

Abstract
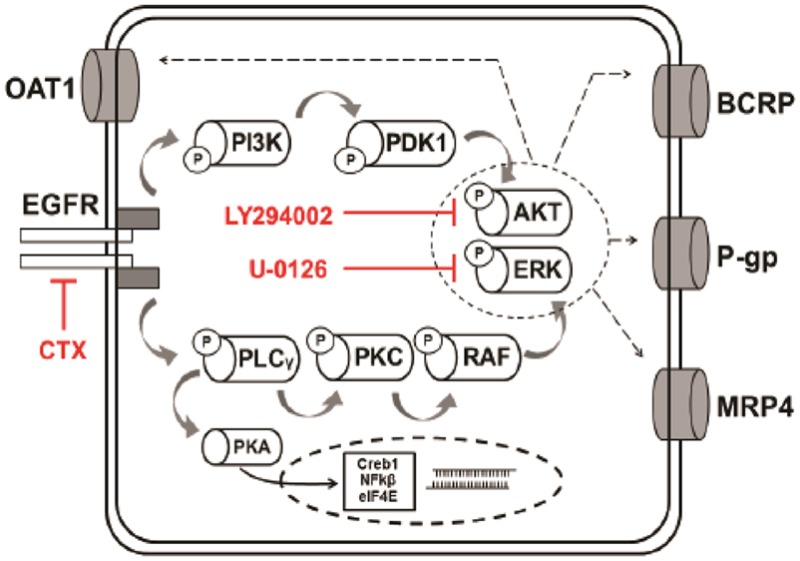
The combination of methotrexate with epidermal growth factor receptor (EGFR) recombinant antibody, cetuximab, is currently being investigated in treatment of head and neck carcinoma. As methotrexate is cleared by renal excretion, we studied the effect of cetuximab on renal methotrexate handling. We used human conditionally immortalized proximal tubule epithelial cells overexpressing either organic anion transporter 1 or 3 (ciPTEC-OAT1/ciPTEC-OAT3) to examine OAT1 and OAT3, and the efflux pumps breast cancer resistance protein (BCRP), multidrug resistance protein 4 (MRP4), and P-glycoprotein (P-gp) in methotrexate handling upon EGF or cetuximab treatment. Protein kinase microarrays and knowledge-based pathway analysis were used to predict EGFR-mediated transporter regulation. Cytotoxic effects of methotrexate were evaluated using the dimethylthiazol bromide (MTT) viability assay. Methotrexate inhibited OAT-mediated fluorescein uptake and decreased efflux of Hoechst33342 and glutathione-methylfluorescein (GS-MF), which suggested involvement of OAT1/3, BCRP, and MRP4 in transepithelial transport, respectively. Cetuximab reversed the EGF-increased expression of OAT1 and BCRP as well as their membrane expressions and transport activities, while MRP4 and P-gp were increased. Pathway analysis predicted cetuximab-induced modulation of PKC and PI3K pathways downstream EGFR/ERBB2/PLCg. Pharmacological inhibition of ERK decreased expression of OAT1 and BCRP, while P-gp and MRP4 were increased. AKT inhibition reduced all transporters. Exposure to methotrexate for 24 h led to a decreased viability, an effect that was reversed by cetuximab. In conclusion, cetuximab downregulates OAT1 and BCRP while upregulating P-gp and MRP4 through an EGFR-mediated regulation of PI3K-AKT and MAPKK-ERK pathways. Consequently, cetuximab attenuates methotrexate-induced cytotoxicity, which opens possibilities for further research into nephroprotective comedication therapies.
Keywords: combination therapy, drug disposition, kinase signaling, drug transporters, renal proximal tubule
Introduction
Current cancer treatments often rely on the administration of multiple chemotherapeutic drugs to improve overall survival. These combination therapies can overcome the limitations of single target drugs, as they exploit different action mechanisms and can be administered under several regimens for curative and palliative purposes. Recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) yields a poor prognosis, with limited treatment options, and a novel phase I/IIb trial (identifier, NCT02054442; https://clinicaltrials.gov/) is set to investigate the efficacy and safety of methotrexate in combination with cetuximab.
Methotrexate is an established antifolate and one of the most extensively used anticancer agents.1,2 It inhibits the enzyme dihydrofolate reductase, disrupting DNA synthesis,3 and can be administered in both high and low doses for the treatment of autoimmune diseases and cancers.4,5 High-dose methotrexate (i.e., 12 000 mg/m2) is used in the treatment of malignancy, while low-dose methotrexate (i.e., 40 mg/m2) is used in metastatic SCCHN and in various nonmalignant immune-mediated disorders.6,7 Cetuximab is an antiepidermal growth factor receptor (EGFR) recombinant monoclonal antibody (IgG1)8 that displays beneficial clinical outcomes in patients with recurrent or metastatic SCCHN and does not exert renal adverse effects.9 It binds to the extracellular domain of EGFR and hinders the ligand-induced tyrosine kinase activation.10 When active, multiple EGFR tyrosine-kinase domains can trigger downstream signal transduction cascades, more specifically the phosphoinositide 3-kinase (PI3K)-AKT and mitogen activated protein kinase (MAPK) pathways.11 These pathways are responsible for a series of intracellular regulatory processes such as cell cycle progression, neovascularization, migration, differentiation, proliferation, and immunogenic responses.12
The kidney, through active tubular secretion, clears a great variety of chemotherapeutics and among those is methotrexate. During this process, methotrexate accumulates within the kidney and, especially at high-dose, the drug can promote extensive necrosis of the proximal tubular epithelial cells (PTEC).13,14 This is due to transporters expressed in PTEC that are involved in the excretion process of methotrexate. The drug is efficiently taken up from the blood compartment by the organic anion transporters 1 and 3 (OAT1–3)15 and excreted into the tubular lumen by the multidrug resistance proteins (MRP) 2 and 416 and breast cancer resistance protein (BCRP).17 In general, transmembrane transporters are key to renal function and their activity and expression can be regulated, among others via EGFR.18 In the kidney, EGFR is expressed in PTEC, and activated by EGF and EGF-like hormones.19 Furthermore, EGFR can stimulate renal epithelial regeneration in response to kidney injury.20
Despite the wide use of methotrexate, a potential pharmacokinetic interaction with cetuximab when combined in therapy is not known. The aim of this study was to investigate the effects of cetuximab on renal methotrexate handling in vitro using conditionally immortalized human PTEC overexpressing either OAT1 or OAT3 (ciPTEC-OAT1/ciPTEC-OAT3).21 Previously, we demonstrated ciPTEC as a representative human proximal tubule cell line with preserved features, including cell polarization, monolayer organization, expression of tight junction proteins, as well as xenobiotic transporter and metabolic enzyme activities.22−24 Using this cell line, we revealed that cetuximab downregulates OAT1 and BCRP while it upregulates MRP4 via EGFR signaling, thereby reducing renal methotrexate uptake as well as its cytotoxic potential. This appeared to be a common regulatory pathway, with effects beyond attenuating methotrexate toxicity, as cisplatin cytotoxicity25 was counteracted as well. This study shows the nephroprotective potential of combination therapy, in vitro and opens possibilities for further research on the use of comedication to manage renal toxicity.
Methods
Chemicals
All chemicals were obtained from Sigma (Zwijndrecht, The Netherlands) unless stated otherwise. Stock solutions of all compounds used for transport assays were prepared according to specification in either dimethyl sulfoxide (DMSO) or dH2O. Cetuximab (Erbitux) was obtained from Merck Serono (Darmstadt, Germany).
