

Summary
Mitochondrial dysfunction occurs during aging, but its impact on tissue senescence is unknown. Here, we find that sedentary but not active humans display an age-related decline in the mitochondrial protein, optic atrophy 1 (OPA1), that is associated with muscle loss. In adult mice, acute, muscle-specific deletion of Opa1 induces a precocious senescence phenotype and premature death. Conditional and inducible Opa1 deletion alters mitochondrial morphology and function but not DNA content. Mechanistically, the ablation of Opa1 leads to ER stress, which signals via the unfolded protein response (UPR) and FoxOs, inducing a catabolic program of muscle loss and systemic aging. Pharmacological inhibition of ER stress or muscle-specific deletion of FGF21 compensates for the loss of Opa1, restoring a normal metabolic state and preventing muscle atrophy and premature death. Thus, mitochondrial dysfunction in the muscle can trigger a cascade of signaling initiated at the ER that systemically affects general metabolism and aging.
Keywords: mitochondria, Opa1, muscle, aging, FGF21, sarcopenia, FoxO, inflammation, oxidative stress
Graphical Abstract
Highlights
-
•
OPA1 is a physical activity sensor that is downregulated during aging sarcopenia
-
•
Muscle OPA1 controls general metabolism and epithelial senescence via FGF21
-
•
Inhibition of muscle OPA1 induces a systemic pro-inflammatory status
-
•
OPA1 controls protein breakdown/synthesis, muscle stem cells and fiber innervation
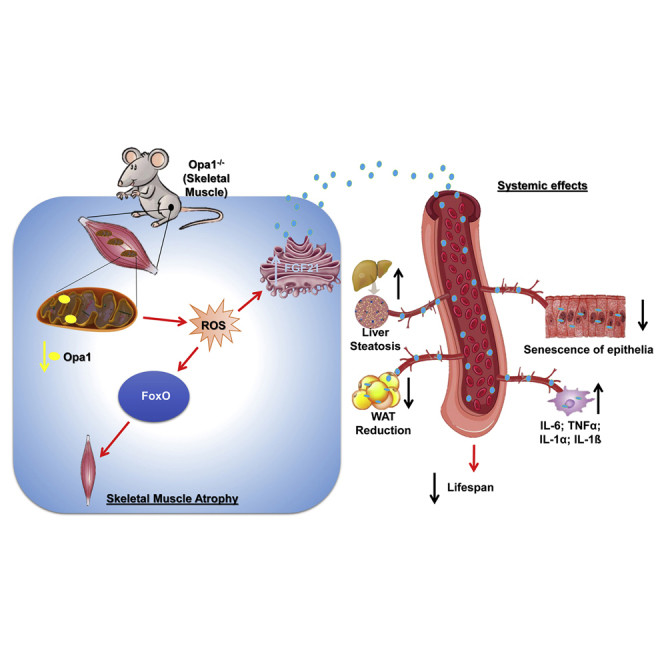
Aging has been reported to be accompanied by changes in mitochondrial dynamics, but the impact on tissue senescence is unknown. Tezze et al. show that deletion of Opa1 impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence and identify FGF21 as the culprit of precocious aging and premature death.
Introduction
Mitochondria are crucial organelles in the production of energy and in the control of signaling cascades. A machinery of pro-fusion and -fission proteins regulates their morphology and subcellular localization. Both mechanisms are evolutionarily conserved from yeast to humans. In mammals, mitofusin (MFN) 1 and 2 and optic atrophy protein 1 (OPA1) are required for fusion of the outer mitochondrial membrane and inner mitochondrial membrane (Romanello and Sandri, 2016). OPA1 requires MFN1 to regulate mitochondrial fusion, but on the other hand, oligomerization of OPA1 regulates apoptosis by controlling cristae remodeling and cytochrome c redistribution (Cipolat et al., 2006, Frezza et al., 2006). The maintenance of a dynamic mitochondrial network is particularly important for cells with a complex and highly structured cytosol, such as neurons and cardiac and skeletal muscles constituted by post-mitotic cells that do not divide. While tissues with high cell turnover can dilute the damaged/altered mitochondria among the dividing cells, the post-mitotic tissues use the fusion/fission machinery to preserve or restore mitochondrial function. Alternatively, post-mitotic organs activate selective autophagy to remove the irreversibly injured mitochondria (mitophagy). A failure of these systems predisposes to tissue dysfunction and degeneration. Mouse knockouts (KO) for each of the pro-fusion genes (Mfn1, Mfn2, and Opa1) are lethal to embryos because of mitochondrial dysfunction (Chen et al., 2003, Davies et al., 2007). Muscle-specific KO mice for Mfn1 and Mfn2 are viable at birth, although they display profound defects of muscle growth characterized by mitochondrial dysfunction, reduction of mitochondrial DNA (mtDNA) in skeletal muscle, and accumulation of point mutations and deletions in the mitochondrial genome that cause death within 6–8 weeks of age (Chen et al., 2010). In humans, mutations in MFN2 and OPA1 genes cause two neurodegenerative diseases, Charcot-Marie-Tooth type 2A (CMT2A) and dominant optic atrophy (DOA), respectively (Alexander et al., 2000, Delettre et al., 2000, Züchner et al., 2004). CMT2A is an inherited neuropathy that is clinically characterized by muscle atrophy. Heterozygous missense recessive mutations of OPA1 genes cause DOA with a muscle involvement revealed as an aspecific myopathy with mitochondrial features (Amati-Bonneau et al., 2008, Schaaf et al., 2011). Interestingly, the first case of homozygous missense mutation has been reported in two sisters that died at 2 and 10 months of age showing myopathy, encephalopathy, and cardiomyopathy (Spiegel et al., 2016). The muscle biopsies of these sisters revealed 50% reduction of OPA1 protein, a decrease in the activity of all the respiratory chain complexes, and an important mtDNA depletion (80%). Therefore, mutations in fusion genes result in brain and muscle dysfunction.
Skeletal muscle is a major site of metabolic activity and the most abundant tissue in the human body, accounting for almost 50% of the total mass. Being the largest protein reservoir, muscle serves as a source of amino acids to be utilized for energy production by various organs during catabolic periods. Importantly, skeletal muscle is an important modulator of general metabolism since it plays an important role in glucose, amino acid, and lipid homeostasis. Recent data in Drosophila show that maintenance of a functional proteostasis specifically in muscles, but not in white adipose tissue, during aging reverberates to the whole organism, leading to an extension of lifespan (Demontis et al., 2014, Demontis and Perrimon, 2010). Therefore, a new concept is emerging from this and other studies that considers the metabolic adaptations occurring in skeletal muscles as disease modifier/controller (Baskin et al., 2015). Indeed, growing evidence suggests that muscle-derived growth factor or cytokines, known as myokines, modulate systemic physiology (Demontis et al., 2013). Importantly, exercise by preserving and ameliorating muscle metabolism is able to counteract the body deterioration by improving function of multiple organs (Neufer et al., 2015). However, the mechanistic insights that link muscle contraction to organ function and longevity are still unknown. Here we show that deletion of Opa1 in skeletal muscle results in a lethal phenotype that is even more severe than Mfn1/2 double KO. This phenotype results from suppression of myogenesis, impaired protein synthesis and activation of protein breakdown. Importantly, several of these features were recapitulated in inducible muscle-specific Opa1 KO mice that additionally show multiple-organ senescence and precocious aging.
Results
Age-Related Muscle Loss in Humans and Mice Is Associated with Decreased OPA1 Expression
Aging has been reported to be accompanied by numerous functional alterations of mitochondria, including changes of mitochondrial dynamics (Ibebunjo et al., 2013). Thus, we tested whether mitochondria-shaping factors are reduced in humans. A decline of Mfn1/2, Opa1, and Drp1 transcripts was found in muscle biopsies of old sedentary (sarcopenic) subjects (Figure 1A and Table S1). Since exercise counteracts most of the age-related features, including the decline in muscle mass/force, we tested whether a long-life regular exercise was able to prevent the reduction of these genes (Figure 1A). Expression of the mitochondria-shaping genes was maintained in muscle biopsies of senior sportsmen. Similar to transcript level, we further confirmed that OPA1, MFN1, and DRP1 proteins were reduced in sedentary subjects and that regular exercise was able to counteract this decline (Figures 1B, S1A, and S1B). When we explored whether the decrease of mitochondrial shaping proteins correlates with muscle mass loss in elderly people, we found a significant correlation only for OPA1 and not for DRP1 or MFN1 (Figure 1C). Similarly, muscle force drop significantly correlates with decrease of OPA1 expression but not of DRP1 or MFN1 (Figure 1D). This association is independent of fiber type (Figures S1C and S1D). Consistently with human data, aged mice displayed a significant reduction of OPA1 in muscles (Figures 1E and S1E). Importantly, 1 week of exercise training is sufficient to reactivate OPA1 expression in muscles of aged mice (Figure 1E). Overall, these findings suggest a causative role of mitochondrial dynamic impairment that correlated with the decreased OPA1 expression and age-related muscle loss and weakness.
Figure 1.

OPA1 Expression Is Reduced during Aging in Sarcopenic Patients
(A) Mitochondria-shaping machinery is downregulated in sarcopenia and maintained by lifelong exercise. Quantitative PCR analysis of mitochondrial dynamics transcripts from human muscle biopsies of 5 young subjects, 7 sedentary seniors, and 11 senior sportsmen is shown. Data represent mean ± SEM and are normalized for GAPDH.
(B) Densitometric analysis of OPA1/Complex II, MFN1/Complex II, and DRP1/Complex II signal ratios revealed by immunoblotting. Data represent mean ± SEM of four independent experiments (young = 5, sedentary seniors = 19, senior sportsmen = 5).
(C) Linear regression analysis including OPA1, DRP1, and MFN1 expression levels and myofiber size in elderly persons (n = 24). Red spots depict senior sportsmen.
(D) Linear regression showing that decrease of OPA1 but not of DRP1 or MFN1 levels correlates with muscle weakness in elderly persons (n = 24). Red spots depict senior sportsmen.
(E) Representative immunoblot and densitometric analysis of OPA1 in homogenates of tibialis anterior muscles from young (6 months old, n = 6), old (18 months old, n = 9) and old trained mice (18 months old, n = 6). Data are expressed as mean ± SEM; ∗p ≤ 0.05, ∗∗p < 0.01.
Muscle-Specific Opa1 Deletion in Newborns Blocks Animal Growth and Is Lethal
Given the important decrease of OPA1 during aging and the fact that there is no animal model of conditional Opa1 KO in post-mitotic tissues, we generated a muscle-specific Opa1 KO mouse line. Muscles in newborns showed a 50% decrease in transcript level and 70% reduction of OPA1 protein (Figures S2A and S2B) specifically in the muscle. Importantly, these mice died within 9 days of postnatal life (Figure 2A) even when they were artificially fed, suggesting that nutrition was not the cause of death. During the first 8 days of postnatal life, the Opa1−/− mice showed an impairment of total body growth, resulting in smaller animals (Figure S2C). These animals displayed hypoglycemia under non-fasting conditions (Figure 2B). The size of the muscles was decreased because of reduced fiber size (Figure S2D) that results in an increased amount of fibers per area (Figure 2C). The Opa1−/− mice did not show decrease of total fiber number (Figure S2E), suggesting that myogenesis and myotube formation are not perturbed, but that postnatal myofiber growth is affected by OPA1 ablation.
