

Abstract
Despite a key role of amyloid-beta (Aβ) in Alzheimer’s disease (AD), mechanisms that link Aβ plaques to tau neurofibrillary tangles and cognitive decline still remain poorly understood. The purpose of this study was to quantify proteins in the sarkosyl-insoluble brain proteome correlated with Aβ and tau insolubility in the asymptomatic phase of AD (AsymAD) and through mild cognitive impairment (MCI) and symptomatic AD. Employing label-free mass spectrometry based proteomics, we quantified 2,711 sarkosyl-insoluble proteins across the prefrontal cortex from 35 individual cases representing control, AsymAD, MCI and AD. Significant enrichment of Aβ and tau in AD was observed, which correlated with neuropathological measurements of plaque and tau tangle density, respectively. Pairwise correlation coefficients were also determined for all quantified proteins to Aβ and tau, across the 35 cases. Notably, 6 of the 10 most correlated proteins to Aβ were U1 small nuclear ribonucleoproteins (U1 snRNPs). Three of these U1 snRNPs (U1A, SmD and U1-70K) also correlated with tau consistent with their association with tangle pathology in AD. Thus, proteins that cross-correlate with both Aβ and tau, including specific U1 snRNPs, may have potential mechanistic roles in linking Aβ plaques to tau tangle pathology during AD progression.
Keywords: mass spectrometry, aggregation, proteostasis, spliceosome, neurodegeneration
1. Introduction
Protein accumulation and aggregation in the brain is one of the major pathologic hallmarks of neurodegenerative diseases [1, 2]. A characteristic of these proteins is their spontaneous conversion from normal functional soluble proteins to pathologic, detergent-insoluble aggregates. These aggregates cause both functional deficits as well as gain-of-function toxicity leading to neurodegeneration [2]. Alzheimer’s disease (AD) is characterized by extracellular deposition of detergent-insoluble β-amyloid (Aβ) as senile plaques, as well as intracellular accumulation of neurofibrillary tangles (NFTs) composed of aggregated tau [3, 4]. Increased deposition of Aβ is thought to precede tangle formation, whereas tau pathology is more closely related to neuronal death and cognitive decline [3–5]. Converging biomarker evidence indicates that the progression of AD is characterized by an extended preclinical phase in which Aβ pathology accumulates years in advance of cognitive symptoms [6]. This has led to the recognition of an asymptomatic phase of AD (AsymAD), whereby individuals exhibit brain Aβ pathology in the absence of cognitive impairment. Thus, while Aβ is proposed to have a central role in disease initiation [7], plaque accumulation is not sufficient to incite tau aggregation and subsequent neurodegeneration. Currently the key protein aggregation events that co-occur with Aβ and underlie conversion from AsymAD to mild cognitive impairment (MCI) and eventually AD are not well established. Defining these changes may aid in our understanding of the link between Aβ deposition, disease-associated aggregation of tau, and ultimately cognitive decline.
The purpose of our study was to map the changes in the aggregated proteome occurring in AsymAD and through the transition to MCI and symptomatic stages of AD. To achieve this goal, we used well characterized postmortem brain tissues derived from the Baltimore Longitudinal Study of Aging (BLSA) [8]. The longitudinal design of this study enabled determination of cognitive status associated with the clinical progression of AD proximate to death. Employing label-free mass spectrometry-based proteomics, we identified and quantified 2,711 detergent-insoluble proteins across the dorsolateral prefrontal cortex from 35 individual brain tissues from control, AsymAD, MCI and AD cases. This revealed significant enrichment of detergent insoluble Aβ and tau, which correlated with neuropathological measurements of plaque and tangle density, respectively. We also confirmed apolipoprotein E (APOE), U1-70K (SNRNP70) and other core U1 small nuclear ribonucleoproteins (snRNPs) as significantly enriched in the sarkosyl-insoluble fractions in AD consistent with previously studies [9, 10]. To examine protein co-aggregation events linked AD pathology pairwise correlation coefficients were also determined for each quantified protein to Aβ and tau, across the 35 individual cases. Notably, 6 of the 10 most correlated proteins to Aβ were U1 small nuclear ribonucleoproteins (U1 snRNPs) even in the asymptomatic phase of AD. Three of these U1 snRNPs (U1A, SmD and U1-70K), also strongly correlated with tau insolubility consistent with their association with tangle pathology in AD [9, 10]. Additional proteins that cross-correlate with both insoluble Aβ and tau included, among others, serum amyloid protein P (APCS) and APOE, which are known to associate with AD pathology in brain. Thus, proteins that cross-correlate with both Aβ and tau identified in this study, including specific U1 snRNPs, may have potential mechanistic roles in linking Aβ plaques to tau tangle pathology during AD progression.
2. Materials and Methods
2.1 Clinical and neuropathological characteristics of cases
All brain tissue used in this analysis was derived from the autopsy collection of the Baltimore Longitudinal Study of Aging (BLSA). Postmortem neuropathological evaluation of amyloid plaque distribution was performed according to the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria [11], while extent of spread of neurofibrillary tangle pathology was assessed in accordance with the Braak staging system [12]. Thirty-five cases were selected for proteomic analysis and sorted into the following four groups: i) cognitively intact individuals without AD pathology (controls), ii) cognitively intact individuals with AD pathology (AsymAD), iii) symptomatic individuals with mild cognitive impairment and AD pathology and iv) symptomatic individuals with severe dementia and AD pathology. The inclusion criteria for each cohort are outlined in Table S1.
2.2 Enrichment of Detergent Insoluble Proteins
All tissue samples were derived from the middle frontal gyrus, corresponding to Brodmann areas 8 and 9, because this region demonstrates cortical thinning during preclinical AD and its CERAD scores tend to mirror the brain as a whole [13]. The enrichment strategy for detergent-resistant fractions was performed as previously described [14]. In brief, frozen post-mortem tissue (200 ± 20 mg) was dounce homogenized in 5 ml/g (20% w/v) of ice-cold homogenization buffer (50 mM HEPES pH 7.0, 250 mM sucrose, 1 mM EDTA and 1× HALT (Pierce) protease inhibitor cocktail). Following homogenization, sarkosyl (N-lauroylsarcosine) and NaCl were added to final concentrations of 1% (w/v) and 0.5 M, respectively (sarkosyl-buffer). This fraction was defined as the total brain homogenate and was sonicated (Sonic Dismembrator System, Fisher Scientific) with 3 × 5 sec pulses at 30% amplitude using a microtip probe to shear nucleic acids. Protein concentrations were determined using the bicinchoninic acid (BCA) method (Pierce). To generate sarkosyl-insoluble fractions, total brain homogenates (5 mg total protein per case) were centrifuged at 180,000 × g for 30 min at 4 °C. For each case, the supernatant (S1) was retained as the sarkosyl-soluble fractions. The pellet (P1) was re-suspended in sarkosyl-buffer and centrifuged at 180,000 × g for an additional 30 min. The final pellet (P2) was solubilized in urea buffer (8M urea and 2% SDS in 50 mM Tris-HCl pH 8.5) to yield the sarkosyl-insoluble fraction.