Cell Culture
The ciPTEC-OAT1 and ciPTEC-OAT3 were cultured in phenol red-free DMEM/F12 (Invitrogen, Breda, The Netherlands), as previously described. Cells were seeded at a density of 63 000 cell/cm2, grown for 24 h at 33 °C and subsequently at 37 °C for 7 days.23,26 ciPTEC required a temperature shift to mature and grow into fully differentiated epithelial cells forming a tight monolayer, prior to each assay. A detailed composition of the ciPTEC medium can be found in the Supporting Information (Table S1).
Cetuximab, Methotrexate, and Cisplatin Treatments
To study the effects of cetuximab, matured ciPTEC were treated for 24 or 48 h by incubation with or without cetuximab (500 μg/mL) in culture medium, in the presence or absence of EGF (10 ng/mL). After cetuximab exposure, cells were incubated with either increasing concentrations of methotrexate or cisplatin (up to 100 μM) for 24 h, using standard ciPTEC culture medium (CM; containing 10% fetal calf serum) or serum-free medium (SFM). Under normal conditions, ciPTEC medium contained EGF, which was considered as control condition.
Fluorescent Functional Assays
The retention of fluorescent substrates was used to determine the changes in activity of membrane transporters.24 Fluorescein, Hoechst33342, calcein-AM, 5-chloromethyl fluorescein diacetate (CMFDA), and (4-(4-(dimethylamino) styryl)-N-methylpyridinium (ASP+) were used to evaluate the function of OAT1, OAT3, BCRP, P-gp, MRP4, and OCT2, respectively, as described.21,24 To study the effect of methotrexate on these transporters, cells were exposed to a single concentration of the substrates (fluorescein and calcein-AM, 1 μM; Hoechst33342 and CMFDA, 1.25 μM), separately, or together with methotrexate. Model inhibitors probenecid, KO143, MK571, and PSC833 were used to validate transporter involved. To investigate cetuximab influence on transport function, a two-fold step dilution was performed to obtain a concentration gradient of the fluorescent substrates with maximum concentrations of 25 μM each. The calcein-AM assay was performed in the presence of 2.0 μM of inhibitor PSC833 and CMFDA in the presence of 5.0 μM of MK571. Cells were washed twice before incubation at 37 °C for 10 min (fluorescein), 30 min (CMFDA, Hoechst33342 and ASP+), or 60 min (calcein-AM), as described previously. Afterward plates were washed twice and cells were lysed with either 0.1 M NaOH (fluorescein) or Triton-X100 1% (calcein-AM and CMFDA). Subsequently, fluorescence was acquired via an Ascent Fluoroskan FL microplate reader (appropriate filter settings: wavelengths, fluorescein, calcein, and glutathione-methyl fluorescein (GS-MF: the end-metabolites of calcein-AM and CMFDA, respectively); excitation, 494 nm; emission, 512 nm. Hoechst33342: excitation, 350 nm; emission, 461 nm. ASP+: excitation, 470 nm; emission, 590 nm). A schematic representation of the interactions between fluorescent substrates and model inhibitors is depicted in the Supporting Information (Figure S1).
Gene Expression
Transporters gene expression profiling was performed by isolating total RNA from cells grown in six-well plates, using an RNeasy Mini kit (Qiagen, Venlo, The Netherlands), according to the manufacturers specifications. Subsequently, cDNA was synthesized using the Omniscript RT-kit (Qiagen). Subsequently, quantitative PCR was performed in a CFX96 real-time PCR detection system (Biorad, Veenendaal, The Netherlands). GAPDH was used as housekeeping gene for normalization, and its expression was not affected by the experimental conditions tested as demonstrated in the Supporting Information (Figure S2). Relative expression levels were calculated as fold change using the 2–ΔΔCT method. The primer-probe sets were obtained from Applied Biosystems: GAPDH - hs99999905_m1; BCRP - hs00184979_m1; MRP4 - hs00195260_m1 and Pgp - hs00184500_m1; OAT1 - hs00537914.
Western Blot Analysis
The protein levels of OAT1 and BCRP were determined by Western blotting using 9% (W/V) sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Cell samples were homogenized in ice-cold Tris-Sucrose (TS) buffer (10 mM Tris-HEPES and 250 mM sucrose, pH 7.4) containing protease inhibitors (PMSF 100 μM, Aprotinin 5 μg/mL, Leupeptin 5 μg/mL, Pepstatin 1 μg/mL, E64 10 μM). Membrane fractions were obtained by high shear passage using a microfluidizer LV1 (Microfluidics, Westwood, AM, USA), and cell lysates were span for 20 and 90 min at 4000 and 25 000 rcf at 4 °C, respectively. Membranes were incubated with mouse anti-BCRP (1:200 dilution; Abcams, Cambridge, UK) and rabbit anti-OAT1 antibody (1:100 dilution; Abcams, Cambridge, UK) overnight at 4 °C. As a loading control, rabbit anti-Na, K-ATPase antibody (α-subunit, 1:4000 dilution, C356-M09,27) was used. Secondary antibodies, Alexa fluor 680 goat antirabbit IgG (1:10 000 dilution; Life Technologies Europe BV), streptavidin Alexa fluor 680 (1:10 000 dilution; Life Technologies Europe BV), and IRDye 800 goat antirabbit IgG (1:10 000 dilution; Rockland, PA). Fluorescence was detected using the Odyssey scanner CLx (Li-Cor Biosciences, USA). Data were normalized to protein expression levels of the loading control using ImageJ software (imagej.nih.gov).
Serine/Threonine and Tyrosine Kinase Microarrays and Pathway Analysis
To investigate downstream cetuximab effects on ciPTEC, the serine/threonine kinase (STK) and protein tyrosine kinase (PTK) activity profiles were determined with 3D dynamic peptide microarrays. Using the PamStation12 platform and PamChip microarrays, containing four identical arrays each, with 142 either STK or PTK immobilized phosphorylation sites, cetuximab treated ciPTEC samples were analyzed as previously described.28 Cells were grown in six-well plates, and after maturation, cells were washed and incubated with cetuximab either in the presence or absence of EGF for 15 min in HBSS. Cell pellets were lysed using mammalian protein extraction reagent (MPER) in the presence of HALT phosphatase and protease inhibitors (Pierce) according to manufacturer instructions. Resulting cell lysates were profiled as described previously.29,30 The data were analyzed using Evolve and BioNavigator (signal acquisition and data analysis software, respectively; Pamgene) to generate heat maps and differential kinase activity hits by comparing ratios over the control condition (+EGF). Significantly relevant peptide phosphorylations (targets/hits) were selected and used to generate canonical pathway signaling hypotheses by performing an unbiased analysis using the Genego MetaCore (thomsonreuters.com) software and database resources.
Intracellular Methotrexate Metabolite Quantification by LC–MS/MS
To determine methotrexate uptake by ciPTEC, the intracellular levels of methotrexate polyglutamates were analyzed following 48 h cetuximab pretreatment and 24 h methotrexate exposure in both CM and SFM. Cells were washed, harvested and cell pellets were frozen in liquid nitrogen and stored at −80 °C prior to analysis. Samples were analyzed as previously described.31 Briefly, 16% perchloric acid was used to precipitate protein, after a centrifugation step, 10 μL (per sample) of supernatant was analyzed by liquid chromatography-electrospray ionization-tandem mass spectrometry (UPLC–ESI–MS/MS) using a Waters Acquity BEH C18 column and a 5–100% gradient of 10 mM ammonium bicarbonate pH10 and methanol on a classic Acquity UPLC (Waters Instruments). Detection was performed during a 6 min run by positive electrospray ionization using a Quattro Premier XE (Waters Instruments).