Figure 2.

OPA1 Deletion Induces a Lethal Phenotype Characterized by Mitochondrial Dysfunction, Reduction of Myogenic Stem Cells, Decreased Protein Synthesis, and Activation of Protein Breakdown
(A) Kaplan-Meier survival curve of Opa1f/f and Opa1−/− littermates (n = 37 for each group) indicates that muscle-specific OPA1 deletion results in lethality by post-natal day 9. ∗∗p < 0.01.
(B) Blood glucose levels in control and knockout mice (Opa1f/f n = 6; Opa1−/− n = 7). Data represent mean ± SEM, ∗∗p < 0.01.
(C) Analysis of the number of fibers normalized per muscle area (μm2). Data represent mean ± SEM for n = 6, ∗∗p < 0.01.
(D) Respiratory control ratio (RCR) of mitochondria energized with 5 mM/2.5 mM glutamate/malate (GLU/MAL) or 10 mM succinate (SUCC/ROTE). Data represent average ± SEM of three independent experiments (n = 9 per each condition), ∗∗p < 0.01.
(E) Densitometric quantification of Blue Native-PAGE of respiratory chain supercomplexes (RCS) that were normalized for individual respiratory chain complexes. Data represent mean ± SEM, n = 3, ∗∗p < 0.01.
(F) BrdU incorporation in Opa1f/f and Opa1−/− muscles. Data represent mean ± SEM, n = 4.
(G) Pax7-positive muscle stem cells and MyoD-positive myoblast were revealed by immunohistochemistry and normalized for muscle area. Data are mean ± SEM for n = 4, ∗∗p < 0.01.
(H) Representative immunoblot of muscle homogenates of three independent experiments. Lamin A/C, MyoD, and myogenin (MyoG) were normalized for GAPDH expression. Statistical significance after densitometric analysis is indicated on the right, ∗∗p < 0.01.
(I) Quantitative RT-PCR of FGF21 expression in Opa1−/− muscles. Data represent mean ± SEM, ∗∗∗p < 0.001, n = 4.
(J) Quantification of puromycin incorporation in skeletal muscles. In vivo SUnSET technique demonstrates a significant reduction of protein synthesis in Opa1−/− muscles. Data are mean ± SEM of three independent experiments (Opa1f/f n = 5; Opa−/− n = 7), ∗∗p < 0.01.
(K) Immunoblots of protein extracts from newborn muscles for the indicated antibodies. Representative immunoblots of three independent experiments are shown. Statistical analysis of the densitometric ratio is indicated on the right, n = 7 per each, ns: not significant, ∗∗p < 0.01.
(L and M) Representative immunoblots (L) and quantitative RT-PCR (M) showing activation of the UPR pathway in Opa1−/−. Data are mean ± SEM of three independent experiments, ∗∗p < 0.01.
(N) Quantitative RT-PCR analysis of FoxO-target genes. Data are mean ± SEM for n = 6, ∗∗p < 0.01.
(O and P) Densitometric quantification of lysine 48 (Lys48; O) and lysine 63 (Lys63; P) poly-ubiquitinated proteins from controls and Opa1−/− muscles. Data are mean ± SEM, n = 4, ∗∗p < 0.01.
(Q) Representative immunoblot of three independent experiments of autophagy-related proteins. Data are mean ± SEM, ∗∗p < 0.01.
Muscle-Specific OPA1 Ablation Alters Mitochondrial Function and Morphology
To understand the molecular mechanisms behind the observed phenotype, we monitored mitochondrial ultrastructure. Electron microscopy revealed a disorganized sarcomere arrangement with Z-disk misalignment, empty spaces between myofibrils, and accumulation of lipid droplets (Figure S2F, left and middle panels). Mitochondria of Opa1−/− were smaller in size than controls, and cristae were dilated (Figure S2F, right panels). However, the mtDNA content per nuclear genome did not differ between the two genotypes (Figure S2H), suggesting that total mitochondrial content was not decreased. Consistent with the mtDNA analyses, the mitochondrial mass revealed by TOM20 and porin proteins was not affected in Opa1−/− (Figure S2I). We then verified whether OPA1 ablation caused compensatory changes in the levels of the other mitochondria-shaping machinery members. PGC1α and Mfn1 transcripts were upregulated, whereas the level of the fission protein DRP1 was not increased (Figures S2I and S2J). At the bioenergetic level, complex I-dependent respiration was reduced, and supercomplexes were destabilized in OPA1-deficient muscles (Figures 2D, 2E, and S2G).
Inhibition of OPA1 Reduces Muscle Stem Cells, Inhibits Protein Synthesis, and Induces Atrophy-Related Genes and Protein Breakdown
Muscle growth during the first 2 weeks of postnatal life depends on fusion of muscle stem cell to growing myotube and on protein synthesis (Schiaffino et al., 2013). First, we checked whether OPA1 deletion in myofiber affected muscle stem cell renewal and myogenic differentiation. Proliferative rate, revealed by BrdU incorporation, did not differ between the two genotypes (Figure 2F). However, immunostaining for Pax7, a marker of the quiescent muscle stem cells, revealed a significant reduction of positive cells in Opa1−/− (Figure 2G). Accordingly, MyoD-positive myoblasts were also decreased (Figure 2G). Western blot analyses confirmed that the markers of myoblast proliferation, MyoD, and differentiation, myogenin, were significantly reduced in KO mice (Figures 2H and S3A). We then explored the involvement of myostatin pathway in stem cell loss, but immunoblotting for pSmad3 ruled out myostatin/TGFβ members (Figure S2K). Since levels of apoptosis were similar in Opa1f/f and Opa1−/− muscles (Figure S2L), we concluded that muscle stem cells were likely depleted and unable to self-renew, suggesting a link between mitochondrial fusion and muscle stem cell maintenance.
The sole muscle stem cell dysfunction could not explain the generalized observed small animal size. When we monitored the growth hormone (GH)/IGF1 axis that controls cellular proliferation in most tissues, including muscle, we found a significant reduction in hepatic expression of one of the most important GH targets, IGF1 (Figure S2M). However, muscle expression of IGF1 was not reduced in Opa1−/− animals (not shown). Alternatively, the cytokine fibroblast growth factor (FGF) 21 could be the culprit, given its ability to induce GH resistance. Indeed, FGF21 transcript was 1,000-fold induced in Opa1−/− muscles compared to Opa1f/f (Figure 2I).
Since GH has an important anabolic function, we checked in vivo whether protein synthesis was altered in Opa1−/−. A SunSET analysis revealed a 50% protein synthesis reduction in Opa1 KO (Figures 2J and S2N). Interestingly, Opa1−/− muscle displayed an increase of total and phospho-Akt (pAkt), which was not sufficient to maintain the phosphorylation of mTOR and of its downstream target S6 (Figures 2K and S3B). Consistent with these data, the downstream target of AMPK, ACC, was hyper-phosphorylated, supporting the concept of an undergoing energy stress that inhibits mTOR. However, when we monitored 4EBP1, a second mTOR downstream target, we found an increase of p4EBP1 in two different sites (Figure 2K). The inconsistency of the Akt/mTOR/4EBP1/S6 axis suggests that other regulators are involved in the observed translation impairment, such as ribosome assembly, controlled by initiation factors, among which eIF2α is the most critical one. When eIF2α is phosphorylated (p-eIF2α), ribosome assembly is blocked and general protein synthesis is shut down. During ER stress, the unfolded protein response (UPR) kinase PERK phosphorylates eIF2α, leading to protein synthesis blockage. Levels of p-eIF2α and of the chaperone Bip/Grp78, a downstream target of UPR, were indeed increased in Opa1−/− (Figures 2L and S3C). Moreover, we observed that PERK-dependent UPR triggered the activation of the transcription factor ATF4 and the transcription of several genes including Bip, GADD34, CHOP, and FGF21 (Figures 2M and 2I). Thus, UPR activation in Opa1−/− muscles accounts for the reduction of translation (Figure 2J) and activates, via FGF21 (Figure 2I), a catabolic condition.
Since cell size is determined by the balance between protein synthesis and degradation, we also monitored the status of the ubiquitin-proteasome and autophagy-lysosome systems. The atrophy-related ubiquitin ligases atrogin-1 and SMART (Milan et al., 2015) were induced (Figures 2N, S2O, and S2P), resulting in an increase of lysine 48 poly-ubiquitinated proteins (Figures 2O and S2Q). Interestingly, lysine 63 poly-ubiquitinated proteins were also significantly increased in Opa1−/− (Figures 2P and S2R). Since lysine 63 poly-ubiquitinated proteins are also autophagy substrates, we checked the autophagy system by monitoring the recruitment of LC3 on autophagosomes (LC3 lipidation) and expression of autophagy-related genes. Despite the induction of p62 and cathepsin L genes and Bnip3 protein (Figures S2S and 2Q), the lipidated LC3 (LC3 II) was not significantly increased, suggesting that autophagosome number is not enhanced at this time point (Figures 2Q and S3D). In conclusion, Opa1 deletion during early postnatal life leads to a defect of muscle growth caused by a block of muscle stem cell renewal and protein synthesis and a concomitant induction of proteasome-dependent protein breakdown.
Acute Opa1 Deletion in the Skeletal Muscle Reverberates to the Whole Body, Leading to a Precocious Aging Phenotype that Culminates with Animal Death
Due to the severe phenotype of conditional Opa1−/− mice, which impaired further analyses in adulthood, we generated a tamoxifen-inducible muscle-specific OPA1 KO mouse. Tamoxifen treatment of 5-month-old mice efficiently reduced OPA1 transcript and protein (Figures S4A and S4B). Importantly, within 50 days from tamoxifen treatment, mice started to lose weight (Figure 3A), and they died within 3 months (Figure 3B), showing a precocious aging phenotype. Mice of 8 months show white hairs and kyphosis (Figure 3C); liver steatosis (Figure 3D); low glucose levels (Figure 3E); high inflammatory cytokines such as IL6, IL1α, IL1β, and TNFα in blood (Figure 3F); loss of myofiber innervation, revealed by myofiber disappearance and increase of NCAM positive fibers (Figure 3G); and disappearance of white adipose tissue (Figure 3H). To further support the aging phenotype, we checked several markers of aging like β-galactosidase and p21. Both are significantly increased in liver, skin, and gut (Figures 3I and 3J). Since the inflammatory cytokine IL6 is also produced by skeletal muscle, we checked whether muscles participate in the systemic inflammation. Interestingly, expression of IL6 and IL1α but not of TNFα or IL1β was strongly upregulated in Opa1−/− mice (Figure S4C). Therefore, acute inhibition of OPA1 in adult muscles leads to metabolic changes, systemic inflammatory response, and precocious epithelial senescence that culminate with animal death.
