

ABSTRACT
Mitotic catastrophe is an oncosuppressive mechanism that targets cells experiencing defective mitoses via the activation of specific cell cycle checkpoints, regulated cell death pathways and/or cell senescence. This prevents the accumulation of karyotypic aberrations, which otherwise may drive oncogenesis and tumor progression. Here, we summarize experimental evidence confirming the role of caspase 2 (CASP2) as the main executor of mitotic catastrophe, and we discuss the signals that activate CASP2 in the presence of mitotic aberrations. In addition, we summarize the main p53-dependent and -independent effector pathways through which CASP2 limits chromosomal instability and non-diploidy, hence mediating robust oncosuppressive functions.
KEYWORDS: Chromosome instability, mitotic slippage, polyploidy, replication stress, spindle assembly checkpoint, targeted cancer therapy
Abbreviations
- BCL9L
B-cell CLL/lymphoma 9-like
- BID
BH3 interacting domain death agonist
- CASP2
caspase 2; CASP3, caspase 3
- CIN
chromosomal instability
- MOMP
mitochondrial outer membrane permeabilization
- PIDD 1
p53-induced death domain protein 1
- PLK1
polo like kinase 1
- RCD
regulated cell death
- SAC
spindle assembly checkpoint
Initially described on the basis of morphological parameters as a failed mitosis1 and later defined functionally as a form of regulated cell death (RCD) distinct from apoptosis,2 mitotic catastrophe is nowadays considered as a prominent oncosuppressive mechanism for preserving genetic integrity.3
The triggering events of mitotic catastrophe are mitotic abnormalities, such as defects in chromosomes, mitotic spindles, or the cytokinesis apparatus. These aberrancies can be generated by perturbations of endogenous or exogenous origin occurring (1) before mitosis, such as DNA replication stress, incomplete DNA replication, and a defective centrosome cycle, which are usually associated with deregulated cell cycle checkpoints; or (2) during mitosis, such as the impairment of the spindle assembly checkpoint (SAC)—which surveys metaphase-anaphase transition4—and dysfunction of the molecular machineries for sister chromatid cohesion, chromosome alignment, or cyto-/karyokinesis.
Once activated, mitotic catastrophe impedes the survival and/or progression of mitotic-defective cells by inducing RCD during a prolonged mitosis (also defined as mitotic death) or, alternatively, by triggering specific cell cycle checkpoints, RCD or cellular senescence during the G1 phase that follows the exit from, or abortion of, an aberrant mitosis.3 Through these complementary strategies, mitotic catastrophe suppresses potentially pro-oncogenic events, such as (1) karyotypic alterations, the most frequent of which are aneuploidy (an unbalanced state arising from the mis-segregation of one or more chromosomes) and tetraploidy (an aberrant form of euploidy resulting from one round of whole-genome doubling); and (2) chromosomal instability (CIN), a condition characterized by the inability to stably maintain the karyotype during cell divisions, which gives rise to, and is fueled by, non-diploidy.5,6 Accordingly, the abrogation of mitotic catastrophe results in the generation and propagation of non-diploid (i.e., aneuploid, tetraploid, or polyploid) cells and has been linked to tumorigenesis.3
The molecular machinery involved in the execution of mitotic catastrophe has been a matter of intense investigation over almost the last 2 decades. In these studies, tumor protein 53 (TP53, best known as p53) has emerged as the major hub of a signaling pathway that directly or indirectly responds to mitotic defects and ploidy changes. Thus, the absence of p53 or its downstream targets was shown to promote both the destabilization of the karyotype and tolerance of non-diploidy.7-9 Nonetheless, the precise signal(s) responsible for p53 activation is(are) debated,10,11 and instances of p53-independent mitotic catastrophe for suppressing non-diploidy have also been described.2 Taking advantage of a mitotic catastrophe model based on the abrogation of the G2/M cell cycle checkpoint in experimentally generated syncytia (which have redoubled their DNA content), we previously demonstrated that the activation of caspase 2 (CASP2) is (one of) the initiating event(s) of mitotic death.12 We also elucidated the cascade ignited by CASP2, which involves the induction of mitochondrial outer membrane permeabilization (MOMP) followed by the activation of caspase 3 (CASP3) independently of p53.2 Of note, in the presence of distinct stress stimuli (e.g., DNA lesions), CASP2 seems to be proteolytically activated by the PIDDosome, a not yet fully characterized multiprotein platform composed of the p53-induced death domain protein 1 (PIDD1, also known as PIDD) and CASP2 and RIPK1 domain containing adaptor with death domain (CRADD, best known as RAIDD).13