Cell Cycle Analysis
For cell cycle analysis, cells were grown in six-well plates, and following treatment cells were harvested using accutase, washed, and centrifuged before fixation with cold 70% (v/v) ethanol for 1 h. Subsequently, the cells, in suspension, were stained with a propidium iodide (PI) solution containing 40 μg/mL PI, 100 μg/mL RNase A, and 0.1% (v/v) Triton X-100 for 45 min. The data were retrieved using a BD FACSCanto II flow cytometer and analyzed using the FlowLogic software (Chromocyte, Sheffield, UK).
Cell Viability
Cell survival upon methotrexate or cisplatin exposure was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were washed twice and incubated with 5 mg/mL MTT (100 μL p/well) for a minimum of 2 h at 37 °C; after washing, wells are dissolved in 200 μL of DMSO (p/well) and absorbance read via a BioRad iMark microplate reader (absorbance: 550–600 nm). Cisplatin toxicity was also evaluated by the Presto-blue (PB) assay, and cells were incubated for 1 h with 1:10 dilution PB solution (100 μL p/well) at 37 °C. Subsequently, the supernatant was transferred to a 96-well plate and absorbance read using Jasco FP8300 Spectrophotometer (excitation wavelength, 560 nm; emission wavelength, 590 nm).
Data Analysis
Transport activity was calculated by normalizing fluorescence intensity, expressed in arbitrary units (a.u.), to baseline values (no inhibitor) after background subtraction, as described.24 Inhibition of efflux activity led to increased total fluorescence; therefore, efflux activity was depicted as the inverse of the fold increase in fluorescence. Nonlinear analysis according to Michaelis–Menten kinetics was performed using GraphPad Prism 5.02 (GraphPad software, San Diego, CA, USA). Differences between groups were considered to be statistically significant when p < 0.05 using a one-tailed Student’s t test. All data are presented as mean ± SEM.
Results
Renal Excretion of Methotrexate Is Mediated by OAT1, OAT3, BCRP, and MRP4
Methotrexate clearance has been studied in humans and rodents in vivo and in cell lines expressing a single transporter; however, its handling in a renal cell model containing multiple relevant transporters has not been performed. We first examined the renal excretion route for methotrexate in ciPTEC-OAT1 and ciPTEC-OAT3 in an indirect way by measuring the interaction of methotrexate with marker substrates fluorescein, GS-MF and Hoechst33342 for OAT1/OAT3, MRP’s and BCRP, respectively.21,24 Model inhibitors were used as positive control and the fluorescence levels without inhibition were normalized to 100%. Upon methotrexate exposure, OAT1, BCRP, and MRP’s activities were significantly reduced in ciPTEC-OAT1, whereas in ciPTEC-OAT3, only OAT3 and MRP4 activities were reduced (Figure 1A–C). BCRP inhibition could not be determined in ciPTEC-OAT3 and P-gp activity was not affected in both cell lines (Figure 1D). These results confirm a renal excretion pathway previously suggested (Figure 1E).
Figure 1.

Methotrexate uptake in ciPTEC-OAT1 and ciPTEC-OAT3. Methotrexate (MTX) significantly reduced the uptake of fluorescein (A) and inhibited the efflux of Hoechst33342, in ciPTEC-OAT1 (B) and GS-MF (C), in both ciPTEC-OAT1 and OAT3. Calcein efflux was not blocked by methotrexate in both cell lines (D), and Hoechst33342 activity in ciPTEC-OAT3 could not be determined. Model inhibitors Probenecid, KO143, MK571, and PSC833 were tested as positive controls. Data are presented as mean values ± SEM. Statistical analysis was performed via unpaired Student’s t test. ∗, p < 0.05 and ∗∗∗, p < 0.01 compared to control (CTRL). A schematic depiction of the potential interactions (E).
Cetuximab Regulates Tubular Xenobiotic Transport
Next, renal drug transporter expression and its activity were determined after treating ciPTEC with cetuximab. Exposure to cetuximab for 48 h resulted in an altered expression and function of all major transport systems present in ciPTEC-OAT1 as compared to the standard culture conditions in the presence of EGF, but not OAT3. Therefore, subsequent experiments were performed with ciPTEC-OAT1. The gene expression of OAT1 was reduced in the absence of EGF or in the presence of cetuximab, which was also observed for BCRP (Figure 2A,C). In agreement, protein expression levels of OAT1 and BCRP were reduced in the absence of EGF (Figure 2A,C). Both MRP4 and P-gp mRNA levels were increased in the absence of EGF as well as in the presence of cetuximab (Figure 2E,G), for which we could not determine quantifiable differences in their expressions using the sample preparation methodology described. The accumulations of fluorescein, Hoechst33342, calcein, and GS-MF were reduced upon cetuximab exposure both in the presence or absence of EGF (Figure 2B,D,F,H). This effect was similar to that observed after culturing cells for 48 h in absence of EGF.
Figure 2.

Expression and function of renal drug transporters upon cetuximab pretreatment. Cells were pretreated with cetuximab (CTX) before gene and protein expression was determined. The activity of renal drug transporter was evaluated using fluorescent substrates (OAT1, fluorescein; BCRP, Hoechst33342; P-gp, calcein and MRP4; GS-MF, respectively). Changes in transport activity are depicted in fluorescent intensity expressed in arbitrary units (a.u). CTX effectively reduced the expression and activity of OAT1 (A, B) and BCRP (C, D), while increasing the expression and activity of P-gp (E, F) and MRP4 (G, H). Protein expression was determined for OAT1 (60 kDa) and BCRP (72 kDa) (A, C lower panel), Na,K-ATPase was used as loading control (112 kDa). Data are presented as mean values ± SEM, n = 3. Statistical analysis was performed via unpaired Student’s t test. ∗, p < 0.05 and ∗∗∗, p < 0.01 compared to control (+EGF condition).
When cetuximab was given in absence of EGF, no further alterations were observed. Functional changes were evidenced from the accumulation of the fluorescent probes, in which a higher intensity yielded more accumulation of the probe. For efflux pumps, a higher activity yielded a reduction in substrate accumulation given an increased rate of removal, as was observed for GS-MF and calcein. The reduction in BCRP was not reflected by an increased Hoechst33342 retention, most likely due to the increased expression in P-gp, which can also secrete Hoechst33342.24 After nonlinear regression analysis according to Michaelis–Menten kinetics, parameters were calculated and presented in Table 1.