2.3 LC-MS/MS analysis and label-free quantification
LC-MS/MS analysis and label free quantification were performed essentially as described [15, 16]. Detergent insoluble fraction (20 μg) of each case was reduced with 5 mM dithiothreitol (DTT) for 15 minutes at 37°C and then alkylated with 20 mM iodoacetamide (IAA) for 30 minutes at 37°C in the dark [17]. The alkylated samples were separated on a 10% SDS gel and stained with Coomassie Blue G-250. Each sample lane was cut into five gel bands corresponding to molecular weight ranges to increase the depth of coverage of the proteome. The gel pieces were then digested overnight with trypsin (12.5 μg/ml) at 37°C. Samples were extracted in a solution of 5% formic acid and 50% acetonitrile (ACN) and following speed-vacuum evaporation an equal amount of each peptide sample was resuspended in loading buffer (0.1% formic acid, 0.03% trifluoroacetic acid, and 1% acetonitrile). The resulting peptides were loaded onto a 20 cm nano-LC column (internal diameter 100 μm) packed with Reprosil-Pur 120 C18-AQ 1.9 μm beads (Dr. Maisch, GmbH) and eluted over a 1 h reverse phase gradient comprised of 4–80% buffer B (Buffer A: 0.1% formic acid, 1% acetonitrile in water; Buffer B: 0.1% formic acid in acetonitrile) generated by a NanoAcquity UPLC system (Waters Corporation). Peptides were ionized with electrospray ionization (2.0 kV) and detected on a hybrid LTQ XL Orbitrap mass spectrometer (Thermo). MS1 spectra (30,000 resolution) were collected in the Orbitrap and data dependent acquisition of MS/MS spectra were obtained in the LTQ by collision induced dissociation (CID). The SageN Sorcerer SEQUEST 3.5 algorithm was used to search and match MS/MS spectra to a complete semi-tryptic human proteome database (NCBI reference sequence revision 54, with 66,652 entries) including pseudo-reversed decoy sequences [18, 19]. Searching parameters included precursor ion mass tolerance (±20 ppm), partial tryptic restriction, fixed mass shift for modification of carbamidomethylated Cys (+57.0215 Da) and dynamic mass shift for oxidized Met (+15.9949). Only b and y ions were considered during the database match. In addition, Xcorr and ΔCn were dynamically increased for groups of peptides organized by a combination of trypticity (fully or partial) and precursor ion charge state to remove false positive hits and decoys until achieving a false discovery rate (FDR) of < 1%. Protein quantification was performed based on the extracted ion current (XIC) measurements of identified peptides as previously reported [15, 20]. Ion intensities for identified peptides were extracted in full-MS survey scans of high-resolution and a ratio of the peak intensities for the peptide precursor ion was calculated using in-house software as previously published [10, 15, 16, 21, 22]. Accurate peptide mass and retention time (RT) was used to derive signal intensity for every peptide across LC-MS/MS runs for each case. For those proteins identified by ≥3 peptides, we averaged the extracted ion intensities for the three most intense tryptic peptides [23].
2.4 Data normalization and statistical and correlation analysis
Protein signal-to-noise (S/N) values for the 5 control cases analyzed in both SDS-PAGE gels was normalized to minimize variance and leverage technical replicate measurements. Briefly, for every protein, which had S/N measurements averaging ≥20, the log2(S/N) average within gel was divided by the log2(S/N) average across both gels to arrive at a gel-specific normalization factor. This within-gel normalization factor was considered as a protein-specific multiplier and applied to each measurement for all samples within that particular gel. This resulted in cross-gel normalized protein measurements. The five control replicates and the AsymAD replicate were averaged, respectively, for subsequent statistical analysis. Differentially enriched or depleted proteins in the insoluble proteome of AsymAD, MCI and AD were found by calculating Student’s t-test p values (unpaired) and fold change difference (±1.5 fold) for the three pair wise comparisons versus control. Volcano plots were generated with the ggplot2 package in R. Venn diagram was plotted with VennDiagram, and vennCounts packages in R. Microsoft Revolution R Open (RRO) 3.2.2 was used to perform the networkScreening function with WGCNA v1.47 package for R. Cytoscape 3.2.1 [24] used to map output Z scores continuously into node sizes, and bicor (biweight midcorrelation) values into edge widths.
2.5 Gene Ontology enrichment analysis
Functional enrichment of the differentially aggregated proteins was determined using the GO-Elite (v1.2.5) package [25]. The set of total proteins identified and quantified (n=2,771) was used as the background. Input lists included proteins either significantly decreased or increased in AsymAD, MCI and AD insoluble proteome, respectively. Z-score determines over-representation of ontologies and permutation (n=2000) P-value was used to assess the significance of the Z-score. For both increased and decreased proteins in the insoluble proteome, a Z-score cut off of 1.96, (P value cut off of 0.05) with a minimum of 3 proteins per category was employed. Horizontal bar graph was plotted in R.
2.6 Western blotting
Western blotting was performed according to standard procedures as reported previously [14]. Sarkosyl-insoluble AsymAD and AD fractions (20 μg) were mixed with Laemmli sample buffer and resolved by SDS-PAGE before an overnight wet transfer to nitrocellulose membranes (BioRad). Membranes were blocked with casein blocking buffer (Sigma B6429) and probed with in-house primary polyclonal antibodies for U1-70K (EM439) [9] and calnexin polyclonal antibody (Enzo Life Sciences, ADI-SPA-860-F) overnight at 4°C. Membranes were incubated with secondary antibodies conjugated to Alexa Fluor 680 (Invitrogen) or IRDye800 (Rockland) fluorophores for one hour at room temperature. Images were captured using an Odyssey Infrared Imaging System (Li-Cor Biosciences).
2.7 Immunofluorescence
Formalin and paraformaldehyde-fixed post-mortem human frontal cortex (50 μm) sections were immunostained essentially as previously described [22]. Sections were incubated with both primary antibodies overnight (EM439 polyclonal for U1-70K and AT8 monoclonal for phosphorylated, paired helical filament tau). Of note, AT8 antibodies detect pSer202/pThr205 phosphorylated tau [26]. Secondary antibodies (Jackson ImmunoResearch, West Grove, PA) were directly conjugated to fluorophores or were biotinylated, followed by avidin–biotin–peroxidase complex and tyramide-conjugated fluorophores (PerkinElmer, Boston, MA) essentially as previously described [27]. Control samples incubated with no or only one primary antibody demonstrated no significant cross-reactivity or background staining. Images were captured using a Nikon Eclispe TE300 widefield microscope.
3.0 Results
3.1 Label-free proteomic analysis of the detergent-insoluble proteome from individual BLSA cases
Our group has previously performed label-free proteomic analysis of detergent-insoluble fractions from control versus AD cases [9, 10]. Although these studies provided new aggregated protein targets in AD, they were conducted on i) pooled samples and ii) were not collected from individuals that had been cognitively assessed longitudinally prior to death. This limited our ability to assess the significance of protein changes with regard to biological variability or to correlate individual proteomic signatures to clinical (e.g., cognitive function) and neuropathological measures of AD (e.g. CERAD and Braak). In this study we collected post-mortem brain tissue from the dorsolateral prefrontal cortex of 35 BLSA cases, which were assessed individually by label-free proteomics (Supplemental Table S1). Control cases (n=5) were defined as cognitively normal prior to death and had low CERAD (0.2 ±0.44) and Braak (2.6 ±0.89), neuropathological measures for amyloid and tau, respectively. In contrast, AD cases (n=13) were demented at the last clinical research assessment, and showed high CERAD (3.0 ±0.00) and Braak (5.6 ±0.77) consistent with severe neuropathological burden. MCI cases (n=11) showed early signs of cognitive decline with brains displaying low-to-moderate CERAD (1.36 ±0.55) and Braak (3.0 ±1.00). Finally, AsymAD cases (n=6) were cognitively normal proximate to death, and had high CERAD (2.16 ±0.75) with moderate Braak (3.8 ±0.421) consistent with a preclinical state. There was no significant difference in age or post mortem interval (PMI) between control, AsymAD, MCI and AD samples and the cases were matched as closely as possible for gender (Supplemental Table S1). Following tissue homogenization and enrichment of sarkosyl-insoluble fractions, an equal amount of protein from all cases was resolved by SDS-PAGE and in-gel digested. The resulting peptides were analyzed by LC-MS/MS and following database searching identified proteins were subsequently quantified based on peptide-ion intensities utilizing the accurate mass and retention time across LC-MS runs. In total, we identified on average 35,706 peptides from a total of 2,711 non-redundant homologous protein groups (henceforth referred to as proteins) mapping to 2,686 gene symbols in each of the 35 cases. For the small minority of proteins identified by a single peptide (n=36, 1.3%), we required a minimum of five peptide spectral counts and a signal/noise (S/N) >5 to be further considered for quantification. The relative abundance (log2 transformed) for all detergent-insoluble proteins, with identified peptide counts and percent coverage, is provided as a master resource in Table S2.