Three recent studies shed light on the mechanism of, and the signal responsible for, CASP2 activation during mitotic catastrophe unveiling the existence of a tight connection between CASP2 and the p53 pathway.14-16 By analyzing colorectal cancers and cell lines displaying high levels of CIN, either at the baseline or upon the pharmacological abrogation of the SAC or increase of replication stress, Lopez-Garcia and colleagues identified a novel mechanism of tolerance to non-diploidy based on the downregulation of CASP2 expression.14 These authors demonstrated that chromosome mis-segregation events specifically promoted the activation of CASP2 in a p53-independent fashion. Activated CASP2 in turn suppressed aneuploidy by (1) stabilizing p53 via a mechanism involving the cleavage of MDM2 proto-oncogene (MDM2) followed by the binding of the MDM2-p60 fragment to p53, or (2) inducing MOMP and CASP3 activation via a p53-independent cascade involving the cleavage of the BH3 interacting domain death agonist (BID) followed by the relocation of the cleaved form of BID (tBID) to the outer mitochondrial membrane (Fig. 1). Through genomic analyses, B-cell CLL/lymphoma 9-like (BCL9L) was identified as a tumor suppressor gene specifically mutated in CIN colorectal cancer. Finally, BCL9L haploinsufficiency was reported to increase the tolerance to aneuploidy by downregulating CASP2 via the inhibition of the transcription factor 4 (TCF4).14
Figure 1.
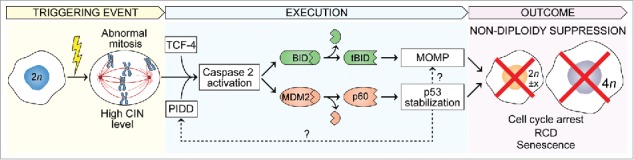
Mitotic catastrophe is a mechanism sensing mitotic alterations provoked by endogenous sources (e.g., dysfunction, deficiency, or downregulation of components or regulators of the machineries involved in DNA replication or chromosome segregation) or exogenous triggers (e.g., pharmacological inhibitors of DNA replication, microtubule polymerization, cell cycle checkpoints, mitotic kinases, or cytokinesis). The key event of mitotic catastrophe is the activation of CASP2, which occurs via a not yet fully elucidated mechanism likely involving the activating platform PIDDosome and the transcription factor TCF4. Activated CASP2 executes mitotic catastrophe by cleaving BID and MDM2 to generate the fragments tBID and MDM2-p60 (p60), which then induce outer mitochondrial membrane permeabilization (MOMP) and p53 stabilization, respectively. These events suppress the generation of non-diploidy (i.e., aneuploidy or tetraploidy) by promoting mitotic death or the activation of cell cycle checkpoints, CASP3-dependent regulated cell death (RCD), and cellular senescence in the G1 phase following mitotic exit.
Fava and colleagues analyzed the cellular response to ploidy changes uncovering a similar surveillance mechanism relying on CASP2 and executed by p53.15 Fava et al. found that cells undergoing whole-genome duplication via cytokinesis failure were arrested in their proliferation by a process that involved the p53-independent activation of CASP2 by the PIDDosome. Activated CASP2 then promoted the stabilization of p53 via MDM2 cleavage, which resulted in p53 accumulation, cyclin-dependent kinase inhibitor 1A (CDKN1A, best known as p21) activation and cell cycle blockade (Fig. 1). These authors also reported that mitotic catastrophe-inducing events distinct from cytokinesis abortion, such as prolonged mitosis or DNA damage, initiated the p53 pathway via a mechanism not requiring CASP2 activity, which is in line with previous studies.11,17,18 Finally, centrosome amplification was identified as the upstream signal triggering CASP2 activation upon cytokinesis abortion.15 It remains to elucidate how and whether BCL9L and other factors responding to non-diploidy, including large tumor suppressor kinase 2 (LATS2) and the Hyppo pathway,19 BCL2 proteins,6 mitogen-activated protein kinase 14 (MAPK14, best known as p38),9 Cyclin D1 (CCND1)20 and ubiquitin specific peptidase 28 (USP28),17,18 may contribute to this CASP2-dependent process.