Table 1. Nonlinear Regression Analysis of Transport Activity of Fluorescent Substrates after 48 h Exposure to Cetuximab in ciPTEC-OAT1a.
| +EGF |
–EGF |
+EGF+CTX |
–EGF+CTX |
|||||
|---|---|---|---|---|---|---|---|---|
| Km (μM) | Vmax (a.u.) | Km (μM) | Vmax (a.u.) | Km (a.u.) | Vmax (a.u.) | Km (μM) | Vmax (a.u.) | |
| Fluorescein | 4.3 ± 0.5 | 14.7 ± 0.5 | 5.6 ± 0.5 | 9.6 ± 0.3 | 5.1 ± 0.6 | 9.4 ± 0.4 | 5.4 ± 0.8 | 8.2 ± 0.5 |
| Hoechst 33342 | 21.3 ± 4.3 | 108.7 ± 8.1 | 23.8 ± 4.6 | 81.0 ± 6.0 | 19.15 ± 4.3 | 82.7 ± 6.6 | 22.4 ± 4.3 | 76.2 ± 5.5 |
| Calcein-AM | 36.8 ± 6.3 | 378.7 ± 43.6 | 38.4 ± 6.7 | 272.8 ± 32.4 | 22.4 ± 3.3 | 209 ± 18.0 | 22.7 ± 3.8 | 178.3 ± 17.5 |
| CMFDA | 20.2 ± 1.5 | 436.5 ± 18.1 | 17.5 ± 1.6 | 296.6 ± 14.8 | 13.0 ± 1.6 | 267.2 ± 15.8 | 13.5 ± 1.4 | 250.4 ± 13.1 |
Data are expressed as mean values ± SEM of a minimum of two independent assays performed in triplicate. Transport activity is depicted in fluorescent intensity, expressed in arbitrary units (a.u).
No significant changes in IC50 values were found for all conditions tested (Table 1), implicating that the results observed relate to changes in expression levels of the transporters rather than changes in affinities.
Cetuximab Activates Serine/Tyrosine Signaling Downstream of EGFR
To investigate the downstream effects of cetuximab-EGFR signaling in ciPTEC-OAT1, kinase activity profiling using peptide microarrays comprising either 144 PTK or STK phosphosite was performed (PamChip) and used to generate an interaction network. Cells were treated with cetuximab for 15 min in the presence and absence of EGF. Overall, cetuximab activated PTK and STK. In the presence of EGF, STK activity decreased, an effect that was counteracted by cetuximab coincubation (Figure 3A). The absence of EGF sustained a similar effect to that of CTX and both conditions did not yield an additive effect in transport activity. This illustrates that the same regulatory mechanism is triggered and involved in the response and maximally affected by either one alone (Figure S3). Subsequently, significantly modulated targets (phosphorylated peptides) were selected and evaluated with the MetaCore software. Analysis provided a list of known pathways that most significantly overlap with the PTK and STK targets determined for cetuximab. Results show that the signaling pathways triggered by cetuximab mainly involve tyrosine kinases, while EGF mostly affects serine kinases, underlining the stark differences between both conditions. An in-depth scrutiny into the predicted pathways (Figure 3C) revealed that in the control condition (+EGF) the key regulatory elements are cAMP-dependent protein kinase A (PKA), serine/threonine specific kinase C (PKC), downstream transcription factor Creb1 (cAMP responsive element binding protein 1), and the nuclear factor kappa light polypeptide gene enhancer (NF-kβ1). On the other hand, cetuximab exposure reduced EGFR, receptor tyrosine-protein kinase (Erbb2) and PKC activity, while enhancing phospholipase C-γ (PLCγ), phosphatidylinositol-4,5-bisphosphate 3-kinase (P13K), and phosphoinositide dependent protein kinase (PDK). There was also a direct link between EGFR inhibition and MRP4 enhanced phosphorylation. Additional results from the kinomic and pathway analyses are presented as Supporting Information. Further down, EGFR signal transduction targeted the AKT and MAPK/ERK pathways implicated in regulating the expression of OAT1, BCRP, P-gp, and MRP4. When cells were exposed to LY294002, an AKT inhibitor, the expression of all transporters was reduced (Figure 3D), whereas the ERK inhibitor U-0126 reduced the expression of OAT1 and BCRP and upregulated the expression of P-gp and MRP4 (Figure 3B).
Figure 3.
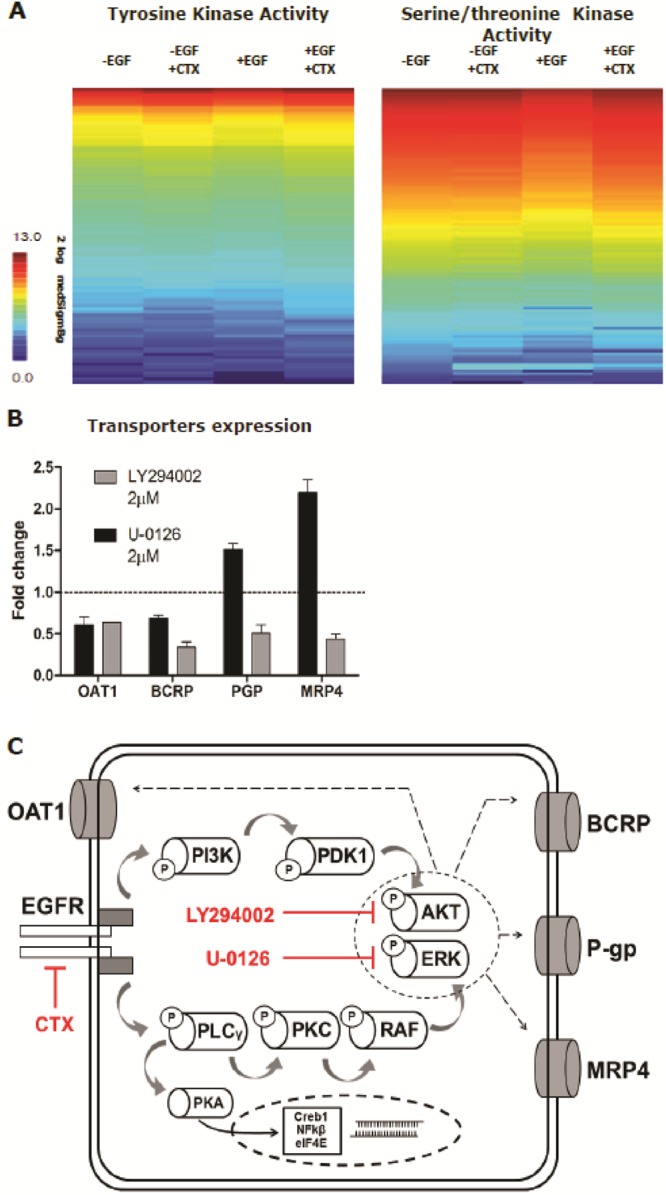
Cetuximab-mediated regulation of EGFR downstream signaling. Cetuximab (CTX) promoted the activation of STK and phospholipases leading to PI3K-AKT and MAPK-ERK regulation of renal drug transporters. Data in panel B is expressed as mean values ± SEM of a minimum of two independent assays performed in triplicate.
Cetuximab Reduces Methotrexate Uptake and Ameliorates Cytotoxicity
Although high-dose methotrexate appears to be nephrotoxic in vivo, ciPTEC-OAT1 were only moderately sensitive to methotrexate, with a reduction in cell viability of about 70–80% at the highest concentration tested (Figure 4). In agreement with its effect on the functional expression of the renal drug transporters, a 24 h cetuximab pretreatment effectively reduced the intracellular concentration of methotrexate-polymglutamate 1 (Figure 4A). Cell cycle analysis showed that after 48 h cetuximab treatment, G0/G1 phase (resting phase) increased from 75 ± 4% to 81 ± 6% (Figure 4B). A similar effect as observed when cells were cultured in the absence of EGF (Figure 4C). When exposed to increasing concentrations of methotrexate, ciPTEC viability reduced as measured by mitochondrial activity (Figure 4D). However, when cells were exposed for 24 h to 100 μM methotrexate followed by a 24 h recovery period, the reduction in cell viability was reversed. This was independent of cetuximab treatment (Figure 4E). In cells treated with cetuximab for 24 h prior to a 48 h methotrexate treatment, the toxic effects of methotrexate could be prevented. A similar effect was observed after 24 h pretreatment with the ERK inhibitor, U-0126 (Figure 4F). This preventive effect of CTX was not observed in the ciPTEC-OAT3 line (Figure 4G), which suggested further that this transporter is not regulated through EGFR signaling.