Figure 3.

Acute Deletion of OPA1 in Adult Muscles Leads to Metabolic Changes, Precocious Senescence, and Degeneration of Multiple Organs
(A) Growth curve of Opa1f/f (black) and Opa1−/− (red) mice after tamoxifen treatment. KO mice start to lose body weight after 30 days from the beginning of the treatment. Data are mean ± SEM (Opa1f/f n = 20, Opa1−/− n = 19), ∗∗p < 0.01.
(B) Kaplan-Meier survival curve of Opa1f/f and Opa1−/− adult animals (n = 10 for each group) indicates that muscle-specific OPA1 deletion results in lethality within 3 months from the beginning of the tamoxifen treatment.
(C) Eight-month-old Opa1−/− mice show a reduction of total body size (upper panel), white hairs (left bottom panel), and kyphosis (right bottom panel).
(D) Lipid content shown by oil red O staining of a representative liver cryosection reveals liver steatosis in Opa1−/− adult animals (left panel). Triglyceride content is increased in liver of Opa1−/−. Data are mean ± SEM for n = 5, ∗∗p < 0.01.
(E) Glycemia is reduced in fed knockout mice (Opa1f/f n = 10, Opa1−/− n = 8). Data are mean ± SEM, ∗∗p < 0.01.
(F) Blood levels of the inflammatory cytokines IL6, IL1α, IL1β, and TNFα (Opa1f/f n = 5, Opa1−/− n = 8). Data are mean ± SEM, ∗∗p < 0.01.
(G) Quantification of myofiber number (left graph) and of denervated NCAM-positive fibers. Data represent mean ± SEM, Opa1f/f n = 3, Opa1−/− n = 4, ∗∗p < 0.01.
(H) Epididymal fat content analysis indicates the reduction of white adipose tissue in OPA1 null animals. Data represent mean ± SEM for n = 4, ∗∗p < 0.01.
(I and J) β-galactosidase (I) and p21 (J) are significantly increased in liver, skin, and gut. Data represent mean ± SEM (n = 10), ∗∗p < 0.01.
Acute Opa1 Deletion Induces Muscle Loss and Weakness
The body weight reduction was secondary to decreased size of glycolytic fibers and adipose tissue loss (Figures 4A, 3H, and S4D). Morphological analyses showed no features of myofiber degeneration such as center-nucleated fibers or inflammation, suggesting that the decrease of muscle mass was due to atrophy (Figure S4E). Quantification of myofiber size indicated that Opa1−/− myofibers were smaller than controls in a time-dependent manner (Figure 4A). Succinate dehydrogenase (SDH) staining was reduced (Figure 4B) and revealed a pattern of localization that resembled central core disease, where regions of myofiber cytoplasm are devoid of mitochondria (Figure 4B). Next we measured the force generated by gastrocnemius muscle in living animals. Maximal absolute force (tetanic force) was significantly reduced (Figure 4C), whereas the specific force (i.e., force normalized for muscle mass) was not significantly affected in KO mice (Figure 4D), indicating an increased loss of sarcomeric proteins. To explain the lipolysis, we checked whether browning of WAT occurred and found an upregulation of UCP1, a well-known marker of brown adipose tissue (Figure S4F). Opa1−/− mice showed an improved glucose tolerance and insulin sensitivity (Figure S4G) because glucose uptake in liver, but not in muscles or WAT, is enhanced (Figures S4H–S4K).
Figure 4.

Acute Deletion of OPA1 in Adult Animals Causes Muscle Atrophy and Weakness and Alters Mitochondrial Morphology and Function
(A) Left panel: representative picture of gastrocnemius muscles from Opa1f/f and Opa1−/− mice. Right panel: myofiber cross-sectional area analysis of controls and Opa1−/− at 30 and at 120 days from the induction, respectively. Values are mean ± SEM from at least five muscles in each group, ∗∗p < 0.01.
(B) SDH staining showed a reduction of mitochondrial content in Opa1−/− that is particularly evident in the center of myofibers (arrow).
(C and D) Force-frequency curves performed in vivo on gastrocnemius muscles. Absence of OPA1 leads to a significant decrease of absolute force (C) but not of maximal specific force (D) generated during tetanic contraction. Data represent mean ± SEM (n = 6), ∗p < 0.05.
(E) Electron micrographs of EDL muscles of controls and Opa1−/−.
(F) Upper panel: representative images of adult isolated myofibers at bright field or immunostained for TOM20 showing decreased mitochondrial content in Opa1−/−. Lower panel: western blot for Tom20 confirms the decrease of mitochondrial mass, ∗∗p < 0.01.
(G) Myofibers were stained with the potentiometric dye TMRM.
(H) Absence of OPA1 induces mitochondrial depolarization. Isolated adult fibers were loaded with TMRM. Oligomycin and the protonophore FCCP were added at the indicated time points. TMRM staining was monitored in at least 40 fibers per condition, data are mean ± SEM, ∗∗p < 0.01.
(I) Mitochondrial respiratory chain (RC) enzymatic activities from muscles of Opa1f/f and Opa1−/−. Data were normalized to citrate synthase (CS) activity and were plotted as percentage of controls, data are mean ± SEM for n = 8, ∗p < 0.05; ∗∗p < 0.01.
(J) Representative Blue Native-PAGE gels showing respiratory chain supercomplexes (RCS). CI subunit NDUFB8 (left panel), CIII-Core2 protein 2 (middle panel), and CIV subunit COXI (right panel). n = 10; ∗p < 0.05; ∗∗p ≤ 0.01. CI, Complex I; CII, Complex II; CIII, Complex III; CIV, Complex IV; CI+CIII, Complex I plus Complex III; CII+CIII, Complex II plus Complex III.
Acute Inhibition of Opa1 Induces Mitochondrial Dysfunction
Analyses of mitochondrial morphology at ultrastructural level confirmed the presence of smaller mitochondria displaying dilated cristae in Opa1−/− muscles (Figure 4E). When we stained isolated single adult myofibers for Tom20 (Figure 4F, upper panels), we found that the typical striated pattern of mitochondrial network was absent in Opa1−/− fibers where areas were completely unstained. Western blot for mitochondrial proteins and real-time PCR for mtDNA indicated a decreased mitochondrial mass (Figure 4F, lower panel; Figures S4L and S5A). Staining with the potentiometric fluorescent dye tetramethylrhodamine methyl ester (TMRM) revealed a mosaic mitochondrial distribution in the cytoplasm of Opa1−/− isolated adult fibers (Figure 4G). Then, we checked the status of the mitochondrial membrane potential (Δψm) in isolated adult fibers of WT and OPA1 KO muscles. As expected, in control mice oligomycin-dependent inhibition of ATP synthase did not alter Δψm (Figure 4H), and mitochondrial depolarization was achieved after membrane permeabilization by the protonophore carbonylcyanide-p-trifluoromethoxy phenylhydrazone (FCCP). Conversely, mitochondria of Opa1−/− fibers underwent a significant depolarization when F1F0-ATPase is inhibited by oligomycin (Grumati et al., 2010) (Figure 4H), suggesting that these fibers were at least in part relying on reverse activity of ATP synthase to preserve their membrane potential as a consequence of proton leak-consuming proton motive force. Mechanistically, this dysfunction was caused by the reduction of respiratory chain enzymatic activity and by a destabilization of the respiratory chain supercomplexes (Figures 4I, 4J, and S4M).
Opa1 Deletion Retrogradely Signals to the Nucleus via UPR and FoxO to Induce a Catabolic Condition
We next checked proteostasis in inducible Opa1−/−. Interestingly, protein synthesis was superimposable in controls and KO mice (Figures S4N and S4O) as well as muscle stem cells and myogenesis (Figures S4P–S4R). Monitoring the signaling pathways related to protein synthesis, we found that pAkt was decreased, whereas pAMPK was reduced, leading to an increase of serine 65 p4EBP1 (Figures 5A and S5B), that was not sufficient to enhance translation because of UPR activation. In fact, p-eIF2α was increased, as was expression of ATF4, GADD34, and CHOP genes (Figures 5B, 5C, and S5C). Importantly, FGF21 was massively upregulated in a time-dependent manner, leading to an important raise of circulating FGF21 in the blood (Figures 5D and 5E). The FGF21 induction occurs specifically in skeletal muscles and not in other tissues such as liver or WAT (Figure S6A). Interestingly, Klotho (KLB) and FGF receptors (FGFR) were expressed in skeletal muscles and were upregulated in Opa1−/− fibers (Figure S6B), but not in the other tissues, with the exception of KLB in the skin (Figures S6C–S6F).
Figure 5.

Acute OPA1 Inhibition Does Not Affect Protein Synthesis but Triggers an Atrophy Program
(A) Total protein extracts from adult muscles were immunoblotted with the indicated antibodies. Representative immunoblots of two independent experiments are shown. Statistical significance after densitometry is indicated on the right. Data are mean ± SEM, ∗p < 0.05, ∗∗p < 0.01.
(B) p-eIF2α and total eIF2α immunoblots of muscle homogenates of three independent experiments. Statistical significance after densitometry is indicated on the right. Data are mean ± SEM, ∗∗p ≤ 0.01.
(C) Quantitative RT-PCR of genes downstream of UPR. Data are mean ± SEM, n = 5, ∗∗p < 0.01.
(D) Quantitative RT-PCR of FGF21 in OPA1-deficient muscles. Data are mean ± SEM, n = 4, ∗∗p < 0.01, ∗∗∗p < 0.001.
(E) FGF21 plasma levels in Opa1−/− mice were determined by ELISA (n = 6). ∗∗p < 0.01.
(F–H) Quantitative RT-PCR of several atrophy-related genes belonging to the ubiquitin proteasome (F and G) and autophagy-lysosome systems (H). Data are mean ± SEM, n = 5, ∗p < 0.05, ∗∗p < 0.01.
(I) Autophagy flux is increased in basal condition in OPA1-deficient muscles. Inhibition of autophagosome-lysosome fusion by colchicine treatment induces higher increase of p62 and LC3II band in OPA1 null than control muscles. Data are mean ± SEM, n = 5, ∗∗p ≤ 0.01.
(J and K) Densitometric quantification of lysine 48 (Lys48) and of lysine 63 (Lys63) poly-ubiquitinated proteins of muscle extracts from Opa1f/f and Opa1−/−. Data are mean ± SEM, n = 8, ∗p < 0.05, ∗∗p ≤ 0.01.
