decline as a clinical trial endpoint, particularly in mild disease. An improved understanding of risk factors for FEV1 decline in a modern cohort with mild disease could help inform the use of rate of decline as a clinical trial endpoint and identify high-risk subgroups in whom more intensive monitoring or treatment might be warranted.
The EPIC Observational Study is a multicenter longitudinal observational study that enrolled Pa negative children ≤12 years of age and evaluated a wide range of demographic, clinical and environmental factors, a number of which have not been previously examined. It thus provides a unique cohort in which to evaluate risk factors for FEV1 decline in young patients with relatively mild disease in the era of widespread Pa eradication therapy. The purpose of the current study was to identify novel risk factors and corroborate previously identified risk factors for FEV1 decline in this well-characterized, young cohort that was Pa-negative at enrollment. Some of the results have previously been presented in abstract form.
Methods
Study Participants
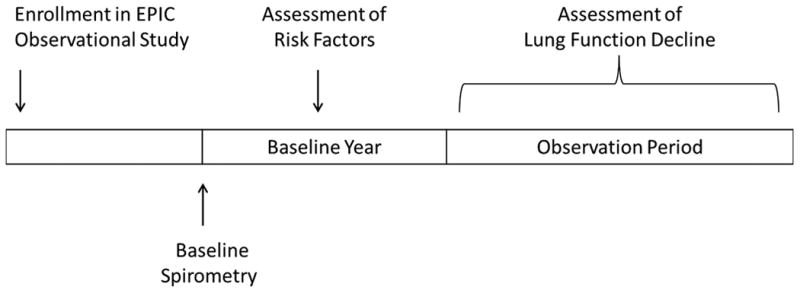
The design of the EPIC Observational Study has been described previously13,14. Children with an established diagnosis of CF15 0 to 12 years of age and with no prior lifetime respiratory isolation of Pa or Pa negative for ≥ 2 years were enrolled at 59 accredited U.S. CF care centers between 2004 and 2006. (Children were also able to enroll in EPIC OBS by virtue of participating in the EPIC Clinical Trial14, nested in the EPIC OBS cohort. Some of these participants were not Pa-negative at enrollment into EPIC OBS and were therefore excluded from the current analysis.) EPIC OBS participants were included in the current analysis if they had at least 3 spirometry measurements obtained after enrollment, as defined below. The first spirometry after enrollment was defined as the baseline spirometry, the 1-year period after the baseline spirometry was defined as the baseline year, and the period starting at the end of the baseline year was defined as the observation period (Figure 1). Spirometry data were only available beginning at age 6. Thus, for children enrolled in the EPIC Observational study prior to age 6, there could be a lag of several years between enrollment into the EPIC Observational Study and the baseline spirometry. Eligible participants were required to have a baseline spirometry and spirometry recorded at least once during the first and fourth years of the observation period. In addition, they were required to have an annual family survey and at least 1 clinical encounter recorded during the baseline year (see Data Collection, below). For families with more than 1 eligible child, a single sibling was randomly selected for inclusion in the analysis. Written informed consent was obtained from the family of each participant, and the study was approved by the Institutional Review Board at each participating site.
Figure 1. Schematic of study flow.
Data collection
EPIC Observational data were collected at each clinical encounter via the Cystic Fibrosis Foundation National Patient Registry (CFFNPR) and study-specific forms, as previously reported13. CFFNPR data included FEV1 for ages 6 and older, results of respiratory cultures obtained as part of routine clinical care, weight and height percentiles, and presence of a pulmonary exacerbation defined as a respiratory illness treated with intravenous antibiotics in either the inpatient or outpatient setting. Respiratory signs and symptoms collected on study-specific encounter forms included: crackles or wheezes on chest auscultation, cough frequency, activity level, and days with activities limited due to cough or shortness of breath. The annual family survey queried families regarding a wide range of exposures including environmental tobacco smoke, daycare, other persons with CF and influenza vaccinations, as well as maternal education and household income13. FEV1 was expressed as a percent of the predicted value based on the reference equations of Wang, et al16 (for males age 6-17 and females age 6-15) and Hankinson, et al17 (for males ≥18 years of age and females ≥16). The data cut off for this analysis was December 31, 2013.
Statistical analysis
Predictor variables were evaluated during the baseline year only, and included demographic characteristics obtained from the CF Foundation National Patient Registry, exposures reported on the annual family survey, and clinical data, including respiratory microbiology. The primary outcome was the annualized rate of change in FEV1 % predicted over the observation period, calculated from a linear model using the best reported values of FEV1 % predicted from each age quarter during the observation period, with age centered at the midpoint of each age quarter. Observation time was calculated from end of the baseline year to the date of the latest spirometry measurement reported in the CFFNPR as of 12/31/2013.