Figure 4.

Cetuximab reduced methotrexate-induced cytotoxicity in ciPTEC-OAT1. Exposure to cetuximab (CTX) for 24 h reduced the uptake of methotrexate (MTX) in both CM (A) and SFM (B). CTX pretreatment effectively rests cell cycle progression (C). For cell viability assessments, cells were exposed to methotrexate (0–100 μM) for 24 h in SFM (D), MTX (100 μM) for 24 h in SFM, followed by 24 h of treatment with CM (E) or CTX. Cells were preconditioned for 24 h in SFM with CTX or U-0126 (2 μM), followed by 48 h of exposure to MTX (100 μM) (F). MTT assay was then performed. CTX preconditioning did not rescue MTX cytotoxicity in ciPTEC-OAT3 (G). Results are expressed in viability (%) compared to control (i.e., SFM-treated cells). CTX pretreatment effectively reduced MTX uptake and toxicity. Values are shown as mean ± SEM of minimally two independent experiments performed in triplicates. ∗, Significantly different from control (p < 0.05); #, significantly different from MTX (100 μM; p < 0.05).
EGFR-Mediated Signaling Is Also Involved in Cisplatin-Induced Cytotoxicity
To investigate whether the attenuation in toxicity by cetuximab was specific for methotrexate or a more common phenomenon in tubule epithelial cells, we investigated the role of EGFR signaling on cisplatin-induced cytotoxicity. Cisplatin is another agent used in SCCHN combination therapy that, unlike methotrexate, is taken up by PTEC via the organic cation transporter 2 (OCT2) and can lead to programmed cell death.32 Similar to the effect observed for methotrexate, pretreatment with cetuximab for 24 h ameliorated cisplatin-induced toxicity. Cisplatin treatment alone reduced viability to 64 ± 5%, and when preceded by cetuximab, viability was maintained at 91 ± 10% (Figure 5A). Increasing concentrations of cisplatin resulted in a severe loss of mitochondrial activity in ciPTEC-OAT1, where a maximum concentration of 100 μM resulted in near total activity loss after 6 h exposure followed by a 72 h recovery period. Cetuximab pretreatment did not exert any tangible effect on cell viability. On the other hand, the absence of EGF resulted in reduced cisplatin toxicity compared to the condition with EGF, revealing TC50 values of 44 ± 10 μM compared to 32 ± 18 μM, respectively (Figure 5B). Furthermore, the uptake of ASP+, a well-known OCT2 model substrate,33 decreased upon cetuximab treatment (Figure 5C), which suggested that the transporter is regulated through EGFR signaling as well.
Figure 5.

Cetuximab reduced cisplatin-induced cytotoxicity. Cell viability in the presence of cisplatin (CDDP) was determined after cetuximab (CTX) pretreatment for 24 h in SFM, followed by exposure to CDDP (25 μM) for 24 h. MTT assay was then performed (A). Preconditioning of cells with CTX decreased CDDP cytotoxicity. Results are expressed in viability (%) compared to control (i.e., SFM-treated cells). CDDP toxicity after 72 h recovery in CM and CTX was determined after 48 h CTX pretreatment followed by 6 h of increasing CDDP concentrations (0–100 μM) (B). CDDP recovery was performed in CM to minimize cell stress. The absence of EGF ameliorated CDDP toxicity. The accumulation of ASP+ (OCT2 substrate) was reduced upon CTX pretreatment (C). Values are shown as mean ± SEM of minimally two independent experiments performed in triplicates. ∗, Significantly different from control (p < 0.05).
Discussion
In the present study, we revealed the mechanistic interactions between methotrexate and cetuximab affecting renal methotrexate transport activity and cytotoxic potency. Our results show that cetuximab changes drug transporter function through EGFR signaling in proximal tubule cells. The changes in transport activity attenuated the toxic effects of methotrexate and cisplatin. These findings shed light on the potential of using EGFR signaling modulation as a target to prevent renal toxicity in combination chemotherapy.
Nephrotoxicity is a major concern in various chemotherapeutic regimens, and often a dose-limiting factor. The delayed elimination of methotrexate in the course of high-dose methotrexate chemotherapy leads to a considerable accumulation of the drug in proximal tubules, greatly exceeding plasma levels up to 100-fold.34 Severe renal toxicity is hence derived from the intratubular precipitation of methotrexate crystals, despite its antifolate properties.35
In proximal tubules, xenobiotic transporters play a key role in drug excretion and disposition. Acting as selective carriers, they remove the majority of xenobiotics and protein-bound drugs from the bloodstream.36 We demonstrated competition between methotrexate and fluorescein for OAT1/OAT3-mediated uptake. Upon entering the cells, methotrexate is metabolized through the addition of glutamate residues by folylpolyglutamate-synthetase,37 of which methotrexate-polyglutamate 1 was measured intracellularly. Furthermore, we confirmed that BCRP as well as MRP4 are involved in cellular excretion of methotrexate, demonstrating that its renal tubular excretion pathway suggested can be mimicked in vitro using ciPTEC.38
Cetuximab influences the activity of renal uptake as well as efflux transporters involved in methotrexate excretion, except for OAT3. Cetuximab downregulated OAT1 and BCRP, while upregulating MRP4 and P-gp, predominantly through ERK mediated signaling downstream EGFR. The receptor is highly expressed in renal tubular cells and key to their physiology, as demonstrated by its involvement in, for example, electrolyte homeostasis by adjusting trans-epithelial resistance and tight-function configuration, and controlling sodium and magnesium reabsorption.39 Changes in gene expression of the transporters after cetuximab exposure are reflected by their protein levels and, subsequently, at the functional level as a result of a transcriptional regulation of the transport systems. Moreover, cetuximab treatments showed parallels with the effects determined in the absence of EGF, further specifying the role of EGFR in renal xenobiotic transporter regulation.
By kinomic and pathway analysis,40 we determined that under standard culturing conditions (in the presence of EGF), the main signaling pathways involve STK activity resulting in the regulation of PKC and PKA. In line with our findings, activation of PKC has previously been shown to affect cellular distribution of OAT1 by promoting ubiquitination of the transport protein. This results in an accelerated internalization of the transporter from cell surface to intracellular compartments and a reduction in Vmax.41−44 Similarly, OCT2 activity can be modulated by a phosphor-tyrosine switch and stimulation by EGF is abolished by MAPK and PKA inhibition.45,46 Moreover, BCRP expression can be regulated by the axis EGFR – AKT – ERK – CREB, whereas P-gp function and membrane traffic are dependent on PKC and PKA-mediated phosphorylation.47,48
Cetuximab treatment led to an extensive shift in the kinase-mediated signaling cascades, most dominantly on PTK activity (thus upstream or initial signaling). Pathway analysis of the significant activity modulations indicated the involvement of PLC and PI3K, underlying the role of phospholipases in this pathway, leading to modulation of PKC and PDK. Here, the PI3K-AKT pathway reporting phosphosites indicated downregulation of the mechanistic target of rapamycin (mTOR) kinase and eukaryotic translation initiation factor 4E (eIF4E). In a seemingly reductant route, PLC-mediated PKC downregulation also results in RAS-mediated ERK and MAPK downregulation. The network of interactions predicted for cetuximab action in ciPTEC-OAT1 are in line with what has been previously described in cancer models.49 Inhibition of AKT and ERK changed the expression of OAT1, BCRP, MRP4, and P-gp in the same fashion as when cells were exposed to cetuximab, further implicating these pathways in regulating the activity of renal drug transporters (Figure 3C). The modulation of transcription factors (Creb1, NFKβ, eIF4E) revealed in our analysis could hypothetically account for the transcriptional changes in expression observed after 48 h exposure to cetuximab.