3.2 Global changes in the insoluble brain proteome correlate with pathological severity and cognitive decline
A total of 265 unique proteins were significantly altered (±1.5 fold and p value <0.05) among the three pairwise comparisons between i) controls vs. AsymAD, ii) controls vs. MCI, and iii) controls vs. AD samples (Fig. 1). The number of significant proteins increased in tandem with disease status in cross-sectional analysis of AsymAD (n=55), MCI (n=81) and AD (n=176), indicating that global changes in the detergent-insoluble proteome and defects in proteostasis correlate with neuropathological burden and cognitive dysfunction. There was also a strong correlation between detergent-insoluble Aβ precursor protein (APP) levels measured by mass spectrometry with CERAD measurements, consistent with the enrichment of aggregated Aβ in AD (p=3.05E-10), MCI (p=0.023) and AsymAD (p=0.024) groups (Fig. 2A and B). Measurements of detergent insoluble APP in this study effectively represents the Aβ peptide, as all fully tryptic APP peptides quantified mapped to the Aβ region (residues 597–638) of the full length 695 residue protein. We also observed strong correlation between detergent-insoluble tau (MAPT) levels with Braak score (Fig. 2C). Tau levels were only significant in AD cases and trended upwards in MCI cases compared to controls consistent with the strong correlation of NFT deposition with cognitive decline (Fig. 2D) and other studies showing that tau lags behind Aβ as a biomarker [3]. Apolipoprotein E (APOE), Amyloid P Component of Serum (APCS), COL25A1 and several members of the U1 snRNP complex (i.e., U1-70K, U1A and SmD corresponding to gene symbols SNRNP70, SNRPA, SNRPD1, respectively) were also increased in AD compared to control cases, consistent with previous proteomic studies of pooled AD cases from a different cohort [9, 10] (Fig. 3A and Table S2).
Figure 1. Significant changes in the detergent insoluble brain proteome increase with pathological severity.
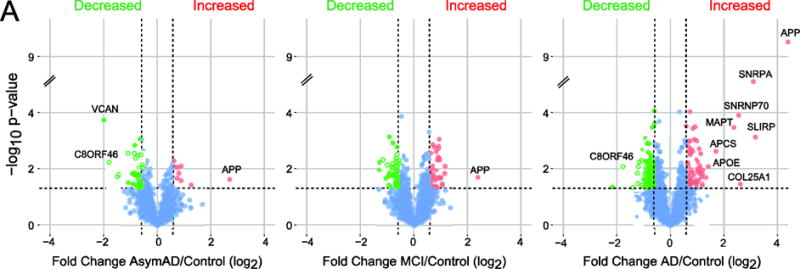
Volcano plots display the protein abundance (log2-fold-change) against the t-statistic (−log10(p-value)) for AsymAD/Control (left), MCI/Control (middle) and AD/Control (right). Red and green dots are differentially enriched or depleted proteins, respectively, whereas blue dots represent proteins that remain unchanged.
Figure 2. Differential enrichment of Aβ and Tau correlate with neuropathological measures.
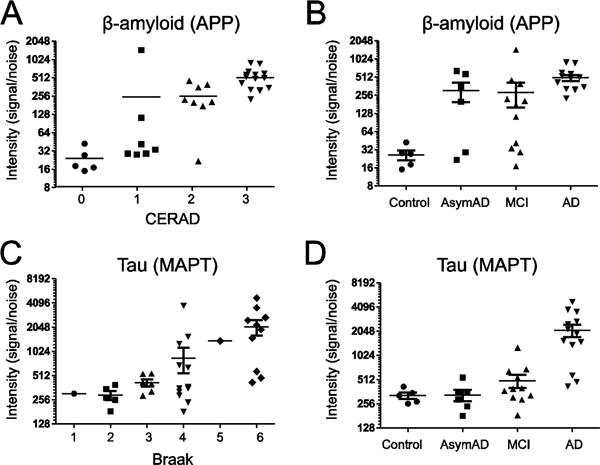
(A) Detergent insoluble β-amyloid (APP) levels measured by label-free MS quantification correlate with CERAD neuropathological plaque severity scores. (B) Detergent insoluble β-amyloid (APP) levels across individual control (n=5), AsymAD (n=6), MCI (n=11) and AD (n=13) cases. (C) Tau (MAPT) measurements measured by label-free MS quantification correlate with Braak neuropathological measures for aggregated tau. (D) Detergent insoluble β-amyloid (APP) levels across control, AsymAD, MCI and AD cases.
Figure 3. Proteins and pathways increased in the detergent insoluble AD proteome.
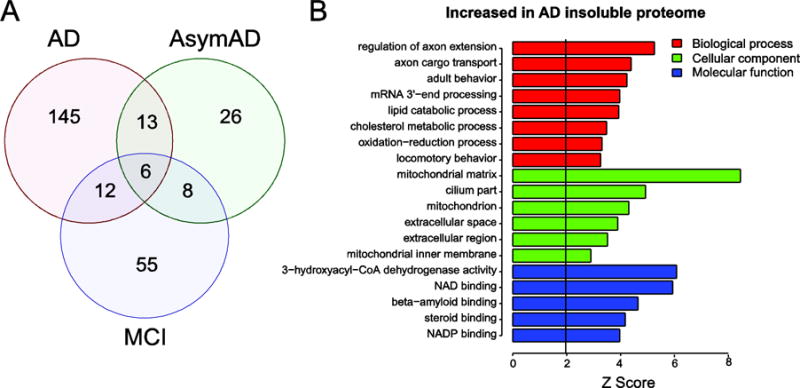
(A) Venn diagram for 265 proteins determined to be significantly altered (±1.5 fold and p value <0.05) among the three pairwise comparisons AsymAD, MCI and AD versus control cases, respectively. (B) Gene ontology (GO) analysis of the significantly enriched proteins for AD/control in the insoluble proteome reveals biological processes (red), cellular components (green) and molecular functions (blue) significantly over-represented (Z>1.96 is equivalent to p<0.05; above black line).