The average annual change in FEV1 % predicted (change in FEV1 % predicted per year of age) was modelled using an extension of the generalized linear model for longitudinal correlated data. Specifically, population-average linear regression models were estimated using generalized estimating equations (GEE) with exchangeable correlation structure and robust variance estimates. A predefined set of predictor variables was evaluated to identify risk factors for decline in lung function. Each risk factor was screened individually in a GEE model that included main effect terms for age in years and the risk factor, and an interaction term between age and the risk factor. The coefficient for each interaction term thus reflects the difference in mean annual change in FEV1 % predicted between participants with the attribute during the baseline year vs. those in the reference category. The significance of each interaction term was assessed by testing the null hypothesis that the coefficient for the interaction term was equal to zero. Risk factors associated with lung function decline when assessed individually (p<0.10) were then evaluated together in multivariable GEE models. Risk factors were retained in the final multivariable model if their interaction with age remained statistically significant (p<0.05) in the presence of other risk factors. All multivariable GEE models were adjusted a priori for Pa status (any positive cultures vs. none), and Medicaid status (receiving Medicaid vs. not) during the baseline year. Because we hypothesized that the association of baseline weight percentile, BMI percentile and FEV1 % predicted with annual FEV1 decline was not linear, we categorized each of these predictors into quartiles based on the observed distribution in the cohort. For simplicity, based on the results of these analyses, we categorize weight and BMI percentiles as above or below the median value in the cohort and FEV1 % predicted as above or below the 75th percentile. Statistical analyses were conducted using Stata Statistical Software (Release 12, StataCorp, 2011, College Station, TX).
Results
Figure 2 illustrates selection of the study cohort. Almost 1,800 participants (N=1797) enrolled in the EPIC Observational Study; 114 were excluded due to being Pa-positive at enrollment or having their CF diagnosis subsequently reversed. Following assignment of exclusion criteria for the current analysis and excluding siblings, the study cohort comprised 946 participants. Characteristics of the study cohort during the baseline year are detailed in Table 1. As the cohort was enrolled between 2004 and 2006 (prior to universal uptake of newborn screening (NBS) in the U.S. in 2006), only 16.5% of participants were identified by prenatal or NBS. The mean age at enrollment into the EPIC Observational Study was 5.7 (SD 3.6) years. Participants were followed during the observation period for an average of 6.2 (SD 1.3) years, during which time they had an average of 31.4 visits with spirometry recorded (SD 14.5).
Figure 2. Selection of study cohort.
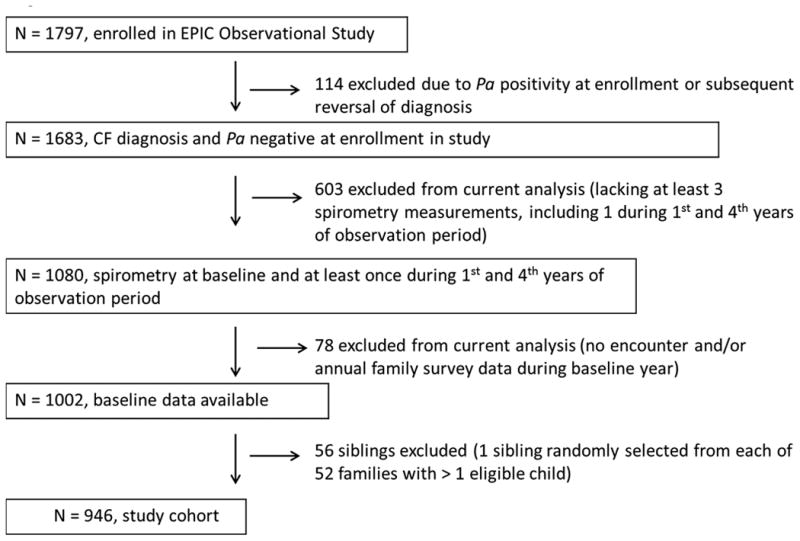
Table 1. Participant characteristics evaluated during the baseline year.
| Total Cohort N=946 |
|
|---|---|
| Age at baseline spirometry, years, mean (SD) | 7.9 (2.0) |
| Female sex, n (%) | 470 (49.7) |
| Caucasian, n (%) | 910 (96.2) |
| Hispanic, n (%) | 31 (3.3) |
| CFTR genotype | |
| F508del homozygous, n (%) | 505 (53.4) |
| F508del heterozygous, n (%) | 358 (37.8) |
| Other/not available, n (%) | 83 (8.5) |
| History of meconium ileus, n (%) | 228 (24.1) |
| Identified by prenatal or newborn screening, n (%) | 156 (16.5) |
| Pancreatic enzyme use, n (%) | 882 (93.2) |
| Household member smokes, n (%)1 | 261 (27.6) |
| Crackles on chest exam, n (%)1 | 209 (22.1) |
| Wheeze on chest exam, n (%)1 | 110 (11.6) |
| Frequent or productive cough, n (%)1 | 545 (57.6) |
| Pulmonary exacerbation(s), n (%)1 | 186 (19.7) |
| P. aeruginosa (Pa) positive, n (%)1 | 211 (22.3) |
| Mucoid Pa positive, n (%)1 | 40 (4.2) |
| Methicillin sensitive S. aureus positive, n (%)1 | 611 (64.6) |
| Methicillin resistant S. aureus positive, n (%)1 | 168 (17.8) |
| S. maltophilia positive, n (%)1 | 137 (14.5) |
| Weight percentile2, mean (SD) | 50.0 (27.6) |
| BMI percentile2, mean (SD) | 62.7 (24.4) |
| FEV1 % predicted2, mean (SD) | 104.4 (15.5) |
Recorded during the baseline year only; does not include data prior to the baseline year.
Best value during the baseline year.