Renal methotrexate uptake is described to be handled primarily by OAT3.50,51 Here we show that OAT1 can also play a role in methotrexate uptake and its cytotoxicity. The role of EGF in regulating both basolateral and apical transporters underlines how the combined activities of different drug transporters impacts methotrexate distribution beyond the activity of a predominant pump. Further, this mechanism may be seen as part of broader physiological response. EGF is an important renal growth factor and can be of both systemic or renal origin.39,52 Modifying renal drug excretion via EGFR signaling could, hypothetically, be promoted by systemic EGF originating from another organ system as response to a toxic event. Regulatory mechanisms involving both SLC and ABC transporters have been proposed to determine the whole-body distribution of metabolites, nutrients and xenobiotics,53−55 and our findings suggest that EGFR regulation could influence drug distribution by modulating both transport systems.
Although high-methotrexate doses have been reported nephrotoxic, methotrexate toxicity in proximal tubule cells in vitro is limited. This is partly explained by the absence of a tubular lumen in which methotrexate can precipitate leading to tubular obstruction and eventually tubulopathy.35 Moreover, ciPTEC-OAT1 are matured differentiated cells that show little proliferation which contributes further to the reduced sensitivity to methotrexate, as the drug preferentially targets proliferating and actively dividing cells. Nonetheless, cetuximab pretreatment prevented methotrexate-induced toxicity in cells grown without serum. Under serum deprivation conditions cell cycle progression can be stimulated,56 which sensitized the cells to methotrexate. Pretreatment with U-0126 also prevented methotrexate-related toxicity, re-enforcing the role EGFR mediated MAPK/ERK signaling plays in nephrotoxicity by regulating transport activity in the proximal tubule. Noteworthy, ciPTEC-OAT1 are immortalized and transduced to overexpress OAT1, which may have implications for its active regulatory pathways in this cell model. Nonetheless, our pathway analysis is consistent with previously reported results with respect to the regulation of drug transporters. Remarkably, in the ciPTEC-OAT3 line, equally sensitive to methotrexate, cetuximab did not recover cytotoxicity in contrast to the observations in ciPTEC-OAT1. This may be indicative of separate regulatory mechanisms for OAT1 and OAT3 or a consequence of the transduction of OAT3 in ciPTEC.21
The consequences of EGFR cascades inhibition with positive outcomes for renal damage have been reported previously in rats in vivo.57 Here, we provide direct evidence implicating EGFR in drug transport regulation through PKA and PKC signaling, thereby suppressing methotrexate cytotoxicity in vitro. This protective effect is not limited to methotrexate, as we show that cisplatin cytotoxicity25 was also suppressed in our model, which further confirms the positive impact of PKA and PKC signaling in drug-induced renal damage. While cetuximab combined with cisplatin improves cancer treatment outcome,58 improved efficacy of cetuximab combined with low-dose methotrexate is under investigation. This study underlined the implications for drug distribution by showing pronounced changes in transport activity promoted by cetuximab, in particular at the efflux side of proximal tubule cells. Further research into the effects of comedication and its influence on the renal excretion of nephrotoxic agents can elucidate about the use of combination therapies to manage and prevent nephrotoxicity.
Acknowledgments
This work was supported by the Nephrotools consortium under the scope of EU FP7 Marie Curie actions (Grant No. 289754) and a Dutch Kidney Foundation grant (Grant No. KJPB 11.023). We thank Milos Mihajlovic and Sam Hariri for their insight and assistance with the FACS analysis performed for this study, and Ola Haj Mustafa for the help in the OTA3 experiments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.7b00308.
ciPTEC culture medium formulation; pathways identified as significant matches to kinase phosphorylation sites obtained from PTK/STK PamChip array; graphic abstract of “Fluorescence-Based Transport Assays Revisited in a Human Renal Proximal Tubule Cell Line”; Ct values for GAPDH in different experimental conditions; cetuximab inhibition of the EGFR phosphosite; metacore knowledge-based pathway analyses of effect of EGF or EGF together with cetuximab on ciPTEC-OAT1; pathway analysis symbols key (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Jolivet J.; Cowan K. H.; Curt G. A.; Clendeninn N. J.; Chabner B. A. The pharmacology and clinical use of methotrexate. N. Engl. J. Med. 1983, 309 (18), 1094–1104. 10.1056/NEJM198311033091805. [DOI] [PubMed] [Google Scholar]
- Bleyer W. A. Methotrexate: clinical pharmacology, current status and therapeutic guidelines. Cancer Treat. Rev. 1977, 4 (2), 87–101. 10.1016/S0305-7372(77)80007-8. [DOI] [PubMed] [Google Scholar]
- Rajagopalan P. T.; Zhang Z.; McCourt L.; Dwyer M.; Benkovic S. J.; Hammes G. G. Interaction of dihydrofolate reductase with methotrexate: ensemble and single-molecule kinetics. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (21), 13481–13486. 10.1073/pnas.172501499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T.; Mori N. Involvement of Multiple Transporters-mediated Transports in Mizoribine and Methotrexate Pharmacokinetics. Pharmaceuticals 2012, 5 (8), 802–836. 10.3390/ph5080802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright R. D. Methotrexate in the treatment of psoriasis. Cutis. 1999, 64 (5), 332–334. [PubMed] [Google Scholar]
- Holmboe L.; Andersen A. M.; Morkrid L.; Slordal L.; Hall K. S. High dose methotrexate chemotherapy: pharmacokinetics, folate and toxicity in osteosarcoma patients. Br. J. Clin. Pharmacol. 2012, 73 (1), 106–114. 10.1111/j.1365-2125.2011.04054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashtari F.; Savoj M. R. Effects of low dose methotrexate on relapsing-remitting multiple sclerosis in comparison to Interferon beta-1alpha: A randomized controlled trial. J. Res. Med. Sci. 2011, 16 (4), 457–462. [PMC free article] [PubMed] [Google Scholar]
- Ciardiello F.; Tortora G. EGFR Antagonists in Cancer Treatment. N. Engl. J. Med. 2008, 358 (11), 1160–1174. 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- Cosmai L.; Gallieni M.; Porta C. Renal toxicity of anticancer agents targeting HER2 and EGFR. J. Nephrol. 2015, 28 (6), 647–57. 10.1007/s40620-015-0226-9. [DOI] [PubMed] [Google Scholar]
