

Genome sequence and ancestry analysis confirm only two canid species in North America.
Abstract
A response to Hohenlohe et al.
INTRODUCTION
Hohenlohe et al. raise a number of concerns about our conclusions. We focus on those involving species status, divergence time, admixture, and D statistics because we regard them as the most consequential points. In general, Hohenlohe et al. largely recapitulate criticisms about sample composition, historic range, and the genuine species status of the eastern and red wolves made during the 25-year history of genetic research on the two forms. With the exception of Wilson et al. (1), which was a preliminary treatment, we know of no convincing genetic arguments for distinct species status of the red wolf. On the contrary, a large body of evidence, including genome-wide studies of large population samples, suggests that it is a hybrid between the coyote and a unique population of the gray wolf (2–8), and our complete sequence data reaffirm these past studies. The “eastern wolf” (or more generally the Great Lakes wolf population) has a controversial taxonomic status, and it has been argued to represent a distinct ecotype of the gray wolf admixed with coyotes (9) or a distinct species centered on Algonquin National Park and surrounded by a large admixture zone of coyotes, gray wolves, and eastern wolf hybrids. Gray wolves and coyotes are verified as interfertile by artificial insemination (10); however, these hybrids then reproduced without assistance in captivity, forming F2s (11). In addition to the empirical population genetic evidence (3, 4, 8, 9, 12–14), this purposeful and subsequent unintentional breeding experiment showed that hybrids of gray wolves and coyotes could readily be formed across two generations and do not follow Haldane’s rule expected for biological species or show any evidence of infertility, confirming that they are very recently diverged, as suggested by the sequence evidence (see below). These data provide suggestive evidence that red and eastern wolves, which are hypothesized to have diverged from the coyote lineage more recently than gray wolf and coyote, must likewise be genetically very similar, reproductively interfertile, and, at best, questionably distinct from coyotes or gray wolves.
In support of this interpretation, we find a low level of unique alleles and genetic divergence among all North American wolf taxa, which requires that species-specific inferences should be made with caution and qualification. Our principal results showed that, with a variety of reference populations, red wolf and wolves from the Great Lakes region, including Algonquin wolves, were genetically very similar to coyotes or gray wolves. Even if a distinct origin is assumed, species status as distinct from either gray wolves or coyotes is questionable. Hohenlohe et al. note that red wolves “exhibit the greatest differentiation from the other groups,” and cite them as comparable to the Mexican wolf. Although FST can be inflated in small populations, we note that FST between the Mexican wolf and Eurasian Gray wolves is 0.416. Furthermore, differentiation between the Mexican wolf and the three coyotes is higher (FST = 0.464); both FST values are more than twice as large as any comparison involving red wolves [see Table 2 in our previous work (8)]. We also note that the maximal FST value involving red wolves (FST = 0.188) is substantially less than the largest value between human populations (FST of up to 0.28) (15). This suggests that genetic differentiation between red wolves and other North American canids is comparable to the amount of genetic differentiation found between different continental human groups, which of course are not considered to be distinct evolutionary lineages.
Hohenlohe et al. also claim that “the observed proportions of unique alleles reveal a higher degree of evolutionary distinctiveness in red and eastern wolves relative to other North American canids.” This statement is factually inaccurate. As stated in our paper (8), the fraction of unique variants in North American canid groups varies from a high estimate of 5.13% in nonreference coyotes to a low estimate of 3.3% in North American gray wolves (that is, the fraction of unique variants is higher in coyotes, even though they are part of the reference group of samples than they are in red or Eastern wolves). Qualitatively, distinct evolutionary history should lead to increased fractions of novel alleles, but that is not what we observed. If we assume, for example, that red wolves were wolf-coyote hybrids with the ancestry proportions estimated [see Table 3 in our previous work (8)], then their expected average fraction of unique variants would be 4.83%, higher than the 4.41% observed. In other words, red wolves have fewer novel variants than expected under our simple two-way admixture model, let alone, any model incorporating substantial ancestry from an additional North American canid lineage. To emphasize this point, we performed a leave-one-out analysis to quantify the expected number of novel alleles separately for high- and low-coverage genomes. For example, for a high-coverage recent hybrid, that is, 75% coyote and 25% wolf (for example, red wolf), we expected ~8.8% of the genome to contain novel alleles, which was comparable to the observed fraction (8.78%) in the red wolf high-coverage sequence [see Table 4 in our previous work (8)]. Similarly, the expected fraction of novel alleles for a recent hybrid with 69% gray wolf ancestry (for example, Great Lakes wolf) is ~6.3%, slightly lower than the observed fraction of 7.13% for the Minnesota wolf. Therefore, the observed fractions of novel alleles in all red and eastern wolf genomes are comparable to, or less than, that expected for a recent wolf-coyote hybrid. We conclude that there is no evidence for an independent ancestry for any of the New World wolves, because this would have led to the observation of more “novel” alleles than what was actually observed.