Mean annualized rate of decline of FEV1 % predicted for the cohort as a whole was 1.01 (95% CI 0.85-1.17) % per year. Figure 3 displays the effect of risk factors on mean annual change in FEV1 % predicted, from univariate models. When compared to the reference group (F508del homozygotes), participants with 1 or no F508del mutations had significantly less mean annual decline in FEV1% predicted. Similarly, participants diagnosed with CF following prenatal or NBS had significantly less mean annual decline in FEV1 % predicted than those diagnosed conventionally. Characteristics in the baseline year associated with a significantly greater annual rate of decline included female sex, pancreatic enzyme use, frequent or productive cough, BMI below the median in the cohort (<66th percentile), 1 or more pulmonary exacerbations and high baseline FEV1 (in the top quartile, ≥115 % predicted). While culturing Pa or mucoid Pa on one or more occasions during the baseline year was not associated with annual FEV1 decline, isolation of MSSA, MRSA, and S. maltophilia were all associated with significantly greater annual decline. A variety of exposures assessed during the baseline year were not significantly associated with mean annual change in FEV1 % predicted, including environmental tobacco smoke exposure, as well as influenza vaccine, wood burning stove use, maternal education, household income and Medicaid status (data not shown).
Figure 3.
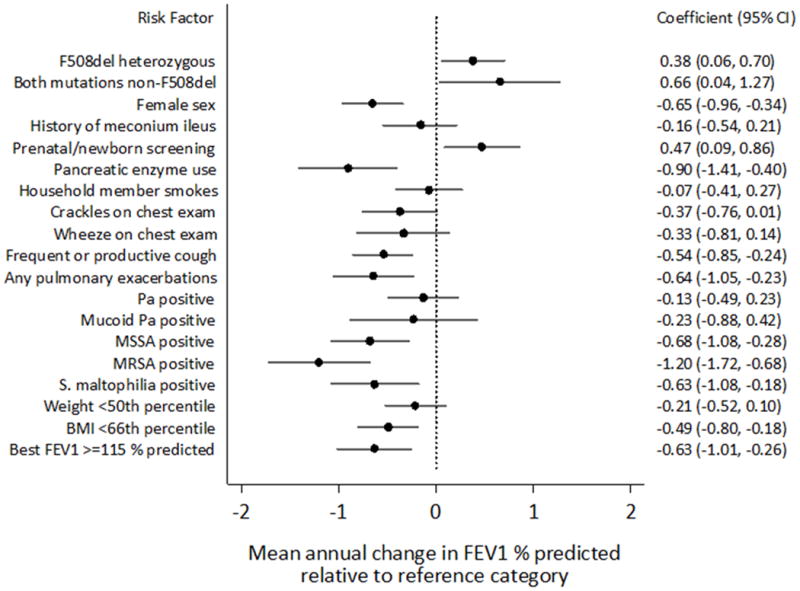
Forest plot of effect of risk factors on annual change in FEV1% predicted, from univariate GEE models. For each risk factor, estimates of mean annual change in FEV1% predicted and associated 95% confidence intervals are displayed for participants with the risk factor relative to participants in the reference category. All risk factors were measured during the baseline year. Based on evaluation of quartiles, best baseline FEV1% predicted was dichotomized at the top quartile value (115%), and best baseline weight and BMI percentiles were dichotomized at the median value (50th and 66th percentile, respectively).
The results of the multivariate model are shown in Table 2. For the risk factors retained in this model, the direction and magnitude of the effect on annual change in FEV1 % predicted was similar to that seen in the univariate models. Note that, for participants in the reference category for all the included characteristics, the mean annualized change in FEV1 % predicted was not significantly different from zero: mean 0.10 (95% CI, -0.33, 0.52) % predicted/year. The strongest risk factors during the baseline year for subsequent annual decline in FEV1 % predicted were isolation of MRSA from a respiratory culture, FEV1 ≥115% predicted, isolation of MSSA, female sex and homozygous F508del CFTR genotype.
Table 2. Multivariable model of risk factors measured during the baseline year for annual change in FEV1 % predicted1.
| Risk Factor | Coefficient | 95% CI | P-value |
|---|---|---|---|
| F508del heterozygous2 | 0.39 | 0.08, 0.70 | 0.01 |
| Both mutations non-F508del2 | 0.55 | -0.07, 1.18 | 0.08 |
| Female sex | -0.60 | -0.90, -0.31 | 0.0001 |
| Frequent or productive cough | -0.31 | -0.61, -0.01 | 0.04 |
| Pulmonary exacerbation | -0.47 | -0.89, -0.06 | 0.02 |
| MSSA3 | -0.68 | -1.05, -0.31 | 0.0004 |
| MRSA3 | -1.00 | -1.51, -0.49 | 0.0001 |
| S. maltophilia positive | -0.50 | -0.93, -0.07 | 0.02 |
| Best BMI <66th percentile | -0.53 | -0.84, -0.23 | 0.001 |
| Best FEV1 ≥115 % predicted | -0.75 | -1.12, -0.38 | 0.0001 |
| Age (yrs) | 0.56 | 0.09, 1.02 | 0.02 |
Results are shown for risk factors that had a significant interaction with age. The coefficient for each risk factor reflects the difference in mean annual change in FEV1 % predicted between participants with the attribute during the baseline year vs. those in the reference category. The coefficient for age reflects the mean annualized change in FEV1 % predicted for participants whose values were in the reference category for all risk factors; coefficients for other main effect terms are not shown. Reference categories included genotype F508del homozygous, male sex, no exacerbations, absence of frequent or productive cough, S. aureus negative, S. maltophilia negative, BMI ≥66th percentile (the median value in the cohort), and FEV1 <115 % predicted (representing the lower 3 quartiles of FEV1 in the cohort). The final model was adjusted for P. aeruginosa status (any positive cultures vs. none) and Medicaid status (on Medicaid vs. not) during the baseline year and reflects data for 931 participants.