- Hynes N. E.; Lane H. A. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5 (5), 341–54. 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Citri A.; Yarden Y. EGF-ERBB signalling: towards the systems level. Nat. Rev. Mol. Cell Biol. 2006, 7 (7), 505–516. 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- Pozzi C.; Cuomo A.; Spadoni I.; Magni E.; Silvola A.; Conte A.; et al. The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat. Med. 2016, 22 (6), 624–631. 10.1038/nm.4078. [DOI] [PubMed] [Google Scholar]
- Zhao S.; Chen C.; Liu S.; Zeng W.; Su J.; Wu L.; et al. CD147 promotes MTX resistance by immune cells through up-regulating ABCG2 expression and function. J. Dermatol. Sci. 2013, 70 (3), 182–189. 10.1016/j.jdermsci.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Dabak D. O.; Kocaman N. Effects of silymarin on methotrexate-induced nephrotoxicity in rats. Renal Failure 2015, 37 (4), 734–739. 10.3109/0886022X.2015.1012984. [DOI] [PubMed] [Google Scholar]
- Takeda M.; Khamdang S.; Narikawa S.; Kimura H.; Hosoyamada M.; Cha S. H.; et al. Characterization of methotrexate transport and its drug interactions with human organic anion transporters. J. Pharmacol. Exp. Ther. 2002, 302 (2), 666–671. 10.1124/jpet.102.034330. [DOI] [PubMed] [Google Scholar]
- Garcia R.; Franklin R. A.; McCubrey J. A. EGF induces cell motility and multi-drug resistance gene expression in breast cancer cells. Cell Cycle 2006, 5 (23), 2820–2826. 10.4161/cc.5.23.3535. [DOI] [PubMed] [Google Scholar]
- zu Schwabedissen H. E. M.; Grube M.; Dreisbach A.; Jedlitschky G.; Meissner K.; Linnemann K.; et al. Epidermal growth factor-mediated activation of the map kinase cascade results in altered expression and function of ABCG2 (BCRP). Drug Metab. Dispos. 2006, 34 (4), 524–533. 10.1124/dmd.105.007591. [DOI] [PubMed] [Google Scholar]
- Liu N.; Wang L.; Yang T.; Xiong C.; Xu L.; Shi Y.; Bao W.; Chin Y. E.; Cheng S. B.; Yan H.; Qiu A.; Zhuang S. EGF Receptor Inhibition Alleviates Hyperuricemic Nephropathy. J. Am. Soc. Nephrol. 2015, 26 (11), 2716–2729. 10.1681/ASN.2014080793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J.; Liu N.; Zhuang S. Role of epidermal growth factor receptor in acute and chronic kidney injury. Kidney Int. 2013, 83 (5), 804–810. 10.1038/ki.2012.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamant M.; Bollee G.; Henique C.; Tharaux P. L. Epidermal growth factor: a new therapeutic target in glomerular disease. Nephrol., Dial., Transplant. 2012, 27 (4), 1297–1304. 10.1093/ndt/gfs030. [DOI] [PubMed] [Google Scholar]
- Nieskens T. T.; Peters J. G.; Schreurs M. J.; Smits N.; Woestenenk R.; Jansen K.; et al. A Human Renal Proximal Tubule Cell Line with Stable Organic Anion Transporter 1 and 3 Expression Predictive for Antiviral-Induced Toxicity. AAPS J. 2016, 18 (2), 465–475. 10.1208/s12248-016-9871-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutsaers H. A.; Wilmer M. J.; Reijnders D.; Jansen J.; van den Broek P. H.; Forkink M.; et al. Uremic toxins inhibit renal metabolic capacity through interference with glucuronidation and mitochondrial respiration. Biochim. Biophys. Acta, Mol. Basis Dis. 2013, 1832 (1), 142–150. 10.1016/j.bbadis.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Jansen J.; Schophuizen C. M.; Wilmer M. J.; Lahham S. H.; Mutsaers H. A.; Wetzels J. F.; et al. A morphological and functional comparison of proximal tubule cell lines established from human urine and kidney tissue. Exp. Cell Res. 2014, 323 (1), 87–99. 10.1016/j.yexcr.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Caetano-Pinto P.; Janssen M. J.; Gijzen L.; Verscheijden L.; Wilmer M. J.; Masereeuw R. Fluorescence-Based Transport Assays Revisited in a Human Renal Proximal Tubule Cell Line. Mol. Pharmaceutics 2016, 13 (3), 933–44. 10.1021/acs.molpharmaceut.5b00821. [DOI] [PubMed] [Google Scholar]
- Manohar S.; Leung N. Cisplatin nephrotoxicity: a review of the literature. J. Nephrol. 2017, 1. 10.1007/s40620-017-0392-z. [DOI] [PubMed] [Google Scholar]
- Wilmer M. J.; Saleem M. A.; Masereeuw R.; Ni L.; van der Velden T. J.; Russel F. G.; et al. Novel conditionally immortalized human proximal tubule cell line expressing functional influx and efflux transporters. Cell Tissue Res. 2010, 339 (2), 449–457. 10.1007/s00441-009-0882-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenderink J. B.; Geibel S.; Grabsch E.; De Pont J. J.; Bamberg E.; Friedrich T. Electrophysiological analysis of the mutated Na,K-ATPase cation binding pocket. J. Biol. Chem. 2003, 278 (51), 51213–51222. 10.1074/jbc.M306384200. [DOI] [PubMed] [Google Scholar]
- Noe G.; Bellesoeur A.; Thomas-Schoemann A.; Rangarajan S.; Naji F.; Puszkiel A.; Huillard O.; Saidu N.; Golmard L.; Alexandre J.; et al. Clinical and kinomic analysis identifies peripheral blood mononuclear cells as a potential pharmacodynamic biomarker in metastatic renal cell carcinoma patients treated with sunitinib. Oncotarget. 2015, 7 (41), 67507–67520. 10.18632/oncotarget.11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilhorst R.; Houkes L.; Mommersteeg M.; Musch J.; van den Berg A.; Ruijtenbeek R. Peptide microarrays for profiling of serine/threonine kinase activity of recombinant kinases and lysates of cells and tissue samples. Methods Mol. Biol. 2013, 977, 259–271. 10.1007/978-1-62703-284-1_21. [DOI] [PubMed] [Google Scholar]
- Lemeer S.; Jopling C.; Naji F.; Ruijtenbeek R.; Slijper M.; Heck A. J.; et al. Protein-tyrosine kinase activity profiling in knock down zebrafish embryos. PLoS One 2007, 2 (7), e581. 10.1371/journal.pone.0000581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boer E.; Meesters R. J.; van Zelst B. D.; Luider T. M.; Hazes J. M.; Heil S. G.; et al. Measuring methotrexate polyglutamates in red blood cells: a new LC–MS/MS-based method. Anal. Bioanal. Chem. 2013, 405 (5), 1673–1681. 10.1007/s00216-012-6581-7. [DOI] [PubMed] [Google Scholar]
- Miller R. P.; Tadagavadi R. K.; Ramesh G.; Reeves W. B. Mechanisms of Cisplatin nephrotoxicity. Toxins 2010, 2 (11), 2490–2518. 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holle S. K.; Ciarimboli G.; Edemir B.; Neugebauer U.; Pavenstadt H.; Schlatter E. Properties and regulation of organic cation transport in freshly isolated mouse proximal tubules analyzed with a fluorescence reader-based method. Pfluegers Arch. 2011, 462 (2), 359–369. 10.1007/s00424-011-0969-7. [DOI] [PubMed] [Google Scholar]