The divergence between the forms is unremarkable, not high enough to justify a revised species status, and is far more recent than originally advanced as 700,000 years ago (1). Our demographic analysis was designed specifically to assess this supposition. The analysis assumes that the red and eastern wolf each has a distinctly divergent origin in the canid phylogeny, and under this assumption, the Generalized Phylogenetic Coalescent Sampler (G-PhoCS) measures rates of gene flow and divergence times. To explain a “distinct origin” model, we have to assume extremely high rates of postdivergence gene flow from gray wolf and coyote, with contributions to each the red and Great Lakes wolf inferred to be >50%. This estimate is an order of magnitude higher than that inferred for species experiencing secondary contact after divergence, such as humans and Neanderthals (16) or dogs and wolves (17). Consequently, these values suggest that a small minority of red and Great Lakes wolf lineages actually trace back through the distinct population in the model. Thus, the divergence time inferred for these two populations cannot be used to argue a distinct origin. At best, it implies that a small fraction of genetic contribution to red wolf comes from a population that diverged from the California coyote population roughly 70,000 years ago. This does not seem sufficient to justify a claim of distinct species or taxon. Although admixture is common in nature, the magnitude of gene flow among North American canids is atypical. We found extensive admixture across genomes in the Great Lakes, with ~50% coyote and gray wolf ancestry in individuals from Algonquin Park, and wolves found elsewhere in the Great Lakes have closer to ~75% gray wolf ancestry, a finding that is consistent with previous studies (6, 9, 13). This complicated evolutionary history presents significant challenges to accurately infer evolutionary history. However, we do agree with Hohenlohe et al. that haplotype analysis would be useful for phased data and that a conservation framework is needed so that effective surrogates, even if admixed, can be preserved on the landscape (18).
Hohenlohe et al. also criticize our interpretation of the D statistic results, arguing that they are uninformative as to the timing of admixture. Although the D statistic does not directly estimate the timing of admixture, it can provide some insight concerning the relative timing of admixture. Gene flow between populations following an admixture event will gradually equilibrate the amount of introgressed ancestry between populations. Uniformity across populations, exemplified by Neanderthal ancestry in non-African humans (16), indicates ancient admixture, whereas variation among populations, as we observed among North American canids, indicates recent or ongoing admixture. There are many possible hypotheses for how this admixture occurred, but given the major anthropogenic environmental disruptions over the last 400 years, we believe that such disruptions are an important cause of hybridization and represent the most parsimonious hypothesis that explains our results.
A goal of the D statistic analysis is to identify introgression from either a coyote or candidate eastern wolf lineage into gray wolves. The eastern wolf is hypothesized to be more closely related to the coyote than to the gray wolf (19). There are three possible outcomes of the analysis: (i) The D statistic values for the eastern wolf introgressor are greater, in which case eastern wolves and coyotes are distinct and eastern wolves were the introgressors; (ii) the D statistic values for the coyote introgressor are greater, in which case the eastern wolves and coyotes are distinct and coyotes were the introgressors; or (iii) the D statistic values are the same, in which case the coyote and eastern wolf are equally related to the introgressor, either because the introgressor is an outgroup to both or because the eastern wolf and the coyote are not distinct. In our previous work (8), the two genomes lacking any detectable gray wolf ancestry are the California coyote, which is unequivocally a coyote, and a canid from Quebec that is a putative eastern wolf. We compare the D statistic values calculated using either the California coyote or the Quebec canid as representative of coyote/eastern wolf and found very similar results (Fig. 1). This leads us to provisionally reject the hypothesis that eastern wolves and coyotes are substantially distinct lineages. Although further sampling could theoretically reveal a cryptic eastern wolf, the simplest conclusion from our data is that only two nonadmixed lineages, gray wolves and coyotes, exist in North America. In summary, we disagree with Hohenlohe et al. and stand by the conclusions in our previous work (8).