(L) Opa1−/− mice and Opa1f/f were transfected in vivo with scramble or siRNA against FoxO1/3. Inhibition of FoxOs by RNAi prevented muscle atrophy in Opa1−/− muscles. n = 6 per each group. Data are mean ± SEM, ∗p < 0.05, ∗∗p < 0.01.
According to muscle loss, several atrophy-related genes were significantly induced (Figures 5F and 5G) and a robust activation of autophagy was observed (Figures 5H, 5I, and S6G), leading to an increase of lysine 48 and lysine 63 poly-ubiquitinated proteins (Figures 5J and 5K). Interestingly, oxidative stress, revealed by the level of carbonylated proteins, was significantly increased in Opa1−/− mice (Figures S6H and S6I). To further support the mitochondrial-dependent oxidative stress, we transfected adult muscles of Opa1−/− and Opa1f/f with a mitochondrial targeted fluorescent ROS sensor (mt-roGFP) (Lo Verso et al., 2014) and found that mitochondria of Opa1−/− produced more ROS than controls (Figure S6J). Our results indicate that acute Opa1 ablation triggers an atrophy program that enhances protein breakdown. Since FoxOs are master regulators of autophagy and ubiquitin-proteasome system (Milan et al., 2015) and are activated by oxidative stress and Akt inhibition, we hypothesized an involvement of these factors in Opa1−/−. Indeed, inhibition of FoxOs, in vivo, by RNAi was sufficient to reduce muscle atrophy in Opa1−/− (Figure 5L).
Chemical Chaperone Treatment Blunts UPR and Prevents Muscle Loss
To explain how an alteration of mitochondrial shape affects a nuclear program of aging and muscle atrophy, we addressed the potential pathogenetic involvement of UPR. We acutely treated our inducible Opa1−/− mice with TUDCA, an established chemical chaperone known to counteract ER stress. TUDCA blunted p-eIF2α and improved pAkt as well as reduced FGF21 expression in Opa1−/− (Figures 6A–6C and S5D). Accordingly, expression of some atrophy-related ubiquitin ligases (Atrogin1, Itch, and SMART) and autophagy-related genes (e.g., LC3, GabarapL, and Bnip3) was reduced by TUDCA (Figures 6D and 6E). When we prolonged the treatment throughout the time during which muscles undergo atrophy, the observed reduction of XBP1 splicing and FGF21 induction (Figure 6F) was accompanied by the maintenance of mitochondrial content (Figures 6G and 6H) but not function (Figure S7A). Body weight loss was partially spared (Figure S7B) because muscle wasting, but not WAT loss, was greatly prevented (Figures 6I and S7C).
Figure 6.

TUDCA and Trolox Treatments Prevent ER Stress, Reduce FGF21 Induction, and Spare Muscle Mass
(A and B) Representative immunoblots of three independent experiments of p-eIF2α (A) and pAkt (B) in KO animals treated with TUDCA compared to untreated OPA1 null mice. Statistical significance after densitometry is indicated on the right. Data are mean ± SEM, n = 6, ∗∗p < 0.01.
(C) Quantitative RT-PCR demonstrated a significant reduction of FGF21 expression in TUDCA-treated (5 days) animals. Data are mean ± SEM, n = 5, ∗∗p < 0.01, ∗∗∗p < 0.001.
(D and E) Quantitative RT-PCR analysis of atrophy-related genes. Data are mean ± SEM, n = 5, ∗p < 0.05, ∗∗p < 0.01.
(F) Quantitative RT-PCR shows the significant reduction of both spliced XBP1 and FGF21 expression when animals were treated with TUDCA for 2 weeks. Data are mean ± SEM, n = 6, ∗∗p < 0.01.
(G) Representative SDH staining shows the maintenance of normal mitochondrial content in TUDCA-treated Opa1−/−.
(H) Representative immunoblots of TOM20.
(I) Quantification of cross-sectional area of Opa1f/f and Opa1−/− myofibers treated or not with TUDCA. Values are mean ± SEM from at least 4 muscles in each group, ∗∗p < 0.01.
(J) Representative immunoblots of p-eIF2α after Trolox treatment in OPA1 null mice. n = 3.
(K) Quantitative RT-PCR of FGF21 expression in Trolox-treated mice. Data are mean ± SEM, n = 7, ∗∗∗p < 0.001.
(L) Growth curve of control and Opa1−/− mice. Trolox prevented the body weight loss of Opa1−/−. Data are mean ± SEM (Opa1f/f n = 20, Opa1−/− n = 19, Opa1−/− + TROLOX n = 10), ∗∗p < 0.01.
(M) Quantification of cross-sectional area of controls and OPA1 knockout myofibers treated or not with Trolox. Values are mean ± SEM from at least four muscles in each group, ∗∗p < 0.01, n.s.: not significant.
(N) Epididymal white adipose tissue content of controls and Opa1−/− mice treated or not with Trolox (n = 6 in each group). Data are mean ± SEM, ∗∗p < 0.01.
Oxidative Stress Links Mitochondria to UPR
Since acute inhibition of OPA1 results in an increased oxidative stress, we reason that mitochondria-derived ROS may connect OPA1 deficiency with ER stress and UPR activation. To test this hypothesis, we treated the inducible KO mice with Trolox, a vitamin E analog with a potent anti-oxidant action. The treatment started 10 days before the beginning of weight loss. Trolox blunted the oxidative stress (Figures S7D and S7E), restored a normal ER function (Figure 6J), and completely prevented FGF21 induction in Opa1−/− mice (Figure 6K). However, mitochondrial dysfunction was not rescued by TROLOX (Figure S7A). Importantly, the antioxidant treatment preserved Opa1−/− from body weight loss (Figure 6L). Both skeletal muscle mass and WAT were maintained by Trolox in Opa1−/− (Figures 6M and 6N). Similar results were obtained by treating mice with a mitochondrial-targeted ROS scavenger named Mito-TEMPO (Lo Verso et al., 2014). Consistent with TUDCA and TROLOX data, Mito-TEMPO prevented protein oxidation, reduced FGF21 induction, blocked the atrophy program, restored an almost normal muscle mass, and partially prevented body weight loss, but did not affect white adipose tissue loss (Figures S7F–S7L). Thus, oxidative stress is the trigger that induces an ER-dependent atrophic program when OPA1 is acutely reduced.
Muscle-Dependent FGF21 Controls General Metabolism and Aging
To address the relevance of muscle-derived FGF21 in the context of Opa1−/− phenotype, we generated double Tamoxifen-inducible muscle-specific OPA1/FGF21 KO (Opa1−/−Fgf21−/−) mice. Acute and simultaneous deletion of Opa1 and Fgf21 restored normal blood levels of FGF21 (Figure 7A). Double KO mice are partially protected from body weight loss (Figures 7B and S7M) because muscle mass and white adipose tissue are, in part, spared (Figures 7C and 7D). The lipolysis is not completely blocked in Opa1−/−Fgf21−/−, because UCP1 is still induced in WAT and muscles are still atrophic, because atrogin1 expression in double KO is less than Opa1−/− but higher than controls (Figures S7N and S7O). Although Complex III activity is improved, mitochondria are still dysfunctional after the deletion of both FGF21 and OPA1, with a significant reduction of Complex I activity and of respiration (Figures S7A and S7P–S7R). Importantly, the aging phenotype was completely reverted by FGF21 deletion. Mice did not show any more white hairs and kyphosis (Figure 7E), liver steatosis was prevented, glycemia was restored to normal level, inflammatory cytokines were now in normal blood range (Figures 7F–7H), and muscle expression of IL6 and IL1α was normalized (Figure S7S). Loss of motorneurons and innervation, revealed by NCAM staining, was prevented (Figure 7I). Additionally, markers of aging such as β-galactosidase activity and p21 expression were normalized to control levels in several tissues (Figures 7J and 7K). Finally, the simultaneous deletion of Opa1 and Fgf21 prevented the precocious death of Opa1−/− mice (Figure 7L).
Figure 7.

Acute Simultaneous Deletion of OPA1 and FGF21 Reverted Precocious Epithelial Senescence, Systemic Inflammation, and Premature Death
(A) FGF21 plasma levels in OPA1/FGF21-KO, determined by ELISA, are significantly decreased compared to OPA1-deficient animals. Data are mean ± SEM, n = 4, ∗∗p < 0.01.
(B) Body weight measured 120 days after the beginning of tamoxifen treatment. Data are mean ± SEM, n = 5, ∗∗p < 0.01.
(C) Muscle weight of controls, Opa1−/−, and Opa1−/−Fgf21−/−. Data are mean ± SEM, n = 4, ∗∗p < 0.01. TA, tibialis anterior; GNM, gastrocnemius.
(D) Epididymal fat is higher in Opa1−/−Fgf21−/− than in Opa1−/− mice. Data are mean ± SEM (controls n = 4, Opa1−/− n = 8, Opa1−/−Fgf21−/− n = 4), ∗∗p < 0.01.
(E) Eight-month-old Opa1−/−Fgf21−/− show reduction of animal size (left panel) but do not show white hairs and kyphosis (right panel).
(F) Left panel: representative images of oil red O staining in liver cryosections. Right panel: Quantification of liver triglyceride content. Data are mean ± SEM, n = 4, ∗∗p < 0.01.
(G) Fasting blood glucose is restored to normal level in Opa1−/−Fgf21−/− mice. Data are mean ± SEM (Opa1f/f n = 4, Opa1−/− n = 8, Opa1−/−Fgf21−/− n = 4), ∗∗p < 0.01.
(H) Blood inflammatory cytokines are back to control level in Opa1−/−Fgf21−/− mice. Data are mean ± SEM, n = 4, ∗∗p < 0.01.
(I) Quantification of fibers number per muscle (left panel) and denervated NCAM-positive fibers (right panel) in Opa1−/−, Opa1−/−Fgf21−/−, and controls. Data are mean ± SEM, n = 5, ∗∗p < 0.01.
(J and K) Quantification of β-galactosidase and p21 levels. Data represent mean ± SEM, n = 4, ∗∗p < 0.01.
(L) Survival curve of control, Opa1−/−, and Opa1−/−Fgf21−/−mice. Data are mean ± SEM (controls n = 19, Opa1−/− n = 20, Opa1−/−Fgf21−/− n = 15), ∗∗p < 0.01.
(M) Regression analysis between age and FGF21 plasma levels of human subjects ranging from 19 to 103 years old. Data represent mean ± SEM, n = 180.
FGF21 Serum Level Increases with Aging in Humans
To corroborate our animal findings, we tested the blood levels of FGF21 in a human cohort ranging from 19 to 103 years old. A highly significant correlation between FGF21 serum levels and age was found (Figure 7M). Thus, muscle-derived FGF21 is an important player in regulation of whole-body metabolism and in senescence but is only partially involved in muscle loss where FoxOs are dominant.