Finally, Dawar and colleagues developed a strategy to boost aneuploidization based on the administration of polo like kinase 1 (PLK1) inhibitors followed or not by drug washout, confirming the key role of CASP2 in eradicating non-diploid cells.16 Dawar et al. performed time-lapse imaging and cytogenetic analyses of primary splenocytes from Casp2−/− mice and CASP2-depleted human cancer cells to show that the absence of CASP2 promoted mitotic slippage as well as the survival of the resulting multinucleated cells that were able to resume proliferation in clonogenic assays. These results suggest that CASP2 may also contribute to SAC signaling, an intriguing hypothesis that requires further confirmation. In this experimental setting, mitotic abnormalities provoked by PLK1 inhibition triggered a mitotic catastrophe pathway relying on the activation of CASP2, which in turn catalyzed the cleavage of BID and subsequent cell death executed via MOMP and CASP3 (Fig. 1).16 It will be important to further explore this model of mitotic catastrophe to elucidate the nature of the signal activating CASP2 and to clarify whether p53 plays a role upstream or downstream of CASP2 activation.
Altogether, these novel findings corroborate our previous results demonstrating that CASP2 acts as a major suppressor of aneuploidy and tetraploidy. Thus, CASP2 adopts its role as the crucial terminator of mitotic catastrophe by setting in motion distinct oncosuppressive networks that may involve p53 activity but also proceed independently from this central oncosuppressor (Fig. 1). Through this activity, CASP2 preserves karyotype stability across consecutive cell divisions in either a p53-proficient or a p53-deficient genetic background, thus limiting the propagation of non-diploid genomes.
In line with a role for CASP2 in maintaining genomic stability, unscheduled activation of CASP2 is prevented during normal mitosis by (1) the inhibitory phosphorylation of CASP2 catalyzed by cyclin-dependent kinase 1 (CDK1) in complex with cyclin B1 (CCNB1),21 and (2) the sequestration of PIDD to kinetochores that is mediated by the SAC player BUB1 mitotic checkpoint serine/threonine kinase B (BUB1B, best known as BUBR1).22 This design likely suppresses mitotic catastrophe in the course of normal cell divisions.
Importantly, increased rate of aneuploidization and CIN has been detected (1) in Casp2−/− mice subjected to chemical carcinogens or oncogenic stress, in which non-diploidy was accompanied by accelerated tumor development and progression,23,24 and (2) in Casp2−/− mouse embryonic fibroblasts (MEFs), in which non-diploidy was associated with escape from senescence, decreased telomere length and increased resistance to antimicrotubular compounds.25,26 Of note, mice deficient for Casp2 also exhibited a reduced lifespan,23,24 further supporting the role of this caspase for normal tissue homeostasis.
Reportedly, most human neoplasms are aneuploid and display a high level of chromosome instability and intratumoral heterogeneity.27 Moreover, it has been estimated that, in approximately 40% of the cases, aneuploid tumors arise from intermediate cells experiencing at least one round of whole-genome doubling followed by repeated chromosome mis-segregation events.28 These observations indicate that mitotic catastrophe control is breached during early phases of oncogenesis by not yet fully elucidated processes. It is tempting to speculate that such oncongenic processes might involve the (transient?) inactivation of the pathway controlled by CASP2.
Moreover, there is accumulating evidence that, beyond their initial oncosuppressive role, the machineries for the maintenance of genetic stability support the progression and evolution of established neoplasms, thereby constituting promising targets for cancer therapy.29 In this context it appears intriguing that non-diploid cancer cells are peculiarly sensitive to mitotic catastrophe inducers, such as inhibitors of checkpoint kinase 1 (CHEK1, best known as CHK1),30 kinesin family member 11 (KIF11, best known as EG5),31 or TTK protein kinase (TTK, best known as MPS1).32