The p-value was 0.02 when both genotype terms were tested simultaneously for the overall effect of genotype on annual change in FEV1 % predicted.
S. aureus status was assigned to one of 3 mutually exclusive categories: MSSA +, MRSA +, or S. aureus negative (reference group). The p-values reflect the fact that the annual change in FEV1 % predicted for both MSSA+ and MRSA+ participants was significantly different than observed for S. aureus negative participants.
Discussion
In a large, contemporary cohort of young CF patients who were Pa negative at enrollment, we identified a number of important risk factors for rate of decline of FEV1 % predicted. Our cohort was healthier at baseline than earlier cohorts; for example, our mean baseline FEV1 was 104% predicted, compared to mid-80 % predicted in the cohort of Konstan, et al 8. Similarly, our cohort had a slower mean annual decline in FEV1: 1.01 % per year compared to 1.1 to 2.4 % per year, depending on age, in the cohort of Konstan, et al. Our cohort was enrolled between 2004 and 2006, when Pa eradication therapy was already in widespread use. In 2006, 97% of the EPIC Observational sites reported prescribing antibiotics for Pa acquisition “often” or “always” on the annual site survey of clinical practices. Nonetheless, we corroborated the work of previous authors in identifying CFTR genotype, pancreatic insufficiency (as measured by pancreatic enzyme use) and pulmonary exacerbations as risk factors for lung function decline. Similar to prior studies, we found parent report of frequent or productive cough and crackles on auscultation of the chest to be risk factors for FEV1 decline8,9. In contrast to some, but not all, prior reports, we did not find meconium ileus to be a risk factor.
There have been conflicting results regarding the negative effects of female sex on lung function decline8,9; in our study, females did have a significantly larger rate of decline than males even though our cohort was young and had relatively mild disease. This was not due to earlier acquisition of Pa among females in our cohort (data not shown). One potential biologic mechanism for poorer lung health in females with CF is upregulation of airway mucus production by estrogen18, 19, which could worsen mucociliary clearance.
In this cohort that was Pa negative at enrollment and among whom Pa eradication treatment was widespread, we found no association between isolation of Pa from a respiratory culture in the baseline year and subsequent rate of FEV1 decline, in contrast to studies of cohorts enrolled prior to widespread uptake of Pa eradication therapy8,9. This result suggests that perhaps chronic Pa infection is necessary to affect rate of lung function decline in the contemporary era. Sanders, et al, in a long term follow up study of 132 participants in the Wisconsin Neonatal Screening Trial, found no association between Pa in general and FEV1, but did find an association between mucoid Pa and FEV19, corroborating their prior work showing a strong impact of mucoid, but not non-mucoid, Pa on lung function and chest radiograph scores20. In our cohort, only 4% of the cohort had mucoid Pa at baseline, limiting our power to detect a significant association.
In our cohort, isolation of MRSA, MSSA and Stenotrophomonas maltophilia during the baseline year were all associated with a greater rate of FEV1 decline. In fact, MRSA was the strongest risk factor in our multivariable model. Although MRSA has previously been reported to be a risk factor for FEV1 decline in CF21, to our knowledge MSSA has only been shown to negatively affect FEV1 decline in infants, in a report from the Australian AREST-CF Study22. In that study, respiratory cultures were obtained by bronchoalveolar lavage, whereas in our cohort almost all cultures were of the posterior oropharynx, making direct comparisons difficult. Our study suggests that MSSA and its role in CF lung disease in the current era needs to be further investigated, particularly in light of the conflicting results of trials of S. aureus prophylaxis 23,24. To our knowledge, no prior studies have shown S. maltophilia to be a risk factor for FEV1 decline. While S. maltophilia has long been considered an organism infecting older persons with CF, recent studies have shown that younger age is actually a risk factor for S. maltophilia acquisition25. In that same study, greater rate of FEV1 decline was found to be a risk factor for S. maltophilia acquisition. Our finding of S. maltophilia as a risk factor for greater rate of FEV1 decline could be a manifestation of this same association. Clearly, the role of S. maltophilia in early CF lung disease deserves further investigation.
The study by Konstan, et al of risk factors for FEV1 decline in the large North American Epidemiologic Study of CF (ESCF) cohort was one of the first to report that a high initial FEV1 (>100% predicted) was associated with a faster rate of decline8. Though the mean baseline FEV1 in our contemporary cohort was 104% predicted compared to 85% predicted in the Konstan cohort, this observation nonetheless held true. When FEV1 was expressed as quartiles in our study, an association was found between baseline FEV1 in the highest quartile (≥115% predicted) and a larger mean annual decline in FEV1. These findings suggest that clinicians may not be consistently recognizing and intervening in early drops in FEV1 in their CF patients with high FEV1, as recently demonstrated by Morgan, et al using data from ESCF26. In regards to nutritional status, while on average the cohort had a normal BMI (62.7% SD (24.4)), a BMI <66th percentile (below the median in the cohort) was negatively associated with FEV1 annual decline, similar to both Konstan, et al8 and Sanders, et al9, who identified poor baseline nutritional status (weight for age and height for age, respectively) as a risk factor for faster rate of decline. Similarly, Yen, et al identified greater weight at age 4 as a predictor of better lung function through age 18 years 27
Although a number of investigators have demonstrated an effect of newborn screening on FEV1, to our knowledge ours is the first study to demonstrate a slower annual decline in FEV1 among children identified by newborn screening than those diagnosed conventionally. While encouraging, these results should be interpreted with caution. Participants were enrolled in our study when only selected U.S. states were screening for CF, so unmeasured confounders in terms of different care practices in these states could have contributed to improved outcomes.