Discussion
Here we have identified how OPA1 in muscles triggers a cascade of signaling that reverberates to the whole body, affecting general metabolism and aging. We have also translated our data to humans and shown that mitochondrial shaping proteins decline during aging in old sedentary people, both as transcripts and proteins. However, among the different fusion and fission genes, only OPA1 expression correlates with age-dependent muscle loss and weakness. Our data confirm previous observations that proteins of mitochondrial dynamics decrease with aging in sedentary rodents and humans (Ibebunjo et al., 2013, Joseph et al., 2012). Furthermore, we have also found that exercise can prevent this decline. Therefore, altogether these findings strongly suggest that a functional mitochondrial network is critical for muscle-mass maintenance and healthy aging. At the moment the question of how mitochondria affect aging and lifespan remains up for debate (Wang and Hekimi, 2015), as does how physical activity counteracts unhealthy aging (Neufer et al., 2015). The generation of inducible muscle-specific OPA1 and double FGF21/OPA1 KO mice enables us to shed a light on these issues. Indeed, we have identified the link that connects mitochondria to systemic metabolic changes, organ function, and longevity. The recent findings that muscle metabolism overcomes a mitochondrial problem in the heart and prevents the onset of dilated cardiomyopathy and heart failure (Wai et al., 2015) suggest that muscles may play a role in general metabolism and tissue function. This is an emerging concept that considers the metabolic adaptations occurring in skeletal muscles as disease modifier/controller (Baskin et al., 2015) and explains why physical activity elicits several beneficial effects in many different diseases. Indeed, exercise by preserving and ameliorating mitochondrial function and muscle metabolism affects the release of myokines and metabolites that systemically counteract organ deterioration. We identified FGF21 as one of these factors that, when chronically expressed at high levels in adulthood, greatly contributes to a pro-senescence metabolic shift. At the moment, whether FGF21 is beneficial or detrimental is debated, especially when related to heart function in humans (Chou et al., 2016, Li et al., 2016). Interestingly, high levels of serum FGF21 have been found in a child affected by progeria (Suomalainen et al., 2011), and circulating levels of FGF21 have been shown to increase with age in humans (Hanks et al., 2015). FGF21 is now recognized and used as a diagnostic marker in patients with mitochondrial myopathy (Lehtonen et al., 2016) who clearly are not longer lived. Importantly, while inhibition of FGF21 greatly ameliorates the precocious aging phenotype of OPA1 KO mice, it shows minor beneficial effects on muscle mass, suggesting that FGF21 mediates systemic metabolic changes while other signaling, e.g., FoxO, is responsible for protein breakdown and muscle loss. The marginal effect on muscle mass explains why FGF21 deletion in newborns did not rescue the lethal phenotype of conditional OPA1 KO mice that die as a result of breathing problems (data not shown). These data also support the concept that FGF21 needs the presence of other factors that synergize to trigger the precocious senescence phenotype of OPA1 KO mice. The finding that OPA1 deletion induces IL6 and IL1α expression in muscles and blood suggests that a concomitant pro-inflammatory status may be permissive for the pro-aging action of FGF21. Since IL6 is known to be detrimental for human health when chronically elevated, the concomitant increase of FGF21 and IL6 can explain the systemic effects on aging that were not reported when FGF21 is expressed from liver or adipose tissue. A further point is that the outcome can be dictated by plasma FGF21 levels. Interestingly, FGF21 blood levels detected in the inducible OPA1 model (Figure 5E) are superimposable with several values found in the blood of very old human subjects (Figure 7M). Indeed, our data in humans do find a highly significant correlation between FGF21 serum levels and age. Importantly, unhealthy aged people that suffer from sarcopenia/frailty syndrome are in a strong catabolic (fasting-like and FGF21-like) condition, with an important anabolic resistance that inhibits muscle recovery after an injury. All together, these observations sustain a detrimental action of muscle-derived FGF21 more than a beneficial effect.
The phenotype of the Opa1−/− described here does not completely match with those of other pro-fusion proteins or with constitutive deletion of the Opa1 gene. Total OPA1 KO mice are lethal, but heterozygous OPA1+/− are viable and have been used to study the role of this gene in exercise performance (Caffin et al., 2013). Hemizygous mice for OPA1 do have problem in training-induced mitochondrial biogenesis but, surprisingly, do not show any problem in physical activity. However, this model suffers from the fact that adaptations and compensations occur in muscle as well as in other tissues consequent to the constitutive reduction of OPA1 from zygote formation. Indeed, mitochondria morphology in these mice is not consistent with a fusion problem, since mitochondria are bigger than wild-type mice, and there are no abnormalities in cristae shape, supporting the concept that something happened during embryological/neonatal development to compensate for the 50% reduction of OPA1 protein (Caffin et al., 2013). More interesting are the data from conditional KO of mitofusin genes. A reduced muscle mass has been also reported in mice ablated for both Mfn1 and Mfn2, although analyses of muscle stem cell and of protein synthesis were not shown (Chen et al., 2010). Importantly, the muscle-specific deletion of Mfn1/Mfn2 genes was performed by using the same driver that we used here for conditional OPA1 ablation, but only some phenotypes appear to be in common. The similarities include a defect of muscle growth, the presence of low blood glucose levels at basal condition, and a reduced mitochondrial function. However, while Chen and colleagues interpret this as a consequence of depleted mtDNA, we observed it only following prolonged fusion inhibition in the model of inducible OPA1 ablation, whereas the impairment in cristae shape and mitochondrial supercomplex assembly was conversely an early consequence and could explain the observed reduced mitochondrial function. We believe that our data corroborate a model in which mtDNA depletion is a consequence of chronic fusion inhibition (Cogliati et al., 2013). Our findings also place autophagic removal of the small abnormal mitochondria in the pathway leading to reduced mtDNA content following fusion ablation, thereby dissociating the myopathic phenotype from mtDNA decrease and suggesting that mitochondrial dysfunction can be amplified by mitochondrial depletion.
In conclusion, our findings point to mitochondria dynamics in muscle as a signaling hub that, by modulating nuclear gene expression programs, not only controls muscle mass but systemically reverberates, affecting lipid homeostasis, inflammation, and the senescence of different tissues.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse anti-Bromodeoxyuridine | Roche | 11170376001 |
| mouse anti-Pax 7 | Hybridoma Bank | Pax7 |
| rabbit anti-phospho-Akt (Ser473) | Epitomics | EP2109Y |
| rabbit anti-total Akt | Cell Signaling | #9272 |
| rabbit anti-phospho-S6 | Cell Signaling | #2215 |
| rabbit anti-total S6 | Cell Signaling | #2217 |
| rabbit anti-LC3 B | Sigma | L7543 |
| rabbit anti-P62 | Sigma | P0067 |
| rabbit anti-phospho-AMPKα (Thr172) | Cell Signaling | #2531 |
| rabbit anti-total AMPKα | Cell Signaling | #2532 |
| rabbit anti-phospho-4EBP1 (Ser65) | Cell Signaling | #9451 |
| rabbit anti-phospho-4EBP1 (Thr37/46) | Cell Signaling | #9459 |
| rabbit anti-total 4EBP1 | Cell Signaling | #9452 |
| rabbit anti-phospho-ACC | Cell Signaling | #3661 |
| rabbit anti-phospho-mTOR | Cell Signaling | #5536 |
| rabbit anti-mTOR | Cell Signaling | #2983 |
| mouse anti-GAPDH | Abcam | ab8245 |
| mouse anti-puromycin | Hybridoma Bank | PMY-2A4 |
| mouse anti-ubiquitinylated proteins (FK2) | Millipore | #04-263 |
| rabbit anti-ubiquitin,lys63-specific (clone Apu3) | Millipore | #05-1308 |
| rabbit anti-MyoD | Santa Cruz | sc-32758 |
| mouse anti-MyoG | Millipore | MAB3876 |
| mouse anti-Lamin A/C (N-18) | Santa Cruz | sc-6215 |
| rabbit anti-phospho-eIF2α | Abcam | ab 32157 |
| rabbit anti-eIF2α | Cell Signaling | #9722 |
| rabbit anti-BIP/GRP78 | BD | 610979 |
| mouse anti-OPA1 | BD | 612606 |
| mouse anti-Drp1 | BD | 611738 |
| rabbit anti-TOM20 | Santa Cruz | Sc-11415 |
| mouse anti-Bnip3 | Sigma | B7931 |
| mouse anti-complexII | Invitrogen | 459200 |
| rabbit anti-phospho-Smad3 | Epitomics | Ep #1880-1 |
| rabbit anti-phospho-smad1/5/8 | Cell Signaling | #9511 |
| mouse anti-Bnip3 | Sigma | B7931 |
| mouse anti-complexII | Invitrogen | 459200 |
| rabbit anti-phospho-Smad3 | Epitomics | Ep #1880-1 |
| rabbit anti-phospho-smad1/5/8 | Cell Signaling | #9511 |
| mouse MFN1 | Abcam | Ab57602 |
| mouse MFN2 | Abcam | Ab56889 |
| Rabbit ACC | Cell Signaling | 3662S |
| mouse NDUF 88 | Abcam | ab110242 |
| mouse CORE2 | Abcam | ab14745 |
| goat anti-mouse Cy3 | Jackson Laboratory | 115-165-003 |
| goat anti mouse IgG | Bio-Rad | 1706516 |
| goat anti rabbit IgG | Bio-Rad | 1706515 |
| Biological Samples | ||
| Human biopsies | EK 07-057-0407; EK 08-102-0608 | |
| Human plasma | EU FP6 Project “GeHA”; EU FP7 Project “MYOAGE” | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen food | Harlan | Td55125 |
| Tudca | Tokyo Chemical Industry | Cas 302-95-4 |
| Trolox | Sigma-Aldrich | Cas 53188-07-1 |
| Mito tempo | Enzolife Science | ALX-400-150-M005 |
| 5-Bromo-20 -deoxyuridine | Sigma-Aldrich | Cat#B9285 |
| Protease Inhibitor | Sigma-Aldrich | Cat#P8340 |
| Phosphatase Inhibitor Cocktail 2 | Sigma-Aldrich | Cat#P5276 |
| Phosphatase Inhibitor Cocktail 3 | Sigma-Aldrich | Cat#P0044 |
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat#23225 |
| SYBR Green Supermix | Bio-Rad | Cat#1725274 |
| Colchicine | Sigma-Aldrich | 09754 |
| Critical Commercial Assays | ||
| Mouse IL6 ELISA Kit | MERCK Millipore | EZMIL6 |
| Mouse TNFα ELISA Kit | MERCK Millipore | EZMTNFA |
| Mouse IL1a ELISA Kit | Thermo Fisher | EMIL1A |
| Mouse IL1b ELISA Kit | Thermo Fisher | EMIL1B |
| Rat/Mouse FGF-21 ELISA Kit | MERCK Millipore | EZRMFGF21-26K |
| Human FGF-21 ELISA Set (10 × 96 T) | Aviscera Bioscience | SK00145-10 |
| Coating buffer, blocking buffer, concentrated diluition buffer, antibody diluent solution, HRP diluent solution | Aviscera Bioscience | CS01, BS-01, BD01, BD18, BD06 |
| 2-Deoxyglucose (2DG) Uptake Measurement Kit | Cosmo Bio | CSR-OKP-PMG-K01TE |
| Experimental Models: Organisms/Strains | ||
| MLC1f-Cre-Opa1f/f /BL6 | Sandri Lab | N/A |
| HSA-Cre-ER-Opa1f/f /BL6 | Sandri Lab | N/A |
| HSA-Cre-ER-Opa1f/f –Fgf21f/f /BL6 | Sandri Lab | N/A |
| Oligonucleotides | ||
| Primers for qPCR, see Table S3 | ||
| Recombinant DNA | ||
| Mt-roGPF1: ro1 with mitochondrial targeting sequence in pEGFP-N1, pRA306 | S.J. Remington (University of Oregon) | N/A |
| pSUPER FoxO1/3 | Mammucari et al., 2007 | N/A |
| Software and Algorithms | ||
| ImageJ Software | N/A | |
| Prism 7 software (GraphPad software) | N/A | |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marco Sandri (marco.sandri@unipd.it).