Fig. 1. Autosomal D statistic values for coyote introgression into North American wolf-like canids.
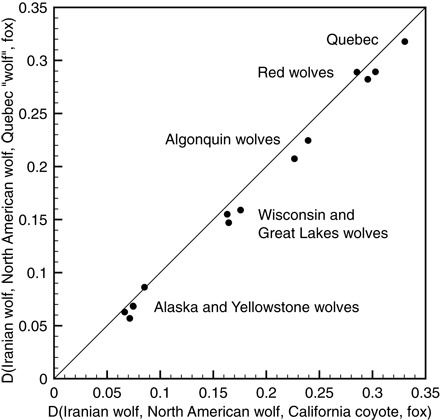
The allopatric gray wolf from Iran is used as a baseline for comparison because of the genome’s complete lack of coyote introgression. Consistent with a two-lineage model but contrary to a three-lineage model, the California coyote and the Quebec canid produce nearly equal D statistic values.
Acknowledgments
Author contributions: All authors contributed to the response equally. Competing interests: The authors declare that they have no competing interests.
REFERENCES
- 1.Wilson P. J., Grewal S., Lawford I. D., Heal J. N. M., Granacki A. G., Pennock D., Theberge J. B., Theberge M. T., Voigt D. R., Waddell W., Chambers R. E., Paquet P. C., Goulet G., Cluff D., White B. N., DNA profiles of the eastern Canadian wolf and the red wolf provide evidence for a common evolutionary history independent of the gray wolf. Can. J. Zool. 78, 2156–2166 (2000). [Google Scholar]
- 2.Wayne R. K., Jenks S. M., Mitochondrial DNA analysis implying extensive hybridization of the endangered red wolf Canis rufus. Nature 351, 565–568 (1991). [Google Scholar]
- 3.Roy M. S., Geffen E., Smith D., Ostrander E. A., Wayne R. K., Patterns of differentiation and hybridization in North American wolflike canids, revealed by analysis of microsatellite loci. Mol. Biol. Evol. 11, 553–570 (1994). [DOI] [PubMed] [Google Scholar]
- 4.Roy M. S., Geffen E., Smith D., Wayne R. K., Molecular genetics of pre-1940 red wolves. Conserv. Biol. 10, 1413–1424 (1996). [Google Scholar]
- 5.Reich D. E., Wayne R. K., Goldstein D. B., Genetic evidence for a recent origin by hybridization of red wolves. Mol. Ecol. 8, 139–144 (1999). [DOI] [PubMed] [Google Scholar]
- 6.vonHoldt B. M., Pollinger J. P., Lohmueller K. E., Han E., Parker H. G., Quignon P., Degenhardt J. D., Boyko A. R., Earl D. A., Auton A., Reynolds A., Bryc K., Brisbin A., Knowles J. C., Mosher D. S., Spady T. C., Elkahloun A., Geffen E., Pilot M., Jedrzejewski W., Greco C., Randi E., Bannasch D., Wilton A., Shearman J., Musiani M., Cargill M., Jones P. G., Qian Z., Huang W., Ding Z.-L., Zhang Y.-p., Bustamante C. D., Ostrander E. A., Novembre J., Wayne R. K., Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 464, 898–902 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.vonHoldt B. M., Pollinger J. P., Earl D. A., Knowles J. C., Boyko A. R., Parker H., Geffen E., Pilot M., Jedrzejewski W., Jedrzejewska B., Sidorovich V., Greco C., Randi E., Musiani M., Kays R., Bustamante C. D., Ostrander E. A., Novembre J., Wayne R. K., A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res. 21, 1294–1305 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.vonHoldt B. M., Cahill J. A., Fan Z., Gronau I., Robinson J., Pollinger J. P., Shapiro B., Wall J., Wayne R. K., Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Sci. Adv. 2, e1501714 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koblmüller S., Nord M., Wayne R. K., Leonard J. A., Origin and status of the Great Lakes wolf. Mol. Ecol. 18, 2313–2326 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Mech L. D., Christensen B. W., Asa C. S., Callahan