Altogether, it appears important for cancer research to further elucidate the molecular mechanisms of mitotic catastrophe involved in the suppression of non-diploidy and how this process (1) is subverted during oncogenesis, (2) contributes to tumor evolution and intra-tumor heterogeneity, (3) confers resistance to cancer therapy, (4) is modulated by the tumor microenvironment, and (5) may be exploited for the eradication of cancer cells. Ever accumulating evidence indicates that CASP2 plays a multipronged role in mitotic catastrophe. The translation of these findings into clinically useful information is urgently awaited.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC, MFAG 2013 grant number 14641 to IV), Ministero Italiano della Salute (grant number RF_GR-2011–02351355 to IV), and Ministero Italiano dell'Istruzione, dell'Università e della Ricerca (MIUR, Programma per i Giovani Ricercatori “Rita Levi Montalcini” 2010 to IV). GK is supported by the Ligue contre le Cancer Comité de Charente-Maritime (équipe labelisée); Agence National de la Recherche (ANR)—Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Institut Universitaire de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière, the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
References
- 1.Andreassen PR, Lacroix FB, Lohez OD, Margolis RL. Neither p21WAF1 nor 14-3-3sigma prevents G2 progression to mitotic catastrophe in human colon carcinoma cells after DNA damage, but p21WAF1 induces stable G1 arrest in resulting tetraploid cells. Cancer Res 2001; 61:7660-8; PMID:11606409 [PubMed] [Google Scholar]
- 2.Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene 2004; 23:2825-37; PMID:15077146; https://doi.org/ 10.1038/sj.onc.1207528 [DOI] [PubMed] [Google Scholar]
- 3.Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol 2011; 12:385-92; PMID:21527953; https://doi.org/ 10.1038/nrm3115 [DOI] [PubMed] [Google Scholar]
- 4.Manic G, Corradi F, Sistigu A, Siteni S, Vitale I. Molecular Regulation of the Spindle Assembly Checkpoint by Kinases and Phosphatases. Int Rev Cell Mol Biol 2017; 328:105-61; PMID:28069132; https://doi.org/ 10.1016/bs.ircmb.2016.08.004 [DOI] [PubMed] [Google Scholar]
- 5.Galluzzi L, Kroemer G. A four-lane highway to cancer. Nat Rev Mol Cell Biol 2016; 17:398; PMID:27220642; https://doi.org/ 10.1038/nrm.2016.73 [DOI] [PubMed] [Google Scholar]
- 6.Vitale I, Manic G, Senovilla L, Kroemer G, Galluzzi L. Karyotypic Aberrations in Oncogenesis and Cancer Therapy. Trends Cancer 2015; 1:124-35; https://doi.org/ 10.1016/j.trecan.2015.08.001 [DOI] [PubMed] [Google Scholar]
- 7.Castedo M, Coquelle A, Vivet S, Vitale I, Kauffmann A, Dessen P, Pequignot MO, Casares N, Valent A, Mouhamad S, et al.. Apoptosis regulation in tetraploid cancer cells. EMBO J 2006; 25:2584-95; PMID:16675948; https://doi.org/ 10.1038/sj.emboj.7601127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vitale I, Senovilla L, Jemaa M, Michaud M, Galluzzi L, Kepp O, Nanty L, Criollo A, Rello-Varona S, Manic G, et al.. Multipolar mitosis of tetraploid cells: inhibition by p53 and dependency on Mos. EMBO J 2010; 29:1272-84; PMID:20186124; https://doi.org/ 10.1038/emboj.2010.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol 2010; 188:369-81; PMID:20123995; https://doi.org/ 10.1083/jcb.200905057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hinchcliffe EH, Day CA, Karanjeet KB, Fadness S, Langfald A, Vaughan KT, Dong Z. Chromosome missegregation during anaphase triggers p53 cell cycle arrest through histone H3.3 Ser31 phosphorylation. Nat Cell Biol 2016; 18:668-75; PMID:27136267; https://doi.org/ 10.1038/ncb3348 [DOI] [PubMed] [Google Scholar]
- 11.Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, Han S, van Deursen JM, Zhang P. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci U S A 2010; 107:14188-93; PMID:20663956; https://doi.org/ 10.1073/pnas.1005960107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castedo M, Perfettini JL, Roumier T, Valent A, Raslova H, Yakushijin K, Horne D, Feunteun J, Lenoir G, Medema R, et al.. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene 2004; 23:4362-70; PMID:15048075; https://doi.org/ 10.1038/sj.onc.1207572 [DOI] [PubMed] [Google Scholar]
- 13.Bock FJ, Peintner L, Tanzer M, Manzl C, Villunger A. P53-induced protein with a death domain (PIDD): master of puppets? Oncogene 2012; 31:4733-9; PMID:22266869; https://doi.org/ 10.1038/onc.2011.639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopez-Garcia C, Sansregret L, Domingo E, McGranahan N, Hobor S, Birkbak NJ, Horswell S, Grönroos E, Favero F, Rowan AJ, et al.. BCL9L Dysfunction Impairs Caspase-2 Expression Permitting Aneuploidy Tolerance in Colorectal Cancer. Cancer Cell 2017; 31:79-93; PMID:28073006; https://doi.org/ 10.1016/j.ccell.2016.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA, et al.. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev 2017; 31:34-45; PMID:28130345; https://doi.org/ 10.1101/gad.289728.