To analyze enrichment of aggregated proteins that participate in the same cellular components, biological processes, or molecular functions, we performed a GO (gene ontology) analysis of only those proteins significantly increased in AD aggregate proteomes versus control (Fig 3A and B). We observed that many of the proteins increased in the insoluble AD proteome were found associated with cellular component and molecular function categories linked to mitochondria. Aggregation of these proteins is consistent with the loss of mitochondrial function and hypometabolic phenotype observed in living patients with AD [28]. Other targets mapping to “steroid binding protein” ontology included 17β-hydroxysteroid dehydrogenase type 10 (HSD17B10), hydroxysteroid dehydrogenase like 2 (HSDL2) and Progesterone Receptor Membrane Component 2 (PGRMC2). Brains of humans with AD and in transgenic AD mice exhibit elevated levels of 17β-HSD10 [29], which has been shown to directly bind Aβ [30]. Furthermore, mutations in the 17β-HSD10 gene results in a progressive infantile neurodegeneration due to mitochondrial dysfunction [31]. This provides a connection between steroid binding and several key proteins in the mitochondrial proteome that were significantly elevated in MCI and AD. Six total proteins including APP (Aβ) were found to be significantly altered in AsymAD, MCI and AD (Fig. 3A). The other five proteins were all decreased in the insoluble fractions, and included syntaxin-binding protein 6 (STXBP6) and ribosome proteins (i.e., RPL12, SBDS, RPLP0, RPL12, RPLP1); the latter group implicates defects in protein translation that occur early and persist into the symptomatic phases of AD [32]. Of note, it is more challenging to interpret the biological significance of proteins found decreased in the AD insoluble proteome. A decrease in protein levels in the insoluble proteome may be due to an array of post-transcriptional and post-translational regulatory mechanisms, the latter of which can have a significant impact on the structure, function, and localization of proteins in a cell. Namely, reduced proteins found in AD insoluble fractions as compared to controls are either (i) more effectively degraded, (ii) subject to preferential protein quality control-mediated reductions in their misfolded state(s), or iii) simply expressed at lower levels; whereas, enhanced insoluble protein signals signify (i) co- or post-translational folding (ii) degradation inefficiencies in disease and/or (iii) an overall increase in protein expression. Taken together, our findings indicate that the detergent-insoluble proteome is a biochemical fraction that harbors robust differences between distinct cognitive and pathological states, providing insights into disease-associated disruptions in proteostasis [33]. Significant proteins as well as GO terms for proteins increased and decreased across all three pairwise comparisons (AsymAD, MCI and AD) versus control are provided in Tables S2 and S3.
3.3 Defining proteins that correlate with Aβ and tau levels in the aggregated brain proteome
A major goal of this study was to define preclinical changes in the AsymAD cases that correlate with detergent insoluble Aβ, which could also be linked to tau aggregation in symptomatic phases of AD. To assess this, we first determined the bi-weight mid-correlation (Bicor) coefficient for all 2,711 pairwise protein comparisons with Aβ and Tau, respectively, across all 35 individual cases (Table 1 and Table S4). Bicor is a median based correlation measure and less susceptible to outliers than the Pearson correlation [34]. Remarkably, 6 of the top 10 most correlated proteins to insoluble Aβ included core members of the U1 snRNP complex (i.e., SNRNP70, SNRPA, SNRPD1, SNRPD2, SNRPB and SNRPD3). The most significant was U1-70K (SNRNP70) with a Bicor=0.79, and p value=1.01×10−8. Others include glycoprotein NMB (GPNMB), norrin (NDP), guanine nucleotide binding protein, beta polypeptide 3 (GNB3) and serum amyloid protein P (APCS). It should be noted that mutations in NDP cause Norrie disease an X-linked recessive disorder [35] and NDP was recently found significantly up-regulated in PSEN1 familial AD iPSC-derived neural progenitors, indicating a potential link between NDP and presenilin-1 function [36].
Table 1.
Proteins most correlated with Aβ (APP) and Tau (MAPT) in the detergent-insoluble human brain proteome
| Proteins most correlated to Aβ (APP) | |||||
|---|---|---|---|---|---|
| RefSeq | Rank | Gene | Bicor | pvalue | qvalue (FDR) |
| NP_003080.2 | 1 | SNRNP70 | 0.80 | 1.01E-08 | 0.001% |
| NP_004587.1 | 2 | SNRPA | 0.69 | 5.41E-06 | 0.33% |
| NP_002501.1 | 3 | GPNMB | 0.61 | 8.96E-05 | 3.6% |
| NP_008869.1 | 4 | SNRPD1 | 0.61 | 9.01E-05 | 3.6% |
| NP_004588.1 | 5 | SNRPD2 | 0.61 | 1.10E-04 | 3.7% |
| NP_937859.1 | 6 | SNRPB | 0.60 | 1.22E-04 | 3.7% |
| NP_000257.1 | 7 | NDP | 0.59 | 1.75E-04 | 4.2% |
| NP_001630.1 | 8 | APCS | 0.58 | 3.02E-04 | 5.6% |
| NP_004166.1 | 9 | SNRPD3 | 0.55 | 6.26E-04 | 9.4% |
| NP_002066.1 | 10 | GNB3 | 0.52 | 1.27E-03 | 15.3% |
| Proteins most correlated to Tau (MAPT) | |||||
| RefSeq | Rank | Gene | Bicor | pvalue | qvalue (FDR) |
| NP_000032.1 | 1 | APOE | 0.77 | 8.65E-08 | 0.008% |
| NP_004587.1 | 2 | SNRPA | 0.74 | 2.95E-07 | 0.02% |
| NP_112487.1 | 3 | SLIRP | 0.73 | 7.54E-07 | 0.03% |
| NP_001630.1 | 4 | APCS | 0.73 | 7.82E-07 | 0.03% |
| NP_942014.1 | 5 | COL25A1 | 0.65 | 2.17E-05 | 0.68% |
| NP_002005.1 | 6 | FKBP4 | 0.61 | 1.07E-04 | 2.3% |
| NP_004386.2 | 7 | DBN1 | 0.60 | 1.28E-04 | 2.4% |
| NP_002766.1 | 8 | HTRA1 | 0.60 | 1.57E-04 | 2.7% |
| NP_001626.1 | 9 | AMPH | 0.58 | 2.78E-04 | 4.0% |
| NP_008877.1 | 10 | SPTBN2 | 0.58 | 2.78E-04 | 4.0% |
Although Aβ aggregation is an early event in AD, the deposition of Aβ does not strongly correlate with the presence of tau NFTs [12, 37]. Consistently, insoluble tau was not significantly correlated to insoluble Aβ (bicor=0.3, p=0.07) across all 35 BLSA cases in this study (Table S4). By contrast, the most correlated proteins to tau in the insoluble proteome were APOE followed by U1A and another RNA binding protein, SRA stem-loop interacting RNA binding protein (SLIRP). Other proteins that were highly correlated to tau included FK506 binding protein 4 (FKBP4) and serine protease HTRA1 (HTRA1), both of which have been reported to associate with tau pathology in animal models and/or humans, respectively [38, 39]. To prioritize the most correlated proteins to both tau and/or Aβ, we generated a cross-correlation network in Cytoscape (Fig 4) [24]. These included SNRPA (U1A) and APCS; the latter directly associates with both Aβ plaques and neurofibrillary tangles [40]. Other U1 snRNPs, U1-70K, SmD, NUDT21, were also correlated with both Aβ and Tau aggregation, but to a lesser degree than U1A (Table S4). Despite their strong cross-correlation with both insoluble Aβ and tau, U1A, U1-70K and SmD associate exclusively with NFTs and not Aβ plaques in AD brain [9, 27].