M., Young J. K., Production of hybrids between western gray wolves and western coyotes. PLOS ONE 9, e88861 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.vonHoldt B. M., Heppenheimer E., Petrenko V., Croonquist P., Rutledge L. Y., Ancestry-specific methylation patterns in admixed offspring from an experimental coyote and gray wolf cross. J. Hered. doi.org/10.1093/jhered/esx004 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Lehman N., Eisenhawer A., Hansen K., Mech L. D., Peterson R. O., Gogan P. J. P., Wayne R. K., Introgression of coyote mitochondrial DNA into sympatric North American gray wolf populations. Evolution 45, 104–119 (1991). [DOI] [PubMed] [Google Scholar]
- 13.Leonard J. A., Wayne R. K., Native Great Lakes wolves were not restored. Biol. Lett. 4, 95–98 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.vonHoldt B. M., Pollinger J. P., Earl D. A., Knowles J. C., Boyko A. R., Parker H., Geffen E., Pilot M., Jedrzejewski W., Jedrzejewska B., Sidorovich V., Greco C., Randi E., Musiani M., Kays R., Bustamante C. D., Ostrander E. A., Novembre J., Wayne R. K., A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res. 21, 1294–1305 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wall J. D., Cox M. P., Mendez F. L., Woerner A., Severson T., Hammer M. F., A novel DNA sequence database for analyzing human demographic history. Genome Res. 18, 1354–1361 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green R. E., Krause J., Briggs A. W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M. H.-Y., Hansen N. F., Durand E. Y., Malaspinas A.-S., Jensen J. D., Marques-Bonet T., Alkan C., Prüfer K., Meyer M., Burbano H. A., Good J. M., Schultz R., Aximu-Petri A., Butthof A., Höber B., Höffner B., Siegemund M., Weihmann A., Nusbaum C., Lander E. S., Russ C., Novod N., Affourtit J., Egholm M., Verna C., Rudan P., Brajkovic D., Kucan Ž., Gušic I., Doronichev V. B., Golovanova L. V., Lalueza-Fox C., de la Rasilla M., Fortea J., Rosas A., Schmitz R. W., Johnson P. L. F., Eichler E. E., Falush D., Birney E., Mullikin J. C., Slatkin M., Nielsen R., Kelso J., Lachmann M., Reich D., Pääbo S., A draft sequence of the Neandertal genome. Science 328, 710–722 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freedman A. H., Gronau I., Schweizer R. M., Ortega-Del Vecchyo D., Han E., Silva P. M., Galaverni M., Fan Z., Marx P., Lorente-Galdos B., Beale H., Ramirez O., Hormozdiari F., Alkan C., Vilà C., Squire K., Geffen E., Kusak J., Boyko A. R., Parker H. G., Lee C., Tadigotla V., Siepel A., Bustamante C. D., Harkins T. T., Nelson S. F., Ostrander E. A., Marques-Bonet T., Wayne R. K., Novembre J., Genome sequencing highlights the dynamic early history of dogs. PLOS Genet. 10, e1004016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wayne R. K., Shaffer H. B., Hybridization and endangered species protection in the molecular era. Mol. Ecol. 25, 2680–2689 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Rutledge L. Y., Devillard S., Bonne J. Q., Hohenlohe P. A., White B. N., RAD sequencing and genomic simulations resolve hybrid origins within North American Canis. Biol. Lett. 11, 20150303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]