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dawar S, Lim Y, Puccini J, White M, Thomas P, Bouchier-Hayes L, Green DR, Dorstyn L, Kumar S. Caspase-2-mediated cell death is required for deleting aneuploid cells. Oncogene 2016; PMID:27991927; https://doi.org/ 10.1038/onc.2016.423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lambrus BG, Daggubati V, Uetake Y, Scott PM, Clutario KM, Sluder G, Holland AJ. A USP28-53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol 2016; 214:143-53; PMID:27432896; https://doi.org/ 10.1083/jcb.201604054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meitinger F, Anzola JV, Kaulich M, Richardson A, Stender JD, Benner C, Glass CK, Dowdy SF, Desai A, Shiau AK, et al.. 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol 2016; 214:155-66; PMID:27432897; https://doi.org/ 10.1083/jcb.201604081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ganem NJ, Cornils H, Chiu SY, O'Rourke KP, Arnaud J, Yimlamai D, Théry M, Camargo FD, Pellman D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 2014; 158:833-48; PMID:25126788; https://doi.org/ 10.1016/j.cell.2014.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crockford A, Zalmas LP, Gronroos E, Dewhurst SM, McGranahan N, Cuomo ME, Encheva V, Snijders AP, Begum J, Purewal S, et al.. Cyclin D mediates tolerance of genome-doubling in cancers with functional p53. Ann Oncol (2017) 28 (1): 149-156; PMID:27864221; https://doi.org/ 10.1093/annonc/mdw612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andersen JL, Johnson CE, Freel CD, Parrish AB, Day JL, Buchakjian MR, Nutt LK, Thompson JW, Moseley MA, Kornbluth S. Restraint of apoptosis during mitosis through interdomain phosphorylation of caspase-2. EMBO J 2009; 28:3216-27; PMID:19730412; https://doi.org/ 10.1038/emboj.2009.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson R, Shah RB, Liu PH, Gupta YK, Ando K, Aggarwal AK, Sidi S. An Inhibitor of PIDDosome Formation. Mol Cell 2015; 58:767-79; PMID:25936804; https://doi.org/ 10.1016/j.molcel.2015.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puccini J, Dorstyn L, Kumar S. Caspase-2 as a tumour suppressor. Cell Death Differ 2013; 20:1133-9; PMID:23811850; https://doi.org/ 10.1038/cdd.2013.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shalini S, Nikolic A, Wilson CH, Puccini J, Sladojevic N, Finnie J, Dorstyn L, Kumar S. Caspase-2 deficiency accelerates chemically induced liver cancer in mice. Cell Death Differ 2016; 23:1727-36; PMID:27518436; https://doi.org/ 10.1038/cdd.2016.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ho LH, Read SH, Dorstyn L, Lambrusco L, Kumar S. Caspase-2 is required for cell death induced by cytoskeletal disruption. Oncogene 2008; 27:3393-404; PMID:18193089; https://doi.org/ 10.1038/sj.onc.1211005 [DOI] [PubMed] [Google Scholar]
- 26.Dorstyn L, Puccini J, Wilson CH, Shalini S, Nicola M, Moore S, Kumar S. Caspase-2 deficiency promotes aberrant DNA-damage response and genetic instability. Cell Death Differ 2012; 19:1288-98; PMID:22498700; https://doi.org/ 10.1038/cdd.2012.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet 2012; 13:189-203; PMID:22269907; https://doi.org/ 10.1038/nrg3123 [DOI] [PubMed] [Google Scholar]
- 28.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH, et al.. Pan-cancer patterns of somatic copy number alteration. Nat Genet 2013; 45:1134-40; PMID:24071852; https://doi.org/ 10.1038/ng.2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pearl LH, Schierz AC, Ward SE, Al-Lazikani B, Pearl FM. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer 2015; 15:166-80; PMID:25709118; https://doi.org/ 10.1038/nrc3891 [DOI] [PubMed] [Google Scholar]
- 30.Vitale I, Galluzzi L, Vivet S, Nanty L, Dessen P, Senovilla L, Olaussen KA, Lazar V, Prudhomme M, Golsteyn RM, et al.. Inhibition of Chk1 kills tetraploid tumor cells through a p53-dependent pathway. PLoS One 2007; 2:e1337; PMID:18159231; https://doi.org/ 10.1371/journal.pone.0001337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rello-Varona S, Vitale I, Kepp O, Senovilla L, Jemaa M, Metivier D, Castedo M, Kroemer G. Preferential killing of tetraploid tumor cells by targeting the mitotic kinesin Eg5. Cell Cycle 2009; 8:1030-5; PMID:19270519; https://doi.org/ 10.4161/cc.8.7.7950 [DOI] [PubMed] [Google Scholar]
- 32.Jemaa M, Manic G, Lledo G, Lissa D, Reynes C, Morin N, Chibon F, Sistigu A, Castedo M, Vitale I, et al.. Whole-genome duplication increases tumor cell sensitivity to MPS1 inhibition. Oncotarget 2016; 7:885-901; PMID:26637805; https://doi.org/ 10.18632/oncotarget.6432 [DOI] [PMC free article] [PubMed] [Google Scholar]