While we had the unique ability to examine a number of environmental risk factors for FEV1 decline, we did not detect an association between any of these characteristics (exposure to cigarette smoke, influenza vaccine or other persons with CF; Medicaid status; household income; maternal education) and FEV1 decline. Our findings may be due at least in part to misclassification (particularly for exposure to cigarette smoke), as data were collected via self-report. Prior authors have found an association between influenza season and pulmonary exacerbations28 rates in CF, and cigarette smoke has recently been shown to produce systemic defects in CFTR function 28. Socioeconomic status has been shown to affect FEV1 in cross-sectional analyses29,30.
Strengths of our study include the ability to focus on young patients with generally mild disease in the era of widespread Pa eradication therapy, the large size of the cohort enrolled at multiple U.S. sites and followed for a relatively long period, and augmentation of CFFNPR data with EPIC Observational study-specific data. Limitations include potential misclassification of environmental risk factors through annual parent self-report and of characteristics recorded in the CFFNPR such as Pa mucoidy. In addition, we were only able to evaluate the association of pulmonary exacerbations requiring intravenous antibiotics on lung function decline; we did not have information on milder exacerbations treated with oral and/or inhaled antibiotics. We chose to evaluate how baseline characteristics predict lung function decline; an alternative and interesting analysis would be to treat each of the predictors as time-varying covariates over the observation period. Finally, as stated above, we relied on oropharyngeal cultures for respiratory microbiology, which have limited diagnostic accuracy compared to lower airway cultures31.
In summary, we have identified a range of risk factors for FEV1 decline in a large cohort of young, Pa negative CF patients with relatively mild disease, including novel as well as previously identified factors. These results could inform the design of a clinical trial in which rate of FEV1 decline is the primary endpoint and identify high-risk groups that may need closer monitoring. Indeed, given that the mean annual rate of FEV1 decline in our contemporary cohort of children was 1.01% predicted per year, perhaps the most relevant conclusion for clinical trial design in this population is that detecting a treatment effect on FEV1 decline would require very large samples and/or long periods of observation.
Acknowledgments
The authors would like to thank the Cystic Fibrosis Foundation for the use of CF Foundation National Patient Registry data to conduct this study. Additionally, we would like to thank the patients, care providers, and clinic coordinators at CF Centers throughout the United States for their contributions to the CF Foundation National Patient Registry.
Specify grants, other financial support received, and granting institutions: Cystic Fibrosis Foundation Therapeutics grants OBSERV04K0 and EPIC0K0 to M. Rosenfeld, Seattle Children's Hospital, Seattle, WA; Nemours Children's Clinic Research Program, Jacksonville, FL
Appendix 1. The EPIC Study Group
Albany Medical College, Albany, NY, Paul Comber MD, PhD and Terese Evens; All Children's Hospital CF Center, St. Petersburg, FL, Magdalen Gondor MD, Kathy Hosler and Stasia Lehmann; Baylor College of Medicine, Houston, TX, Peter Hiatt MD, Omalee Lopez, and Charlene Hallmark; Brown University Medical School – Rhode Island Hospital, Providence, RI, Karen Daigle MD and Camille White; Cardinal Glennon Children's Medical Center, St. Louis, MO, Blakeslee Noyes MD and Vikki Kociela; Children's Hospital, Boston, MA, Henry Dorkin MD and Jonathan Greenberg, Ashish George, Erin Leone Thakkallapalli, and Jane Solomon; Children's Hospital & Clinics, Minneapolis, MN, John McNamara MD, Sandy Landvik, and Mary Sachs; Children's Hospital Colorado, Aurora, CO, Frank Accurso MD, Meg Anthony, and Shelley Mann; Children's Hospital of Los Angeles, Los Angeles, CA, Arnold Platzker MD; Children's Hospital Medical Center of Akron, Akron, OH, Gregory Omlor MD and Deborah Ouellette; Children's Hospital of Michigan, Detroit, MI, Ibrahim Abdulhamid MD and Catherine Van Wagnen; Children's Hospital of Milwaukee, Milwaukee, WI, Diana Quintero MD, MaryEllen Freeman, and Tami Miller; Children's Hospital – University of Birmingham, Birmingham, AL, Hector Gutierrez MD, Heather Hathorne, and Gina Sabbatini; Children's Hospital of Pittsburgh, Pittsburgh, PA, David