Figure 4. Network analysis of protein co-aggregation as specified by correlation with Aβ and Tau in detergent insoluble AD proteome.
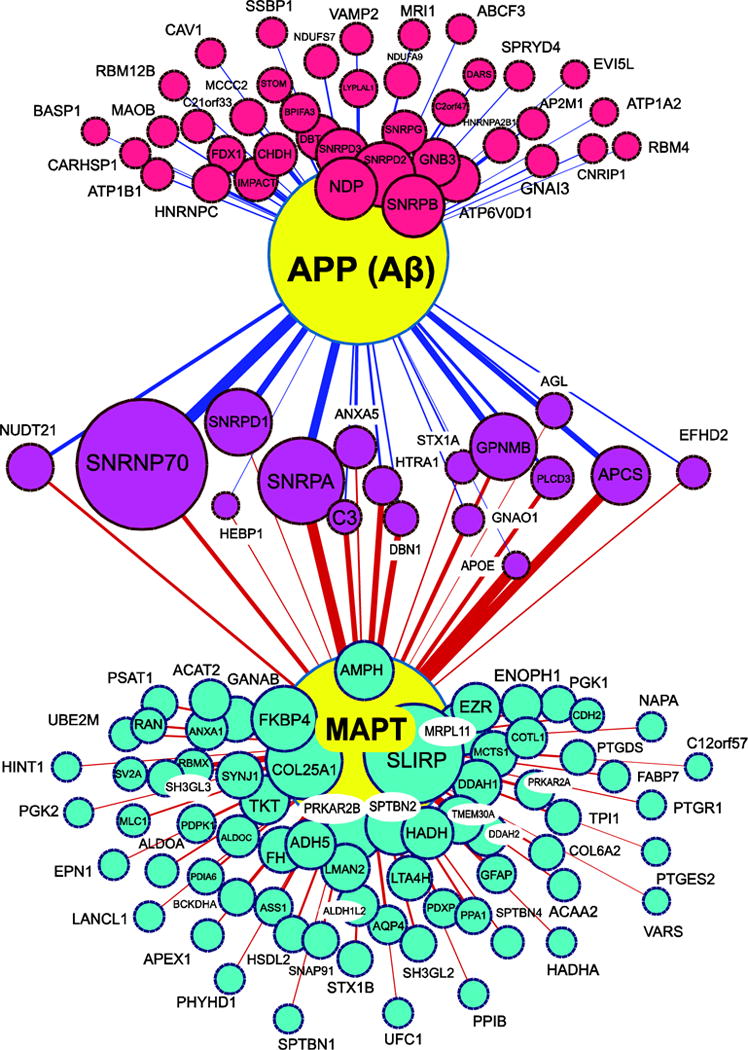
Aβ (APP) and MAPT (large yellow nodes) correlate with various proteins measured in the insoluble proteome from the 35 cases analyzed and quantified by label-free MS quantification. Node size is proportional to significance (Z score) and edge weight is proportional to the bicor correlation value for each inferred pairwise interaction. A minimum bicor correlation of +0.40 to either tau or Aβ was required for consideration. Proteins most correlated to Aβ or tau are represented by red and turquoise nodes, respectively. Node sizes for the 17 magenta nodes reflecting proteins with significant cross-correlation to both Aβ and MAPT are based on the Z score of correlation with Aβ.
3.4 U1 snRNPs aggregate in AsymAD consistent with their strong correlation with insoluble Aβ levels
The strong correlation with U1 snRNPs and Aβ described above is consistent with the co-enrichment of U1-70K, U1A and SmD in individual AsymAD and MCI cases (Fig 5A and B). Furthermore, an increase in U1-70K levels was confirmed in certain AsymAD cases by western blot analysis (Fig. 5C). Although U1-70K associates with NFTs in AD frontal cortex, immunohistochemical studies showed some discordant distributions of U1-70K and tau aggregates in certain brain regions [9], suggesting that the aggregation of U1-70K could occur independently. To test this possibility, we performed double-labeled immunofluorescence microscopy on AD cortical sections using antibodies against U1-70K and phosphorylated tau (AT8) (Fig. 5D). U1-70K and phosphorylated tau co-aggregated in cells consistent with previous reports [9], whereas certain cells displayed cytoplasmic tangle-like structures were either immunoreactive for U1-70K or AT8 antibodies independent of each other. This observation coupled with the label-free proteomics, highlighting U1-70K insolubility in AsymAD and strong correlation to Aβ prior to significant tau deposition (Fig 5A), suggests that U1-70K can aggregate in the absence of tau deposition. Additional studies will be needed to resolve the temporal sequence of events and mechanisms linking Aβ and tau with U1-70K aggregation events.
Figure 5. U1 snRNP components correlate with Aβ detergent insolubility and aggregate in AsymAD cases (.
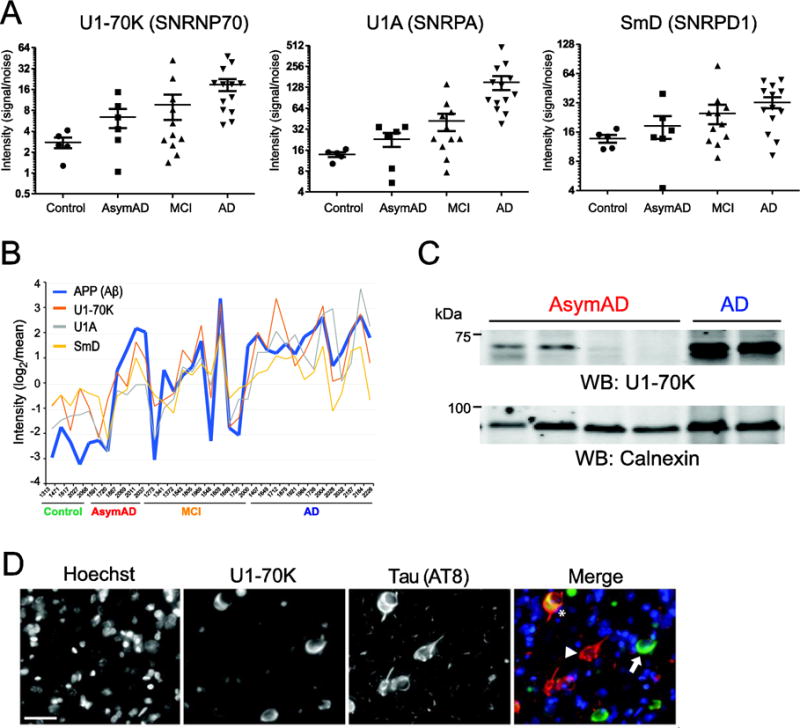
A) Relative quantification of U1-70K (left), U1A (middle) SmD (right) in the detergent-insoluble proteome across individual control, AsymAD, MCI and AD cases. (B) Strong correlation was observed between Aβ and the U1 snRNPs U1-70K, U1A and SmD across all 35 samples analyzed by label-free MS quantification. Log2-transformed quantification data (signal/noise), centered on the geometric mean is plotted on the y-axis and each individual case on the x-axis. (C) Western blot analysis of two AsymAD cases with high levels of U1-70K (left lanes) and two AsymAD cases that had low levels of U1-70K (middle lanes). Two AD cases served as positive control of U1-70K enrichment (right lanes). Calnexin was used as a loading control (bottom). (D) Double-labeled immunofluorescence of cells in AD prefrontal cortex displaying cytoplasmic aggregates of U1-70K alone (green, arrow), tau alone (red, arrowhead), and both (asterisk). Hoescht dye was used to stain nuclei (blue). Scale bar is 20 μm.