Experimental Model and Subject Details
Human subjects
All participants were healthy volunteers and declared not to have any specific physical/disease issues. The specific exclusion criteria were body mass index ≥ 40kg/m2; previous knee replacement; recent deep vein thrombosis or any infection; myopathy; neurologic, pulmonary, or symptomatic cardiovascular diseases; vertigo or impaired cognitive function; recent or past cancer; rheumatism; or any other relevant limitations of the musculoskeletal system. All participants were informed about the testing procedures and the possible risks, and written informed consent had to be signed prior to inclusion in this study. In addition, ethical approval was obtained from the ethical commission of the municipality of Vienna (EK 07-057-0407) and (EK08-102-0608).
Nutritional status before skeletal muscle biopsy consisted in daily routine. Muscle biopsy collection was performed quite at the same time in the morning between 10.00 and 11.00 from the Vastus Lateralis of three different groups of male subjects as described (Zampieri et al., 2015): Young subjects (23 to 34 years of age) performing moderate training activities; sedentary seniors (53 to 82 years of age), either inactive healthy subjects performing only routine daily activities or seniors with knee arthropathy, and senior sportsmen: (66 to 72 years of age) who routinely practiced (lifelong) sport activities more 3 to 5 times a week (one –to –two hours per session). 15 mg of muscle tissue were sufficient to perform both, proteins and RNA extraction.
Plasma Human samples
The human samples used in this study were obtained from volunteers of different ages, ranging from 19-103 years-old. recruited in the framework of the EU FP6 Project “GeHA – Genetics of Healthy Ageing”; the EU FP7 Project “MYOAGE – Understanding and combating age-related muscle weakness”; and the Italian National Project “PRIN2009CB4C9F - Ruolo della epigenetica e della genetica del DNA mitocondriale nella longevità: studi su soggetti con più di 105 anni di età (semi-supercentenari).”
The study protocols were approved by the local Ethical Committees responsible for the Bologna unit, respectively:
- for the GeHA study: Sant’Orsola-Malpighi University Hospital (Bologna), ethical clearance n. 118/2004/U/Tess issued on 20/07/2004;
- for the MYOAGE study: Istituto Ortopedico Rizzoli (Bologna) ethical clearance n. 10823 issued on 26/04/2010;
- for the PRIN study: Sant’Orsola-Malpighi University Hospital (Bologna) ethical clearance n. 22/2007/U/Tess issued on 27/02/2007, amendment n. EM 157/2011/U issued on 25/11/2011.
Inclusion/ Exclusion criteria
MYOAGE samples from 24-96 years-old subjects: Healthy subjects. Exclusion criteria were: to be non-autonomous in the dayly living activities, to be unable to walk a distance of 250 m, presence of morbidity (neurologic disorders, metabolic diseases, rheumatic diseases, recent malignancy, heart failure, severe chronic obstructive pulmonary disease (COPD), haemocoagulative syndromes), use of medication (immunosuppressive drugs, insulin), immobilization for 1 week during last 3 months, and orthopedic surgery during the last 2 years or still causing pain or functional limitation.
Hip replacement subjects: Subjects who underwent surgery for hip dysplasia. Age (> 20 years) and the ability to provide informed consent and to cooperate with the study personnel were inclusion criteria, while exclusion criteria were presence of diseases, i.e., chronic kidney and/or liver diseases, bleeding disorders, severe type 2 diabetes, rheumatic diseases, osteoarthritis, neuromuscular disorders, malignancies and systemic infections, other than chronic steroid use, major psychological problems or history of alcohol or drug abuse, evidence of prior surgery in the involved hip.
GEHA samples: Inclusion criteria of participants: i) they should be at least 90 years of age (a nonagenarian); ii) they should have a living sibling who also is a nonagenarian; iii) they should be able to understand the scope of the project.
PRIN2009CB4C9F samples: Inclusion criteria: 100+ years old subjects
For FGF21 detection the HUMAN FGF-21 IMMMUNOASSAY CAT NUM DF2100 (R&D SYSTEMS) kit was used. To obtain sufficient technical replicates for the Elisa assay, at least 300 ul of plasma samples were needed.
Mouse models
Animals were handled by specialized personnel under the control of inspectors of the Veterinary Service of the Local Sanitary Service (ASL 16 - Padova), the local officers of the Ministry of Health. All procedures are specified in the projects approved by the Italian Ministero Salute, Ufficio VI (authorization number 1060/2015 PR). Muscles were removed at various time periods and frozen in liquid nitrogen for subsequent analyses.
To generate constitutive muscle-specific OPA1 knockout (KO) animals, mice bearing Opa1 floxed alleles (Opa1f/f) (Cogliati et al., 2013) were crossed with transgenic mice expressing Cre under the control of a Myosin Light Chain 1 fast promoter (MLC1f-Cre) and kept in a homozygous mating system. Experiments were performed on newborns at post-natal day 7 or 8. Cre-negative littermates were used as controls.
A second KO model with inducible muscle-specific deletion of OPA1 was obtained by crossing the Opa1f/f line with mice carrying Cre-ER driven by human skeletal actin promoter (HSA). Tamoxifen-induced Cre LoxP recombination was activated by oral administration of tamoxifen-containing chow (Tam400/Cre ER Harlan) which was provided ad libitum for 5 weeks. A third KO model was obtained by crossing HSA- Opa1f/f line with Fgf21f/f. Tamoxifen-induced CreLoxP recombination was activated by oral administration of tamoxifen-containing chow (Tam400/Cre ER Harlan) which was given ad libitum for 5 weeks. Animals with food intake received indicatively, 1 mg of tamoxifen per day. Mice were housed in individual cages in an environmentally controlled room (23°C, 12-h light–dark cycle) with ad libitum access to food and water. All experiments were performed on 3- to 18-month-old male (17–32 g) and female mice (16–26 g). Mice of the same sex and age were used for each individual experiment. Muscles were collected at different times: 30, 50 or 120 days after the beginning of the tamoxifen diet. Cre-negative littermates, also receiving tamoxifen treatment were used as controls. Fgf21 floxed (Fgf21f/f) were purchased by The Jackson Laboratory and crossed with HSA- Opa1f/f Cre positive.
PCR genotyping was performed with the following primers:
OPA1 Fw: CAGTGTTGATGACAGCTCAG
OPA1 Rv: CATCACACACTAGCTTACATTTGC
FGF21 Fw: AGGAGGCTAGGGCTTGACTCT
FGF21 Rw: TGACAGGGTCTCAGGTTCAA
Cre Fw: CACCAGCCAGCTATCAACTCG
Cre Rv: TTACATTGGTCCAGCCACCAG
Method Details
Gene expression analyses
Total RNA was prepared from muscles using TRIzol (Invitrogen). Complementary DNA was generated from 0.4 μg of RNA reverse-transcribed with SuperScript III Reverse Transcriptase(Invitrogen). Duplicates of cDNA samples were then amplified on the 7900HT Fast Real-Time PCR System (Applied Biosystems) using the Power SYBR Green RT-PCR kit (Applied Biosystems). All data were normalized to GAPDH and to actin expression and plotted in arbitrary units as mean ± SEM. The oligonucleotide primers used are shown in table S3.
Immunoblotting
Muscles were lysed and immunoblotted as previously described (Mammucari et al., 2007). Blots were stripped using Restore western blotting stripping buffer (Pierce) according to the manufacturer’s instructions and probed again when necessary. List of antibodies is indicated in the key resources table
β-galactosidase assay
In vitro β-galactosidase measurement was performed on different tissues (liver, skin and gut). Lysates were prepared using T-PER Tissue Protein Extraction Reagent (cat n78510, Thermofisher) then Pierceβ-Galactosidase Assay Reagent (Product No. 75705) and β-Galactosidase Assay Stop Solution (Product No. 75706) to develop the reaction.
Imaging and Transmission Electron Microscopy
Cryosections of both P8 newborns hindlimb cross-sections and adult TA were stained for H&E and SDH. Total myofiber number was calculated from entire hindlimb cross-section based on assembled mosaic image (20X magnification). CSA was performed on adult TA as described (Mammucari et al., 2007). ATPase staining: Serial cryosections (8 μm) from human muscle biopsies were mounted on polysine glass slides, air-dried, and stained either with hematoxylin and eosin (H&E) or conventional techniques for myofibrillar ATPases to evaluate muscle fiber type. For ATPase stains, slow-type fibers are dark, whereas fast-type fibers are lightly stained following preincubation at pH 4.35. Fiber type grouping is identified on the basis that one myofiber is surrounded by fibers of the same phenotype. Morphometric analyses were performed on stained cryosections using Scion Image for Windows version Beta 4.0.2 (2000 Scion Corporation) (Zampieri et al., 2015). For immunostaining, we used antibodies specific for Pax7 (1:20, Hybridoma Bank), BrdU (1:40, Roche) and TOM20 (1:100, Santa Cruz). The secondary antibodies, goat anti-mouse Cy3 were obtained from Jackson lab. Hoechts co-staining, allowed to identify subsarcolemmal position of myonuclei.
For electron microscopy, we used conventional fixation-embedding procedures based on glutaraldehyde-osmium fixation and Epon embedding.
Measurement of MtDNA Copy Number
Total gastrocnemius DNA was isolated using Puregene Cell and Tissue Kit(QIAGEN) and was amplified using specific primers for mtCOXII and 18S by real-time PCR using the Power SYBR Green RT-PCR kit (Applied Biosystems). The mtDNA copy number was calculated using 18S amplification as a reference for nuclear DNA content.