Orenstein MD, Caitlin Clarke, Judy Fulton, Elizabeth Hartigan and Sandra Hurban; Children's Hospital at Westchester Medical Center, Valhalla, NY, Allen Dozor MD, Ingrid Gherson, and Madelint Heydendael; Children's Medical Center of Dayton, Dayton, OH, Robert Fink MD and Sandy Bartosik; Children's Mercy Hospital, Kansas City, MO, Philip Black MD, Rose Thompson, Karie Robinson, and Candy Schmoll; University of Vermont Children's Hospital, Burlington, VT, Thomas Lahiri MD, Joan Lippmann, and Sandra Diehl; Cohen Children's Hospital, Great Neck, NY, Joan DeCelie-Germana MD, Lynn Bonitz, and Susan Galvin; Cook Children's Medical Center, Fort Worth, TX, Maynard Dyson MD, Trudy Morris, and Sara Scott; Dartmouth Hitchcock Clinic – Lebanon, Lebanon, NH, H. Worth Parker MD, Alix Ashare, Jennifer Helm, Dana Dorman, Pamela Hofley, and Lisa Moulton; Alfred I. DuPont Hospital for Children, Wilmington, DE, Aaron Chidekel MD and Sandra Budd; Emory University School of Medicine, Atlanta, GA, Michael Schechter MD, Arlene Stecenko MD, Monica Haughton, Eric Hunter, Jeannette Peabody, and Joy Dangerfield; Helen DeVos Women & Children's Center, Grand Rapids, MI, Susan Millard MD, Mary Flanagan, Hollie Bonnema, and Teri Crumb; Intermountain CF Center, Salt Lake City, UT, Barbara Chatfield MD, Heather Oldroyd, and Jane Vroom; Kaiser Permanente Medical Care Program, Oakland, CA, Gregory Shay MD, Julie Lee, and Erika Marmolejo; Le Bonheur Children's Medical Center, Memphis, TN, Robert Schoumacher MD and Barbara Culbreath; Medical College of Georgia, Augusta, GA, Kathleen McKie MD and Heidi Stapp; Milton S. Hershey Medical Center – Penn State University, Hershey, PA, Gavin Graff MD, Lisa Allwein, and Diane Kitch; Maine Medical Center Cystic Fibrosis Center, Portland, ME, AnneMarie Cairns DO, Harmony Renna, Mary Ellen Corrigan, and Carrie Milliard; Monmouth Medical Center, Long Branch, NJ, Robert Zanni MD and Bridget Marra; Nationwide Children's Hospital, Columbus, OH, Karen McCoy MD, Barbara Butera, Diana Gilmore, Mary Terri Johnson, Veronica Lowell, Patricia Olson, and Laura Raterman; Nemours Children's Clinic, Jacksonville, FL, David Schaeffer MD, Elizabeth DeLuca, and Rena Sprinkle; Northwestern University, Chicago, IL, Adrienne Prestridge MD, Jennifer Milam, and Catherine Powers; Oregon Health Sciences University, Portland, OR, Michael Wall MD, Alexandra Cornell, Aaron Guzik, and Ben McCullar; Rainbow Babies and Children's Hospital, Cleveland, OH, Michael Konstan MD, Colette Bucur, Ellen Divoky, Kate Hilliard, Jeanne Krenicky, and Cheryl Velotta; Riley Hospital for Children, Indianapolis, IN, Michelle Howenstine MD, Terry Barclay, and Lisa Bendy; Seattle Children's Hospital, Seattle, WA, Margaret Rosenfeld MD, MPH, Sharon McNamara, Judy Gabrysiak, and Alan Genatossio; St. Christopher's Hospital for Children; Philadelphia, PA, Laurie Varlotta MD and Marcella Aramburo; St. Louis Children's Hospital, St. Louis, MO, Thomas Ferkol MD, Mary Boyle, Patricia Burks, and Jane Quante; Stanford University Medical Center, Palo Alto, CA, Richard Moss MD, Zoe Davies, Colleen Dunn, and Cassie Everson; SUNY Upstate Medical University, Syracuse, NY, Ran Anbar MD, Donna Lindner, and Valoree Suttmore; UMass Memorial Medical Center, Worcester, MA, Brian O'Sullivan, MD and Karen Longtine; University of California at San Francisco, San Francisco, CA, Dennis Nielson MD and Courtney Moreno; University of Iowa Hospital and Clinics, Iowa City, IA, Richard Ahrens MD, Jean Frauenholtz, Tom Santacroce, and Mary Teresi; University of Kentucky, Lexington, KY, Jamshed Kanga MD, Brittany Fuller, Tammy Taylor, and Tammy Watts; University of Michigan Health System, Ann Arbor, MI, Samya Nasr MD and Dawn Kruse; University of Mississippi Medical Center, Jackson, MS, Suzanne Miller MD, Michelle King, Joseph Majure, Kim Adcock, and Linda Bonham; University of Nebraska Medical Center, Omaha, NE, John Colombo MD, Diane Acquazzino, Shandalle Fertig, and Sandra Strizek; University of North Carolina at Chapel Hill, Chapel Hill, NC, George Retsch-Bogart MD, Carol Barlow, Paul Jones, Kelly Moormann, and Caroline LaFave; University of Rochester Medical Center, Rochester, NY, Clement Ren MD, Nancy Jenks, and Mary Platt; University of Virginia Health System, Charlottesville, VA, Deborah Froh MD, Mary Alice Blackwell, and Patricia Moss; University of Wisconsin, Madison, WI, Michael Rock MD and Linda Makholm; Vanderbilt Children's Hospital, Nashville, TN, Rebekah Brown MD and Alice Bray; Women & Children's Hospital of Buffalo, Buffalo, NY, Daniel Sheehan MD, Jameelah Ali, and Nadine Caci.