4. Discussion
In this study, we utilized label free mass spectrometry (MS)-based proteomics to quantify 2,711 detergent-insoluble proteins across the dorsolateral prefrontal cortex from 35 individuals representing control, AsymAD, MCI and AD cases. As expected, Aβ was significantly increased in AsymAD, MCI and AD cases, which correlated with CERAD, whereas insoluble tau levels correlated with Braak, and significantly increased in symptomatic stages of disease. To examine protein co-aggregation events linked AD pathology pairwise correlation coefficients were also determined for each quantified protein to Aβ and tau, across all cases. Notably, 6 of the 10 most correlated proteins to Aβ were U1 small nuclear ribonucleoproteins (U1 snRNPs). Three of these U1 snRNPs (U1A, SmD and U1-70K), also strongly correlated with tau insolubility consistent with their association with tangle pathology in AD. Additional proteins that cross-correlate with both insoluble Aβ and tau included, among others, serum amyloid protein P (APCS), APOE and complement C3 each with roles in AD pathophysiology [40–42]. Together this these findings support the utility of label-free proteomics to identify targets in the detergent-insoluble proteome linked to AD neuropathology and clinical progression.
We have previously reported an association of cytoplasmic aggregated U1 snRNPs with tau tangles in both sporadic [9, 14] and familial cases of AD [43], but not in other tauopathies. However, this is the first report of U1 snRNP aggregation in the AsymAD cases, defined by significant Aβ deposition in the absence of significant cortical tau deposition and cognitive impairment. Although the mechanisms underlying the relationship between Aβ, Tau, and U1 snRNP aggregation are unknown our proteomic findings are potentially consistent with a model in which detergent-insoluble Aβ levels influence U1 snRNP aggregation in brain, which may precede and possibly influence subsequent tau deposition into insoluble aggregates. Importantly, Aβ is secreted and predominantly aggregates in the extracellular space; whereas tau is intracellular, and is mislocalized from the axonal to the cytoplasmic and dendritic compartments in AD [44]. Prior to nuclear import, U1 snRNP biogenesis occurs in the cytoplasm [45], potentially bringing U1 snRNP components into contact with Tau and/or under the influence of intracellular signaling events triggered by extracellular Aβ. It has recently been recognized that numerous RNA-binding proteins— including many components of the spliceosome—are enriched for prion-like, low complexity (LC) domains that may predispose to fibrillization [46, 47], including co-aggregation with established neurodegenerative disease proteins such as FUS [48], hnRNPA1 [49] and even Tau [50]. In our prior work, we discovered evidence for splicing impairment in AD potentially consistent with spliceosomal functional disruption [9]. We also showed that LC domains in U1-70K were important for aggregation [14] suggesting that the aggregation properties of U1 snRNPs in AD are similar to the aforementioned RNA binding proteins in other neurodegenerative diseases. Furthermore, in two independent screens performed in Drosophila models, genetic manipulation of selected spliceosome components (including the homolog of SNRPB) modulated Tau-induced neurotoxicity in vivo [51, 52]. Separately, in a RNA-interference screen conducted in Drosophila cell culture, manipulation of multiple orthologs of U1 snRNP components (i.e., SNRPD2, SNRPD3, SNRNP70 and SNRPB2) influenced Aβ production [53], suggesting the possibility of reciprocal interactions. Thus, additional investigation is needed to resolve the precise sequence and mechanisms linking Aβ and Tau deposition to U1 snRNP aggregation and the potential connection to neuronal dysfunction and death in AD pathogenesis.
Other notable proteins that cross-correlated with Aβ and Tau insolubility included those with previously described roles in AD (i.e., APCS, NDP, PLCD3, ANXA5, HTRA1, and complement C3). For example, APCS has a binding site in fibrillar Aβ, and has been shown to promote Aβ accumulation via inhibition of its degradation [54]. Another protein that may be derived from either serum or CSF which co-aggregated with both Aβ and tau based on correlation analysis was ANXA5. This protein was decreased specifically in the CSF-producing choroid plexus in early AD stages [55], but increased in blood serum of AD patients [56]. HTRA1, high-temperature requirement serine protease A1, has been linked to inflammation and increases significantly with marked frailty in aged individuals [57]. Phospholipase C may under normal circumstances play a protective role by releasing membrane-associated Aβ that is more likely to aggregate and promote neuronal loss [58, 59]. Complement C3 in the brain is primarily expressed by microglia [60], and is essential in the process of microglial-mediated early synaptic loss in AD models [61]. Thus, our strategy of identifying cross-correlated proteins with enhanced aggregation in AD brain is valid for identifying proteins with roles in pathological aggregation of Aβ. Moreover, the proteins which cross-correlate with both Aβ and tau such as the proteins pictured in magenta in Fig. 4 are likely to have yet to be determined mechanistic roles in bridging the formation of plaques to the onset of clinical dementia symptoms, which coincides with NFT formation [3]. Modulation of these proteins, and perhaps some of those which correlate exclusively with tau insolubility (turquoise nodes in Fig. 4), are therefore excellent targets to assess direct associations with Aβ and/or tau in model systems.
Finally, a special class of proteins involved in proteostasis, chaperones, should be particularly scrutinized in our correlation analysis (Table S2), where depleted chaperones may also indicate a failure to engage amyloid or tau in attempts at refolding or protein quality control. Within that list, HSPA12A and prefoldins stand out as excellent candidates for mitigation of both Aβ and tau aggregation in normal, healthy age-matched controls. We have also shown previously that co-aggregation can be mediated by post-translational modifications [62] including ubiquitination [63] or cleavage [64, 65]. Thus, further investigation of co-aggregated and depleted proteins involved in post-translational modification may point to other interesting avenues of exploration for AD mechanistic insight. Our results highlight the utility of label-free mass spectrometry proteomics to identify disease-specific protein aggregation event that co-occur in different stages of AD, including AsymAD and MCI. Mechanistic inquiry of these new targets may enhance our understanding of potential pathways linking Aβ to tau aggregation and subsequent cognitive decline in AD.
Supplementary Material
Statement of Significance.
Alzheimer’s disease (AD) is characterized by the extracellular deposition of detergent-insoluble β-amyloid (Aβ) as senile plaques and intracellular accumulation of aggregated tau neurofibrillary tangles. Strong evidence indicates that Aβ deposition starts years prior to clinical symptoms, which has prompted widespread recognition of an asymptomatic phase of AD (AsymAD). Currently the key protein aggregation events that co-occur with Aβ and tau and associate with conversion from AsymAD to mild cognitive impairment (MCI) and eventually symptomatic AD are not well established. To fill this gap in knowledge we employed label-free mass spectrometry proteomics to quantify sarkosyl-insoluble proteins across the prefrontal cortex of 35 individual cases representing control, AsymAD, MCI and AD. Pairwise correlation analysis revealed that core members of the U1 small nuclear ribonucleoprotein (U1 snRNP) complex were some of the most highly correlated proteins to Aβ, even in the asymptomatic stage of disease. Three U1 snRNPs (U1A, SmD and U1-70K) also cross-correlated with insoluble tau, consistent with their association with tangle pathology in AD brain. Our findings support a hypothesis that specific members of the U1 snRNP complex have a potential mechanistic role in linking Aβ plaques to tau tangle pathology during AD progression.