Mitochondrial Assays
Muscle mitochondria from the indicated genotype were isolated as described (Frezza et al., 2007). To detect RCS, Blue Native PAGE was performed by resuspending mitochondrial pellets in Native Buffer (Invitrogen) plus 4% Digitonin (SIGMA) to a final concentration of 10 μg/μl and incubated for 1 hr on ice. After 20 min of centrifugation at 16,000 g, the supernatant was collected and one-third of digitonin percentage of Sample Buffer 5% G250 (Invitrogen) was added. Then 50 μg of mitochondrial membrane proteins was loaded and run on a 3%–12% Bis-Tris gel (Invitrogen) as described in the NativePAGE Novex® Bis-Tris Gel System manual. Western blot was performed using a Trans-Blot transfer cell (Bio-Rad) at 4°C O/N. List of antibodies is depicted in Table S2.
To measure respiration, mitochondria (1 mg/ml) were incubated in Experimental Buffer (EB: 150 mM KCl, 10 mM Tris Mops,10 mM EGTA-Tris, 10mM ATP). When indicated, mitochondria were transferred into a Clark’s type oxygen electrode chamber and 5mM glutamate/2.5 mM malate or 2 mM rotenone/10 mM succinate were added. Basal O2 consumption was recorded (state 2) and after 2 min 100 mMADP was added (state 3), followed by 2.5 mg/ml oligomycin (state 4) and 200 mMFCCP (state 3u). Assesment of mitochondrial respiratory chain enzymatic activities on muscles was determined as described (Spinazzi et al., 2012). Results were normalized to citrate synthase enzymatic activity.
Protein Carbonyls Detection
Carbonylation of muscle proteins were detected by using the OxyBlot Protein Oxidation Detection Kit (Millipore, s7150) (Masiero et al., 2009). Quantification analysis was performed with ImageJ Software and all values were normalized for the housekeeping GAPDH.
Torque measurement and testing
An isometric measurement on a dynamometer (S2P Ltd., Lubljana, Slovenia) with 90° hip flexion and 60° knee flexion (full knee extension = 0°) was performed as described (Šarabon et al., 2013a, Šarabon et al., 2013b) three times at each leg to assess the maximal isometric torque of the left and right knee extensors. The mean of the best values of each leg were taken for further analyses.
The time up and go test (TUGT) was performed to evaluate mobility and function in activities of daily living. Briefly, the subjects were asked to stand up from a standard chair, walk 3 m, turn around, walk back to the chair, and sit down again all as fast as possible.
VAS-48h (visual analog scale in rest and during activity) and WOMAC (Western Ontario and McMaster Universities Arthritis Index) questionnaire were performed to evaluate pain in the activities of daily living in the last 48h prior to the assessment.
Mouse Studies
Mitochondrial oxidative stress measurement
Mt-roGFP1, which measures the thiol/disulfide equilibrium in the mitochondrial matrix, was used as an indicator of mitochondrial redox status, as previously described (Lo Verso et al., 2014). Briefly, Adult FDB muscles were transfected by electroporation with mt-roGFP1 plasmid. After 7 d of transfection single muscle fibers were isolated from control and knock-out mice. Mt-roGFP1 fluorescence (excitation: 405 and 480 nm, emission: 535 nm, 20 × objective) was measured for 5 min every 10 s and upon H2O2. The ratio of fluorescence intensities (exc 405/480) was determined by ImageJ Software.
In vivo protein synthesis measurements
In vivo protein synthesis was measured by using the SUnSET technique (Goodman et al., 2011, Schmidt et al., 2009). Mice were anesthetized and then given an intraperitoneal injection of 0.040 μmol/g puromycin dissolved in 100 μL of PBS. At exactly 30 min after injection, muscles were collected and frozen in liquid N2 for WB analysis using a mouse IgG2a monoclonal anti-puromycin antibody (clone 12D10, 1:5000).
In vivo drug treatments
KO and control mice received TUDCA (Tokyo Tanabe, Tokyo, Japan) at a dose of 500 mg/kg twice a day (8:00 AM and 8:00 PM) by oral administration for 5 days or 2 weeks.
Mice were intraperitoneally injected daily for 5 days or 2 weeks with 30 mg/kg of TROLOX (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid).
Mito-TEMPO (Enzo Life Science, ALX-430-150-M005) at a dose of 1.4 mg/kg was administered through an intraperitoneal injection every day for 5 days or 2 weeks
Force measurements
In vivo force measurements were performed as described previously (Blaauw et al., 2009). Briefly, mice were anesthetized and stainless steel electrodes wires were placed on either side of the sciatic nerve. Torque production of the plantar flexors was measured using a muscle lever system (Model 305c; Aurora Scientific, Aurora ON, Canada). The force–frequency curves were determined by increasing the stimulation frequency in a stepwise manner, pausing for 30 s between stimuli to avoid effects due to fatigue. Force was normalized to the muscle mass as an estimate of specific force. Following force measurements, animals were sacrificed by cervical dislocation and muscles were dissected and weighed.
Single Fibers Isolation and Mitochondrial Membrane Potential analyses
Mitochondrial membrane potential was measured in isolated transfected fibers from flexor digitorum brevis (FDB) muscles as previously described (Mammucari et al., 2007, Zhao et al., 2007). Briefly, FDB myofibers were placed in 1 mL Tyrode’s buffer and loaded with 2.5 nM TMRM (Molecular Probes) supplemented with 1μM cyclosporine H (a P-glycoprotein inhibitor) for 30 min at 37°C. Myofibers were then observed at Olympus IMT-2 inverted microscope (Melville, NY) equipped with a CellR imaging system. Sequential images of TMRM fluorescence were acquired every 60 s with a 20X 0.5, UPLANSL N A objective (Olympus). At the times indicated by arrows, oligomycin (Olm, 5 μM) (Sigma) or the protonophore carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP, 4 μM) (Sigma) were added to the cell culture medium. Images were acquired, stored and analysis of TMRM fluorescence over mitochondrial regions of interest was performed using ImageJ software (https://imagej.nih.gov/ij/). When stated, isolated FDB fibers were stained with a rabbit polyclonal anti-TOM20 (Santa Cruz 1:200) and were further analyzed by confocal microscopy.
Plasma measurements
Plasma was obtained from blood collected from controls and OPA1 null mice. Blood FGF21 levels were determined using Rat/Mouse FGF21 Enzime-linked Immunosorbent ELISA-Kit (Millipore, EZRMFGF21-26K). OPA1 KO mice FGF21 relative quantification was normalized to controls. Data are expressed as fold increase of controls. Blood IL-6 (Millipore, EZMIL6), TNF-α (Millipore, EZMTNFA), IL-1α (Thermofisher, EMIL1A) IL-1β (Thermofisher, EM2IL1B) were measured following the manufacturer’s instructions. Data are expressed in pg/mL. Glucose measurement was performed at 9 a.m. in fed condition, with the One Touch Ultra Easy glucose meter (LifeScan Inc.). Data are expressed in mg/dL.
Exercise experiments
In this study 18 months-old mice performed concentric exercise on a treadmill (Biological Instruments, LE 8710 Panlab Technology 2B), with 10° incline. Mice were trained for one week, 1 hr per day at constant speed of 18 cm/s
Glucose and insulin tolerance tests
Mice were fasted for 8 hr before analysis during their normal rest/fasting phase. Blood glucose was measured using a Glucocard G+ meter (Arkray Factory, Shiga, Japan). Glucose tolerance tests (GTT) and insulin tolerance tests (ITT) were performed at ZT12, For GTT, mice were injected i.p. with glucose (2 g/kg body weight). Blood glucose was measured at 0, 10, 20, 30, 60 and 120 min via the tail vein. For ITT, mice were injected i.p. with human insulin (0.75 U/kg body weight; Sigma) and blood glucose was measured at 10, 0, 10, 20, 30, 60, and 90 min via the tail vein.
Autophagic flux quantification
Autophagic flux was monitored in fed condition using colchicine (C9754, Sigma-Aldrich) as previously described (Milan et al., 2015). Briefly, HSA OPA1−/− and OPA1f/f mice were treated, by i.p. injection, with vehicle or with 0,4mg/ kg colchicine. The treatment was administered twice, at 24 hr and at 12 hr before muscle collection.
In vivo Glucose uptake
Glucose uptake was measured using a non-radioactive colorimetric method (Saito K and Minokoshi Y et al. Analytical Biochem 412: 9-17, 2011), by means of the 2-deoxyglucose uptake measurement kit (Cosmo Bio Co., LTD, Tokyo, Japan). Mice were fasted 5 hr before the beginning of the experiment. Mice were then injected intraperitoneally with a solution containing D-glucose (1 g/Kg) and 2-deoxy-D-glucose (0.027 g/Kg). One hour after injection, mice were killed by cervical dislocation, and tissues (soleus, tibialis anterior, liver, epidydimal white adipose tissue) were rapidly removed, weighed and placed in ice cold Tris-HCl (10mM, pH8) buffer. Muscles from left and right legs were separately processed. Entire soleus and tibialis anterior muscles, 50-100 mg of liver and 100-200 mg of adipose tissue were used for the assay. Tissues were subsequently homogenized in 1ml of Tris-HCl (10mM, pH8) with TissueLyser II (QIAGEN), by shaking at maximum speed for 3 min, and heated at 95°C for 15 min. Samples were then centrifuged at 16000 g for 15 min at 4°C to remove tissue debris. Supernatants were finally collected, and a portion was diluted in 20μl (final volume; 2μl for soleus, 5μl for tibialis anterior, 1μl for liver and 10μl for WAT) with the sample dilution buffer provided with the kit. The assay was then performed following manufacturer’s instructions.
Quantification and Statistical Analysis
Statistical analysis
All data are expressed as means ± SEM. Statistical analysis was performed using one-tailed or two-tailed Student’s t test. Normal distribution of the variables of interest was checked using the Shapiro-Wilk test. For experiments in which more than two groups should be compared, 2-way analysis of variant (ANOVA) was used. When ANOVA revealed significant differences, further analysis was performed using Bonferroni’s multiple comparison test. Differences between groups were considered statistically significant for p ≤ 0.05 or p ≤ 0.01. Linear regression models were used to verify the linear correlation between variables. t test were performed on the slopes of the regressions to verify their significance (GraphPad). Statistical significance was set at p < 0.05 were considered statistically significant for p ≤ 0.05 or p ≤ 0.01. Regression linear analysis was done using an F test (GraphPad). n, mean and SEM are reported within the figure legends.
Author Contributions
C.T. and V.R. generated the inducible and muscle-specific knockout mice, performed experiments, and analyzed and interpreted data. C.T., V.R., L. Salviati, L. Scorrano, and M.S. designed experiments, discussed data, and wrote the manuscript. M.A.D., M.A., G.F., S.C., M.E.S., V.M., C.C., M.C., B.B., and S.Z. contributed to experiments and data collection. G.P.F., S.L., H.K., C.F., S.S., and L. Salviati provided reagents and materials and discussed data. M.S. and L. Scorrano supervised the work.