Footnotes
Identify meetings (if any) at which the paper was presented: North American Cystic Fibrosis Conference, Salt Lake City, UT, October 17-19th, 2013
References
- 1.Corey M, Edwards L, Levison H, Knowles M. Longitudinal analysis of pulmonary function decline in patients with cystic fibrosis. J Pediatr. 1997;131(6):809–814. doi: 10.1016/s0022-3476(97)70025-8. [DOI] [PubMed] [Google Scholar]
- 2.Schluchter MD, Konstan MW, Davis PB. Jointly modelling the relationship between survival and pulmonary function in cystic fibrosis patients. Statistics in medicine. 2002;21(9):1271–1287. doi: 10.1002/sim.1104. [DOI] [PubMed] [Google Scholar]
- 3.Konstan MW, Byard PJ, Hoppel CL, Davis PB. Effect of high-dose ibuprofen in patients with cystic fibrosis. N Engl J Med. 1995;332(13):848–854. doi: 10.1056/NEJM199503303321303. [DOI] [PubMed] [Google Scholar]
- 4.Ren CL, Pasta DJ, Rasouliyan L, Wagener JS, Konstan MW, Morgan WJ, Scientific Advisory G the Investigators, Coordinators of the Epidemiologic Study of Cystic F. Relationship between inhaled corticosteroid therapy and rate of lung function decline in children with cystic fibrosis. J Pediatr. 2008;153(6):746–751. doi: 10.1016/j.jpeds.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 5.Konstan MW, Wagener JS, Pasta DJ, Millar SJ, Jacobs JR, Yegin A, Morgan WJ, Scientific Advisory G Investigators, Coordinators of Epidemiologic Study of Cystic F. Clinical use of dornase alpha is associated with a slower rate of FEV1 decline in cystic fibrosis. Pediatr Pulmonol. 2011;46(6):545–553. doi: 10.1002/ppul.21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McPhail GL, Acton JD, Fenchel MC, Amin RS, Seid M. Improvements in Lung Function Outcomes in Children with Cystic Fibrosis are Associated with Better Nutrition, Fewer Chronic Pseudomonas aeruginosa Infections, and Dornase Alfa Use. J Pediatr. 2008;153(6):752–757. doi: 10.1016/j.jpeds.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 7.Schaedel C, de Monestrol I, Hjelte L, Johannesson M, Kornfalt R, Lindblad A, Strandvik B, Wahlgren L, Holmberg L. Predictors of deterioration of lung function in cystic fibrosis. Pediatr Pulmonol. 2002;33(6):483–491. doi: 10.1002/ppul.10100. [DOI] [PubMed] [Google Scholar]
- 8.Konstan MW, Morgan WJ, Butler SM, Pasta DJ, Craib ML, Silva SJ, Stokes DC, Wohl ME, Wagener JS, Regelmann WE, Johnson CA. Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis. J Pediatr. 2007;151(2):134–139. doi: 10.1016/j.jpeds.2007.03.006. 139 e131. [DOI] [PubMed] [Google Scholar]
- 9.Sanders DB, Li Z, Laxova A, Rock MJ, Levy H, Collins J, Ferec C, Farrell PM. Risk factors for the progression of cystic fibrosis lung disease throughout childhood. Ann Am Thorac Soc. 2014;11(1):63–72. doi: 10.1513/AnnalsATS.201309-303OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welsh L, Robertson CF, Ranganathan SC. Increased rate of lung function decline in Australian adolescents with cystic fibrosis. Pediatr Pulmonol. 2014;49(9):873–877. doi: 10.1002/ppul.22946. [DOI] [PubMed] [Google Scholar]
- 11.Waters V, Stanojevic S, Atenafu EG, Lu A, Yau Y, Tullis E, Ratjen F. Effect of pulmonary exacerbations on long-term lung function decline in cystic fibrosis. Eur Respir J. 2012;40(1):61–66. doi: 10.1183/09031936.00159111. [DOI] [PubMed] [Google Scholar]
- 12.Vandenbranden SL, McMullen A, Schechter MS, Pasta DJ, Michaelis RL, Konstan MW, Wagener JS, Morgan WJ, McColley SA Investigators, Coordinators of the Epidemiologic Study of Cystic F. Lung function decline from adolescence to young adulthood in cystic fibrosis. Pediatr Pulmonol. 2012;47(2):135–143. doi: 10.1002/ppul.21526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Treggiari MM, Rosenfeld M, Mayer-Hamblett N, Retsch-Bogart G, Gibson RL, Williams J, Emerson J, Kronmal RA, Ramsey BW. Early anti-pseudomonal acquisition in young patients with cystic fibrosis: rationale and design of the EPIC clinical trial and observational study'. Contemp Clin Trials. 2009;30(3):256–268. doi: 10.1016/j.cct.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenfeld M, Emerson J, McNamara S, Joubran K, Retsch-Bogart G, Graff GR, Gutierrez HH, Kanga JF, Lahiri T, Noyes B, Ramsey B, Ren CL, Schechter M, Morgan W, Gibson RL. Baseline characteristics and factors associated with nutritional and pulmonary status at enrollment in the cystic fibrosis EPIC observational cohort. Pediatr Pulmonol. 2010;45(9):934–944. doi: 10.1002/ppul.21279. [DOI] [PubMed] [Google Scholar]