Acknowledgments
We are grateful to participants in the Baltimore Longitudinal Study of Aging (BLSA) and Emory brain bank donors for their invaluable contribution. Funding: Support was provided by the Accelerating Medicine Partnership AD grant U01AG046161-02, the NINDS Emory Neuroscience Core (P30NS055077), and the Emory Alzheimer’s Disease Research Center (P50AG025688). This research was also supported in part by the Intramural Research Program of the NIH, National Institute on Aging. CMH is supported by NINDS K08NS087121 and by the American Brain Foundation. NTS is supported in part by an Alzheimer’s Association (ALZ), Alzheimer’s Research UK (ARUK), The Michael J. Fox Foundation for Parkinson’s Research (MJFF), and the Weston Brain Institute Biomarkers Across Neurodegenerative Diseases Grant (11060).
Footnotes
Conflict of Interest: The authors have no conflicts of interest to report.
References
- 1.Taylor JP, Hardy J, Fischbeck KH. Toxic Proteins in Neurodegenerative Disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- 2.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nature medicine. 2004;10(Suppl):S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 3.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harbor Perspectives in Medicine. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends in Pharmacological Sciences. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 5.Bierer LM, Hof PR, Purohit DP, et al. NEocortical neurofibrillary tangles correlate with dementia severity in alzheimer's disease. Archives of Neurology. 1995;52:81–88. doi: 10.1001/archneur.1995.00540250089017. [DOI] [PubMed] [Google Scholar]
- 6.Sperling RA, Aisen PS, Beckett LA, Bennett DA, et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s and Dementia. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardy J, Selkoe DJ. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 8.O’Brien RJ, Resnick SM, Zonderman AB, Ferrucci L, et al. Neuropathologic Studies of the Baltimore Longitudinal Study of Aging (BLSA) Journal of Alzheimer’s disease : JAD. 2009;18:665–675. doi: 10.3233/JAD-2009-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai B, Hales CM, Chen PC, Gozal Y, et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2013;110:16562–16567. doi: 10.1073/pnas.1310249110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gozal YM, Duong DM, Gearing M, Cheng D, et al. Proteomics analysis reveals novel components in the detergent-insoluble subproteome in Alzheimer’s disease. Journal of proteome research. 2009;8:5069–5079. doi: 10.1021/pr900474t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mirra SS, Heyman A, McKeel D, Sumi SM, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 12.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 13.Dickerson BC, Bakkour A, Salat DH, Feczko E, et al. The cortical signature of Alzheimer’s disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex. 2009;19:497–510. doi: 10.1093/cercor/bhn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diner I, Hales CM, Bishof I, Rabenold L, et al. Aggregation Properties of the Small Nuclear Ribonucleoprotein U1-70K in Alzheimer Disease. Journal of Biological Chemistry. 2014;289:35296–35313. doi: 10.1074/jbc.M114.562959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dammer EB, Duong DM, Diner I, Gearing M, et al. Neuron enriched nuclear proteome isolated from human brain. Journal of proteome research. 2013;12:3193–3206. doi: 10.1021/pr400246t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dammer EB, Lee AK, Duong DM, Gearing M, et al. Quantitative Phosphoproteomics of Alzheimer’s Disease Reveals Crosstalk between Kinases and Small Heat Shock Proteins. Proteomics. 2015;15:508–519. doi: 10.1002/pmic.201400189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seyfried NT, Gozal YM, Donovan LE, Herskowitz JH, et al. Quantitative Analysis of the Detergent-Insoluble Brain Proteome in Frontotemporal Lobar Degeneration Using SILAC Internal Standards. Journal of proteome research. 2012;11:2721–2738. doi: 10.1021/pr2010814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Meth. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 19.Xu P, Duong DM, Peng J. Systematical Optimization of Reverse-Phase Chromatography for Shotgun Proteomics. Journal of proteome research. 2009;8:3944–3950. doi: 10.1021/pr900251d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donovan LE, Higginbotham L, Dammer EB, Gearing M, et al. Analysis of a membrane-enriched proteome from postmortem human brain tissue in Alzheimer’s disease. Proteomics Clinical applications. 2012;6:201–211. doi: 10.1002/prca.201100068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donovan L, Dammer E, Duong D, Hanfelt J, et al. Exploring the potential of the platelet membrane proteome as a source of peripheral biomarkers for Alzheimer’s disease. Alzheimer’s Research & Therapy. 2013;5:32. doi: 10.1186/alzrt186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seyfried NT, Gozal YM, Donovan LE, Herskowitz JH, et al. Quantitative analysis of the detergent-insoluble brain proteome in frontotemporal lobar degeneration using SILAC internal standards. Journal of proteome research. 2012;11:2721–2738. doi: 10.1021/pr2010814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silva JC, Gorenstein MV, Li GZ, Vissers JPC, Geromanos SJ. Absolute Quantification of Proteins by LCMSE : A Virtue of Parallel ms Acquisition. Molecular & Cellular Proteomics. 2006;5:144–156. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 24.Shannon P, Markiel A, Ozier O, Baliga NS, et al. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Research. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zambon AC, Gaj S, Ho I, Hanspers K, et al. GO-Elite: a flexible solution for pathway and ontology over-representation. Bioinformatics. 2012;28:2209–2210. doi: 10.1093/bioinformatics/bts366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neuroscience Letters. 1995;189:167–170. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- 27.Hales CM, Dammer EB, Diner I, Yi H, et al. Aggregates of small nuclear ribonucleic acids (snRNAs) in Alzheimer’s disease. Brain pathology. 2014;24:344–351. doi: 10.1111/bpa.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexander GE, Chen K, Pietrini P, Rapoport SI, Reiman EM. Longitudinal PET Evaluation of Cerebral Metabolic Decline in Dementia: A Potential Outcome Measure in Alzheimer’s Disease Treatment Studies. American Journal of Psychiatry. 2002;159:738–745. doi: 10.1176/appi.ajp.159.5.738. [DOI] [PubMed] [Google Scholar]
- 29.Yang SY, He XY, Isaacs C, Dobkin C, et al. Roles of 17β-hydroxysteroid dehydrogenase type 10 in neurodegenerative disorders. The Journal of Steroid Biochemistry and Molecular Biology. 2014;143:460–472. doi: 10.1016/j.jsbmb.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 30.Lustbader JW, Cirilli M, Lin C, Xu HW, et al. ABAD Directly Links Aß to Mitochondrial Toxicity in Alzheimer’s Disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 31.Zschocke J, Ruiter JPN, Brand J, Lindner M, et al. Progressive Infantile Neurodegeneration Caused by 2-Methyl-3-Hydroxybutyryl-CoA Dehydrogenase Deficiency: A Novel Inborn Error of Branched-Chain Fatty Acid and Isoleucine Metabolism. Pediatr Res. 2000;48:852–855. doi: 10.1203/00006450-200012000-00025. [DOI] [PubMed] [Google Scholar]