- Yang S. L.; Zhao F. Y.; Song H.; Shen D. Y.; Xu X. J. Methotrexate Associated Renal Impairment Is Related to Delayed Elimination of High-Dose Methotrexate. Sci. World J. 2015, 2015, 751703. 10.1155/2015/751703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagos Y.; Wolff N. A. Assessment of the role of renal organic anion transporters in drug-induced nephrotoxicity. Toxins 2010, 2 (8), 2055–2082. 10.3390/toxins2082055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J.; Wang J. Renal drug transporters and their significance in drug-drug interactions. Acta Pharm. Sin. B 2016, 6 (5), 363–373. 10.1016/j.apsb.2016.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeling H.; Horneff G.; Benseler S. M.; Fritzler M. J. Pharmacogenetics: can genes determine treatment efficacy and safety in JIA?. Nat. Rev. Rheumatol. 2014, 10 (11), 682–690. 10.1038/nrrheum.2014.140. [DOI] [PubMed] [Google Scholar]
- Morrissey K. M.; Stocker S. L.; Wittwer M. B.; Xu L.; Giacomini K. M. Renal transporters in drug development. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 503–29. 10.1146/annurev-pharmtox-011112-140317. [DOI] [PubMed] [Google Scholar]
- Melenhorst W. B.; Mulder G. M.; Xi Q.; Hoenderop J. G.; Kimura K.; Eguchi S.; et al. Epidermal growth factor receptor signaling in the kidney: key roles in physiology and disease. Hypertension 2008, 52 (6), 987–993. 10.1161/HYPERTENSIONAHA.108.113860. [DOI] [PubMed] [Google Scholar]
- Ferguson B. D.; Tan Y. H.; Kanteti R. S.; Liu R.; Gayed M. J.; Vokes E. E.; Ferguson M. K.; Iafrate A. J.; Gill P. S.; Salgia R.; et al. Novel EPHB4 Receptor Tyrosine Kinase Mutations and Kinomic Pathway Analysis in Lung Cancer. Sci. Rep. 2015, 5, 10641. 10.1038/srep10641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Hong M.; Duan P.; Pan Z.; Ma J.; You G. Organic anion transporter OAT1 undergoes constitutive and protein kinase C-regulated trafficking through a dynamin- and clathrin-dependent pathway. J. Biol. Chem. 2008, 283 (47), 32570–32579. 10.1074/jbc.M800298200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros S. A.; Srimaroeng C.; Perry J. L.; Walden R.; Dembla-Rajpal N.; Sweet D. H.; et al. Activation of protein kinase Czeta increases OAT1 (SLC22A6)- and OAT3 (SLC22A8)-mediated transport. J. Biol. Chem. 2009, 284 (5), 2672–2679. 10.1074/jbc.M808078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Zhang Q.; You G. Three ubiquitination sites of organic anion transporter-1 synergistically mediate protein kinase C-dependent endocytosis of the transporter. Mol. Pharmacol. 2013, 84 (1), 139–146. 10.1124/mol.113.086769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D.; Huang H.; Toh M. F.; You G. Serum- and glucocorticoid-inducible kinase sgk2 stimulates the transport activity of human organic anion transporters 1 by enhancing the stability of the transporter. Int. J. Biochem. Mol. Biol. 2016, 7 (1), 19–26. [PMC free article] [PubMed] [Google Scholar]
- Soodvilai S.; Chatsudthipong A.; Chatsudthipong V. Role of MAPK and PKA in regulation of rbOCT2-mediated renal organic cation transport. Am. J. Physiol. Renal Physiol. 2007, 293 (1), F21–F27. 10.1152/ajprenal.00043.2007. [DOI] [PubMed] [Google Scholar]
- Sprowl J. A.; Ong S. S.; Gibson A. A.; Hu S.; Du G.; Lin W.; et al. A phosphotyrosine switch regulates organic cation transporters. Nat. Commun. 2016, 7, 10880. 10.1038/ncomms10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y.; Nakanishi T.; Natarajan K.; Safren L.; Hamburger A. W.; Hussain A.; et al. Functional cyclic AMP response element in the breast cancer resistance protein (BCRP/ABCG2) promoter modulates epidermal growth factor receptor pathway- or androgen withdrawal-mediated BCRP/ABCG2 transcription in human cancer cells. Biochim. Biophys. Acta, Gene Regul. Mech. 2015, 1849 (3), 317–327. 10.1016/j.bbagrm.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtal K. A.; de Vries E.; Hoekstra D.; van Ijzendoorn S. C. Efficient trafficking of MDR1/P-glycoprotein to apical canalicular plasma membranes in HepG2 cells requires PKA-RIIalpha anchoring and glucosylceramide. Mol. Biol. Cell 2006, 17 (8), 3638–3650. 10.1091/mbc.E06-03-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegeman H.; Kaanders J. H.; Wheeler D. L.; van der Kogel A. J.; Verheijen M. M.; Waaijer S. J.; Iida M.; Grenman R.; Span P. N.; Bussink J. Activation of AKT by hypoxia: a potential target for hypoxic tumors of the head and neck. BMC Cancer 2012, 12, 463. 10.1186/1471-2407-12-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki Y.; Kusuhara H.; Kondo T.; Hasegawa M.; Shiroyanagi Y.; Nakazawa H.; et al. Characterization of the uptake of organic anion transporter (OAT) 1 and OAT3 substrates by human kidney slices. J. Pharmacol. Exp. Ther. 2007, 321 (1), 362–369. 10.1124/jpet.106.113076. [DOI] [PubMed] [Google Scholar]
- Wang L.; Sweet D. H. Renal organic anion transporters (SLC22 family): expression, regulation, roles in toxicity, and impact on injury and disease. AAPS J. 2013, 15 (1), 53–69. 10.1208/s12248-012-9413-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher D. A.; Salido E. C.; Barajas L. Epidermal growth factor and the kidney. Annu. Rev. Physiol. 1989, 51, 67–80. 10.1146/annurev.ph.51.030189.000435. [DOI] [PubMed] [Google Scholar]
- Ahn S. Y.; Nigam S. K. Toward a systems level understanding of organic anion and other multispecific drug transporters: a remote sensing and signaling hypothesis. Mol. Pharmacol. 2009, 76 (3), 481–490. 10.1124/mol.109.056564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.; Dnyanmote A. V.; Nigam S. K. Remote communication through solute carriers and ATP binding cassette drug transporter pathways: an update on the remote sensing and signaling hypothesis. Mol. Pharmacol. 2011, 79 (5), 795–805. 10.1124/mol.110.070607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigam S. K. What do drug transporters really do?. Nat. Rev. Drug Discovery 2014, 14 (1), 29–44. 10.1038/nrd4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langan T. J.; Chou R. C. Synchronization of mammalian cell cultures by serum deprivation. Methods Mol. Biol. 2011, 761, 75–83. 10.1007/978-1-61779-182-6_5. [DOI] [PubMed] [Google Scholar]
- Wada Y.; Iyoda M.; Matsumoto K.; Shindo-Hirai Y.; Kuno Y.; Yamamoto Y.; et al. Epidermal growth factor receptor inhibition with erlotinib partially prevents cisplatin-induced nephrotoxicity in rats. PLoS One 2014, 9 (11), e111728. 10.1371/journal.pone.0111728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco A. G.; Cohen E. E. Current Treatment Options for Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 2015, 33 (29), 3305–13. 10.1200/JCO.2015.62.0963. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.