- 15.Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, Durie PR, Legrys VA, Massie J, Parad RB, Rock MJ, Campbell PW., 3rd Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr. 2008;153(2):S4–S14. doi: 10.1016/j.jpeds.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Dockery DW, Wypij D, Gold DR, Speizer FE, Ware JH, Ferris BG., Jr Pulmonary function growth velocity in children 6 to 18 years of age. Am Rev Respir Dis. 1993;148(6 Pt 1):1502–1508. doi: 10.1164/ajrccm/148.6_Pt_1.1502. [DOI] [PubMed] [Google Scholar]
- 17.Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999;159(1):179–187. doi: 10.1164/ajrccm.159.1.9712108. [DOI] [PubMed] [Google Scholar]
- 18.Tam A, Morrish D, Wadsworth S, Dorscheid D, Man SF, Sin DD. The role of female hormones on lung function in chronic lung diseases. BMC women's health. 2011;11:24. doi: 10.1186/1472-6874-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeitlin PL. Cystic fibrosis and estrogens: a perfect storm. J Clin Invest. 2008;118(12):3841–3844. doi: 10.1172/JCI37778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z, Kosorok MR, Farrell PM, Laxova A, West SE, Green CG, Collins J, Rock MJ, Splaingard ML. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA. 2005;293(5):581–588. doi: 10.1001/jama.293.5.581. [DOI] [PubMed] [Google Scholar]
- 21.Dasenbrook EC, Merlo CA, Diener-West M, Lechtzin N, Boyle MP. Persistent methicillin-resistant Staphylococcus aureus and rate of FEV1 decline in cystic fibrosis. Am J Respir Crit Care Med. 2008;178(8):814–821. doi: 10.1164/rccm.200802-327OC. [DOI] [PubMed] [Google Scholar]
- 22.Pillarisetti N, Williamson E, Linnane B, Skoric B, Robertson CF, Robinson P, Massie J, Hall GL, Sly P, Stick S, Ranganathan S. Infection, inflammation, and lung function decline in infants with cystic fibrosis. Am J Respir Crit Care Med. 2011;184(1):75–81. doi: 10.1164/rccm.201011-1892OC. [DOI] [PubMed] [Google Scholar]
- 23.Weaver LT, Green MR, Nicholson K, Mills J, Heeley ME, Kuzemko JA, Austin S, Gregory GA, Dux AE, Davis JA. Prognosis in cystic fibrosis treated with continuous flucloxacillin from the neonatal period. Arch Dis Child. 1994;70(2):84–89. doi: 10.1136/adc.70.2.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stutman HR, Lieberman JM, Nussbaum E, Marks MI. Antibiotic prophylaxis in infants and young children with cystic fibrosis: a randomized controlled trial. J Pediatr. 2002;140(3):299–305. doi: 10.1067/mpd.2002.121930. [DOI] [PubMed] [Google Scholar]
- 25.Stanojevic S, Ratjen F, Stephens D, Lu A, Yau Y, Tullis E, Waters V. Factors influencing the acquisition of Stenotrophomonas maltophilia infection in cystic fibrosis patients. J Cyst Fibros. 2013;12(6):575–583. doi: 10.1016/j.jcf.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 26.Morgan WJ, Wagener JS, Yegin A, Pasta DJ, Millar SJ, Konstan MW Scientific Advisory Group i, coordinators of the Epidemiologic Study of Cystic F. Probability of treatment following acute decline in lung function in children with cystic fibrosis is related to baseline pulmonary function. J Pediatr. 2013;163(4):1152–1157 e1152. doi: 10.1016/j.jpeds.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yen EH, Quinton H, Borowitz D. Better nutritional status in early childhood is associated with improved clinical outcomes and survival in patients with cystic fibrosis. J Pediatr. 2013;162(3):530–535 e531. doi: 10.1016/j.jpeds.2012.08.040. [DOI] [PubMed] [Google Scholar]
- 28.Ortiz JR, Neuzil KM, Victor JC, Wald A, Aitken ML, Goss CH. Influenza-associated cystic fibrosis pulmonary exacerbations. Chest. 2010;137(4):852–860. doi: 10.1378/chest.09-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schechter MS, Shelton BJ, Margolis PA, Fitzsimmons SC. The association of socioeconomic status with outcomes in cystic fibrosis patients in the United States. Am J Respir Crit Care Med. 2001;163(6):1331–1337. doi: 10.1164/ajrccm.163.6.9912100. [DOI] [PubMed] [Google Scholar]
- 30.Taylor-Robinson DC, Thielen K, Pressler T, Olesen HV, Diderichsen F, Diggle PJ, Smyth R, Whitehead M. Low socioeconomic status is associated with worse lung function in the Danish cystic fibrosis population. Eur Respir J. 2014;44(5):1363–1366. doi: 10.1183/09031936.00063714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenfeld M, Emerson J, Accurso F, Armstrong D, Castile R, Grimwood K, Hiatt P, McCoy K, McNamara S, Ramsey B, Wagener J. Diagnostic accuracy of oropharyngeal cultures in infants and young children with cystic fibrosis. Pediatr Pulmonol. 1999;28(5):321–328. doi: 10.1002/(sici)1099-0496(199911)28:5<321::aid-ppul3>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]