- 32.Ding Q, Markesbery WR, Chen Q, Li F, Keller JN. Ribosome dysfunction is an early event in Alzheimer’s disease. J Neurosci. 2005;25:9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting Proteostasis for Disease Intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 34.Langfelder P, Horvath S. Fast R Functions for Robust Correlations and Hierarchical Clustering. Journal of statistical software. 2012;46:i11. [PMC free article] [PubMed] [Google Scholar]
- 35.Braunger BM, Tamm ER. In: Retinal Degenerative Diseases. LaVail MM, Ash DJ, Anderson ER, Hollyfield GJ, Grimm C, editors. Springer US; Boston, MA: 2012. pp. 679–683. [Google Scholar]
- 36.Sproul AA, Jacob S, Pre D, Kim SH, et al. Characterization and Molecular Profiling of PSEN1 Familial Alzheimer’s Disease iPSC-Derived Neural Progenitors. PLoS ONE. 2014;9:e84547. doi: 10.1371/journal.pone.0084547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karran E, Mercken M, Strooper BD. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 38.Tennstaedt A, Pöpsel S, Truebestein L, Hauske P, et al. Human High Temperature Requirement Serine Protease A1 (HTRA1) Degrades Tau Protein Aggregates. The Journal of Biological Chemistry. 2012;287:20931–20941. doi: 10.1074/jbc.M111.316232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giustiniani J, Chambraud B, Sardin E, Dounane O, et al. Immunophilin FKBP52 induces Tau-P301L filamentous assembly in vitro and modulates its activity in a model of tauopathy. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:4584–4589. doi: 10.1073/pnas.1402645111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalaria RN, Grahovac I. Serum amyloid P immunoreactivity in hippocampal tangles, plaques and vessels: implications for leakage across the blood-brain barrier in Alzheimer’s disease. Brain Research. 1990;516:349–353. doi: 10.1016/0006-8993(90)90941-4. [DOI] [PubMed] [Google Scholar]
- 41.Richey PL, Siedlak SL, Smith MA, Perry G. Apolipoprotein E Interaction with the Neurofibrillary Tangles and Senile Plaques in Alzheimer Disease: Implications for Disease Pathogenesis. Biochemical and Biophysical Research Communications. 1995;208:657–663. doi: 10.1006/bbrc.1995.1389. [DOI] [PubMed] [Google Scholar]
- 42.Eikelenboom P, Hack CE, Rozemuller JM, Stam FC. Complement activation in amyloid plaques in Alzheimer’s dementia. Virchows Archiv B. 1988;56:259–262. doi: 10.1007/BF02890024. [DOI] [PubMed] [Google Scholar]
- 43.Hales C, Seyfried N, Dammer E, Duong D, et al. U1 small nuclear ribonucleoproteins (snRNPs) aggregate in Alzheimer’s disease due to autosomal dominant genetic mutations and trisomy 21. Mol Neurodegener. 2014;9:15. doi: 10.1186/1750-1326-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spillantini MG, Goedert M. Tau pathology and neurodegeneration. The Lancet Neurology. 12:609–622. doi: 10.1016/S1474-4422(13)70090-5. [DOI] [PubMed] [Google Scholar]
- 45.BATTLE DJ, KASIM M, YONG J, LOTTI F, et al. The SMN Complex: An Assembly Machine for RNPs. Cold Spring Harbor Symposia on Quantitative Biology. 2006;71:313–320. doi: 10.1101/sqb.2006.71.001. [DOI] [PubMed] [Google Scholar]
- 46.Couthouis J, Hart MP, Shorter J, DeJesus-Hernandez M, et al. A yeast functional screen predicts new candidate ALS disease genes. Proceedings of the National Academy of Sciences. 2011;108:20881–20890. doi: 10.1073/pnas.1109434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim HJ, Kim NC, Wang YD, Scarborough EA, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murakami T, Qamar S, Lin Julie Q, Schierle Gabriele SK, et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron. 88:678–690. doi: 10.1016/j.neuron.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Molliex A, Temirov J, Lee J, Coughlin M, et al. Phase Separation by Low Complexity Domains Promotes Stress Granule Assembly and Drives Pathological Fibrillization. Cell. 163:123–133. doi: 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vanderweyde T, Apicco Daniel J, Youmans-Kidder K, Ash Peter EA, et al. Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Reports. 15:1455–1466. doi: 10.1016/j.celrep.2016.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shulman JM, Imboywa S, Giagtzoglou N, Powers MP, et al. Functional screening in Drosophila identifies Alzheimer’s disease susceptibility genes and implicates Tau-mediated mechanisms. Human Molecular Genetics. 2014;23:870–877. doi: 10.1093/hmg/ddt478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butzlaff M, Hannan SB, Karsten P, Lenz S, et al. Impaired retrograde transport by the Dynein/Dynactin complex contributes to Tau-induced toxicity. Human Molecular Genetics. 2015;24:3623–3637. doi: 10.1093/hmg/ddv107. [DOI] [PubMed] [Google Scholar]
- 53.Page RM, Münch A, Horn T, Kuhn PH, et al. Loss of PAFAH1B2 Reduces Amyloid-β Generation by Promoting the Degradation of Amyloid Precursor Protein C-Terminal Fragments. The Journal of Neuroscience. 2012;32:18204–18214. doi: 10.1523/JNEUROSCI.2681-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tennent GA, Lovat LB, Pepys MB. Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proc Natl Acad Sci U S A. 1995;92:4299–4303. doi: 10.1073/pnas.92.10.4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krzyzanowska A, García-Consuegra I, Pascual C, Antequera D, et al. Expression of Regulatory Proteins in Choroid Plexus Changes in Early Stages of Alzheimer Disease. Journal of Neuropathology & Experimental Neurology. 2015;74:359–369. doi: 10.1097/NEN.0000000000000181. [DOI] [PubMed] [Google Scholar]
- 56.Sohma H, Imai S, Takei N, Honda H, et al. Evaluation of annexin A5 as a biomarker for Alzheimer’s disease and dementia with lewy bodies. Frontiers in aging neuroscience. 2013;5:15. doi: 10.3389/fnagi.2013.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lorenzi M, Lorenzi T, Marzetti E, Landi F, et al. Association of frailty with the serine protease HtrA1 in older adults. Experimental gerontology. 2016;81:8–12. doi: 10.1016/j.exger.2016.03.019. [DOI] [PubMed] [Google Scholar]
- 58.Nagarathinam A, Hoflinger P, Buhler A, Schafer C, et al. Membrane-anchored Abeta accelerates amyloid formation and exacerbates amyloid-associated toxicity in mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:19284–19294. doi: 10.1523/JNEUROSCI.2542-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peters C, Espinoza MP, Gallegos S, Opazo C, Aguayo LG. Alzheimer’s Abeta interacts with cellular prion protein inducing neuronal membrane damage and synaptotoxicity. Neurobiology of aging. 2015;36:1369–1377. doi: 10.1016/j.neurobiolaging.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Y, Chen K, Sloan SA, Bennett ML, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ren RJ, Dammer EB, Wang G, Seyfried NT, Levey AI. Proteomics of protein post-translational modifications implicated in neurodegeneration. Translational neurodegeneration. 2014;3:23. doi: 10.1186/2047-9158-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dammer EB, Fallini C, Gozal YM, Duong DM, et al. Coaggregation of RNA-binding proteins in a model of TDP-43 proteinopathy with selective RGG motif methylation and a role for RRM1 ubiquitination. PloS one. 2012;7:e38658. doi: 10.1371/journal.pone.0038658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Herskowitz JH, Gozal YM, Duong DM, Dammer EB, et al. Asparaginyl endopeptidase cleaves TDP-43 in brain. Proteomics. 2012;12:2455–2463. doi: 10.1002/pmic.201200006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Z, Song M, Liu X, Kang SS, et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nature medicine. 2014;20:1254–1262. doi: 10.1038/nm.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.