Acknowledgments
This work was supported by CARIPARO Foundation 2010 Excellence Project to M.S. and to L. Scorrano; by ERC (282310-MyoPHAGY), AFM-Telethon (19524), Italian Ministry of Education (MiUR) (PRIN 2010/2011), Foundation Leducq, AIRC (17388), and RISE (645648) to M.S.; by CARIPARO Foundation Starting Grant to V.R. and M.S.; by Telethon Italy (GPP10005, GGP12162, GGP14187), ERC (FP7-282280), EU FP7 (CIG PCIG13-GA-2013-618697), and MiUR (FIRB AUTOMED RBAP11Z3YA_005) to L. Scorrano; and by Telethon Italy (GGP13222, GGP14187) and Fondazione CARIPARO to L. Salviati. We acknowledge Prof. Chiara Romualdi (Department of Biology, University of Padua) for help with statistical analyses.
Published: May 25, 2017
Footnotes
Supplemental Information includes seven figures and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2017.04.021.
Contributor Information
Luca Scorrano, Email: luca.scorrano@unipd.it.
Marco Sandri, Email: marco.sandri@unipd.it.
Supplemental Information
References
- Alexander C., Votruba M., Pesch U.E., Thiselton D.L., Mayer S., Moore A., Rodriguez M., Kellner U., Leo-Kottler B., Auburger G. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- Amati-Bonneau P., Valentino M.L., Reynier P., Gallardo M.E., Bornstein B., Boissière A., Campos Y., Rivera H., de la Aleja J.G., Carroccia R. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain. 2008;131:338–351. doi: 10.1093/brain/awm298. [DOI] [PubMed] [Google Scholar]
- Baskin K.K., Winders B.R., Olson E.N. Muscle as a “mediator” of systemic metabolism. Cell Metab. 2015;21:237–248. doi: 10.1016/j.cmet.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaauw B., Canato M., Agatea L., Toniolo L., Mammucari C., Masiero E., Abraham R., Sandri M., Schiaffino S., Reggiani C. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J. 2009;23:3896–3905. doi: 10.1096/fj.09-131870. [DOI] [PubMed] [Google Scholar]
- Caffin F., Prola A., Piquereau J., Novotova M., David D.J., Garnier A., Fortin D., Alavi M.V., Veksler V., Ventura-Clapier R., Joubert F. Altered skeletal muscle mitochondrial biogenesis but improved endurance capacity in trained OPA1-deficient mice. J. Physiol. 2013;591:6017–6037. doi: 10.1113/jphysiol.2013.263079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Detmer S.A., Ewald A.J., Griffin E.E., Fraser S.E., Chan D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Vermulst M., Wang Y.E., Chomyn A., Prolla T.A., McCaffery J.M., Chan D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou R.H., Huang P.H., Hsu C.Y., Chang C.C., Leu H.B., Huang C.C., Chen J.W., Lin S.J. Circulating Fibroblast Growth Factor 21 is Associated with Diastolic Dysfunction in Heart Failure Patients with Preserved Ejection Fraction. Sci. Rep. 2016;6:33953. doi: 10.1038/srep33953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S., Rudka T., Hartmann D., Costa V., Serneels L., Craessaerts K., Metzger K., Frezza C., Annaert W., D’Adamio L. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163–175. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Cogliati S., Frezza C., Soriano M.E., Varanita T., Quintana-Cabrera R., Corrado M., Cipolat S., Costa V., Casarin A., Gomes L.C. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies V.J., Hollins A.J., Piechota M.J., Yip W., Davies J.R., White K.E., Nicols P.P., Boulton M.E., Votruba M. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 2007;16:1307–1318. doi: 10.1093/hmg/ddm079. [DOI] [PubMed] [Google Scholar]
- Delettre C., Lenaers G., Griffoin J.M., Gigarel N., Lorenzo C., Belenguer P., Pelloquin L., Grosgeorge J., Turc-Carel C., Perret E. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- Demontis F., Perrimon N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell. 2010;143:813–825. doi: 10.1016/j.cell.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis F., Piccirillo R., Goldberg A.L., Perrimon N. The influence of skeletal muscle on systemic aging and lifespan. Aging Cell. 2013;12:943–949. doi: 10.1111/acel.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis F., Patel V.K., Swindell W.R., Perrimon N. Intertissue control of the nucleolus via a myokine-dependent longevity pathway. Cell Rep. 2014;7:1481–1494. doi: 10.1016/j.celrep.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C., Cipolat S., Martins de Brito O., Micaroni M., Beznoussenko G.V., Rudka T., Bartoli D., Polishuck R.S., Danial N.N., De Strooper B., Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Frezza C., Cipolat S., Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- Goodman C.A., Mabrey D.M., Frey J.W., Miu M.H., Schmidt E.K., Pierre P., Hornberger T.A. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J. 2011;25:1028–1039. doi: 10.1096/fj.10-168799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumati P., Coletto L., Sabatelli P., Cescon M., Angelin A., Bertaggia E., Blaauw B., Urciuolo A., Tiepolo T., Merlini L. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 2010;16:1313–1320. doi: 10.1038/nm.2247. [DOI] [PubMed] [Google Scholar]
- Hanks L.J., Gutiérrez O.M., Bamman M.M., Ashraf A., McCormick K.L., Casazza K. Circulating levels of fibroblast growth factor-21 increase with age independently of body composition indices among healthy individuals. J. Clin. Transl. Endocrinol. 2015;2:77–82. doi: 10.1016/j.jcte.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibebunjo C., Chick J.M., Kendall T., Eash J.K., Li C., Zhang Y., Vickers C., Wu Z., Clarke B.A., Shi J. Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol. Cell. Biol. 2013;33:194–212. doi: 10.1128/MCB.01036-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph A.M., Adhihetty P.J., Buford T.W., Wohlgemuth S.E., Lees H.A., Nguyen L.M., Aranda J.M., Sandesara B.D., Pahor M., Manini T.M. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell. 2012;11:801–809. doi: 10.1111/j.1474-9726.2012.00844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtonen J.M., Forsström S., Bottani E., Viscomi C., Baris O.R., Isoniemi H., Höckerstedt K., Österlund P., Hurme M., Jylhävä J. FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology. 2016;87:2290–2299. doi: 10.1212/WNL.0000000000003374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Zhang Y., Ding D., Yang Y., Chen Q., Su D., Chen X., Yang W., Qiu J., Ling W. Association Between Serum Fibroblast Growth Factor 21 and Mortality Among Patients With Coronary Artery Disease. J. Clin. Endocrinol. Metab. 2016;101:4886–4894. doi: 10.1210/jc.2016-2308. [DOI] [PubMed] [Google Scholar]
- Lo Verso F., Carnio S., Vainshtein A., Sandri M. Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy. 2014;10:1883–1894. doi: 10.4161/auto.32154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C., Milan G., Romanello V., Masiero E., Rudolf R., Del Piccolo P., Burden S.J., Di Lisi R., Sandri C., Zhao J. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Masiero E., Agatea L., Mammucari C., Blaauw B., Loro E., Komatsu M., Metzger D., Reggiani C., Schiaffino S., Sandri M. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–515. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Milan G., Romanello V., Pescatore F., Armani A., Paik J.H., Frasson L., Seydel A., Zhao J., Abraham R., Goldberg A.L. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015;6:6670. doi: 10.1038/ncomms7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufer P.D., Bamman M.M., Muoio D.M., Bouchard C., Cooper D.M., Goodpaster B.H., Booth F.W., Kohrt W.M., Gerszten R.E., Mattson M.P. Understanding the Cellular and Molecular Mechanisms of Physical Activity-Induced Health Benefits. Cell Metab. 2015;22:4–11. doi: 10.1016/j.cmet.2015.05.011. [DOI] [PubMed] [Google Scholar]
- Romanello V., Sandri M. Mitochondrial Quality Control and Muscle Mass Maintenance. Front. Physiol. 2016;6:422. doi: 10.3389/fphys.2015.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šarabon N., Loefler S., Cvecka J., Sedliak M., Kern H. Strength training in elderly people improves static balance: a randomized controlled trial. Eur. J. Transl. Myol. 2013;23:85–89. [Google Scholar]
- Šarabon N., Rošker J., Fruhmann H., Burggraf S., Loefler S., Kern H. Reliability of maximal voluntary contraction related parameters measured by a portable isometric knee dynamometer. Phys. Med. Rehabil. Kuror. 2013;23:22–27. [Google Scholar]
- Schaaf C.P., Blazo M., Lewis R.A., Tonini R.E., Takei H., Wang J., Wong L.J., Scaglia F. Early-onset severe neuromuscular phenotype associated with compound heterozygosity for OPA1 mutations. Mol. Genet. Metab. 2011;103:383–387. doi: 10.1016/j.ymgme.2011.04.018. [DOI] [PubMed] [Google Scholar]
- Schiaffino S., Dyar K.A., Ciciliot S., Blaauw B., Sandri M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013;280:4294–4314. doi: 10.1111/febs.12253. [DOI] [PubMed] [Google Scholar]
- Schmidt E.K., Clavarino G., Ceppi M., Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods. 2009;6:275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- Spiegel R., Saada A., Flannery P.J., Burté F., Soiferman D., Khayat M., Eisner V., Vladovski E., Taylor R.W., Bindoff L.A. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J. Med. Genet. 2016;53:127–131. doi: 10.1136/jmedgenet-2015-103361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinazzi M., Casarin A., Pertegato V., Salviati L., Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012;7:1235–1246. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- Suomalainen A., Elo J.M., Pietiläinen K.H., Hakonen A.H., Sevastianova K., Korpela M., Isohanni P., Marjavaara S.K., Tyni T., Kiuru-Enari S. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol. 2011;10:806–818. doi: 10.1016/S1474-4422(11)70155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T., García-Prieto J., Baker M.J., Merkwirth C., Benit P., Rustin P., Rupérez F.J., Barbas C., Ibañez B., Langer T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science. 2015;350:aad0116. doi: 10.1126/science.aad0116. [DOI] [PubMed] [Google Scholar]
- Wang Y., Hekimi S. Mitochondrial dysfunction and longevity in animals: Untangling the knot. Science. 2015;350:1204–1207. doi: 10.1126/science.aac4357. [DOI] [PubMed] [Google Scholar]
- Zampieri S., Pietrangelo L., Loefler S., Fruhmann H., Vogelauer M., Burggraf S., Pond A., Grim-Stieger M., Cvecka J., Sedliak M. Lifelong physical exercise delays age-associated skeletal muscle decline. J Gerontol A Biol Sci Med Sci. 2015;70:163–173. doi: 10.1093/gerona/glu006. [DOI] [PubMed] [Google Scholar]
- Zhao J., Brault J.J., Schild A., Cao P., Sandri M., Schiaffino S., Lecker S.H., Goldberg A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Züchner S., Mersiyanova I.V., Muglia M., Bissar-Tadmouri N., Rochelle J., Dadali E.L., Zappia M., Nelis E., Patitucci A., Senderek J. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.